

Abstract
Recent work has stimulated interest in the use of exosomes as nanocarriers for delivery of small drugs, RNAs, and proteins to the central nervous system (CNS). To overcome the blood-brain barrier (BBB), exosomes were modified with brain homing peptides that target brain endothelium but likely to increase immune response. Here for the first time we demonstrate that there is no need for such modification to penetrate the BBB in mammals. The naïve macrophage (Mϕ) exosomes can utilize, 1) on the one hand, the integrin lymphocyte function-associated antigen 1 (LFA-1) and intercellular adhesion molecule 1 (ICAM-1), and, 2) on the other hand, the carbohydrate-binding C-type lectin receptors, to interact with brain microvessel endothelial cells comprising the BBB. Notably, upregulation of ICAM-1, a common process in inflammation, promotes Mϕ exosomes uptake in the BBB cells. We further demonstrate in vivo that naïve Mϕ exosomes, after intravenous (IV) administration, cross the BBB and deliver a cargo protein, the brain derived neurotrophic factor (BDNF), to the brain. This delivery is enhanced in the presence of brain inflammation, a condition often present in CNS diseases. Taken together, the findings are of interest to basic science and possible use of Mϕ-derived exosomes as nanocarriers for brain delivery of therapeutic proteins to treat CNS diseases.
Keywords: BBB, Endocytosis, Inflammation, Macrophage exosomes, Pharmacokinetics, Protein delivery
TABLE OF CONTENTS GRAPHIC
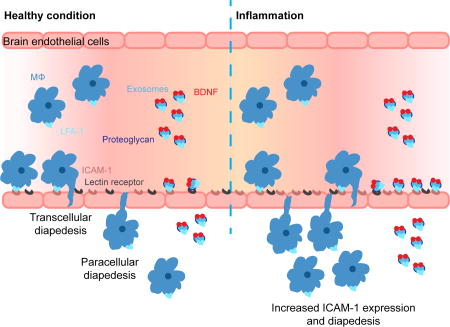
INTRODUCTION
Exosomes are 40 to 200 nm membrane-encased vesicles secreted by cells via fusion of multivesicular bodies with cell plasma membranes. They contribute to intercellular communication by carrying proteins and RNAs between neighboring cells or even to distant organs, and were clinically evaluated as cancer vaccines. Tumor-derived exosomes loaded with curcumin were previously shown to accumulate in microglial cells and alleviate brain inflammation [1]. Moreover, Mϕ-derived exosomes loaded with catalase produce neuroprotection in a Parkinson’s disease (PD) mouse model [2]. Both studies employed an intranasal route of administration, which in theory allows substances to bypass the BBB and directly enter the brain with minimum blood exposure. However, it is unclear if these exosomes access the brain directly or indirectly via absorption into blood and subsequent blood to brain transport. Nonetheless, the percentage of injected dose reaching the brain following intranasal delivery is usually lower than 1% and can be as low as 0.1% or even less [3]. Furthermore, this administration route has high variability in the dose reaching the brain due to variability in drug disposition and is limited by the drug amounts that can be delivered in humans [4]. A systemic, for example, IV administration route enables direct access to the BBB and uniform brain distribution through the brain capillaries. However, brain bioavailability of ~ 98% of small- molecule drugs and almost all biomacromolecules is very poor [5, 6]. To address BBB penetration, exosomes from dendritic cells (DCs) [7] and human embryonic kidney 293T cells [8] were decorated with the brain targeting peptide (rabies virus glycoprotein). These modified exosomes delivered siRNA and silenced in the brain a gene of relevance to Alzheimer’s disease (AD) [7] and morphine relapse [8]. Unfortunately, such peptide-decorated exosomes can cause immune response, especially during chronic treatments. The immunogenicity of the targeting peptide alone is likely low. However, exosomes present major histocompatibility complex (MHC) molecules and a co-stimulatory molecule CD86 on the surface, which can potentially boost the immune response, especially with chronic exposure [9, 10]. Naïve exosomes released from brain endothelial cells were shown to penetrate the BBB, delivering a fluorescent marker and a chemotherapeutic agent, doxorubicin, to the brain in Zebrafish [11]. Therefore, it is crucial to investigate whether naïve exosomes could penetrate the BBB in preclinical animal models, such as rodents and primates.
We examined the possibility of utilizing naïve Mϕ exosomes as drug carriers for CNS. Our rationale is based on prior experience using Mϕs as cell carriers to treat CNS disorders [12–14]. Pathology of many neurologic disorders, including multiple sclerosis, AD, PD, stroke, brain tumors, traumatic brain injuries, and others can result in BBB dysfunction [15]. The inflammatory processes associated with some of these pathologies can increase diapedesis of peripheral immune cells across the BBB [16]. Gendelman’s group used Mϕs as carriers for nanoformulated antiretroviral therapy for human immunodeficiency virus type 1 (HIV-1)-associated neurocognitive disorders [17]. We used Mϕs as carriers for systemic delivery of the nanoformulated therapeutic proteins and genes to the inflamed brain in the PD models [12–14]. The functional activity of Mϕs in these applications can be in part mediated by exosomes that share common elements, such as LFA-1 involved in the diapedesis of Mϕs across endothelial barriers [18]. The Mϕ exosomes can mediate the transfer of their cargo to other brain resident cells [12, 14, 19, 20], or facilitate spreading of viruses and virus proteins from periphery to and within the brain [21, 22].
MATERIALS and METHODS
Materials
Fetal bovine serum (FBS), penicillin-streptomycin, Dulbecco’s modified eagle medium (DMEM), 0.25 % trypsin/EDTA, chemically defined lipid concentrate, HEPES (1M), CM-DiI, protein G magnetic beads, Illustra Nap-5 columns, G418 sulfate, Alexa Fluor 488-Transferrin(Tf), Alexa Fluor 488-Cholera toxin subunit B (CTB), and SlowFade® Gold antifade mountant were from Life Technologies (Grand Island, NY). Rat collagen I (lower viscosity) and Anti-Human DEC-205 antibodies were from R&D systems (Minneapolis, MN). Endothelial cell growth EBM-2medium was from Lonza (Basel, Switzerland). Goat anti-Alix, goat anti-Tsg 101, goat anti-LFA-1 and rabbit anti-ICAM-1antibodies were from Santa Cruz (Dallas, Texas). Rat anti-LAMP-2, rabbit anti-clathrin heavy chain antibodies was from Abcam (Cambridge, MA). Rabbit anti-caveolin-1 antibodies were from Cell Signaling (Danver, MA). Na125I and Na131I were from Perkin-Elmer Life Sciences (Boston, MA). Formvar coated copper grid (200 meshes), M-per mammalian protein extraction reagent, Micro BCA protein assay kit, agarose, HALT™ proteinase and phosphatase inhibitor cocktail and ECL western blotting substrate were purchased from Thermo Fisher Scientific (Rockford, IL). BDNF was purchased from PeproTech (Rocky Hill, NJ). 4–15% polyacrylamide gel, native sample buffer, and laemmli sample buffer were purchased from Bio-Rad (Hercules, CA). Human basic fibroblast growth factor (bFGF), hydrocortisone, FITC-dextran conjugates (70 kDa) and lipopolysaccharides (LPS) and all other chemicals were from Sigma-Aldrich (St-Louis, MO).
Cell Culture
Raw 264.7 Mϕs (RAW Mϕs, American Type Culture Collection ATCC® TIB-71TM, Rockville, MD) between passage 1 and 30 were used. The cells were grown in DMEM medium plus 10% FBS and 1% penicillin-streptomycin, and subcultured by scraping. The conditioned medium for exosome collection was DMEM plus 1% penicillin-streptomycin and 10% FBS pre-centrifuged at 120 kg for 140 min to remove serum exosomes. hCMEC/D3 cells (a kind gift from Dr. Pierre-Olivier Couraud in Cochin Institute, France) between passage 30 and 35 were used. All cell cultureware for hCMEC/D3 cells was coated with 0.15 mg/ml rat collagen I. The cells were grown in EBM-2 containing 5% FBS, 1% Penicillin-Streptomycin, 1.4 µM hydrocortisone, 5µg/ml acid ascorbic, 100× diluted chemically defined lipid concentrate, 10 mM HEPES and 1 ng/ml bFGF. NIH-3T3 cells stably transfected with TrkB receptors (a kind gift from Dr. David Kaplan in University of Toronto, Canada) was cultured in DMEM supplemented with 10% Colorado Calf serum and 100 µg/ml of G418 sulfate.
Animals
All animal experiments were conducted under the approval of the University of North Carolina Institutional Animal Care and Use Committee. Six to eight weeks old male CD-1 mice were purchased from Charles River Laboratories.
Purification of Exosomes
Exosomes were purified by the common sequential centrifugation method [7, 23]. Raw Mϕs were grown in 7 T75 flasks to reach 70–80% confluence. Following two phosphate-buffered saline (PBS) washes, the cells were cultured in 10 ml conditioned medium for 2 days. The medium was then collected and centrifuged sequentially at 300g for 15 min, 3,000g for 15 min, 20,000g for 70 min, and filtered through 0.2 µm membrane filters to remove cells and large particles. Exosomes were pelleted at 120,000g for 70 min, washed by PBS to remove proteins, pelleted again, and then resuspended in 1 ml PBS. For cell uptake study, the exosomes were labeled with CM-DiI dyes (2 µg/ml) added to the medium before the first 120,000g pelleting step. The exosome suspension was filtered through 0.22 µm membrane filters to sterilize and remove the dye precipitate and stored in −80 °C for at most 3 weeks. Each batch of exosomes contained around 65 µg exosomal proteins as determined by microBCA and 3×1011 exosomes as determined by nanoparticle tracking analysis (NTA).
Characterization of Exosomes
Exosomes were characterized by dynamic light scattering (DLS) for intensity-weighted z-average diameter, polydispersity index (PDI), and zeta potential, by NTA for number-weighted diameter and particle concentration, and by transmission electron Microscopy (TEM) for morphology. For DLS, the size was measured in PBS, and the zeta- potential was measured in 10 mM NaCl at 23 °C with a 173° scattering angle using Zetasizer Nano-ZS instrument (Malvern, UK) in at least triplicates. For NTA, each sample was diluted 500 times in PBS and loaded into Nanosight NS500 (Malvern, UK). Three videos of 60s with a sample advance in between were recorded with the minimal expected particle size, minimum track length and blur setting all set to automatic. For TEM, exosomes were adsorbed onto Formvar coated copper grid (200 mesh), stained with 2% uranyl acetate and characterized using Zeiss TEM 910 Transmission Electron Microscope (Jena, Germany) at 80 kV accelerating voltage.
Protein Composition and Exosomal Markers
Mϕs and Mϕ exosomes were lysed with RIPA buffer mixed with proteinase and phosphatase inhibitor cocktail. Protein composition and exosomal markers were detected by standard sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting under reducing condition [23].
Cell Viability
Cell viability was determined by 3-(4, 5-dimethylthiazol-2-yl)-2, 5- diphenyltetrazolium bromide (MTT) assay. hCMEC/D3 cells were seeded in 96-well plates at 2000 cells/well in culture medium. After overnight incubation, the cells were treated with test agents in culture medium for time durations indicated in figure legend, and let grown in fresh culture medium (200 µl) for another 72 h. 20 µl of MTT in PBS (5 mg/ml) was added to each well. After 4h incubation at 37 °C, the formed formazan precipitate was dissolved in 150 µ l of DMSO. Absorbance at 570 nm (A) was read on a microplate reader SpectraMax M5 (Molecular devices). Blanks (wells without cells) that account for solvent adsorption and controls (wells with cells without test agents) for 100% viability were treated similarly. Cell viability (%) was calculated as . Data are means ± SD of 6 replicate wells.
Flow Cytometry
hCMEC/D3 cells were grown in 24-well plate at 5×104 cells/well for 4–5 days to reach confluence. In the uptake mechanism studies, the cells were pretreated with endocytosis inhibitors, carbohydrates or EGTA for 0.5 h, and then co-treated with CM-DiI labeled exosomes (0.6×1010 exosomes/ml) for 4 h. The inhibition of endocytosis markers uptake was studied similarly. The endocytosis markers used were Alexa Fluor 488-Transferrin (10 µg/ml) for clathrin mediated endocytosis, Alexa Fluor 488-CTB (5 µg/ml) for caveolae mediated endocytosis, and FITC-Dextran (70 kDa (10 mg/ml) for macropinocytosis. The antibody block assays were done by co-incubating exosomes with antibodies or isotope controls at 100 µg/ml for 4 h. The cells were washed thrice by PBS, detached by 0.25% trypsin/EDTA, collected by centrifugation at 100 g for 10 min, fixed with 4% paraformaldehyde for 10 min, and then resuspended in 0.35 ml PBS. Viable singlets were gated based on forward scatter and side scatter. 5,000–10,000 viable singlets were recorded for each sample on Becton Dickinson LSRII (BD Biosciences) using 488 nm and 532 nm lasers. Unless otherwise noted in figure legend, data are not normalized and reported as mean fluorescence intensity (MFI) ± SD of 3 replicate wells.
Laser scanning confocal microscopy (LSCM)
hCMEC/D3 cells were cultured in 35 mm glass bottom dishes at 1×105 cells/well for 5–6 days to reach confluence. In endocytosis pathway studies, the cells were treated with CM-DiI labeled exosomes (1×1011 exosomes/ml) and Alexa Fluor 488-Transferrin (25 µ g/ml) or Alexa Fluor 488-CTB (5 µg/ml) for 0.5 h, and then fixed before imaging. In the immunofluorescence studies, the cells were treated with CM-DiI labeled exosomes (1×1011 exosomes/ml) for 0.5 h, fixed by 4% paraformaldehyde, blocked with 10% goat serum/0.3% Triton® X-100 in PBS at 23 °C for 1 h, and incubated with anti-clathrin heavy chain or anti-caveolin 1 antibodies in 1% goat serum/1% BSA/0.3% Triton® X-100 in PBS at 4 °C overnight. Followed three washes using 0.1% BSA in PBS, the cells were incubated with Alexa Fluor 488 conjugated secondary antibodies, washed trice and mounted in SlowFade® Gold antifade mountant (Life technologies). Images were collected by Zeiss CLSM 700/710 spectral confocal laser scanning microscope (Jena). Mander’s colocalization coefficients were calculated using Image J and JACoP plugin [24, 25].
Native gel electrophoresis
Protein samples were mixed with equal volume of native sample buffer (Bio-Rad), and then resolved on 4–15% polyacrylamide gel (Bio-Rad) in 25 mM Tris-Cl/250 mM glycine, pH 8.5 at 120 v for 55 min, or on 0.5% horizontal agarose gel in 40 mM Tris/20 mM acetic acid/1 mM EDTA, pH 7.6 at 80 v for 50 min. The gels were stained with coomassie blue G250, and scanned using FluorChem E System (ProteinSimple).
Radioactive Labeling
Exosomes and BDNF/BSA proteins were labeled with iodine by chloramine-T method [26]. For this study, we collected Mϕ exosomes after 12 h incubation of Mϕ in DMEM to exclude iodination of serum proteins that could be co-precipitated during isolation of exosomes by ultracentrifugation. Briefly, exosomes or proteins were mixed with 1 mCi of Na125I or Na131I (Perkin Elmer) and 10 µ g of chloramine-T in phosphate buffer (0.25 M, pH 7.5) for 60 s. Labeled exosomes and proteins were purified by Illustra Nap-5 columns (Life technologies) and collected in tubes pretreated with 1% BSA in PBS to prevent nonspecific adsorption. The iodine association (iodine in labeled sample/total iodine) was determined by trichloroacetic acid precipitation method [26]. Briefly, 1 µl of purified samples was mixed with 0.5 ml of 1% BSA in PBS and 0.5 ml of 30% TCA, and then centrifuged at 5400 g for 10 min. The resulted pellet and supernatant were counted on r-counter (PerkinElmer). The iodine association was calculated as the percentage of pellet radioactivity to total radioactivity. The iodine association for exosomes and BSA/BDNF was higher than 85% and 98%, respectively. In the brain perfusion study, BSA was labeled with Technetium-99m (99mTc). In detail, 120 µ g of stannous tartrate and 1 mg of BSA were dissolved in 500 µl of water. The pH was adjusted to 2.5-3.3 by adding 20 µl of 0.2 M HCl, and then 1 mCi of 99mTc was added to the solution. The mixture was incubated at 23 °C for 20 min and then purified similarly as the iodinated proteins.
Animal Procedure
Mice were anesthetized with 40% urethane (4 g/kg) by intraperitoneal injection. 125I-labeled substances (4×105 cpm) and 131I-labeled BSA were co-injected to the right jugular vein. At each time point, blood was collected from the left carotid artery, allowed to clot and then centrifuged at 5400 g for 10 min to collect serum. The mouse was immediately decapitated after collection of the arterial blood and the whole brain and peripheral organs removed and weighed immediately after decapitation. The radioactivity of serum and tissues were counted and normalized to injected dose (ID) by volume (ml) or weight (g) (%ID/ml or %ID/g). An injection check representing ID was also counted (n=3).
Pharmacokinetic analysis
The noncompartmental PK parameters - the volume of distribution at steady state Vss (ml), clearance CL (ml/min), the mean residence time from the time of dosing to last detectable concentration MRTlast (h) and the mean residence time from the time of dosing to infinity MRTinf (h) were estimated using Phoenix®WinNonlin® 6.3 (Pharsight).
Multiple-time regression analysis of influx into the brain
The Ki (slope) and Vi (y-intercept) were calculated from the linear portion of multiple-time regression analysis [27]. Brain/serum ratios ( , ml/g) of co-injected BSA were used to correct for the vascular space or leakage [28], and subtracted from that of the test substance to yield the delta brain/serum ratios. The delta brain/serum ratios were plotted against their respective exposure times using the equation
, where the exposure time was the trapezoidal integral of serum cpm at time t (Cpt) from time 0 to time t divided by Cpt.
Brain accumulation
Brain accumulation of test substance (%ID/g) was calculated by multiplying the delta brain/serum ratio (ml/g) with serum concentration (%ID/ml) at selected time point.
Capillary depletion
Capillary depletion was performed to determine whether the injected substances cross the capillary walls and enter the brain parenchyma as previously reported [28]. 10 min after injection, serum and brain samples were collected as mentioned above. The brain was homogenized in 1.25 ml of physiological buffer containing 10 mM HEPES, 141 mM NaCl, 4 mM KCl, 2.8 mM CaCl21 mM MgSO41 mM NaH2PO4and 10 mM D-glucose, pH 7.4, and then mixed with equal volume of 40 % Dextran (70 kDa, Sigma) by vortex. The mixture was centrifuged at 3500 g for 20 min at 4 °C. The radioactivity of the capillary pellet, parenchyma supernatant, and serum was counted. The delta capillary/serum ratio and parenchyma/serum ratio of 125I-labeled test substance was corrected using 131I-BSA data, and then multiplied by the serum concentration to calculate the uptake (%ID/g) in brain capillary and parenchyma.
Brain perfusion
The mice were treated with intraperitoneal injection of saline (healthy mice) or LPS in saline (3 mg/kg, a brain inflammation model) at 0, 6, and 24 h. At 28 h, the mice were anesthetized by intraperitoneal injection of urethane. The brain perfusion was done by clamping the descending aorta, cutting the jugular vein, and then injecting the perfusion solution into the heart. The mouse brains were perfused with 125I-labeled exosomes and 99mTc-labeled BSA (as a vascular marker) in lactated ringer’s buffer containing 1% unlabeled BSA (to prevent nonspecific binding) at 2 ml/min for 2.5 min. The radioactivity in brain and in perfusion buffer was counted simultaneously and normalized by weight and by volume. The brain/perfusion ratio of BSA was subtracted from the ratio of exosomes to account for BBB leakiness.
Statistical analysis
Statistical analysis was performed using Prism 6.0 (GraphPad Software Inc.) unpaired two-tailed student t-test (# p < 0.05, ## p < 0.01, and ### p < 0.001), paired two- tailed student t-test(& p < 0.05, && p < 0.01, and &&& p < 0.001), or one-way ANOVA with post Newman-Keuls multiple comparison test (* p < 0.05, ** p < 0.01, and *** p < 0.001) as indicated in the figure legend.
RESULTS AND DISCUSSION
Mϕ exosomes are natural nanoparticles
We hypothesized that Mϕ-derived exosomes can naturally bind with brain endothelial cells, pass across the BBB and enter the brain. To test this hypothesis, we collected exosomes secreted by RAW Mϕs by the sequential centrifugation method [7, 23]. We characterized their size distribution and zeta potential by DLS (Figure S1a, b) and NTA (Figure 1a), morphology by TEM (Figure S1c), and protein composition by SDS- PAGE (Figure S1d) and western blotting (Figure 1b). Consistent with published results [7, 29], Mϕ exosomes were heterogeneous in size with an intensity-weighted z-average diameter of 149 ± 10 nm, a relatively small PDI of 0.134 ± 0.048 as determined by DLS, and a number-weighted mean diameter of 130 ± 49 nm, a mode diameter of 90 ± 2 nm by NTA. Mϕ exosomes were negatively charged (zeta potential −18 ± 1 mV) in 10 mM NaCl. TEM showed spherical morphology as published [23]. Occasionally, we observed aggregation and cup-shaped nanovesicles (insert of Figure S1c) as an artifact of sample drying [23]. Based on SDS-PAGE, the protein composition of exosomes differed from both the parent Mϕs and FBS [23]. As revealed using western blotting, compared to Mϕs, exosomes were enriched with apoptosis- linked-gene-2 interacting protein X (Alix) and tumor susceptibility gene 101 protein (Tsg 101), two exosomal markers related to the biogenesis of multivesicular bodies [21]. Mϕ exosomes also contained a transmembrane protein lysosome-associated membrane protein 2 (LAMP 2) and a cytosolic protein β-actin that are frequently detected in exosomes [21].
Figure 1. Characterization of Mϕ exosomes.

(a) Number-weighted size distribution of exosomes by NTA (different colors show three repeated measurements). (b) Western blotting of RAW Mϕs and exosome lysates at comparable protein loading amounts showing exosomal markers and conserved proteins. (c) Concentration-dependent uptake of CM-DiI labeled exosomes at 4 h. (d) Inhibition of uptake of CM-DiI labeled exosomes (0.6×1010 exosomes/ml at 4 h) by non-labeled exosomes. *** p < 0.001 vs untreated cells background (bkg) or indicated groups. Exosomes were purified by sequential centrifugation of RAW Mϕs-conditioned medium. Cell uptake was determined by Flow cytometry. Data are mean fluorescence intensity (MFI) ± SD of 5000–10000 live singlets, n = 3, * p < 0.05 and *** p < 0.001 vs. untreated cells or indicated groups by one-way ANOVA and post Newman-Keuls multiple comparison test.
Uptake of Mϕ exosomes in human cerebral microvascular endothelial cells
Next, we characterized the interactions of Mϕ exosomes with human cerebral microvascular endothelial cells (hCMEC/D3) as an in vitro BBB model. The MTT assay showed that the viability of these cells was not affected by their exposure to Mϕ exosomes for 24 h up to the highest tested concentration of 2×1011 exosomes/ml (Figure S2a, b). All subsequent studies involving hCMEC/D3 cells used lower concentrations of exosomes. We labeled exosomes with the lipophilic dye CM-DiI to monitor their uptake by hCMEC/D3 cells. CM-DiI labeling did not significantly change the size distribution and zeta-potential of exosomes as determined by DLS (the intensity-based diameter of CM-DiI labeled exosomes was 166 ± 18 nm, PDI 0.139 ± 0.03, and zeta potential −18 ± 5 mV). After an initial lag period of 4 h, the amount of Mϕ exosomes in hCMEC/D3 cells increased in a linear fashion over 48 h (Figure S2c). Incubation at 4 °C completely blocked the uptake of Mϕ exosomes (Figure S3a), suggesting that their internalization in hCMEC/D3 cells is an energy-dependent process.
To dissect the endocytosis pathways, we pre-incubated hCMEC/D3 cells with endocytosis inhibitors for 0.5 h, and then co-incubated with fresh inhibitors and Mϕ exosomes for another 4 h. We selected hyperosmolar sucrose [30], nystatin [30], and 5-(N-Ethyl-N-isopropyl) amiloride (EIPA) [31] as the inhibitors for clathrin-mediated endocytosis, caveolae-mediated endocytosis, and macropinocytosis, respectively. Each inhibitor, at non-toxic concentrations, diminished uptake of a corresponding endocytosis marker (transferrin, CTB, and dextran) (Figure S3 b, c, d, and S4). Each of these inhibitors also decreased the uptake of Mϕ exosomes (Figure S3a), suggesting that these exosomes, similar to exosomes derived from tumor or immune cells [32], utilize multiple pathways (including clathrin, caveolae, and macropinocytosis) to enter the endothelial cells. Involvement of clathrin and caveolae pathways was further supported by colocalization of Mϕ exosomes with transferrin, anti-clathrin heavy chain antibodies, CTB, and anti-caveolin 1 antibodies (Figure S5). The cellular uptake of Mϕ exosomes displayed saturation at high exosome concentrations (Figure 1c) and was inhibited by unlabeled exosomes in a concentration-dependent fashion (Figure 1d). This suggested a possibility of saturable receptor interactions and we therefore examined the molecular mechanisms involved.
ICAM-1/LFA-1 mediated uptake of Mϕ exosomes in hCMEC/D3 cells
The interactions between an integrin LFA-1 and ICAM-1 were previously implicated in the uptake of Mϕ exosomes in umbilical vein endothelial cells [33]. The LFA-1 and ICAM-1 were indeed expressed in our Mϕ exosomes and hCMEC/D3 cells, respectively (Figure 2a, b). The ICAM-1 was upregulated by treating hCMEC/D3 cells with LPS (Figure 2b), which mimics the response of brain endothelial cells to inflammation [34], and was accompanied by the increased accumulation of exosomes in the cells (Figure 2c). Moreover, each of anti-ICAM-1 or anti-LFA- 1 antibodies and their combination inhibited the exosome uptake (Figure 2d), suggesting that both LFA-1 and ICAM-1 play an important role in the uptake of Mϕ exosomes in brain endothelial cells.
Figure 2. ICAM-1/LFA-1 mediate uptake of Mϕ exosomes in hCMEC/D3 cells.

(a) Expression of LFA-1 in Raw Mϕs and Mϕ-derived exosomes by western blotting at equal protein loading. (b) Expression of ICAM-1 in hCMEC/D3 cells in response to 3 or 6 h of stimulation with LPS (100 ng/ml). (c) Uptake of exosomes in hCMEC/D3 cells with or without 3 or 6 h of LPS stimulation. (d) Effect of co-incubation with anti-ICAM-1 or anti-LFA-1 antibodies (100 µg/ml) on cell uptake of exosomes in hCMEC/D3 cells. Cell uptake was determined by flow cytometry after 4 h incubation with CM-DiI labeled exosomes (0.6×1010 exosomes/ml). Data are MFI ± SD of 5000–10000 live singlets, n = 3, * p < 0.05, ** p < 0.01 and *** p < 0.001 vs. indicated groups by one-way ANOVA and post Newman-Keuls multiple comparison test.
C-type lectin receptors mediated uptake of Mϕ exosomes in hCMEC/D3 cells
During the endocytosis inhibition studies (Figure S3), we noted that sucrose had a more profound inhibitory effect on the cell uptake of Mϕ exosomes than that of transferrin. Therefore, we hypothesized that some additional carbohydrate binding receptors may be involved in the uptake of exosomes. The possible candidates are the C-type lectin receptors that require binding of calcium for their carbohydrate-binding activity [35]. It was reported that DCs-derived exosomes are internalized into recipient cells partially by mannose/glucosamine-binding C-type lectin receptors, as was demonstrated by a blocking assay using DEC205 antibodies, the calcium chelator ethylenediaminetetraacetic acid (EDTA), and a panel of monosaccharides, including mannose and glucosamine [35]. We confirmed the presence of lectin receptors in hCMEC/D3 cells by western blotting using DEC205 antibodies (Figure 3a and Figure S6). Furthermore, the accumulation of Mϕ-derived exosomes in hCMEC/D3 cells was decreased by a panel of carbohydrates, the calcium chelator ethylene glycol-bis(2-aminoethylether)-N, N, N’, N’- tetraacetic acid (EGTA), and DEC205 antibodies (Figure 3b, c, d). Among the carbohydrates, glucosamine inhibited the uptake of exosomes at much lower concentrations, suggesting that the inhibitory effect differed from the hyperosmotic effect of sucrose that blocked endocytosis [36]. It was reported that some carbohydrates, for example glucosamine, inhibit ICAM-1 expression in rat cardiomyocytes [37] and human retinal pigment epithelial cells [38]. We tested this possibility and demonstrated that even at the highest concentrations used, glucose and glucosamine did not inhibit the expression of ICAM-1 in our cell model (Figure S7). Therefore, in addition to ICAM-1, specific carbohydrate binding receptors, especially glucosamine-binding C-type lectin receptors, mediate the accumulation of Mϕ-derived exosomes in hCMEC/D3 cells. The LPS stimulation did not enhance expression of lectin receptors in hCMEC/D3 cells (Figure S8), suggesting that, in contrast to ICAM-1, these receptors are not involved in the increased uptake of Mϕ exosomes in response to LPS stimulation.
Figure 3. C-type lectin receptors mediate uptake of Mϕ-derived exosomes in hCMEC/D3 cells.

(a) Expression of lectin receptors in hCMEC/D3 cells. Raw Mϕs and Mϕ-derived exosomes examined by western blotting at equivalent protein loading for C-type lectin receptor (DEC205) at non-reducing condition and β-actin at reducing condition. (b) Effect of carbohydrates on the uptake of exosomes. The carbohydrates were added to cells 0.5 h before and during incubation with exosomes. The data are normalized to the control cells treated with exosomes only. (c, d) Accumulation of exosomes in hCMEC/D3 cells in the presence of (c) EGTA and (d) DEC205 antibody (100 µg/ml). In (a, c, d) cells were exposed to CM-DiI labeled exosomes (0.6×1010 exosomes/ml) for 4 h and then analyzed by flow cytometry. Data are MFI ± SD of 5000–10000 live singlets, n = 3, ***p < 0.001 by one-way ANOVA and post Newman- Keuls multiple comparison tests. The used concentrations of carbohydrates and EGTA ensured at least 80% cell viability (Figure S4).
Pharmacokinetics (PK) and distribution of Mϕ exosomes in healthy mice
Next, we characterized the PK and distribution of Mϕ exosomes in healthy CD-1 mice after IV injection of 125I-radiolabeled Mϕ exosomes along with 131I-labeled bovine serum albumin (BSA) as a vascular marker [39]. The labeled exosomes and BSA were separated from free iodine in a Nap- 5 column. We characterized the size distribution of exosomes before and after elution to evaluate if the labeling procedure changes the size distribution. The z-average diameter of exosomes was decreased to 116.2 ± 6.2 nm from 147 ± 8.3 nm after elution, suggesting that the size exclusion column removed the large particles. The clearance of both Mϕ exosomes and BSA showed a two-phase decay (Figure 4a). Their PK parameters are presented in Table S1. Similar to tumor-derived exosomes [40], Mϕ exosomes mainly accumulated in the liver and spleen at 10 min, 4 h, and 24 h (Figure 4b), suggesting entrapment of exosomes in the mononuclear phagocyte system (MPS) [41] or their binding with ICAM-1[42] and lectin receptors [43] expressed in liver and spleen. Accumulation of Mϕ exosomes in the brain at 10 min and 4 h was 0.093%ID/g, 0.088%ID/g, and decreased to 0.05%ID/g at 24 h, suggesting a slow rate of clearance from the brain. Importantly, 94% of exosomes penetrated the brain accumulated in the parenchyma fraction in the capillary depletion assay (Figure S9).
Figure 4. PK and distribution of Mϕ exosomes in healthy and brain-inflamed CD-1 mice.

The brain inflammation in CD-1 mice was induced by intracranial injection of 10 µg LPS 24 h before the exosome administration. The mice were co-injected with 125I-exosomes and 131I-BSA via jugular vein. (a) Clearance of exosomes and BSA in healthy mice. (b) Distribution of exosomes in healthy mice. (c) Multiple-time regression analysis of exosomes for brain influx rate in healthy and brain-inflamed mice. Delta brain/serum ratios were calculated by subtracting the brain/serum ratios of BSA from those of exosomes to correct for vascular space [28]. (d) Distribution of exosomes at 10 min. Tissue accumulation was corrected for vascular space using BSA data. Data are means ± SEM, n = 3-6. # p < 0.05, ## p < 0.01, and ### p < 0.001 vs. indicated group by unpaired two-tailed t-test.
Neuroinflammation increased brain influx rate and brain accumulation of Mϕ exosomes
To examine whether the inflammation increases the homing of Mϕ-derived exosomes to the brain, we compared the PK of Mϕ exosomes in healthy mice and in mice with LPS- induced encephalitis, a mouse model of brain inflammation [44]. The clearances of exosomes and co-injected BSA in the brain-inflamed mice resembled those in the healthy mice (Figure S10a). Both healthy and brain-inflamed mice showed a significant net brain influx of exosomes (Figure 4c): the slopes (Ki) of the delta brain/serum ratios plotted against exposure time significantly deviated from zero (p < 0.05), thus demonstrating net brain influx. The unidirectional brain influx rate (Ki) and initial volume of brain distribution (Vi) of exosomes in the brain-inflamed mice were respectively 3- and 2-fold higher than those in the healthy mice. The plot of the delta brain/serum ratios against serum concentrations directly demonstrated increased accumulation of Mϕ-derived exosomes in the inflamed brains at similar serum concentrations of exosomes as in the healthy mice (Figure S10b). In addition, the brain influx rates of BSA in the brain-inflamed and healthy mice were comparable (p = 0.076, Figure S10c), suggesting that the increase in the brain influx of exosomes under inflammation was not caused by BBB disruption. Consistent with the increased brain influx rate, the brain accumulation of Mϕ exosomes in the inflamed brain at 10 min was 5.8 fold higher than that in the healthy mice (Figure 4d). (The percentage of injected dose at 10 min is 0.093 ± 0.02 %D/g brain in healthy brain and 0.538 ± 0.315 %ID/g brain in inflamed brain. This corresponds to the percentage of injected dose of ~0.0372 ID% in healthy brain and ~0.215 ID% in inflamed brain calculated using the average mouse brain weight of 0.4 g.) Consistent with our in vitro finding of an increased accumulation of exosomes in LPS treated brain endothelial cells in response to increased ICAM-1 expression (Figure 2c), the in vivo brain penetration of exosomes increased during inflammation when ICAM-1 expression was upregulated (Figure S11). Due to the weak fluorescence of exosomes in brain, we were not able to confirm the colocalization of exosomes with ICAM-1. However, murine ICAM-1 and LFA-1 are highly homologous to human ICAM-1 [45, 46] and LFA-1 [47], thus the interaction of exosomal LFA-1 with human ICAM-1 demonstrated in vitro using human brain endothelial cells can be indicative of the in vivo interaction of exosomal LFA-1 with murine ICAM-1. The finding of exosome brain penetration mediated by an ICAM-1/LFA-1 interaction is likely to stand independently of the species and murine strain of the exosomes, endothelial cell model, and the animal model.
The increased brain penetration of exosomes under neuroinflammation has some commonality with the increased brain infiltration of Mϕs upon inflammation [16, 48]. To confirm that exosomes can penetrate the BBB independently of brain-infiltrating immune cells as we showed in vitrowe perfused the mouse brain with exosomes in physiological buffer in healthy mice and brain inflamed mice. In this brain perfusion study, we used another brain inflammation model, which was established by three intraperitoneal injections of LPS a day before the PK study as reported previously [28]. Peripheral inflammation which may divert exosomes to inflamed peripheral organs due to upregulation of ICAM-1 is not a concern as only the brain is perfused. The brain perfusion study excludes the potential involvement of an immune cell-mediated pathway, which involves endocytosis by immune cells and subsequent brain penetration via the cell carriers. In this setting, exosomes still showed higher brain/perfusion ratio than a co-administered vascular marker BSA in both healthy mice and the brain inflammation model (delta brain/perfusion ratio is positive), suggesting that exosomes can penetrate the brain independently of infiltrating immune cells (Figure S12). In addition, the delta brain/perfusion ratio of exosomes in the brain inflammation model was significantly higher than that in the healthy mice, further supporting the aforementioned increased brain penetration of exosomes under neuroinflammation.
The mouse model of intracranial LPS-induced encephalitis also displayed significantly higher accumulation of Mϕ exosomes in the heart (1.6 fold), lungs (7.1 fold) and kidneys (3.9 fold) (Figure 4d). This could be explained by peripheral inflammation resulting from the absorption of LPS from the brain cerebrospinal fluid (CSF) to the blood [28] and/or activation of microglia and release of other inflammatory cytokines to the peripheral [49, 50]. Zhou et al. showed that intracerebroventricular injection of LPS at a lower dose (2 µg) activates microglia as well as peritoneal macrophage, clearly suggesting that a peripheral inflammation process does occur after brain local injection of LPS [50].
The inflammation-responsive brain distribution of Mϕ exosomes is remarkable and provides a strong rationale for their potential application as natural nanocarriers for neurodegenerative disorders commonly accompanied with brain inflammation.
Mϕ exosomes delivered a protein cargo to the brain
To determine whether Mϕ exosomes could deliver cargo to the brain, we loaded them with the BDNF as a model protein by simply mixing exosomes and BDNF in 10 mM phosphate buffer (pH 7.4). The binding of BDNF (isoelectric point 9.99) [51] with the negatively charged exosomes (zeta potential −18 mV) by electrostatic and polysaccharide interactions [52, 53] formed a BDNF-exosome complex (ExoBDNF), which was confirmed by demonstrating 1) that exosomes are precipitated by Protein G-magnetic beads coupled with BDNF antibodies (Figure 5a) and 2) that exosomes prevent migration of BDNF into a native polyacrylamide gel (Figure 5b). The latter indicated that Mϕ-derived exosomes can capture as much as 20 % by weight of BDNF relative to its own protein. Characterized by DLS, the z-average diameter of exosomes, ExoBDNF, and BDNF in one batch were 147.0 ± 8.3 nm, 209.5 ± 4.7 nm, and 36.1 ± 13.5 nm. The increase in diameter, the pull-down assay, and the native electrophoresis all support that BDNF can associate with exosomes by simple mixing. Interestingly, ExoBDNF released BDNF upon binding with the BDNF receptor TrkB, and the released neurotrophin was able to induce phosphorylation of receptor and downstream Akt signaling in NIH-3T3-TrKB cells (Figure S13).
Figure 5. Formation and brain delivery of ExoBDNF complex.

(a) ExoBDNF were isolated using protein G magnetic beads modified with BDNF-specific antibodies. The presence of LAMP 2 in the magnetic bead-separated fractions indicated that the exosomes were captured on the beads. A control group of exosomes without BDNF treatment was used to account for nonspecific binding. (b) Native PAGE of the mixtures of Mϕ exosomes and BDNF at different protein ratios. Exosomes prevented the neurotrophic factor migration in the gel toward the cathode up to a BDNF: exosomal protein weight ratio of 1:5. (c) Healthy mice were co-injected with 131I-BSA and 125I-BDNF with or without Mϕ exosomes via jugular vein. The brain/serum ratios at time points of 1, 2, 3, 4, 5, 6, 7.5, 10, 15, 20, 30 min were averaged. ### p < 0.001 by unpaired two-tailed t-test. (d) Multiple-time regression analysis of co-injected BSA in healthy mice. Both slopes are comparable to 0 (p=0.064 for BSA and p=0.09 for BSA + Exosomes). (e) Brain accumulation of naked BDNF or BDNF in ExoBDNF in healthy or brain-inflamed mice at 10 min. Data are means ± SEM, n = 4, ** p < 0.01 indicated group by one-way ANOVA and post Newman-Keuls multiple comparison test. (f) Distribution of BDNF or BDNF in ExoBDNF in brain parenchyma at 10 min in healthy mice determined by capillary depletion assay. Data are means ± SEM, n = 5, && p < 0.01 and &&& p < 0.001 by paired two-tailed t-test, ### p < 0.001 by unpaired two-tailed t-test.
We further determined whether Mϕ-derived exosomes facilitate BDNF transfer to the brain. In this experiment, we co-injected 131I- BSA and 125I-BDNF with or without exosomes into the jugular vein of healthy CD-1 mice. The brain/serum ratios of BDNF loaded onto exosomes were significantly higher than those BDNF alone (Figure 5c). To the contrary, the brain influx rates of co-injected BSA in both groups were comparable and did not differ from 0 (Figure 5d). The brain/serum ratios of BSA in both groups were similar as well (Figure S14). These data also indirectly suggested that BDNF at least in part remained with exosomes after administration in vivo. We also found that ExoBDNF complex was stable in the presence of NaCl up to the highest tested concentration of 1 M (Figure S15). We further compared the brain accumulation of naked and exosome-formulated BDNF in healthy and brain-inflamed mice (Figure 5e). The brain accumulation of exosome-formulated BDNF in healthy mice was slightly but not significantly increased compared to BDNF alone (p = 0.63). Brain inflammation resulted in a trend to increase the brain accumulation of free BDNF but the difference was not significant when compared to the healthy mice (p = 0.11). In contrast, accumulation of exosome-formulated BDNF (ExoBDNF) in the brain-inflamed mice was significantly increased compared to the same formulation in the healthy animals (3.6 fold). Moreover, the brain accumulation of BDNF with this formulation was also superior to that of the BDNF alone in the inflamed brain (2.2 fold). Importantly in the capillary depletion experiment, 89% and 95% of the BDNF accumulated in the brain distributed to the brain parenchyma in the BDNF alone group and ExoBDNF group (Figure 5f), suggesting that both BDNF alone and BDNF in ExoBDNF group can penetrate the BBB. In addition, brain accumulation of ExoBDNF in the parenchyma fraction was significantly higher than BDNF alone in healthy mice. Note that we subtracted the serum contamination from both capillary and parenchyma fractions using co-injected BSA as in the tissue distribution study, however, the percentages of injected dose accumulated in parenchyma fractions (Figure 5f) were higher than the percentages in whole brain (Figure 5e), which may suggest mass transfer of iodinated BDNF from serum fractions to parenchyma fractions during homogenization in the capillary depletion experiment likely due to enhanced adsorption to distorted cell membrane and/or extracellular matrix in brain parenchyma that is otherwise separated from blood by the brain endothelium. Note that BDNF can bind to extracellular matrix [52, 53]. Thus, we calculated the percentage of injected dose per gram of brain in the parenchyma fractions by subtracting the value in the capillary fractions (in Figure 5f, 0.014%ID/g for both groups) from the value in total brain (in Figure 5e) as a rough estimation, which yielded 0.024%ID/g for BDNF alone group and 0.038%ID/g for ExoBDNF group. This calculation indicated that 63.2% and 73.1% of iodinated BDNF in brain distributed to the parenchyma in BDNF and ExoBDNF group. Nonetheless, the increased whole brain accumulation of BDNF upon mixing with exosomes suggests that even though the cargo was loaded onto the surface of exosomes, exosomes still preserve their brain penetration property.
CONCLUSION AND FUTURE DIRECTIONS
To summarize, we isolated exosomes from Raw Mϕs and characterized their interaction with immortalized human brain endothelial cells (hCMEC/D3 cells) in vitroand their PK in mice with or without brain inflammation. In vitro, we first showed that hCMEC/D3 cells internalized Mϕ exosomes in a saturable manner using multiple pathways (clathrin-/caveolae-mediated endocytosis, and macropinocytosis), implying a possible receptor-mediated pathway. We then confirmed that Mϕ-derived exosomes inherited LFA-1 from their parental cells, a protein that interacts with endothelial ICAM-1 and mediates the lateral migration and diapedesis of Mϕs across the BBB [18]. More importantly, the LFA-1 and ICAM-1 also mediated cellular uptake of Mϕ-derived exosomes in hCMEC/D3 cells. The cellular uptake of exosomes increased significantly under inflammation conditions in response to overexpression of ICAM-1 receptors on endothelial cells, resembling the increased diapedesis of Mϕs across the BBB under inflammation. We also identified that cellular uptake of Mϕ-derived exosomes in hCMEC/D3 cells involves C-type lectin receptors, because various carbohydrates, a specific antibody, and calcium chelators blocked the accumulation. However, the C-type lectin receptors were not responsible for the increased cellular uptake under inflammation. In vivo, we demonstrated that IV administered Mϕ-derived exosomes penetrated the BBB to the brain parenchyma in healthy mice. Although these exosomes mainly distributed in the liver and spleen, they also accumulated in brain at a level as high as 0.093 ID%/g, and were slowly cleared from the brain. Consistent with increased cellular uptake under inflammation, Mϕ-derived exosomes entered the brain 3.1-fold faster and accumulated 5.8-fold greater in the inflamed brain than in the healthy brain, without increased accumulation in liver and spleen. We also demonstrated that Mϕ-derived exosomes can enter the brain independently without the help from brain infiltrating immune cells and that the increased accumulation in inflamed brain was due to enhanced exosome-brain endothelium interaction. Strategies to reduce uptake by the MPS organs such as PEGylation can be explored in the future to shield exosomes and extend circulation time in order to further improve brain accumulation. Furthermore, we loaded a model cargo protein radiolabeled BDNF onto Mϕ exosomes and showed that they successfully delivered BDNF to the brain parenchyma in healthy mice, and even at greater extent to the brain with inflammation (0.19 ID%/g).
Noteworthy, our findings contradict with the results reported by Hwang et al [54]. This report indicated that technetium-radiolabeled Mϕ exosomes and the Mϕ extruded membrane vesicles showed no detectable signal in the brain region of BALB/c mice by single photon emission computed tomography (SPECT) [54]. This discrepancy can be possibly explained by the higher sensitivity of iodine detection method and difference in size distribution. Compared to exosomes utilized in this investigation (130 nm), the Mϕ-derived extruded membrane vesicles had larger average diameter (218 nm), which may result in increased sequestration in peripheral organs causing lower brain transport. Besides, our study was consistent with the study showing that fluorescently labeled exosomes released from autologous dendritic cells was detectable in mouse brains 24 h after IV injection [55], and other recent study showing that HEK293T exosomes can penetrate the Transwell model of BBB transcellularly under inflamed conditions (TNF-α activated) [56].
Although we did not specifically evaluate the blood-CSF barrier (choroid plexus or ependymal barrier), our in vivo model can reflect an influence of that barrier. The vascular barrier is usually thought to be the preferred route for drug delivery because of its proximity to the neurons and the blood-CSF barrier is generally assumed to primarily inventory the vascular BBB [57–59], however, such a view can underestimate the importance of the choroid plexus. We showed in vitro that exosomes can be internalized into human brain endothelial cells. Therefore, we conclude that exosomes can penetrate the vascular barrier, but can make no conclusions regarding their ability to penetrate the blood-CSF barrier.
The Raw 264.7 Mϕ exosomes, which originate from BALB/c mice, were tested in a different murine strain (CD-1 mice). Possibly, these exosomes are cleared faster in CD-1 mice than in autologous BALB/c mice due to immune response. However, recent publications demonstrate that exosomes from different species (human, rat, or mouse) and exosomes from different mouse strains all show rapid clearance and sequestration by MPS organs such as liver, spleen, and lung in mice [40, 54, 55, 60–62]. These data indicate that exosomes may not readily avoid the MPS sequestration as assumed from their wide existence in biological fluids and their “self” nature. If accelerated clearance in CD-1 mice did occur, we would expect higher brain uptake of these exosomes in BALB/c mice as a result of longer exposure to the BBB. As gender difference in inflammatory response [63] and in the brain transduction of adeno-associated virus serotype 9 has been reported, it would be interesting to investigate the potential gender influence in brain penetration of exosomes.
The ability of Mϕ exosomes to cross the BBB and ferry a cargo to the brain parenchyma especially under brain inflammation, a common condition associated with many CNS disorders [65], can be explored to deliver various disease-modulating proteins to the brain. The dose reaching the brain is generally low when compared to peripheral organs and tumor [66]. The brain bioavailability is often below 0.2 %ID/g brain and can be as low as 0.01 %ID/g brain [66]. Considering the low brain bioavailability of exosomes under neuroinflammation, a choice of a highly potent therapeutic protein is beneficial. For instance, although the brain bioavailability of an anti-transferrin receptor/β-secretase bispecific antibody is less than 1 %ID/g brain, this bispecific antibody efficiently reduces the brain burden of amyloid β peptides [67]. Moreover one could expect that exosomes are safer than synthetic drug delivery systems due to the biological origin of exosomes. Another potential benefit of natural exosomes over artificial targeting drug carriers is the ease of manufacturing and scalability. One can engineer the genetically modified parent cells to continuously secret exosomes containing the desired therapeutic proteins. In principle, one could also implant the engineered parent cells in vivo to reduce the injection frequency. Moreover, exosomes mediated cargo transfer from transplanted Mϕs to contiguous brain resident cells such as endothelial cells and neurons [12, 14, 19, 20], and facilitated spreading of disease-relevant proteins inside the brain and into CSF [21, 22], suggesting exosomes may facilitate drug distribution in the brain upon penetrating the BBB.
Taken together, Mϕ-derived exosomes are promising nanocarriers for brain delivery of therapeutic proteins. These findings are of interest to basic understanding of possible role of exosomes in transfer and distribution of exogenous biological molecules as well as of significance to practical applications in drug delivery to CNS.
Supplementary Material
Acknowledgments
We gratefully acknowledge Xiang Yi for her expertise in PK study. We also gratefully acknowledge Robert Bagnell and Victoria J. Madden from UNC microscopy services laboratory and Evan Trudeau from UNC flow cytometry core facility for their expertise. This work was supported in parts by the National Institutes of Health awards 1R21 NS088152 and RO1 NS36229, the Rettsyndrome.org HeART Award #3112, and the Carolina Partnership, a strategic partnership between the UNC Eshelman School of Pharmacy and The University Cancer Research Fund. The UNC flow cytometry core facility is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST
The authors declare no other competing financial interest, except for the patent(s) filed by their institution on behalf of several co-authors (A. K., D. Y., E. B., and M. H.).
AUTHOR CONTRIBUTIONS
A. K. and D. Y. conceived the entire study and wrote the paper. D. Y. carried out the experiments in the paper. Y. Z. took part in PK experiments. W. B. designed PK study and assisted in data interpretation. M. H. and E. B. assisted in exosome experimental protocols and interpretation of the results.
APPENDIX A. SUPPLEMENTARY DATA
Supplementary data related to this article can be found at
References
- 1.Zhuang X, Xiang X, Grizzle W, Sun D, Zhang S, Axtell RC, Ju S, Mu J, Zhang L, Steinman L, Miller D, Zhang HG. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol Ther. 2011;19:1769–1779. doi: 10.1038/mt.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haney MJ, Klyachko NL, Zhaoa YL, Gupta R, Plotnikova EG, He ZJ, Patel T, Piroyan A, Sokolsky M, Kabanov AV, Batrakova EV. Exosomes as drug delivery vehicles for Parkinson's disease therapy. Journal of Controlled Release. 2015;207:18–30. doi: 10.1016/j.jconrel.2015.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Djupesland PG, Messina JC, Mahmoud RA. The nasal approach to delivering treatment for brain diseases: an anatomic, physiologic, and delivery technology overview. Ther Deliv. 2014;5:709–733. doi: 10.4155/tde.14.41. [DOI] [PubMed] [Google Scholar]
- 4.Djupesland PG. Nasal drug delivery devices: characteristics and performance in a clinical perspective-a review. Drug Deliv Transl Res. 2013;3:42–62. doi: 10.1007/s13346-012-0108-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1959–1972. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pardridge WM. The Blood-Brain Barrier: Bottleneck in Brain Drug Development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Li D, Liu Z, Zhou Y, Chu D, Li X, Jiang X, Hou D, Chen X, Chen Y, Yang Z, Jin L, Jiang W, Tian C, Zhou G, Zen K, Zhang J, Zhang Y, Li J, Zhang CY. Targeted exosome-mediated delivery of opioid receptor Mu siRNA for the treatment of morphine relapse. Sci Rep. 2015;5:17543. doi: 10.1038/srep17543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. 2014;14:195–208. doi: 10.1038/nri3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 11.Yang TZ, Martin P, Fogarty B, Brown A, Schurman K, Phipps R, Yin VP, Lockman P, Bai SH. Exosome Delivered Anticancer Drugs Across the Blood-Brain Barrier for Brain Cancer Therapy in Danio Rerio. Pharm Res-Dordr. 2015;32:2003–2014. doi: 10.1007/s11095-014-1593-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haney MJ, Zhao Y, Harrison EB, Mahajan V, Ahmed S, He Z, Suresh P, Hingtgen SD, Klyachko NL, Mosley RL, Gendelman HE, Kabanov AV, Batrakova EV. Specific transfection of inflamed brain by macrophages: a new therapeutic strategy for neurodegenerative diseases. PLoS One. 2013;8:e61852. doi: 10.1371/journal.pone.0061852. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Brynskikh AM, Zhao Y, Mosley RL, Li S, Boska MD, Klyachko NL, Kabanov AV, Gendelman HE, Batrakova EV. Macrophage delivery of therapeutic nanozymes in a murine model of Parkinson's disease. Nanomedicine (Lond) 2010;5:379–396. doi: 10.2217/nnm.10.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Y, Haney MJ, Gupta R, Bohnsack JP, He Z, Kabanov AV, Batrakova EV. GDNF-transfected macrophages produce potent neuroprotective effects in Parkinson's disease mouse model. PLoS One. 2014;9:e106867. doi: 10.1371/journal.pone.0106867. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–1596. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dou H, Grotepas CB, McMillan JM, Destache CJ, Chaubal M, Werling J, Kipp J, Rabinow B, Gendelman HE. Macrophage delivery of nanoformulated antiretroviral drug to the brain in a murine model of neuroAIDS. J Immunol. 2009;183:661–669. doi: 10.4049/jimmunol.0900274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller WA. Getting leukocytes to the site of inflammation. Vet Pathol. 2013;50:7–22. doi: 10.1177/0300985812469883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haney MJ, Suresh P, Zhao Y, Kanmogne GD, Kadiu I, Sokolsky-Papkov M, Klyachko NL, Mosley RL, Kabanov AV, Gendelman HE, Batrakova EV. Blood-borne macrophage-neural cell interactions hitchhike on endosome networks for cell-based nanozyme brain delivery. Nanomedicine (Lond) 2012;7:815–833. doi: 10.2217/nnm.11.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haney MJ, Zhao Y, Li S, Higginbotham SM, Booth SL, Han HY, Vetro JA, Mosley RL, Kabanov AV, Gendelman HE, Batrakova EV. Cell-mediated transfer of catalase nanoparticles from macrophages to brain endothelial, glial and neuronal cells. Nanomedicine (Lond) 2011;6:1215–1230. doi: 10.2217/nnm.11.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fais S, Logozzi M, Lugini L, Federici C, Azzarito T, Zarovni N, Chiesi A. Exosomes: the ideal nanovectors for biodelivery. Biological chemistry. 2013;394:1–15. doi: 10.1515/hsz-2012-0236. [DOI] [PubMed] [Google Scholar]
- 22.Gupta A, Pulliam L. Exosomes as mediators of neuroinflammation. Journal of Neuroinflammation. 2014;11:68. doi: 10.1186/1742-2094-11-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current protocols in cell biology / editorial board, Juan S. Bonifacino … [et al.], Chapter 3. 2006 doi: 10.1002/0471143030.cb0322s30. Unit 3 22. [DOI] [PubMed] [Google Scholar]
- 24.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc-Oxford. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 26.Yi X, Yuan D, Farr SA, Banks WA, Poon CD, Kabanov AV. Pluronic modified leptin with increased systemic circulation, brain uptake and efficacy for treatment of obesity. Journal of controlled release : official journal of the Controlled Release Society. 2014 doi: 10.1016/j.jconrel.2014.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patlak CS, Blasberg RG, Fenstermacher JD. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab. 1983;3:1–7. doi: 10.1038/jcbfm.1983.1. [DOI] [PubMed] [Google Scholar]
- 28.Banks WA, Robinson SM. Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav Immun. 2010;24:102–109. doi: 10.1016/j.bbi.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh PP, Smith VL, Karakousis PC, Schorey JS. Exosomes isolated from mycobacteria-infected mice or cultured macrophages can recruit and activate immune cells in vitro and in vivo. J Immunol. 2012;189:777–785. doi: 10.4049/jimmunol.1103638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen CL, Hou WH, Liu IH, Hsiao G, Huang SS, Huang JS. Inhibitors of clathrin-dependent endocytosis enhance TGFbeta signaling and responses. J Cell Sci. 2009;122:1863–1871. doi: 10.1242/jcs.038729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng D, Zhao WL, Ye YY, Bai XC, Liu RQ, Chang LF, Zhou Q, Sui SF. Cellular Internalization of Exosomes Occurs Through Phagocytosis, Traffic. 2010;11:675–687. doi: 10.1111/j.1600-0854.2010.01041.x. [DOI] [PubMed] [Google Scholar]
- 32.Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014;3 doi: 10.3402/jev.v3.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jang SC, Kim OY, Yoon CM, Choi DS, Roh TY, Park J, Nilsson J, Lotvall J, Kim YK, Gho YS. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS nano. 2013;7:7698–7710. doi: 10.1021/nn402232g. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Ye L, Wang X, Liu J, Wang Y, Zhou Y, Ho W. (-)-Epigallocatechin gallate inhibits endotoxin-induced expression of inflammatory cytokines in human cerebral microvascular endothelial cells. J Neuroinflammation. 2012;9:161. doi: 10.1186/1742-2094-9-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hao S, Bai O, Li F, Yuan J, Laferte S, Xiang J. Mature dendritic cells pulsed with exosomes stimulate efficient cytotoxic T-lymphocyte responses and antitumour immunity. Immunology. 2007;120:90–102. doi: 10.1111/j.1365-2567.2006.02483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oka JA, Christensen MD, Weigel PH. Hyperosmolarity inhibits galactosyl receptor-mediated but not fluid phase endocytosis in isolated rat hepatocytes. J Biol Chem. 1989;264:12016–12024. [PubMed] [Google Scholar]
- 37.Zou L, Yang S, Champattanachai V, Hu S, Chaudry IH, Marchase RB, Chatham JC. Glucosamine improves cardiac function following trauma-hemorrhage by increased protein O-GlcNAcylation and attenuation of NF-{kappa}B signaling, American journal of physiology. Heart and circulatory physiology. 2009;296:H515–523. doi: 10.1152/ajpheart.01025.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen CL, Liang CM, Chen YH, Tai MC, Lu DW, Chen JT. Glucosamine modulates TNF-alpha-induced ICAM-1 expression and function through O-linked and N-linked glycosylation in human retinal pigment epithelial cells. Investigative ophthalmology & visual science. 2012;53:2281–2291. doi: 10.1167/iovs.11-9291. [DOI] [PubMed] [Google Scholar]
- 39.Banks WA, Niehoff ML, Ponzio NM, Erickson MA, Zalcman SS. Pharmacokinetics and modeling of immune cell trafficking: quantifying differential influences of target tissues versus lymphocytes in SJL and lipopolysaccharide-treated mice. J Neuroinflammation. 2012;9:231. doi: 10.1186/1742-2094-9-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smyth T, Kullberg M, Malik N, Smith-Jones P, Graner MW, Anchordoquy TJ. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J Control Release. 2015;199:145–155. doi: 10.1016/j.jconrel.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hume DA. The mononuclear phagocyte system. Curr Opin Immunol. 2006;18:49–53. doi: 10.1016/j.coi.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 42.Aoudjit F, Potworowski EF, Springer TA, St-Pierre Y. Protection from lymphoma cell metastasis in ICAM-1 mutant mice: a posthoming event. J Immunol. 1998;161:2333–2338. [PubMed] [Google Scholar]
- 43.Vasta GR, Ahmed H. Animal lectins : a functional view. CRC Press; Boca Raton: 2009. [Google Scholar]
- 44.Zhao Y, Haney MJ, Mahajan V, Reiner BC, Dunaevsky A, Mosley RL, Kabanov AV, Gendelman HE, Batrakova EV. Active Targeted Macrophage-mediated Delivery of Catalase to Affected Brain Regions in Models of Parkinson's Disease. J Nanomed Nanotechnol. 2011;S4 doi: 10.4172/2157-7439.S4-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collins T. Leukocyte recruitment, endothelial cell adhesion molecules, and transcriptional control : insights for drug discovery. Kluwer Academic Publishers; Boston: 2001. [Google Scholar]
- 46.Horley KJ, Carpenito C, Baker B, Takei F. Molecular cloning of murine intercellular adhesion molecule (ICAM-1) EMBO J. 1989;8:2889–2896. doi: 10.1002/j.1460-2075.1989.tb08437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Li D, Nurieva R, Yang J, Sen M, Carreno R, Lu S, McIntyre BW, Molldrem JJ, Legge GB, Ma Q. LFA-1 affinity regulation is necessary for the activation and proliferation of naive T cells. J Biol Chem. 2009;284:12645–12653. doi: 10.1074/jbc.M807207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carman CV. Mechanisms for transcellular diapedesis: probing and pathfinding by 'invadosome-like protrusions'. J Cell Sci. 2009;122:3025–3035. doi: 10.1242/jcs.047522. [DOI] [PubMed] [Google Scholar]
- 49.Dickens AM, Vainio S, Marjamaki P, Johansson J, Lehtiniemi P, Rokka J, Rinne J, Solin O, Haaparanta-Solin M, Jones PA, Trigg W, Anthony DC, Airas L. Detection of microglial activation in an acute model of neuroinflammation using PET and radiotracers 11C-(R)-PK11195 and 18F-GE-180. J Nucl Med. 2014;55:466–472. doi: 10.2967/jnumed.113.125625. [DOI] [PubMed] [Google Scholar]
- 50.Zhou H, Lapointe BM, Clark SR, Zbytnuik L, Kubes P. A requirement for microglial TLR4 in leukocyte recruitment into brain in response to lipopolysaccharide. J Immunol. 2006;177:8103–8110. doi: 10.4049/jimmunol.177.11.8103. [DOI] [PubMed] [Google Scholar]
- 51.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 52.Kanato Y, Kitajima K, Sato C. Direct binding of polysialic acid to a brain-derived neurotrophic factor depends on the degree of polymerization. Glycobiology. 2008;18:1044–1053. doi: 10.1093/glycob/cwn084. [DOI] [PubMed] [Google Scholar]
- 53.Kanato Y, Ono S, Kitajima K, Sato C. Complex formation of a brain-derived neurotrophic factor and glycosaminoglycans. Biosci Biotechnol Biochem. 2009;73:2735–2741. doi: 10.1271/bbb.90637. [DOI] [PubMed] [Google Scholar]
- 54.Hwang do W, Choi H, Jang SC, Yoo MY, Park JY, Choi NE, Oh HJ, Ha S, Lee YS, Jeong JM, Gho YS, Lee DS. Noninvasive imaging of radiolabeled exosome-mimetic nanovesicle using (99m)Tc-HMPAO. Sci Rep. 2015;5:15636. doi: 10.1038/srep15636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wiklander OP, Nordin JZ, O'Loughlin A, Gustafsson Y, Corso G, Mager I, Vader P, Lee Y, Sork H, Seow Y, Heldring N, Alvarez-Erviti L, Smith CI, Le Blanc K, Macchiarini P, Jungebluth P, Wood MJ, Andaloussi SE. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles. 2015;4:26316. doi: 10.3402/jev.v4.26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen CC, Liu L, Ma F, Wong CW, Guo XE, Chacko JV, Farhoodi HP, Zhang SX, Zimak J, Segaliny A, Riazifar M, Pham V, Digman MA, Pone EJ, Zhao W. Elucidation of Exosome Migration across the Blood-Brain Barrier Model In Vitro. Cell Mol Bioeng. 2016;9:509–529. doi: 10.1007/s12195-016-0458-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rip J, Schenk GJ, de Boer AG. Differential receptor-mediated drug targeting to the diseased brain. Expert Opin Drug Deliv. 2009;6:227–237. doi: 10.1517/17425240902806383. [DOI] [PubMed] [Google Scholar]
- 58.Banks WA. Developing drugs that can cross the blood-brain barrier: applications to Alzheimer's disease. BMC Neuroscience. 2008;9:S2–S2. doi: 10.1186/1471-2202-9-S3-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen Y, Liu L. Modern methods for delivery of drugs across the blood–brain barrier. Advanced Drug Delivery Reviews. 2012;64:640–665. doi: 10.1016/j.addr.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 60.Takahashi Y, Nishikawa M, Shinotsuka H, Matsui Y, Ohara S, Imai T, Takakura Y. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J Biotechnol. 2013;165:77–84. doi: 10.1016/j.jbiotec.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 61.Imai T, Takahashi Y, Nishikawa M, Kato K, Morishita M, Yamashita T, Matsumoto A, Charoenviriyakul C, Takakura Y. Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. J Extracell Vesicles. 2015;4:26238. doi: 10.3402/jev.v4.26238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morishita M, Takahashi Y, Nishikawa M, Sano K, Kato K, Yamashita T, Imai T, Saji H, Takakura Y. Quantitative analysis of tissue distribution of the B16BL6-derived exosomes using a streptavidin-lactadherin fusion protein and iodine-125-labeled biotin derivative after intravenous injection in mice. J Pharm Sci. 2015;104:705–713. doi: 10.1002/jps.24251. [DOI] [PubMed] [Google Scholar]
- 63.Casimir GJ, Duchateau J. Gender differences in inflammatory processes could explain poorer prognosis for males. J Clin Microbiol. 2011;49:478. doi: 10.1128/JCM.02096-10. author reply 478-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maguire CA, Crommentuijn MH, Mu D, Hudry E, Serrano-Pozo A, Hyman BT, Tannous BA. Mouse gender influences brain transduction by intravascularly administered AAV9. Mol Ther. 2013;21:1470–1471. doi: 10.1038/mt.2013.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Batrakova EV, Kabanov AV. Cell-mediated drug delivery to the brain. Journal of Drug Delivery Science and Technology. 2013;23:419–433. [Google Scholar]
- 66.van Rooy I, Cakir-Tascioglu S, Hennink WE, Storm G, Schiffelers RM, Mastrobattista E. In vivo methods to study uptake of nanoparticles into the brain. Pharm Res. 2011;28:456–471. doi: 10.1007/s11095-010-0291-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y, Atwal J, Elliott JM, Prabhu S, Watts RJ, Dennis MS. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med. 2011;3:84ra44. doi: 10.1126/scitranslmed.3002230. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.