

Abstract
Purpose of review
Histone H3, lysine 4 methylation (H3K4me) is one chromatin modification that defines distinct regulatory states of euchromatin. Mammals express 6 main histone methyltransferase enzymes (HMTs) that modify H3K4 by mono-, di-, or tri-methylation. Recent studies examine roles of some of these HMTs and their cofactors in hematopoiesis and leukemia. We discuss these emerging studies together with prior embryonic stem (ES) data revealing how these enzymes function.
Recent findings
Murine models have been employed to conditionally or constitutively knock out HMTs (MLL1/KMT2A, MLL2/KMT2B, MLL3/KMT2C, MLL4/KMT2D, SETD1A/KMT2F and SETD1B/KMT2G) as well as specific domains or partners of these enzymes in normal hematopoietic populations and in the context of hematologic malignancies. These studies demonstrate that global or gene-specific changes in H3K4 modification levels can be attributed to particular enzymes in particular tissues.
Summary
Loss-of-function studies indicate largely non-overlapping roles of the six H3K4 HMTs. These roles are not all necessarily due to differences in enzymatic activity and are not always accompanied by large global changes in histone modification. Both gain- and loss-of-function mutations in hematologic malignancy are restricted to MLL1 and MLL3/MLL4, but emerging data indicate that SETD1A/SETD1B and MLL2 can be critical in leukemia as well.
Keywords: Epigenetic regulation, H3K4, HMT, histone methyltransferase, bivalent, poised
Introduction
Hematopoiesis represents an ongoing re-iteration of developmental processes, depending on the ability of stem cells and progenitors to both retain their identity and resist differentiation but also when appropriate, to respond to extracellular cues to change their identity. Identity can be defined as the transcriptome status plus the potential to express particular genes. The fidelity of gene expression patterns is critically dependent on chromatin regulation, which can either resist or enable a response to a given developmental cue. Here we focus on a particular group of enzymes that perform H3K4 mono-, di- or tri-methylation, as these modifications are associated with active enhancers, lineage-specific gene expression, or active/poised genes, respectively.
Roles of H3K4 methylation states
Actively transcribed euchromatin is characterized by relative enrichment for certain histone modifications, H3K4 methylation being prominent among these. H3K4 tri-methylation (H3K4me3) typically peaks over transcription start sites (TSS) with a nucleosome-free region centered over the TSS. Recent studies have illustrated correlations between the breadth H3K4me3 enrichment with transcriptional fidelity and enhanced elongation rates [1,2**]. H3K4 di-methylation (H3K4me2) has a more complex distribution, but can be enriched over TSS’s, gene bodies and enhancers. Five patterns of enrichment relative to the TSS have been bioinformatically identified [3,4] and the nature of the genes characterized by each of the patterns is very distinct, suggesting a particular tissue-specific role for this mark. Genes enriched for TSS H3K4me2 without concordant H3K4me3 enrichment have also been identified in hematopoietic progenitors as a type of “poised” gene, since this group marks a particular subset of developmental regulators and genes that are upregulated upon erythroid differentiation [4]. Since methylation of H3K4 does not change the structure or charge of the histone octamer, the outcome of these modifications is thought to attract transcriptional effector complexes through subunits that harbor H3K4 binding proteins, or “chromatin reader” domains. These chromatin reader domains can be part of basal transcription factors, repressive, or activating chromatin modifying complexes (reviewed in [5]).
H3K4me3 promoter enrichment is also associated with “bivalent” promoters. In ES cells, bivalent genes were described as harboring both H3K4me3 and H3K27me3 modifications at TSS-proximal histones, the latter mark conferred by repressive Polycomb complexes [6]. Bivalent genes are typically expressed only at low/undetectable levels, but upon induction of ES differentiation to neuronal precursor cells, genes resolve into predominantly H3K27me3- or H3K4me3-enriched and become silenced or more highly expressed, respectively [7]. This concept of developmentally bivalent genes has since been molecularly characterized further and shown to be relevant in a variety of pluripotent cell types, including hematopoietic progenitors [4,8].
Use of H3K4 methylation status to categorize enhancers in hematopoietic cells
Enhancer histone composition is so connected to cell identity that H3K4 mono-methylation (H3K4me1) (enriched at enhancers) can accurately cluster hematopoietic stem/progenitor/differentiated populations by cell type [9]. Recent studies provide evidence for both the de novo generation of lineage-specific enhancers as well as the priming-generated acquisition of lineage-specific enhancers, possibly depending on the particular lineage studied [9–11]. H3K4me1 enrichment can be used in combination with other histone modifications to define enhancers of distinct transcriptional outputs and capabilities (Figure 1). Active enhancers are typically enriched for H3K4me1 and H3K27-acetyl (ac) marks and lack H3K4me3 enrichment (Figure 1A). Primed enhancers are similarly marked, but lack H3K27ac and poised enhancers have been defined as enriched for H3K4me1 but marked with H3K27me3. Therefore these two categories differ in the enzymatic requirements for their activation (reviewed in [12]). An interesting functional capacity defined by H3K4me1 enrichment involves enhancers that became active in macrophages upon treatment with an inflammatory activators or cytokines (Figure 1B). Upon stimulation, these enhancers typically become enriched for H3K4me1 and H3K27ac nucleosomes in the time-frame of ~4 hours in synchrony with induction of expression the associated gene. After 48-hour withdrawal of the inducing signal, these enhancers lost transcription factor occupancy and H3K27ac modification, but approximately 30% of enhancers retained their H3K4me1 enrichment. Remarkably, the kinetics of induction of the genes controlled by the H3K4me1-retaining enhancers was much more rapid upon re-stimulation with the same signal [13]. These data suggest that H3K4me1 at enhancers functions to remember the prior stimulus and more rapidly respond to re-organize the active enhancer. Given these important functional correlations, it is important to determine how these defining histone marks are enzymatically regulated. Building on prior biochemical knowledge, the following loss-of-function studies begin to resolve which enzymes perform which modifications in distinct tissues.
Figure 1. Enhancer categories defined based on enrichment for histone modifications.
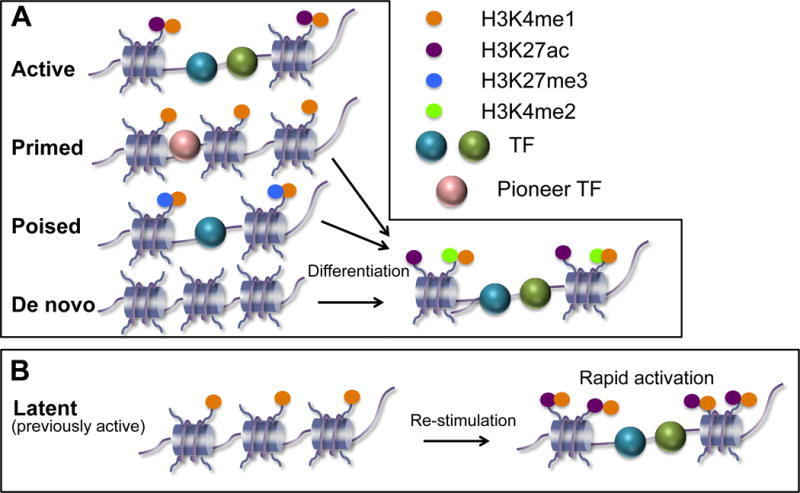
A) Enhancers and their propensity for becoming active can be defined by combinations of H3K4me1 and other modifications and transcription factor (TF) occupancies. Pioneer TFs are those who may bind to enhancers prior to any histone modifications to recruit subsequent enzymes and remodeling complexes. Active enhancers are open chromatin domains enriched minimally for H3K4me1 and H3K27ac. Primed enhancers are pre-marked by H3K4me1 and can be further activated at upon a differentiation signal. Poised enhancers are enriched for H3K4me1 and repressive H3K27me3, and are thus are “poised” to become active upon removal of the H3K27me3 mark and differentiation [9]. De novo activated enhancers are inferred from the lack of modifications at a prior differentiation stage [10,11]. B) In the context of macrophage activation [13], latent enhancers may have been previously activated by transcription factors but retain H3K4me1 as “memory” and are more rapidly activated upon re-stimulation relative to those lacking H3K4me1.
Six major H3K4 methyltransferases in mammals
Mammals have diversified the number and roles of dedicated H3K4 methyltransferases relative to yeast and flies. There are three pairs of SET/MLL paralogs in mammals: SETD1A/SETD1B, MLL1/MLL2, and MLL3/MLL4. We would like to emphasize that there is confusion in the literature in nomenclature for MLL2 and MLL4: here we use MLL2 for the protein encoded by the murine Wbp7 or Kmt2b gene.
All six proteins exhibit H3K4me activity when in complex with their protein partners, which include WDR5, RbBP5, ASH2L and DPY30 (“WRAD”), critical for the enzymatic activity of the Su(var)3–9/Enhacer of Zeste/Trithorax (SET), or catalytic domain of MLL1-4 and the SET proteins [14,15]. In addition to sharing WRAD components, there are protein partners that are specific to the related pairs. For example, Menin and LEDGF/PSIP1 interact exclusively with MLL1 and MLL2, whereas WDR82 and CFP1/CXXC1 interact with the SETD1A/SETD1B complexes [16–18]. In addition to these core component differences, there are differences in transiently interacting proteins among these family members, due to the unique sequences of each of the six enzymes. The expression of MLL1-4 and SET proteins are all fairly ubiquitous, resulting in co-expression of most enzymes in most tissues (http://biogps.org).
MLL1 and MLL2
Due to its discovery in chromosomal translocations of childhood leukemia, MLL1 was the first of this family to be studied extensively in the hematopoietic system using loss-of-function approaches. Multiple groups demonstrated that Mll1 knockout was embryonic lethal and affected development of the hematopoietic system [19–24]. Specifically, Mll1 in germ-line knock-out embryos is required to produce transplantable hematopoietic stem cells (HSCs) from the aorta-gonad-mesonephros region [19]. When specifically deleted in the hematopoietic lineage, Mll1 is not required for fetal liver hematopoietic cell expansion [22,25], suggesting that there were cell-extrinsic contributions to the germ-line knock-out phenotype. Nonetheless, Mll1−/− fetal liver hematopoietic cells also do not engraft irradiated adult recipients [22,25]. Similarly, adult HSCs (generated through conditional knockout) are severely defective in transplantation assays, likely due to the loss of expression of transcriptional factors including Mecom/Evi-1, Hoxa9, Prdm16, Pbx1 and other important HSC regulators [26]. Interestingly, maintaining the expression of these genes in HSCs did not require the histone methyltransferase (HMT) domain of Mll1, rather, correlated with the ability of the MLL1 complex to impart Histone H4, lysine 16 (H4K16) acetylation. This acetylation activity is not encoded by Mll1 itself but was attributed to recruited acetyltransferases [27].
MLL2 is the paralog of MLL1 and is highly similar in the SET domain and in primary structure [28]. Unlike MLL1, MLL2 is not involved in chromosomal translocations. In fact, the N-terminus of MLL2 cannot replace MLL1 in leukemia oncoproteins, likely due to a lower affinity for CpG sequences [29,30]. Germ-line deletions of Mll2 result in delayed development early in embryogenesis, neural tube defects and widespread apoptosis [28]. Surprisingly, Mll2 deletion results in global H3K4me2/3 loss in oocytes [31]. In contrast, complete deletion of Mll2 mid-gestation had no effect on global H3K4 methylation, hematopoietic or other organ homeostasis [32].
One hematopoietic cell type in which Mll2 does play a role is macrophages. An inducible Cre system was used to generate Mll2−/− macrophages from bone marrow, which appear grossly normal. However, these cells exhibited a specific defect in the induction of NFκB target genes through toll receptor 4 (TLR4) stimulation due to the failure to express MLL2 target genes critical for this signaling pathway. Several direct MLL2 target genes were identified in this study and these all exhibited reduced TSS H3K4me3 peaks with a corresponding increase in H3K27me3, a mark associated with repressed or bivalent genes. This observation suggests that MLL2’s role in maintaining expression of these target genes is to maintain H3K4me3 promoter enrichment and resist invading repression complexes. Interestingly, many other genes that were hypo-H3K4 methylated in Mll2−/− macrophages exhibited no change in expression level [33]. These data demonstrate that some genes may not be as sensitive to H3K4me3 promoter depletion than others. Nonetheless, at least some MLL2-dependent genes appear to be regulated by promoter-targeted H3K4me3/me2 modification.
Although MLL1 was known to be a proto-oncogene for more than 25 years [34], it was not clear whether it participated in non-MLL-translocation hematologic malignancies. Recently, deletion of the endogenous Mll1 gene in NUP98-HOXA9 or MN1-driven acute myelogenous leukemia (AML) demonstrated that endogenous MLL1 activity does contribute to leukemia maintenance in each of these distinct cytogenetic AML models [35**,36]. The role of endogenous MLL1 in MLL-driven leukemia is evolving, with some studies suggesting that it is required and some suggesting that it is not [27,37–39]. Furthermore, emerging data suggests that MLL2 may play a more important role in MLL-fusion-driven leukemia (Yufei Chen, University of Colorado, personal communication) as well as a role in solid tumors (reviewed in [40]).
MLL3 and MLL4
MLL3/4 in mammals were identified as HMTs in a co-activator complex associated with nuclear receptors [41–44] and are also recruited by other sequence-specific activators [43,45,46]. Complete knockout of either gene is embryonic or perinatal lethal and there is also evidence for redundancy between these genes, as co-deletion of both MLL3 and MLL4 was required to observe a global decrease in H3K4me1 in adipocytes [43]. MLL3 and MLL4 in vivo predominantly maintain H3K4me1 levels on enhancers [43,47] and recruit CBP/p300 to enhancers [47,48*].
Both MLL3 and MLL4 act as tumor supressors in leukemia, and have also been implicated in sold tumors [40]. MLL3 resides in the 7q region deleted in AML and knockdown of Mll3 collaborates with Nf1 knockdown in mouse models to produce an aggressive AML [49]. MLL4 is mutated in 30–90% of human diffuse large B-cell lymphomas (DLBCL) and follicular lymphomas (FL) [50, 51**, 52]. Genomic alterations are predicted to encode loss-of-function proteins, with about half mono-allelic and half bi-allelic. The direct role of MLL4 as a tumor suppressor was confirmed in murine leukemia models. Knock-down of Mll4 in a Vav-Bcl2 transgenic model or deletion of Mll4 in an activation-induced cytidine deaminase transgenic lymphoma model accelerated lymphomagenesis [53**]. In wild type animals, B cell-specific deletion of Mll4 increased steady-state transitional B cell numbers, enhanced germinal center formation and enhanced proliferation in response to CD40 stimulation. Loss of MLL4 was also sufficient for lymphoma development with a mean survival of just under 1 year [53**] and to observe a global reduction in H3K4me3/2/1 [51**]. Therefore MLL4 functions in a non-redundant manner to suppress mature B-cell proliferation and act as a tumor suppressor in FL and DLBCL.
The role of the MLL3/MLL4 proteins in normal hematopoiesis appears generally similar as either knockdown of Mll3 or knockout of Mll4 in hematopoietic stem/multipotent progenitor cells (HSPCs) results in impaired differentiation of HSPCs and increased HSPC numbers [49,54]. Mll4−/− HSPCs exhibit reduced engraftment in secondary recipients, accompanied by increased reactive oxygen species [49,54]. In the case of the Mll4 knockout, these defects were attributed to reduced expression of MLL4-dependent genes that protect from oxidative stress [54]. In contrast to the lymphoma context described above, deletion of Mll4 in an MLL-AF9-driven AML model system limited leukemia progression [54]. These distinct tumor-promoting or tumor-suppressor roles of MLL4 most likely reflect the different cellular contexts and target genes in AML versus lymphoma.
SETD1A and SETD1B
These two proteins are highly related overall and in SET domain sequence, but are each independently required for embryogenesis. Setd1a-/- blastocysts do not gastrulate and exhibit a defective inner cell mass and ES cells cannot be derived from knockout blastocysts. Setd1b-/- embryos exhibit growth defects from embryonic day (E) 7.5 and die before E11.5. In ES cells engineered with inducible (Rosa-ERT2) Cre, SETD1A loss is more severe, with growth arrest and cell death occurring upon protein reduction, as well as a global reduction in H3K4me3/2/1. Parallel experiments to delete Setd1b did not result in global H3K4 methylation changes, but it is not yet clear if redundancy between these two SET proteins accounts for this observation [55].
The conditional deletion of Setd1a in bone marrow hematopoietic cells revealed a unique role in B cell differentiation from the pro-B to pre-B cell stage. Global H3K4 methylation levels were not reported in these studies, but reduction of H3K4me3 at important B-cell regulators and the immunoglobulin heavy chain locus was suggested to underlie the block in differentiation [56]. Similarly, erythroid-specific Setd1a deletion partially blocked erythropoiesis, resulting in mild anemia. Reduced expression of lineage regulators such as Gata1 and Tal1 was noted, as was reduced H3K4me3 at the promoters of these genes [57*].
An interesting comparison to the Setd1a/Setd1b knockouts described above are knockouts of two genes encoding components of the SET complexes, CFP1/CXXC1 and DPY30. CFP1 provides CpG-element and chromatin targeting to the SET complexes. Cxxc1 germ-line deletion results in a pre-gastrulation phenotype very similar to Setd1a-/- embryos [58]. In Cxxc1−/− ES cells, about half of all H3K4me3 peaks at promoters are reduced, particularly from the highly expressed genes. In addition, H3K4me3 is ectopically-acquired in new nuclear territories, resulting in ectopic transcription of formerly silenced genes [59,60]. These cells can be maintained in pluripotent conditions, but fail to differentiate. Amazingly, the most highly expressed genes suffered the greatest H3K4me3 loss, yet did not exhibit reduced expression, similar to the Mll2 observations discussed above [60]. Therefore, CFP1 is critical for directing SET1 protein-mediated H3K4 methylation activity to proper genomic locations. DPY30, in contrast to CFP1, participates in all H3K4 methyltransferase complexes. In ES cells, depletion of Dpy30 does result in a global reduction in H3K4me3/me2. Similar to Cxxc1−/− ES cells, Dpy30-depleted ES cells can be maintained as pluripotent cells but fail to differentiate [61]. These ES studies suggested that 1) reduction in H3K4me3/2 level at the TSS does not necessarily result in reduced expression of the corresponding gene, and 2) H3K4me3/2 change is required for differentiation.
Within the hematopoietic system, conditional deletion of Setd1a, Cxxc1, or Dpy30 results in the accumulation of short-term HSCs and multi-potent progenitors but these cells do not engraft secondary recipients in the case of Setd1a and Dpy30 knockouts [62**,63]. These data suggest that the loss of Setd1a-mediated H3K4 modifications may dominate the phenotype in the Dpy30 knockout, but more detailed side-by-side comparisons will be required to determine this. Genes deregulated in HSPCs are very distinct between the Dpy30/Setd1a as compared to Mll1 and Mll2 knockouts, further underscoring the unique functions of each of the HMTs in the hematopoietic system ([62**,63], Waskow C, personal communication, TU Dresden).
Conclusions
Significant progress has occurred in the last several years toward understanding both the roles of H3K4me1/me2/me3 enriched genomic regions and in unraveling which enzymes are responsible for which modifications in specific tissues. Hematopoietic differentiation has provided a very well-characterized biological system for understanding the dynamics of these modifications and their relationship to differentiation. One principle emerging from murine knockout and human leukemia/lymphoma studies is that despite similarity between paralogs and co-expression in many of the same tissues, each H3K4 HMT has mainly unique, non-redundant functions, although one can find examples of redundancy. The first observation underscores the importance of the molecules that recruit the particular HMT complexes to particular loci in the genome, which is currently poorly understood. Most H3K4 HMTs play multiple and significant roles in the hematopoietic system and likely also play important roles in hematologic malignancies. Determining in what setting and how to target these enzymes with small molecule inhibitors represents an exciting new frontier.
Key points.
H3K4me1, in combination with other marks, defines several enhancer states
H3K4me3 enrichment can be uncoupled from active gene expression
Most H3K4 HMTs play important roles in leukemia/lymphoma and MLL1, MLL3 and MLL4 are mutated in leukemia
All six mammalian H3K4 HMTs perform unique functions in the hematopoietic system
Acknowledgments
We thank Yufei Chen and Claudia Waskow for sharing unpublished work. We are grateful to Kai Ge for critical review.
Financial Support and Sponsorhip
This work was partially supported by funds from the NIH (OD019716, HL90036).
Footnotes
Conflicts of Interest
PE owns Amgen stocks. No other conflicts were declared.
References
- 1.Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E, et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell. 2014;158:673–688. doi: 10.1016/j.cell.2014.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2**.Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, Rodriguez B, Xi Y, Xia Z, Chen X, et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet. 2015;47:1149–1157. doi: 10.1038/ng.3385. This study identified a novel connection between broad H3K4me3 with increased transcriptional elongation and strong enhancer activity. Specifically, it underlined the observation that tumor suppressors are marked by broad H3K4me3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pekowska A, Benoukraf T, Ferrier P, Spicuglia S. A unique H3K4me2 profile marks tissue-specific gene regulation. Genome Res. 2010;20:1493–1502. doi: 10.1101/gr.109389.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orford K, Kharchenko P, Lai W, Dao MC, Worhunsky DJ, Ferro A, Janzen V, Park PJ, Scadden DT. Differential H3K4 methylation identifies developmentally poised hematopoietic genes. Dev Cell. 2008;14:798–809. doi: 10.1016/j.devcel.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19:1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuettengruber B, Martinez AM, Iovino N, Cavalli G. Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Biol. 2011;12:799–814. doi: 10.1038/nrm3230. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 8.Abraham BJ, Cui K, Tang Q, Zhao K. Dynamic regulation of epigenomic landscapes during hematopoiesis. BMC Genomics. 2013;14:193. doi: 10.1186/1471-2164-14-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, et al. Immunogenetics. Chromatin state dynamics during blood formation. Science. 2014;345:943–949. doi: 10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choukrallah MA, Song S, Rolink AG, Burger L, Matthias P. Enhancer repertoires are reshaped independently of early priming and heterochromatin dynamics during B cell differentiation. Nat Commun. 2015;6:8324. doi: 10.1038/ncomms9324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luyten A, Zang C, Liu XS, Shivdasani RA. Active enhancers are delineated de novo during hematopoiesis, with limited lineage fidelity among specified primary blood cells. Genes Dev. 2014;28:1827–1839. doi: 10.1101/gad.240101.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–154. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 14.Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 15.Shinsky SA, Monteith KE, Viggiano S, Cosgrove MS. Biochemical reconstitution and phylogenetic comparison of human SET1 family core complexes involved in histone methylation. J Biol Chem. 2015;290:6361–6375. doi: 10.1074/jbc.M114.627646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Nuland R, Smits AH, Pallaki P, Jansen PW, Vermeulen M, Timmers HT. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol Cell Biol. 2013;33:2067–2077. doi: 10.1128/MCB.01742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JH, Skalnik DG. CpG-binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3-Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J Biol Chem. 2005;280:41725–41731. doi: 10.1074/jbc.M508312200. [DOI] [PubMed] [Google Scholar]
- 18.Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ernst P, Fisher JK, Avery W, Wade S, Foy D, Korsmeyer SJ. Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev Cell. 2004;6:437–443. doi: 10.1016/s1534-5807(04)00061-9. [DOI] [PubMed] [Google Scholar]
- 20.Hess JL, Yu BD, Li B, Hanson R, Korsmeyer SJ. Defects in yolk sac hematopoiesis in Mll-null embryos. Blood. 1997;90:1799–1806. [PubMed] [Google Scholar]
- 21.Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378:505–508. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- 22.McMahon KA, Hiew SY, Hadjur S, Veiga-Fernandes H, Menzel U, Price AJ, Kioussis D, Williams O, Brady HJ. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell. 2007;1:338–345. doi: 10.1016/j.stem.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Ayton P, Sneddon SF, Palmer DB, Rosewell IR, Owen MJ, Young B, Presley R, Subramanian V. Truncation of the Mll gene in exon 5 by gene targeting leads to early preimplantation lethality of homozygous embryos. Genesis. 2001;30:201–212. doi: 10.1002/gene.1066. [DOI] [PubMed] [Google Scholar]
- 24.Yagi H, Deguchi K, Aono A, Tani Y, Kishimoto T, Komori T. Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood. 1998;92:108–117. [PubMed] [Google Scholar]
- 25.Gan T, Jude CD, Zaffuto K, Ernst P. Developmentally induced Mll1 loss reveals defects in postnatal haematopoiesis. Leukemia. 2010;24:1732–1741. doi: 10.1038/leu.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P. Unique and Independent Roles for MLL in Adult Hematopoietic Stem Cells and Progenitors. Cell Stem Cell. 2007;1:324–337. doi: 10.1016/j.stem.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mishra BP, Zaffuto KM, Artinger EL, Org T, Mikkola HK, Cheng C, Djabali M, Ernst P. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep. 2014;7:1239–1247. doi: 10.1016/j.celrep.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glaser S, Schaft J, Lubitz S, Vintersten K, van der Hoeven F, Tufteland KR, Aasland R, Anastassiadis K, Ang SL, Stewart AF. Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development. Development. 2006;133:1423–1432. doi: 10.1242/dev.02302. [DOI] [PubMed] [Google Scholar]
- 29.Bach C, Mueller D, Buhl S, Garcia-Cuellar MP, Slany RK. Alterations of the CxxC domain preclude oncogenic activation of mixed-lineage leukemia 2. Oncogene. 2009;28:815–823. doi: 10.1038/onc.2008.443. [DOI] [PubMed] [Google Scholar]
- 30.Risner LE, Kuntimaddi A, Lokken AA, Achille NJ, Birch NW, Schoenfelt K, Bushweller JH, Zeleznik-Le NJ. Functional specificity of CpG DNA-binding CXXC domains in mixed lineage leukemia. J Biol Chem. 2013;288:29901–29910. doi: 10.1074/jbc.M113.474858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andreu-Vieyra CV, Chen R, Agno JE, Glaser S, Anastassiadis K, Stewart AF, Matzuk MM. MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glaser S, Lubitz S, Loveland KL, Ohbo K, Robb L, Schwenk F, Seibler J, Roellig D, Kranz A, Anastassiadis K, et al. The histone 3 lysine 4 methyltransferase, Mll2, is only required briefly in development and spermatogenesis. Epigenetics Chromatin. 2009;2:5. doi: 10.1186/1756-8935-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Austenaa L, Barozzi I, Chronowska A, Termanini A, Ostuni R, Prosperini E, Stewart AF, Testa G, Natoli G. The histone methyltransferase Wbp7 controls macrophage function through GPI glycolipid anchor synthesis. Immunity. 2012;36:572–585. doi: 10.1016/j.immuni.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 34.Li BE, Ernst P. Two decades of leukemia oncoprotein epistasis: the MLL1 paradigm for epigenetic deregulation in leukemia. Exp Hematol. 2014;42:995–1012. doi: 10.1016/j.exphem.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35**.Xu H, Valerio DG, Eisold ME, Sinha A, Koche RP, Hu W, Chen CW, Chu SH, Brien GL, Park CY, et al. NUP98 Fusion Proteins Interact with the NSL and MLL1 Complexes to Drive Leukemogenesis. Cancer Cell. 2016;30:863–878. doi: 10.1016/j.ccell.2016.10.019. This study showed a mechanism by which MLL1 is required in NUP98-fusion-driven (non-MLL rearranged) leukemogenesis through physical interaction with NUP98 fusion proteins and potentially regulate Hox gene expression by co-localize with NUP98 fusion proteins on promotor regions of Hox gene. Also endogenous Mll1 deletion in contrast did not affect MLL-AF9 driven leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36*.Riedel SS, Haladyna JN, Bezzant M, Stevens B, Pollyea DA, Sinha AU, Armstrong SA, Wei Q, Pollock RM, Daigle SR, et al. MLL1 and DOT1L cooperate with meningioma-1 to induce acute myeloid leukemia. J Clin Invest. 2016;126:1438–1450. doi: 10.1172/JCI80825. This study suggested assessed genetic requirements for another subset of Hox-high AML driven by MN-1. They show how MLL1 through interaction with other histone methyltransferease could enhance MN-1-driven leukemogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33:661–667. doi: 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, Liu L, Chen Y, Ouillette P, Zhu J, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell. 2014;53:247–261. doi: 10.1016/j.molcel.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thiel AT, Blessington P, Zou T, Feather D, Wu X, Yan J, Zhang H, Liu Z, Ernst P, Koretzky GA, et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell. 2010;17:148–159. doi: 10.1016/j.ccr.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ford DJ, Dingwall AK. The cancer COMPASS: navigating the functions of MLL complexes in cancer. Cancer Genet. 2015;208:178–191. doi: 10.1016/j.cancergen.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 41.Goo YH, Sohn YC, Kim DH, Kim SW, Kang MJ, Jung DJ, Kwak E, Barlev NA, Berger SL, Chow VT, et al. Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol Cell Biol. 2003;23:140–149. doi: 10.1128/MCB.23.1.140-149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mo R, Rao SM, Zhu YJ. Identification of the MLL2 complex as a coactivator for estrogen receptor alpha. J Biol Chem. 2006;281:15714–15720. doi: 10.1074/jbc.M513245200. [DOI] [PubMed] [Google Scholar]
- 43.Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife. 2013;2:e01503. doi: 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jozwik KM, Chernukhin I, Serandour AA, Nagarajan S, Carroll JS. FOXA1 Directs H3K4 Monomethylation at Enhancers via Recruitment of the Methyltransferase MLL3. Cell Rep. 2016;17:2715–2723. doi: 10.1016/j.celrep.2016.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M, et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem. 2007;282:20395–20406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patel SR, Kim D, Levitan I, Dressler GR. The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev Cell. 2007;13:580–592. doi: 10.1016/j.devcel.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu D, Gao X, Morgan MA, Herz HM, Smith ER, Shilatifard A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol. 2013;33:4745–4754. doi: 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48*.Wang C, Lee JE, Lai B, Macfarlan TS, Xu S, Zhuang L, Liu C, Peng W, Ge K. Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc Natl Acad Sci U S A. 2016;113:11871–11876. doi: 10.1073/pnas.1606857113. This study thoroughly investigated whether and how MLL4 is required at enhancers for cell fate maintenance and transition in multiple models. The authors showed that MLL4 is dispensable for cell fate maintenance but essential for cell fate transition by controling p300 binding to enhancers activated during cell differentiation, rather than through its enzymatic (H3K4me1) activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, Shroff AS, Dickins RA, Vakoc CR, Bradner JE, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 2014;25:652–665. doi: 10.1016/j.ccr.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43:830–837. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51**.Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015;21:1190–1198. doi: 10.1038/nm.3940. This publication revealed MLL4’s role in normal murine B cell development and GC formation. The tumor suppressor role of MLL4 was further demonstrated by crossing to a Vav-Bcl2 model of folicular lymphoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53**.Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21:1199–1208. doi: 10.1038/nm.3943. This study showed that Mll4 deletion accelerates B cell lymphoma in the Vav-Bcl2 model and AID transgenic model as well as without a driver mutation. Comprehensive genome-wide profilling of MLL4 target genes in the knockout model and human samples identified common MLL4-regulated genes in normal and malignant B cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Santos MA, Faryabi RB, Ergen AV, Day AM, Malhowski A, Canela A, Onozawa M, Lee JE, Callen E, Gutierrez-Martinez P, et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature. 2014;514:107–111. doi: 10.1038/nature13483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bledau AS, Schmidt K, Neumann K, Hill U, Ciotta G, Gupta A, Torres DC, Fu J, Kranz A, Stewart AF, et al. The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development. 2014;141:1022–1035. doi: 10.1242/dev.098152. [DOI] [PubMed] [Google Scholar]
- 56.Tusi BK, Deng C, Salz T, Zeumer L, Li Y, So CW, Morel LM, Qiu Y, Huang S. Setd1a regulates progenitor B-cell-to-precursor B-cell development through histone H3 lysine 4 trimethylation and Ig heavy-chain rearrangement. FASEB J. 2015;29:1505–1515. doi: 10.1096/fj.14-263061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57*.Li Y, Schulz VP, Deng C, Li G, Shen Y, Tusi BK, Ma G, Stees J, Qiu Y, Steiner LA, et al. Setd1a and NURF mediate chromatin dynamics and gene regulation during erythroid lineage commitment and differentiation. Nucleic Acids Res. 2016;44:7173–7188. doi: 10.1093/nar/gkw327. This study generated a erythroid-specific knockout model of Setd1a and demonstrated partially blocked erythropoiesis upon Setd1a loss. Mechanisms proposed include that SETD1A reuglates H3K4me3 at promoters of key erythropoiesis regulators Gata1 and Tal1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carlone DL, Skalnik DG. CpG binding protein is crucial for early embryonic development. Mol Cell Biol. 2001;21:7601–7606. doi: 10.1128/MCB.21.22.7601-7606.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tate CM, Lee JH, Skalnik DG. CXXC finger protein 1 restricts the Setd1A histone H3K4 methyltransferase complex to euchromatin. FEBS J. 2010;277:210–223. doi: 10.1111/j.1742-4658.2009.07475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clouaire T, Webb S, Skene P, Illingworth R, Kerr A, Andrews R, Lee JH, Skalnik D, Bird A. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 2012;26:1714–1728. doi: 10.1101/gad.194209.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang H, Shukla A, Wang X, Chen WY, Bernstein BE, Roeder RG. Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell. 2011;144:513–525. doi: 10.1016/j.cell.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62**.Yang Z, Shah K, Khodadadi-Jamayran A, Jiang H. Dpy30 is critical for maintaining the identity and function of adult hematopoietic stem cells. J Exp Med. 2016 Oct 17;213(11):2349–2364. doi: 10.1084/jem.20160185. 2016. This is the first published Dpy30 conditional knock-out mouse model. Detailed characterization of role of DPY30 in the maintenance of adult HSCs is presented. The authors show globally reduced levels of all H3K4me marks in lineage-depleted bone marrow cells and a significant accumulation of early HSPCs, distinguishing this knock-out from previously published MLL family knock-out phenotypes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chun KT, Li B, Dobrota E, Tate C, Lee JH, Khan S, Haneline L, HogenEsch H, Skalnik DG. The epigenetic regulator CXXC finger protein 1 is essential for murine hematopoiesis. PLoS One. 2014;9:e113745. doi: 10.1371/journal.pone.0113745. [DOI] [PMC free article] [PubMed] [Google Scholar]