

Abstract
Adoptive immunotherapy with chimeric antigen receptor-modified T (CAR-T) cells is a rapidly growing therapeutic approach to treating patients with refractory cancer, with over 100 clinical trials in various malignancies in progress. The enthusiasm for CAR-T cells has been driven by the clinical success of CD19-targeted CAR-T therapy in B-cell acute lymphoblastic leukemia, and the promising data in B-cell non-Hodgkin’s lymphoma and chronic lymphocytic leukemia. Despite the success of targeting CD19 with CAR-T cells in early clinical studies, many challenges remain to improve outcomes, reduce toxicity, and determine the appropriate settings for CAR-T cell immunotherapy. Reviewing the lessons learned thus far in CD19 CAR-T cell trials and how some of these challenges may be overcome will help guide the development of CAR-T cell therapy for malignancies of B-cell origin, as well as for other hematopoietic and non-hematopoietic cancers.
1 Introduction
1.1 The Rationale for CD19 CAR-T Cell Immunotherapy for B Cell Malignancies
A component of the adaptive immune system, T cells are effectors of cell-mediated immunity. In response to engagement of the T cell receptor by a cognate peptide antigen presented in the context of a specific major histocompatibility complex (MHC) molecule, T cells exert effector functions and induce lysis of antigen-bearing target cells. T cells were noted to have anti-tumor effects during studies of T cell-depleted hematopoietic stem cell transplantation (HSCT), in which patients who received grafts depleted of T cells had a higher risk of disease relapse compared to their counterparts who received T-cell replete grafts.[1] Early approaches to generate large numbers of tumor-reactive T cells for adoptive transfer to cancer patients involved repetitive in vitro stimulation with antigen, were cumbersome, and infrequently met with clinical success.[2] More recent efforts have taken advantage of genetic modification strategies to rapidly redirect the specificity of polyclonal T cells by introduction of a tumor-targeted recombinant antigen receptor, such as a chimeric antigen receptor (CAR). A CAR comprises an extracellular antibody-derived single chain variable fragment (scFv) specific for a target antigen that is linked to one or more intracellular T cell-derived signaling sequences (Fig 1), which enables T cell activation on ligation of the scFv with its target antigen. Limited therapeutic activity was noted in clinical trials using T cells engineered to express first generation CARs, which contained an intracellular T cell signaling sequence (e.g. CD3ζ) in the absence of a costimulatory molecule sequence.[3–5] Clinical activity has been markedly improved by T cell products that incorporate second generation CARs that include costimulatory sequences derived, for example, from 4-1BB or CD28.[6–12] Third and fourth generation CARs, which contain multiple co-stimulatory domains and/or other signals are in development, but clinical experience with these constructs in B cell malignancies so far is limited.[13, 14]
Fig. 1.

Chimeric antigen receptor (CAR) design. A first generation CAR incorporates a CD19-specific single chain variable fragment (scFv) fused through linker sequences to CD3ζ. When introduced into a T cell by genetic modification, the CAR allows redirection of T cell specificity to CD19. Second and third generation CARs incorporate additional costimulatory domains.
CD19 is a very good target antigen for CAR-T cell immunotherapy of B cell malignancies, as it is expressed at high and stable levels on tumor tissue from most patients with B cell acute lymphoblastic leukemia (B-ALL), non-Hodgkin’s lymphoma (NHL), and chronic lymphocytic leukemia (CLL). It is also expressed on normal B cells, but not on other tissues outside the B cell lineage, limiting known “on-target off-tumor” toxicities to B cell aplasia, a condition that can be managed with immunoglobulin replacement.[15]
1.2 Lymphodepletion Chemotherapy, CAR-T Cell Manufacturing, and Infusion
Approaches for CAR-T cell production differ at each center, but typically involve isolation of autologous T cells from the patient using leukapheresis, followed by in vitro stimulation with anti-CD3 or anti-CD3/anti-CD28 beads, genetic modification by transduction with a retroviral or lentiviral vector to express a CAR, and subsequent culture for approximately 2–3 weeks. After leukapheresis and while CAR-T cells are being manufactured, patients in most protocols will receive lymphodepleting chemotherapy, which creates a favorable immune environment for adoptively transferred CAR-T cells, improving their in vivo expansion, subsequent persistence, and clinical activity (Fig 2).[16] During the acute phase of in vivo CAR-T cell expansion, patients are monitored closely for the development of adverse effects of CAR-T cell immunotherapy, such as cytokine release syndrome (CRS) and neurotoxicity. CRS is associated with immune T cell activation and is characterized by fevers, hypotension, capillary leak and coagulopathy.[17] Neurotoxicity commonly presents as delirium, but can be manifest as focal neurological deficits, seizures or coma. Neurotoxicity usually occurs in association with CRS, but its pathogenesis is unclear. Although in a majority of cases CRS and neurotoxicity are self-limited, the IL-6-receptor antibody, tocilizumab, and/or corticosteroids have been used to treat serious cases. Toxicity grading and therapy algorithms are still under development.[7, 17–19]
Fig. 2.
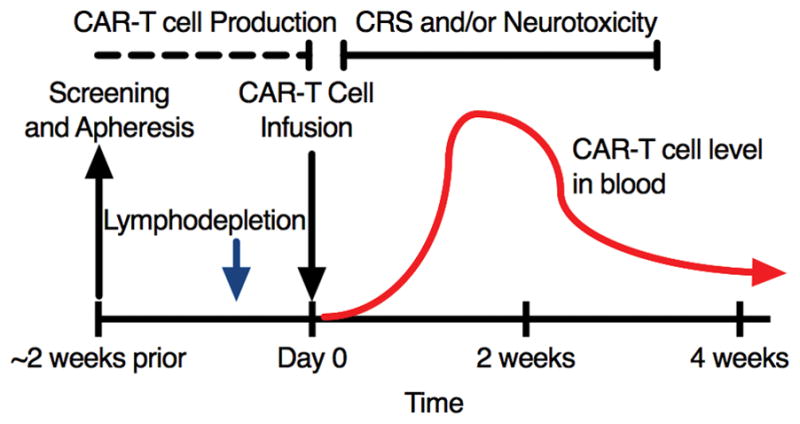
Timeline of a typical course for a patient undergoing CAR-T cell immunotherapy. After leukapheresis to isolate T cells, CAR manufacturing takes approximately 1–3 weeks. The patient usually receives lymphodepletion chemotherapy shortly before CAR-T infusion. Over 1–3 weeks after infusion the CAR-T cells proliferate in vivo (red line) then contract, leaving a fraction of persistent CAR-T cells. Patients are closely monitored for cytokine release syndrome (CRS) and neurotoxicity during the first 3–4 weeks after infusion.
2 CD19 CAR-T Cell Clinical Trials in B-cell Malignancies
A majority of the published clinical experience has come out of four centers, each using a distinct CAR design and manufacturing approaches (Table 1). Clinical trial data from these centers that have been published or recently presented in abstract form at the annual meetings of the American Society of Clinical Oncology (ASCO) or American Society of Hematology (ASH) are presented in this review.
Table 1.
Design and formulation of four different chimeric antigen receptor (CAR) constructs in published clinical trials.
| Institution | scFv | Costimulatory Domain | Vector | Formulation |
|---|---|---|---|---|
| MSKCC | Murine (SJ25C1) | CD28 | Gamma-retrovirus | not pre-defined |
| NCI | Murine (FMC63) | CD28 | Gamma-retrovirus | not pre-defined |
| UPENN / CHOP | Murine (FMC63) | 4-1BB | Lentivirus | not pre-defined |
| FHCRC | Murine (FMC63) | 4-1BB | Lentivirus | 1:1 ratio of CD4:CD8 CAR-T cells |
MSKCC, Memorial Sloan Kettering Cancer Center; NCI, National Cancer Institute; UPenn, University of Pennsylvania; CHOP, Children’s Hospital of Philadelphia; FHCRC, Fred Hutchinson Cancer Research Center; scFv, single chain variable fragment.
2.1 B-ALL
The group at Memorial Sloan Kettering Cancer Center (MSKCC) initially published their experience with 5 adult B-ALL patients in 2013, followed by a second manuscript in 2014.[18, 20] Park et al updated their data to include a total of 51 patients at ASCO 2016.[21] Following either cyclophosphamide (Cy) or Cy with fludarabine (Flu) lymphodepletion, 1 × 106 to 3 × 106 CAR-T cells/kg were infused. On restaging after CAR-T cell infusion, of 50 evaluable patients 41 (82%) had a morphologic complete remission (CR) (Table 2). Thirty-nine patients with morphologic CR were evaluated with marrow flow cytometry and 33 were in minimal residual disease (MRD)-negative CR. Thus, the overall MRD-negative CR rate in evaluated patients was approximately 69%. The authors identified a relationship between toxicity and tumor burden; 13 of 31 patients with morphologic disease (>5% blasts) developed severe CRS requiring mechanical ventilation or vasopressors, compared to only one of 20 with MRD.[22, 23] Neurotoxicity occurred in 15 of 51 patients. Three patients with morphologic disease died after receiving 3 × 106 CAR-T cells/kg, leading to a risk-adapted therapy, in which patients without morphologic disease receive 3 × 106 cells/kg, whereas patients with morphologic disease receive a 1 × 106 cell/kg dose.
Table 2a.
Reports on CD19-targeted CAR-T cells in the treatment of B-ALL.
| Disease | Institution | Patients | Lymphodepletion | CAR-T Cell Dose | Response, Survival, and Relapse | Reference |
|---|---|---|---|---|---|---|
| ALL | MSKCC | 51 adults (22–74 years) | Cy (n=42) or Cy/Flu (n=9)* | 1 × 106 or 3 × 106 CAR-T cells/kg | 41/50 (82%) achieved a CR, and 33/39 patients with CR who underwent flow cytometry assessment being MRD-negative. 15/33 (45%) of MRD-negative CR patients subsequently relapsed. | [18, 20, 21] |
| UPenn / CHOP | 59 children and young adults (<24 years) | Investigator’s choice, commonly Cy-based with or without Flu | 1 × 107 to 1 × 108 TNC/kg** | 55/59 (93%) achieved a morphologic CR, with 52/59 (88%) having a MRD-negative CR. At 12 months, 34 patients had ongoing CR with a RFS of 55% and OS 79%. | [6, 7, 27] | |
| UPenn | 27 adults (21–72 years) | Investigator’s choice | 5 × 107 or 5 × 108 CAR-T cells◆ | 15/24 (62.5%) evaluable patients achieved CR, with 9/12 (75%) of patients in the high dose fractionated group achieving CR. | [28] | |
| NCI | 38 children and young adults (1–30 years) | <25% blasts, Flu 25 mg/m2/d days 1–3 + Cy 900 mg/m2 on day 3; >25% blasts, ‘high intensity regimen’, most commonly FLAG | 1 × 106 or 3 × 106 CAR-T cells/kg | 23/38 (61%) achieved a CR; 13/16 (81%) patients achieved CR in the low burden disease group, and 10/22 (45%) achieved CR in the high burden group; 20/38 (53%) patients achieved an MRD-negative CR. Among MRD-negative, RFS was 45.5% at 18 months. | [9, 24] | |
| NCI | 5 adults (20–68 years) | None | 4.2 × 106 to 7.0 × 106 CAR-T cells/kg◆◆ | 4/5 patients (80%) developed MRD-negative CR | [25] | |
| FHCRC | 30 adults (20–73 years) | Initially Cy-based (e.g. Cy 2–4 g/m2 day 1 ± VP 100 mg/m2 days 1–3; subsequently Cy/Flu combination (e.g. Cy 30–60 mg/kg on day 1 + Flu 25 mg/m2/d on days 2–4 or 2–6) | 2 × 105, 2 × 106, or 2 × 107 CAR-T cells/kg | 27/29 (93%) of patients achieved flow cytometry MRD-negative CR, 2 of the patients with flow-negative CR had MRD on molecular testing. Addition of Flu improved CAR-T persistence and disease free survival. | [12, 35] |
Cy, cyclophosphamide; Flu, fludarabine; FLAG, fludarabine and high dose cytarabine based regimen; CR, complete remission; MRD, minimal residual disease; HSCT, hematopoietic stem cell transplant; RFS, relapse free survival; TNC, total nucleated cells.
Doses not reported in ASCO 2016 abstract, in previous papers reported Cy 1.5 – 3 g/m2 for Cy alone group;
reported as total number of nucleated cells given, with a transduction efficacy of 2.3–45%;
total number of CAR-T cells given;
CAR-T cells manufactured from the allogeneic donor.
In 2015, Lee et al. from the National Cancer Institute (NCI) reported 21 patients, and updated their B-ALL data at ASH 2015 to a total of 38 patients.[9, 24] Lymphodepletion intensity was adjusted according to tumor burden; patients with ≥25% blasts in the marrow received a variety of high-intensity regimens, while patients with <25% blasts received a lower intensity combination of Cy and Flu. In the initial cohort of 20 patients it was determined that the maximum tolerated dose was 1 × 106 CAR-T cells/kg; therefore, this dose was used in the second cohort of 18 patients. MRD-negative CRs were seen in 20 of 38 patients (53%). In those who achieved an MRD-negative CR, leukemia-free survival was 45.5% at 18 months. 16% of patients in the first cohort and 5.6% in the second cohort developed grade 4 CRS.[17] Neurotoxicity was not reported.
Another report from the NCI outlined treatment of allogeneic HSCT recipients with CAR-T cells manufactured from the HSCT donor and administered without antecedent lymphodepletion chemotherapy.[25, 26] Four of 5 patients obtained a CR without evidence of acute graft versus host disease.
The University of Pennsylvania (UPenn) and Children’s Hospital of Philadelphia (CHOP) reported their initial results in 2013 and 2014, with updates at ASCO 2016.[6, 7, 27]. 59 children and young adults (≤24 years old) were treated with 1 – 10 × 107 total T cells/kg with a CAR transduction efficacy of 2.3 – 45% following a variety of lymphodepletion regimens. Fifty-five (93%) patients achieved a negative marrow by flow cytometry. Twenty patients relapsed, 13 of them with CD19 negative disease, giving a relapse free survival (RFS) of 55% and overall survival (OS) of 79% at 12 months. CRS (any grade) developed in 88% of patients, with severe CRS occurring in 27% and being more frequent in those with high tumor burden.
At ASCO 2016, Frey et al. reported on the UPenn experience with 27 adults in B-ALL, using fixed doses of 5 × 107 versus 5 × 108 total CAR-T cells given, as a single infusion or in split fractions.[28] Of 9 patients treated with 5 × 107 CAR-T cells administered as a single dose, only 3 obtained a CR, while of the 6 patients treated with 5 × 108 CAR-T cells as a single dose, 3 achieved CR and 3 died of severe CRS. After introduction of a fractionated schedule to administer 5 × 108 CAR-T cells, the CR rate was 75% (9 of 12). While 75% of patients developed grade 3–4 CRS, no patients died of acute toxicity.
Relationships between infused CAR-T cell dose and clinical outcomes were difficult to define in early studies of CAR-T cell therapy, potentially a result of variability in the T cell subset composition of the infused CAR-T cell products, which can affect the potency of a CAR-T cell product.[29] In an effort to manufacture a more uniform CAR-T cell product that could assist in defining relationships between infused CAR-T cell dose and clinical outcomes, we developed an approach at Fred Hutchinson Cancer Research Center (FHCRC) in which patients received CAR-T cells formulated in a defined 1:1 ratio of CD4+:CD8+ CAR-T cells. The defined composition product was infused at set dose levels of 2 × 105, 2 × 106 or 2 × 107 CD19 CAR-T cells/kg following lymphodepletion chemotherapy with Cy-based regimens with or without Flu. We recently published our findings in 30 B-ALL patients in early 2016.[12] Twenty-seven of 29 evaluable patients (94%) achieved a flow cytometry-negative CR; two of these patients were found to have MRD by molecular testing. Twenty-five of the 30 developed CRS with 7 cases being severe enough to require ICU care. Severe toxicity was encountered in 2 patients treated at the highest dose level; therefore, no further patients were treated with this dose. Similar to other groups, we identified that the burden of CD19+ cells in the marrow prior to therapy was a risk factor for subsequent toxicity, leading to a risk-adapted therapy approach in which patients with >20% marrow involvement received 2 × 105 cells/kg while those with ≤20% received 2 × 106 cells/kg. After adoption of this approach, only one of 10 patients developed severe CRS requiring ICU care. In the early part of the study, we used Cy-based lymphodepletion without Flu. Although CR rates in this cohort were robust, early relapse was noted in a subset of patients, associated with loss of CAR-T cells in blood due to an anti-CAR-T cell immune response directed at epitopes in the murine scFv. This mechanism may contribute to early loss of CAR-T cells in some patients in trials that use a CAR containing a murine scFv (Table 1). Addition of Flu to Cy in the lymphodepleting regimen minimized the effect of transgene immunogenicity, improving CAR-T cell expansion, persistence, and clinical outcomes.
2.2 B-NHL and CLL
In 2015, the NCI group reported treatment of 11 B-NHL patients and 4 CLL patients.[30] Nine of the 11 NHL patients had aggressive disease on histology (4 with diffuse large B-cell lymphoma (DLBCL), NOS, 4 with primary mediastinal B-cell NHL, one with Richter’s transformation to DLBCL after CLL) and 2 had indolent disease. Patients received lymphodepletion with high dose Cy (60–120 mg/kg) followed by 5 days of Flu 25mg/m2, with infusion one day later of 1 – 5 × 106 CAR-T cells/kg. Of the 9 patients with aggressive histology, 7 were evaluable, with 4 patients achieving a CR and 2 achieving a partial response (PR). Three of the 4 CLL patients achieved a CR. Adverse events included grade ≥3 hypotension in 4 of 15 (27%) patients and neurotoxicity in 6 of 15 (40%) patients. One patient died on day 16 from an unclear etiology.
At ASCO 2016, Kochenderfer et al. reported outcomes in 22 patients given low-dose Cy (300mg/m2-500mg/m2) for 3 days, with concurrent Flu 30mg/m2 administration as lymphodepletion.[31] Eight of 19 DLBCL patients achieved a CR with an overall response rate (ORR) in DLBCL of 68% (13 of 19 patients). One MCL patient and two FL patients obtained CR.
Brudno et al. reported data from treatment of allogeneic HSCT recipients with CAR-T cells that were manufactured from T cells directly isolated from the HSCT donor and administered without antecedent lymphodepletion chemotherapy in 2015.[25, 26] Ten B-NHL and 5 CLL patients were treated, with responses observed in 2 of 10 B-NHL patients and 2 of 5 CLL patients.
At ASH 2015, UPenn reported treatment of 24 NHL patients with 3.08 × 106 to 8.87 × 106 CAR-T cells/kg following a range of lymphodepletion regimens.[32] Eight of 11 patients with follicular lymphoma, 7 of 15 patients with DLBCL and one of 2 patients with mantle cell NHL responded, with an ORR of 68%. Sixteen of 24 patients developed CRS and 3 patients developed neurotoxicity.
The UPenn group also reported success in CLL, with an ORR of 57% (4 of 14 CR, 4 of 14 PR).[8] Results from a subsequent phase II dose optimization study in 35 patients were reported at ASCO 2016. [33]. Patients received a high (5 × 108) or low (5 × 107) total CD19 CAR-T cell dose. Stage I of the study demonstrated a higher response rate in the high dose cohort, leading to expansion of this dose level in the stage II group. Nine of the 17 evaluable patients at the high dose level responded, with an ORR of 53%. Nineteen of 35 patients (48%) developed CRS, of which 7 were grade 3–4.
At ASCO 2016, Geyer et al from MSKCC reported on treatment of 8 patients with refractory CLL after first line pentostatin, cyclophosphamide, and rituximab.[34] After Cy 600mg/m2, patients were given 3 × 106, 1 × 107, or 3 × 107 CAR-T cells/kg. Two patients obtained a CR, with an ORR of 50% (4 of 8). Despite progressive disease in 3 patients, 2 had evidence of a marrow response.
We recently reported treatment of 32 patients with a variety of B-NHL histologic types (11 de novo DLBCL, 10 transformed DLBCL, 5 FL, and 4 MCL).[11] Of the 30 evaluable patients, 10 (33%) had a CR and 9 (30%) a PR, giving an ORR of 63%. Severe CRS requiring ICU care was seen in 4 of 32 (12.5%) of patients, and grade ≥3 neurotoxicity was noted in 9 of 32 (28%) patients. As observed in our studies in B-ALL patients, addition of Flu to Cy-based lymphodepletion improved CAR-T cell expansion, persistence, and clinical outcomes in NHL patients. Patients treated at the highest CAR-T cell dose (2 × 107 cells/kg) after Cy and Flu lymphodepletion experienced more toxicity; therefore, 2 × 106 cells/kg was deemed the maximum tolerated dose. Infusion of this dose after Cy and Flu lymphodepletion to 11 patients resulted in a CR rate of 64% and an ORR of 82%.
At ASCO 2016, we reported 13 CLL patients who were treated with lymphodepletion chemotherapy and CD19 CAR-T cells at FHCRC.[35] All patients had previously received ibrutinib. Of the 12 restaged patients, 10 (83%) achieved clearance of the marrow by flow cytometry and 6 (50%) achieved CR by CT+/−PET imaging.
3 Challenges in CD19 CAR-T Cell Immunotherapy
Outcomes in relapsed/refractory patients with B cell malignancies are promising, but many challenges remain.
3.1 Toxicities and Management
CRS arises from the activation of CAR-T cells, leading to inflammatory cytokine release, and is manifest as a spectrum of findings, including fever, constitutional symptoms, hypotension, capillary leak, coagulopathy, and organ dysfunction, usually presenting in the first 1–2 weeks after CAR-T cell infusion. Neurotoxicity can occur with or after the onset of CRS, and in some cases can present after resolution of CRS. The pathogenesis is poorly understood. Presentations include delirium, speech disturbances, focal neurological deficits, seizures, and occasionally coma. Both CRS and neurotoxicity are reversible in the majority of cases; however, fatalities may occur. Although tocilizumab and corticosteroids are used to treat severe CRS and neurotoxicity, the roles of these drugs in treatment of neurotoxicity and prophylaxis of CRS or neurotoxicity are unclear. Early detection testing may be able to identify patients at risk of severe toxicity who might benefit from early intervention.[11, 12] Grading systems for CRS and neurotoxicity have been proposed, but none is currently universally accepted.[8, 17, 18]
3.2 Failure of CAR-T Cell Immunotherapy
The success of CAR-T cell therapy is associated with the capacity of infused CAR-T cells to proliferate and induce effector function on encounter with antigen-expressing tumor. In most cases, CAR-T cell therapy is delivered as an autologous, patient specific product, and the outcomes are in part dependent on the quality of the collected T cells and the manufactured product. Approaches to T cell collection and CAR-T cell manufacturing methods are actively being investigated by many groups in an effort to allow delivery of more consistent and potent products.[36–38] Even after manufacturing of CAR-T cells with robust in vitro functional capacity, CAR-T cell activation in vivo may be inhibited by a suppressive tumor microenvironment established by the expression of inhibitory molecules and receptors (e.g. PD-1/PD-L1) by the tumor or stromal cells. Combination therapies to limit immune suppression and allow unrestrained activation of CAR-T cells in the tumor microenvironment are in development.[39, 40]
Despite good in vivo CAR-T cell expansion and achievement of CR, relapses can occur after CD19 CAR-T cell immunotherapy. Two categories of relapse can be identified. Relapse of tumor that remains CD19-positive can occur due to a suppressive tumor microenvironment, but can also occur in association with loss of CAR-T cell persistence. The reasons for loss of CAR-T cell persistence are complex and may be difficult to determine in individual patients. In a subset of patients an immune response to the CAR transgene can lead to CAR-T cell rejection and loss of persistence. Modification of lymphodepletion regimens to suppress an anti-CAR immune response or use of less immunogenic CAR designs might minimize the impact of immune-mediated CAR-T cell rejection. In other patients, activation induced cell death (AICD) or senescence may contribute to loss of CAR-T cells. In these situations, strategies to optimize CAR signaling to minimize AICD or improve manufacturing to produce less differentiated CAR-T cells might improve outcomes. Relapse of CD19-negative tumor is a distinct category that involves a change in tumor phenotype to escape an active CD19-directed anti-tumor immune response, and may occur despite robust CAR-T cell persistence. It appears to be more common in ALL than NHL or CLL. A variety of mechanisms of CD19 loss in tumors have been described, including phenotypic lineage switch and alternative splicing.[6, 41, 42] Targeting additional tumor antigens in combination with CD19 (e.g. CD20 or CD22) is being investigated as a strategy to reduce the risk of CD19-negative escape.[43, 44]
3.3 Role of HSCT after CAR-T Cell Therapy
Despite the potential for durable responses in response to CD19 CAR-T cell immunotherapy, it is unclear currently whether additional consolidation approaches such as allogeneic HSCT should be used to maintain remission for a subset of patients who might be at increased risk of relapse. A personalized approach is currently warranted, with more definitive guidelines to be determined by future studies.
4 Conclusions
CAR-T cell therapy is an effective novel therapeutic with outstanding success in B-ALL and promising results in B-NHL and CLL, signaling a new era of cancer treatment. Understanding CAR-T cell immunotherapy for B cell malignancies will assist in broadening the field to provide more effective therapies for other malignancies.
Table 2b.
Reports on CD19 targeted CAR-T cells in the treatment of B-NHL and CLL
| Disease | Institution | Patients | Lymphodepletion | CAR-T Dose | Response, Survival, and Relapse | Reference |
|---|---|---|---|---|---|---|
| NHL | NCI | 11 adults (30–64 years) | Cy 60–120 mg/kg day 1 + Flu 25mg/m2/d days 2–6 | 1 × 106, 2.5 × 106, or 5 × 106 CAR-T cells/kg | 4/7 evaluable patients with refractory DLBCL subtypes achieved a CR with an ORR of 6/7; in the 2 patients with low grade NHL 1 achieved a CR and 1 achieved a PR | [30] |
| NCI | 22 adults, age not reported | Concurrent Cy 300–500 mg/m2/d and Flu 30mg/m2/d × 3 days | Not reported in abstract | 8/19 DLBCL patients achieved a CR, with an additional 5 having a PR giving an ORR of 13/19 (68%); 3 remaining patients (1 with MCL and 2 with FL) developed a CR. | [31] | |
| NCI | 10 adults (44–63 years) | None | 0.7 × 106 to 8.2 × 106 CAR-T cells/kg* | 1 patient with DLBCL achieved CR, 1 patient with MCL achieved PR. | [25] | |
| UPenn | 24 adults (25–77 years) | Investigator’s choice | 3.08 × 106 to 8.87 × 106 CAR-T cells/kg | 15/22 (68%) ORR (7/13 DLBCL, 7/7 FL, and 1/2 MCL). PFS at 11.7 months follow up was 62%. | [32] | |
| FHCRC | 32 adults (36–70 years) | Initially Cy-based (e.g. Cy 2–4 g/m2 day 1 ± VP 100–200 mg/m2 days 1–3; subsequently Cy/Flu combination (e.g. Cy 60 mg/kg on day 1 + Flu 25 mg/m2/d on days 2–4 or 2–6) | 2 × 105, 2 × 106, or 2 × 107 CAR-T cells/kg | 19/30 (64%) ORR (7/11 DLBCL, 5/6 transformed LBCL, 4/5 FL, and 1/4 MCL). In patients treated with Cy/Flu at the maximum tolerated dose of 2 × 106, the CR rate was 64% (ORR 82%). | [11] | |
| CLL | UPenn | 14 adults (51–78 years) | Investigator’s choice | 1.4 × 107 to 11 × 108, CART cells◆ | 4/14 (29%) patients achieved a MRD-negative CR, with an ORR of 8/14 (57%). Median PFS was 7 months, OS 29 months. Two patients with CR have ongoing remissions more than 4 years. | [8] |
| UPenn | 35 adults (median age 62) | Investigator’s choice | 5 × 107 or 5 × 108, CART cells◆ | In stage I, 6/11 patients in the high dose group responded (with 4 CRs) versus 4/13 in the low dose group (with 1 CR); therefore, in stage II all patients were treated with the high dose. Of all 21 patients treated at higher dose (in both stage I and II), 17 evaluable for response with 9/17 (53%) ORR, 6 of whom achieved CR. 5 remain in CR at 26 months’ median follow-up. | [33] | |
| NCI | 5 adults (44–66 years) | None | 0.4 × 106 to 3.1 × 106 CAR-T cells/kg* | 1/5 patients achieved CR (was durable for 30+ months) and 1/5 achieved PR | [25] | |
| NCI | 4 adults (48–68 years) | Cy 60–120 mg/kg day 1 + Flu 25mg/m2/d days 2–6 | 1 × 106, 2.5 × 106, or 4 × 106 CAR-T cells/kg | 3/4 patients achieved a CR | [30] | |
| MSKCC | 8 adults (45–68 years) | Cy 600mg/m2 | 3 × 106, 1 × 107, or 3 × 107 CAR-T cells/kg | 2 patients achieved a CR, with an ORR of 4/8 (50%). 2 patients had marrow response but progressive disease in lymph nodes. | [34] | |
| FHCRC | 13 adults (40–73 years) | Predominantly Cy 30–60 mg/kg on day 1 + Flu 25 mg/m2/d on days 2–4 | 2 × 105, 2 × 106, or 2 × 107 CAR-T cells/kg | 10/12 restaged patients (83%) eliminated CLL from the marrow and 6 (50%) achieved a CR. | [35] |
Cy, Cyclophosphamide; Flu, Fludarabine; DLBCL, diffuse large B cell lymphoma; FL, follicular lymphoma; MCL, mantle cell lymphoma; CR, complete remission; ORR, overall response rate; PFS, progression free survival.
CAR-T cells manufactured from the allogeneic donor;
total number of CAR-T cells given.
Key Points.
Chimeric antigen receptor-modified T (CAR-T) cell therapy is an effective novel therapeutic with outstanding success in B-ALL and promising results in B-NHL and CLL, signaling a new era of cancer treatment.
Understanding the challenges in CAR-T cell immunotherapy for B cell malignancies will assist in broadening the field to provide more effective therapies for other malignancies.
Footnotes
Compliance with Ethical Standards
KAH is supported by the University of British Columbia, Clinical Investigator Program Fellowship. KAH declares no conflict of interest. CJT receives research funding from Juno Therapeutics and has received payment for participation on advisory boards and for speaking at educational events. CJT has patents pending related to CAR-T cells.
References
- 1.Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, Rimm AA, Ringdén O, Rozman C, Speck B. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990;75:555–562. [PubMed] [Google Scholar]
- 2.Turtle CJ. Chimeric antigen receptor modified T cell therapy for B cell malignancies. Int J Hematol. 2014;99:132–140. doi: 10.1007/s12185-013-1490-x. [DOI] [PubMed] [Google Scholar]
- 3.Till BG, Jensen MC, Wang J, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–2271. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, Forman SJ. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grupp SA, Kalos M, Barrett DM, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porter DL, Hwang W-T, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139–303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turtle CJ, Hanafi L-A, Berger C, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor–modified T cells. Sci Transl Med. 2016;8:355ra116–355ra116. doi: 10.1126/scitranslmed.aaf8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turtle CJ, Hanafi L-A, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Till BG, Jensen MC, Wang J, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119:3940–3950. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15:1145–1154. doi: 10.1517/14712598.2015.1046430. [DOI] [PubMed] [Google Scholar]
- 15.Stamenkovic I, Seed B. CD19, the earliest differentiation antigen of the B cell lineage, bears three extracellular immunoglobulin-like domains and an Epstein-Barr virus-related cytoplasmic tail. J Exp Med. 1988;168:1205–1210. doi: 10.1084/jem.168.3.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brentjens RJ, Riviere I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA, Mackall CL. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25–224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teachey DT, Lacey SF, Shaw PA, et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016;6:664–679. doi: 10.1158/2159-8290.CD-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38–177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park JH, Riviere I, Wang X, Purdon T, Sadelain M, Brentjens RJ. Impact of disease burden on long-term outcome of 19–28z CAR modified T cells in adult patients with relapsed B-ALL. J Clin Oncol. 2016;34(suppl) (abstr 7003) [Google Scholar]
- 22.Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127:3312–3320. doi: 10.1182/blood-2016-02-629063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geyer MB, Brentjens RJ. Review: Current clinical applications of chimeric antigen receptor (CAR) modified T cells. Cytotherapy. 2016;18:1393–1409. doi: 10.1016/j.jcyt.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee DW, Stetler-Stevenson M, Yuan CM, et al. Safety and Response of Incorporating CD19 Chimeric Antigen Receptor T Cell Therapy in Typical Salvage Regimens for Children and Young Adults with Acute Lymphoblastic Leukemia. Blood. 2015;126:684. [Google Scholar]
- 25.Brudno JN, Somerville R, Shi V, et al. Allogeneic T-Cells Expressing an Anti-CD19 Chimeric Antigen Receptor Cause Remissions of B-Cell Malignancies after Allogeneic Hematopoietic Stem Cell Transplantation without Causing Graft-Versus-Host Disease. Blood. 2015;126:99. doi: 10.1200/JCO.2015.64.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brudno JN, Somerville RPT, Shi V, et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. Journal of Clinical Oncology. 2016;34:1112–1121. doi: 10.1200/JCO.2015.64.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maude SL, Teachey DT, Rheingold SR, et al. Sustained remissions with CD19-specific chimeric antigen receptor (CAR)-modified T cells in children with relapsed/refractory ALL. J Clin Oncol. 2016;34 (suppl–abstr 3011) [Google Scholar]
- 28.Frey NV, Shaw PA, Hexner EO, et al. Optimizing chimeric antigen receptor (CAR) T cell therapy for adult patients with relapsed or refractory (r/r) acute lymphoblastic leukemia (ALL) J Clin Oncol. 2016;34 doi: 10.1200/JCO.19.01892. (suppl–abstr 7002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, Riddell SR. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33:540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kochenderfer J, Somerville R, Lu T, et al. Anti-CD19 chimeric antigen receptor T cells preceded by low-dose chemotherapy to induce remissions of advanced lymphoma. J Clin Oncol. 2016 (suppl–abstr LBA3010) [Google Scholar]
- 32.Schuster SJ, Svoboda J, Dwivedy Nasta S, et al. Sustained Remissions Following Chimeric Antigen Receptor Modified T Cells Directed Against CD19 (CTL019) in Patients with Relapsed or Refractory CD19+ Lymphomas. Blood. 2015;126:183. [Google Scholar]
- 33.Porter DL, Frey NV, Melenhorst JJ, et al. Randomized, phase II dose optimization study of chimeric antigen receptor (CAR) modified T cells directed against CD19 in patients (pts) with relapsed, refractory (R/R) CLL. J Clin Oncol. 2016;34 (suppl–abstr 3009) [Google Scholar]
- 34.Geyer MB, Park JH, Riviere I, Wang X, Purdon T, Sadelain M, Brentjens RJ. Updated results: phase I trial of autologous CD19-targeted CAR T cells in patients with residual CLL following initial purine analog-based therapy. J Clin Oncol. 2016;34 doi: 10.1016/j.ymthe.2018.05.018. (suppl–abstr 7526) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turtle CJ, Hanafi L-A, Berger C, et al. Rate of durable complete response in ALL, NHL, and CLL after immunotherapy with optimized lymphodepletion and defined composition of CD19 CAR-T cells. J Clin Oncol. 2016;34 (suppl–abstr 102) [Google Scholar]
- 36.Gattinoni L, Lugli E, Ji Y, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cieri N, Camisa B, Cocchiarella F, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121:573–584. doi: 10.1182/blood-2012-05-431718. [DOI] [PubMed] [Google Scholar]
- 38.Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, Klebanoff CA, Rosenberg SA, Leonard WJ, Restifo NP. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111:5326–5333. doi: 10.1182/blood-2007-09-113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130–3144. doi: 10.1172/JCI83092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, Lewis ID, Brenner MK, Brown MP. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol Ther. 2016;24:1135–1149. doi: 10.1038/mt.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sotillo E, Barrett DM, Black KL, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015;5:1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gardner R, Wu D, Cherian S, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127:2406–2410. doi: 10.1182/blood-2015-08-665547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haso W, Lee DW, Shah NN, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zah E, Lin M-Y, Silva-Benedict A, Jensen MC, Chen YY. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol Res. 2016;4:498–508. doi: 10.1158/2326-6066.CIR-15-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]