

Abstract
Tumor-associated fibrosis is characterized by unchecked pro-fibrotic and pro-inflammatory signaling. The components of fibrosis including significant numbers of cancer-associated fibroblasts, dense collagen deposition, and extracellular matrix stiffness, are well appreciated regulators of tumor progression but may also be critical regulators of immune surveillance. While this suggests that the efficacy of immunotherapy may be limited in highly fibrotic cancers like pancreas, it also suggests a therapeutic opportunity to target fibrosis in these tumor types to reawaken anti-tumor immunity. This review discusses the mechanisms by which fibrosis might subvert tumor immunity and how to overcome these mechanisms.
Keywords: Fibrosis, Extracellular matrix, Tumor microenvironment, Tumor immunity, Pancreas cancer, Regulatory myeloid suppressor cells
Introduction
It is becoming clear that tumor immunity and the response to immunotherapy are affected by many factors besides the antigenicity and/or mutational burden of cancer. This wide array of parameters includes the competency of the host immune system, the origin and current tissue location of the malignant lesion, the underlying genetic and epigenetic programs in the malignant cells, the metabolic profile of the target lesion, and the composition of the tumor-associated stroma. Many of these parameters are affected by fundamental biological processes that are commonly activated in response to tissue injury but are co-opted by the growing malignancy. One such co-opted biological process that affects tumor immunity is pathological fibrosis.
Fibrotic responses to malignancies are common in many cancers, and are important features in cancers such as pancreatic adenocarcinoma (PDAC), esophageal squamous cell carcinoma, prostate cancer, colon cancer, and some subsets of breast cancer (Fig. 1). Although this characteristic is well known, how fibrosis affects tumor immunity and/or the response to immunotherapy is unclear. Part of this challenge is due to the organ-specific complexity involved in fibrosis. Important players in fibrosis include diverse subsets of activated fibroblasts and components of the extracellular matrix (ECM). This complex milieu is further complicated by the diverse outcomes possible from even the simplest evaluation of a single ECM component. For example, Type I collagen can be integrated into diverse superstructures, has ligand-dependent activity on a significant number of receptors including integrins, and has the ability to regulate the bioavailability of growth factors and cytokines based on physical stiffness or organization. All these parameters, even for a single molecule, can exert differential effects on tumor immunity. Based upon such complexity, in the following review we will emphasize some of the general processes by which fibrotic tumor microenvironments (TME) might impair tumor immunity and will discuss how these potential barriers can be targeted for possible therapeutic benefit.
Fig. 1.

Fibrosis in cancer. Representative trichrome (blue) staining for collagen in normal pancreas and different cancerous tissues. Magnification ×20
The role of activated fibroblasts in tumor immunity
Fibrosis is characterized by the presence of excess of fibrous connective tissue in an organ, often involving excessive deposition of various forms of extracellular collagen [1]. Fibroblasts have been reported to be major drivers of fibrotic responses. They often deposit most of the extracellular collagen and in many cases also secrete pro-inflammatory mediators necessary to sustain fibrotic pathology [2]. The heterogeneity of fibroblasts depends on their dynamic status in cancer. Many different markers can be used to identify activated fibroblasts such as fibroblast-specific protein 1 (FSP1/S100A4), vimentin, α-smooth muscle actin (α-SMA), fibroblast activation protein (FAP), PDGF receptor-α (PDGFR-α), desmin, and discoidin domain-containing receptor 2 (DDR2) [3, 4]. It is important to emphasize that none of these markers are exclusive for activated fibroblasts [4], a fact that has made their study more complicated. The heterogeneity of fibroblasts also depends on their tissue of origin. Activated fibroblasts are reported to arise from bone marrow-derived precursors, mesenchymal stem cells (MSCs), endothelial cells, liver and pancreas stellate cells, resting tissue fibroblasts and possibly from certain types of epithelial cell [5]. While an overwhelming abundance of literature supports a tumor-promoting role of cancer-associated fibroblasts (CAFs), some studies also suggest that certain fibroblast subsets may have tumor-restraining activity. For example, the deletion of α-SMA+ myofibroblasts in pancreatic cancer leads to invasive and undifferentiated tumors [6, 7]. For simplicity, we will focus on tumor-promoting CAF subsets unless otherwise stated. When compared with homeostatic tissue fibroblasts, CAFs typically exhibit higher proliferation rates, can express “activation” markers (such as FAP or FSP1), and can activate pro-inflammatory pathways including signal transducer and activator of transcription 3 (STAT3) and NF-κB, which can then induce pro-inflammatory cytokine production, ECM deposition, and ECM-modifying enzyme secretion. CAFs themselves can be activated by tumor cells and immune cells during tumor progression. For instance, IL-1β secreted from resident immune cells can reprogram normal fibroblasts into pro-inflammatory CAFs. These activated CAFs further mediate tumor-enhancing inflammation by recruiting TAMs and promoting angiogenesis [8]. Furthermore, cancer cells can produce mitogenic and fibrogenic factors which promote the activation of pancreatic stellate cells (PSCs), such as platelet-derived growth factor (PDGF), transforming growth factor β (TGFβ), and sonic hedgehog (SHH) [9]. While CAFs are present in non-fibrotic tumors as well, high CAF density concomitant with extensive ECM deposition is characteristic of fibrotic tumors. Pancreatic cancer is perhaps the “flagship” for tumor-associated fibrosis, with an estimated 60–70% of tumor tissue composed of desmoplastic stroma characterized by extensive extracellular collagen deposition and activated CAFs (Fig. 1) [10]. The role of fibrosis in tumor cell proliferation, invasion, and metastasis has been well-defined. However, our understanding of how this level of fibrosis affects tumor immunity is currently limited.
CAFs as regulators of T cell function
CAFs can suppress cytotoxic T lymphocyte (CTL)-driven antitumor immunity by multiple mechanisms (Fig. 2). CAFs have been shown to be suppressive, and using in vitro assays, some of these subsets like MSCs have been shown to exhibit direct immunosuppressive activities in a manner similar to regulatory T cells (Treg) [11, 12]. CAFs can impair CTL activation, cytokine production, and cytotoxicity through the production of soluble factors such as IL10, TGF-β, or VEGF, as well as through metabolic reprograming via prostaglandin E2 (PGE2), indoleamine 2,3-dioxygenase (IDO), and arginase [2, 13] or expression of T cell checkpoint molecules like programmed death-ligand 1 (PD-L1) [2, 13]. Additionally, CAFs can produce factors like CXCL12, which can limit T cell movement and/or recruitment into tumor tissue [14]. Thus, when CAFs are highly abundant, as in PDAC, their combined action can slow T cell movement into tumor tissue while simultaneously impairing CTL activity and survival through multiple immunosuppressive processes. Although these processes are probably important for successful wound healing and/or protective scar tissue formation, they likely drive immune suppression in various types of fibrotic tumors. Consistent with this possibility, depletion of CAFs by genetic ablation has been reported to restore antitumor immunity and potentiate immunotherapy in mouse models of cancer [14–17].
Fig. 2.
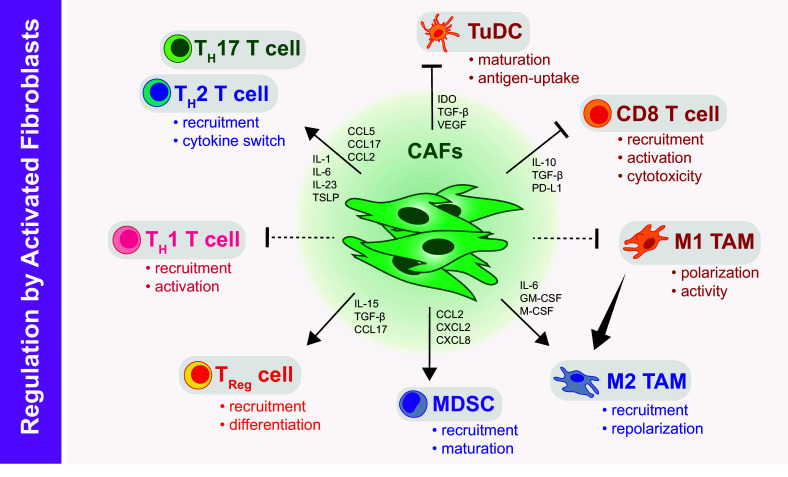
The impact of CAFs on immune cell regulation and function. CAF subsets can impair CD8+ CTL activation, cytokine production, and cytotoxicity; CAFs also affect CD4+ T cells to favor tumor-promoting TH2 and TH17 responses through production of chemoattractants and polarizing cytokines. Likewise, CAFs can induce suppressive Treg differentiation and recruitment. CAFs can limit maturation of myeloid derived suppressors (MDSCs), putatively suppress M1-like macrophage activity, and/or switch monocyte differentiation programs towards a tumor-promoting M2 macrophage lineage. Also, CAFs can influence maturation and activity of DCs in the tumor microenvironment (TuDCs) to undermine CD8 T cell activation and function
In addition to directly inactivating CTL responses, CAFs can also suppress T cell immunity by affecting T helper (TH) cell responses. Pathological fibrosis has been associated with both TH2 and TH17 responses. These responses are thought to be important in disease pathology. Notably, the TH2 cytokines IL-4 and IL-13 can induce both fibroblast proliferation and fibroblast ECM deposition, while TH17 cytokines may regulate associated inflammatory pathways during pulmonary fibrosis [18]. Pathological fibrosis can further enhance TH effects via a feed-forward loop. In tumor-associated fibrosis, the presence of activated CAFs produces high levels of TH cell chemoattractants such as CCL5, CCL17, CCL2, and polarizing cytokines such as IL-1, IL-6, IL-23, IL-13, and thymic stromal lymphopoietin (TSLP), which can favor tumor-promoting TH2 and TH17 responses over tumor protective TH1 immunity [19–23]. In addition to affecting effector cells, CAF production of CCL17, IL-15, and TGF-β can promote Treg recruitment and differentiation. Consistent with this possibility, depletion of FAP+ CAFs in mammary tumors has been reported to shift the balance towards a TH1 phenotype with enhanced CTL recruitment and activation, reduction of Treg cells, and other suppressive cells in mammary tumor models [17].
CAFs as regulators of immunosuppressive myeloid cells
Activated CAFs in the tumor microenvironment can also mediate immune suppression by modulating myeloid cells (Fig. 2), including tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and dendritic cells (DCs). TAMs derive from either circulating monocyte or yolk sac-derived tissue resident pools and can be broadly classified into two phenotypes: macrophages exhibiting M1 signatures (tumor-suppressing) and M2 signatures (tumor-promoting). In general, majority of tumor-associated macrophages demonstrate an M2-like phenotype, expressing markers such as CD206, Relm-α, and YM1 [24]. Initial studies reported that IL-6 produced by CAFs switched monocyte differentiation programs towards a tumor-promoting M2 lineage [25]. Subsequent in vitro studies of tumor cell–CAF interactions and proteomic profiles of CAFs isolated from tumor models identified numerous pro-inflammatory cytokines such as GM-CSF, CSF-1, CCL2, CCL7, CXCL1, CXCL2, and CXCL8 that are involved in MDSC recruitment and differentiation of macrophages into a pro-tumor M2 phenotype [26–28], which is likely due to suppression of M1-promoting pathways. Activated fibroblasts have also been reported to directly regulate myeloid cell maturation. Pancreatic CAF production of high levels of IL-6 has been reported to drive monocyte precursors towards an MDSC phenotype via STAT3 and induce immune suppression [29, 30]. Additionally, CAFs can also affect DCs in the immune response. CAF-derived IDO, TGF-β, and VEGF inhibit maturation and antigen uptake activity of tumor-infiltrating DCs (TuDCs) thus inducing a tolerogenic phenotype in T cells [31, 32]. Alternatively, in PDAC and breast cancer, CAF-derived TSLP and IL-13 production can polarize DCs to favor pro-fibrotic and pro-tumorigenic TH2 immune responses instead of tumor protective TH1 immunity [23].
Taken together, these reports suggest that CAFs can be potent modulators of immune surveillance by directly modulating T cell activity, as well as by reprogramming myeloid responses to further induce immunosuppression. Despite previous studies describing the mechanisms by which CAFs can mediate immune suppression, it is still unclear how these mechanisms might differ based on the distinct subsets of CAFs that exist in tumor-associated fibrosis, or the extent to which the origin of CAFs (i.e. locally or bone marrow-derived) might differentially affect immune regulatory phenotypes and/or tumor progression.
The effect of the fibrotic ECM on T cell function
In addition to the cellular components of tumor-associated fibrosis, the dense collagen-rich ECM has both direct and indirect effects on T cell infiltration and function (Fig. 3). It has been proposed that when fibrosis is as extensive as seen in PDAC, the scar-like ECM may act as a physical barrier to CTL infiltration into tumors. While mouse models of PDAC have reported that this fibrotic barrier effect is not insurmountable [33], several studies have also reported that the ability of T cells to infiltrate from the stroma into close proximity to PDAC cell nests is impaired when the ECM density is high [34, 35]. In addition to physical exclusion, matrix density may result in preferential localization of T cells. In lung cancer models, matrix density and architecture induced the localization and migration of T cells into the tumor stroma rather than into tumor cell nests [36]. Additionally, while loose areas of fibronectin and collagen tend to facilitate T cell motility, dense and stiff ECMs impede T cell velocity/migration [37]. Apart from the rigidity, the propensity of certain ECM components can also play a role in modulating T cell activation and proliferation [38–40], although our understanding of this process in the context of tumor-associated ECMs is somewhat limited. The presence of stiff and dense ECM, therefore, may retard CTL infiltration and likely regulate their functionality in fibrotic tumors. However, the molecular mechanism(s) of this process and its impact on the therapeutic aspects of the immune response are still not well understood and need further study.
Fig. 3.
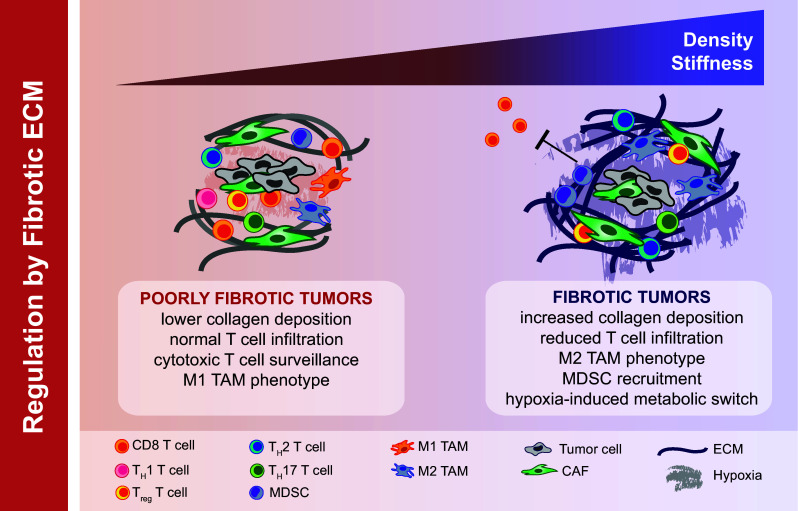
The impact of ECM on immune cell function. The dense collagen-rich ECM of fibrotic tumors can act as a physical barrier to CTL infiltration into tumors. Increased ECM stiffness and aligned structural barriers in a fibrotic tumor also influence the localization and migration of T cells into the tumor stroma. Tumor-associated ECM components as well as sequestered chemokine factors can influence macrophage polarization (towards an M2 signature) and affect the maturation and migration of monocytes and MDSCs. Dense ECM architecture can also prompt changes in availability of oxygen and micronutrients, inducing hypoxia and thereby influencing the immune contexture of the tumor microenvironment
The impact of fibrotic ECM on innate immunity
In addition to influencing T cell responses, fibrotic ECMs can significantly impact tumor immunity by affecting the localization and activity of tumor infiltrating myeloid cells. Numerous in vitro and in vivo reports have established the importance of ECM stiffness, compaction, and plasticity on the differentiation and function of macrophages [41, 42]. However, the effect of these changes on immune suppression is less well understood. Tumor matrix components such as HA can influence macrophage polarization in mammary tumor models [43]. In lung inflammation models, ECM turnover products have been reported to affect the migration of monocytes and neutrophils [44, 45] and thus may play a role in fibrotic tumors. In human pancreatic and breast cancers, extensive deposition of Type I collagen correlates with robust TAM infiltration, suggesting that fibrosis and macrophage infiltration are functionally coupled. This process can be explained by observations that key monocyte chemoattractants CCL2 and CSF-1 are overexpressed by tumor cells or CAFs in response to stiff collagen-rich ECMs [46, 47]. Moreover, increasing evidence reports that ECM physical properties directly affect macrophage phenotype and/or polarization [48, 49]. For example, a collagen-rich ECM favors a protumorigenic polarization phenotype, whereas a fibronectin-rich ECM promotes the anti-tumorigenic activity of macrophages [50–53]. Furthermore, the increased presence of Type I collagen in fibrotic tumors can directly activate inhibitory receptors such as LAIR-1 on immune cells [54], or act as a reservoir for secreted suppressive factors such as TGF-β.
It is presently unclear whether the regulation of macrophage polarization by ECM stiffness/composition is functionally important in tumor immunity and whether this interaction can be used for therapeutic intervention. Similarly, the understanding of these effects on MDSCs or DCs is incomplete. Also, it is unclear how or if dense ECM networks cause dysfunctional antigen presentation in the tumor microenvironment through their regulation of TAMs and DCs.
The impact of hypoxia driven by the fibrotic microenvironment on immune suppression
In addition to regulation of anti-tumor immunity by CAF and the ECM in the TME, tumor-associated fibrosis can create hypoxia which may also play a role in preventing immune surveillance (Fig. 3). When fibrosis is extensive, tumor tissue is often poorly innervated with blood vessels resulting in a highly hypoxic tumor microenvironment with limited access to nutrients and significant alterations in cellular metabolism. Thus, hypoxia induced by tumor-associated fibrosis may be an important modulator of tumor immunity. There is also evidence that hypoxia can induce deposition of ECM in hypoxic tumor regions, indicating a positive-feedback loop. Recent studies have uncovered mechanisms wherein hypoxia induces fibrosis by causing an increase expression of collagen genes [55] and intracellular/extracellular collagen-modifying enzymes [56]. Together these data suggest the existence of a feed-forward loop between fibrosis and hypoxia in certain tumor types.
Hypoxia is a well-established regulator of tumor immunity. Hypoxia-induced chemoattractant secretion within the TME promotes the accumulation of TAMs and MDSCs. For instance, it has been shown that the activation of HIF-1α is essential for myeloid cell infiltration and activation in vivo through a mechanism independent of VEGF [57]. Another potential mechanism of macrophage recruitment into hypoxic regions is that hypoxia-induced Semaphorin 3A acts as an attractant for TAMs by triggering VEGFR1 phosphorylation [58]. Hypoxia has been implicated in regulating the function and differentiation of MDSC in the tumor microenvironment. HIF-1α has been reported to influence MDSC function by regulating PD-L1 expression, suggesting blockade of PD-L1 along with inhibition of HIF-1α may represent a novel approach to target MDSCs as part of cancer immunotherapy in fibrotic tumors [59].
Fibrosis-induced hypoxia may also further suppress T cell infiltration and function in tumors. One of the mechanisms of hypoxia-mediated T cell suppression is that constant activation of HIF-1α negatively regulates T cell receptor signal transduction partially because of increased NF-κB activation [60]. Likewise, accumulation of extracellular adenosine within the TME can trigger immunosuppressive signaling via intracellular cyclic AMP-elevating A2A adenosine receptors (A2AR) on antitumor T cells [61]. Together, these data suggest that fibrosis-induced hypoxia may also impair T cell-mediated immune surveillance.
Overall, tumor-associated fibrosis creates a very challenging TME for sustained tumor immunity. However, despite the fact that fibrosis is a major suppressant of tumor immunity, there is a distinct possibility that it could also act as a double-edged sword. Recently, the notion that tumor-associated fibrosis has a purely tumor-promoting activity has been challenged by suggestions that some components of fibrosis may also have tumor-restraining activity [6, 7]. Thus, it is logical to assume that some discrete elements of fibrosis in specific contexts or tumor types might actually provide critical support for tumor immunity and enhance tumor responses to immunotherapy. At this point this perspective is purely speculative.
Targeting fibrosis to improve immunotherapy
In recent years, several approaches to limit tumor-associated fibrosis have moved from pre-clinical testing to clinical evaluation. These developments have mainly been driven by the potential of these therapies to increase the delivery of cytotoxic agents into highly fibrotic and poorly vascularized tumors. However, based on the potential immunosuppressive and/or “immune-protective” role of tumor-associated fibrosis, it seems plausible that these approaches could also facilitate responses to immunotherapy in highly fibrotic tumors.
Targeting ECM and ECM crosslinking
Abundant collagen deposition in the stroma is a key feature of tumor-associated fibrosis and thus several investigators have tested targeting collagen synthesis and crosslinking. Halofuginone, which inhibits Type I collagen synthesis, has demonstrated efficacy in reducing fibrosis in pancreas and liver fibrosis [62, 63] and has shown promising in murine melanoma models [64]. In addition to targeting collagen synthesis, targeting collagen crosslinking may reduce the pro-tumorigenic effects of fibrosis. Lysyl oxidase (LOX) initiates the process of covalent intra- and inter-molecular crosslinking of collagen by oxidative deamination of specific lysine and hydroxylysine residues. LOX activity is frequently elevated in fibrotic tumor types [65]. Reduction of lysyl oxidase-mediated collagen crosslinking has been shown to prevented mammary tumor-induced fibrosis and tumor progression [66–68]. LOX inhibition in PDAC mouse models also leads to stromal depletion and this in turn results in increased drug delivery [69]. Interestingly, LOX inhibition paradoxically also results in increased macrophage and granulocyte infiltration into PDAC tumors. It is unclear if this is further enforcement of immune suppression in this model, or an invigoration of immunologically cold tumor. Nonetheless, LOX inhibitors have moved to clinical testing. Similarly, all-trans retinoic acid (ATRA) [70] and vitamin D receptor (VDR) [9] have been reported to regulate ECM remodeling and activation of pancreatic stellate cells (PSCs), suggesting ATRA and VDR could be potential targets to selectively deplete stroma.
Another targetable ECM component is HA which is abundant in tumor-associated fibrosis in a number of cancers [71]. This has been an attractive target because accumulation of HA in tumor stroma may increase tumor interstitial pressure, thereby blocking drug delivery in fibrotic tumors [72]. There has been great interest in developing therapeutic strategies targeting HA. Three different therapeutic approaches may be identified: (1) inhibiting HA synthesis, (2) blocking HA signaling, and (3) depleting stromal HA. 4-methylumbelliferone (4-MU) inhibits HA synthesis by downregulating HA synthases HAS2 and HAS3, and in mouse models has resulted in inhibited HA synthesis and reduced tumor progression [73, 74]. Perhaps more therapeutically relevant is the delivery of hyaluronidase to tumors to degrade existing HA. The systemic administration of PEG-fused hyaluronidase (PEGPH20) has been shown to downregulate HA levels in murine PDAC models, and improve the delivery and efficacy of chemotherapy by normalizing interstitial fluid pressures as well as re-expanding the microvasculature [72]. This has been translated into successful phase I trials in combination with Gemcitabine and nab-paclitaxel in PDAC patients and is now in phase II testing (NCT02487277). However, the effects of this agent on immunotherapy are unknown.
Reprogramming the immune system to target fibrosis
Another approach to overcoming fibrosis-induced immune suppression is to reprogram the immune responses to combat activated CAFs and dense ECM. This strategy would have significant benefit over directly targeting fibrotic molecules if the strategy employed bolsters T cell responses while simultaneously stripping fibrosis. Recent data in PDAC models have shown that tumor-specific T cells when given in sufficient numbers by adoptive cell therapy can infiltrate into fibrotic tumors, and in turn result in reduced overall fibrosis [33]. This suggests that when immune surveillance is dominant, the TME is polarized away from tumor-promoting fibrosis. However, adoptive T cell therapy has been used in human pancreatic cancer with only rare success, suggesting this approach alone may not be sufficient for most patients. Another approach is to make CAFs one of the targets of immunotherapy. This has the advantage of initiating a T cell response, while simultaneously attacking an immunosuppressive cell type. To this effect, several approaches have been tested in animal models, including adoptive transfer of FAP-specific T cells, DNA vaccines targeting FAP peptides, and whole-cell tumor vaccine modified to express FAP [75–78]. In total, these approaches can induce T cell-mediated killing of FAP+ CAFs which results in decreased collagen density and increased T cell-mediated control of tumor progression [79]. However, the efficacy of these approaches in combination with other immunotherapeutics has not been established. A third possible approach is to reprogram myeloid response to degrade dense ECMs. It is well appreciated that macrophages are not only critical players in promoting pathologic fibrosis, but can also resolve fibrosis by degrading excess ECM [80]. Recent work in PDAC mouse models has shown that CD40 agonist antibodies lead to the degradation of the fibrotic ECM through MMP activation in monocytes and macrophages [81]. This approach is particularly exciting as CD40 agonists have already been shown to reprogram myeloid and other cell types to favor tumor responsiveness to immune checkpoint inhibitors [82, 83].
Targeted therapies to reduce fibrosis and reestablish tumor immunity
Several pathway-targeted inhibitors have been evaluated for their ability to reduce fibrosis and enhance the efficacy of immunotherapy. One of the first examples of this approach was targeting of the Hedgehog signaling pathway. Indeed, work from Ken Olive and colleagues demonstrated that depletion of the stroma by administration of IPI-926, an inhibitor of the Hedgehog signaling pathway, improved delivery of gemcitabine by increasing intratumoral vascular density in genetically engineered mouse models of PDAC, However, these effects led only to transient responses in preclinical models and did not translate to successful clinical outcomes [84]. More recent work has suggested that direct stromal depletion can lead to more poorly differentiated tumors that are more aggressive [6, 7]. This duplicity of stromal desmoplasia is still highly debated. Nonetheless, stromal depletion strategies may still render tumor more susceptible to immune attack if they can concomitantly restrain tumor progression [6]. To this effect, targeting CXCR4 signaling has shown ability to slow tumor progression in PDAC mouse models while simultaneously decreasing CAF numbers [14]. As such, CXCR4 inhibition improved short-term responses to checkpoint inhibition [14]. While the durability of this therapeutic approach was not tested in animal models, these studies have led to clinical evaluation of CXCR4 inhibitors in combination with PD-1 blockade.
Recently, our group has focused on using targeted inhibition of focal adhesion kinase (FAK) to reduce fibrosis and unlock immunotherapeutic responsiveness in fibrotic tumors [35]. FAK signaling has long been studied for its well-appreciated tumor cell-intrinsic pro-survival and pro-invasive properties. As such, FAK inhibitors have been clinically developed and tested in cancer patients [85]. Outside its tumor-intrinsic effects, FAK signaling has a significant role in pathological fibrosis [86, 87] and recently this role has been shown to be conserved in tumor-associated fibrosis in a subset of cancer types [85, 88, 89]. In addition to regulating fibrosis, FAK signaling in cancer cells may regulate immune surveillance. In squamous carcinoma models, loss of FAK signaling in malignant cells resulted in decreased FOXP3+ Treg recruitment and consequently, increased T cell-mediated tumor control [90]. Our own data in PDAC mouse models have shown that inhibition of FAK simultaneously results in reduced stromal collagen density and decreased tumor infiltration by potentially immunosuppressive TAMs, MDSCs, and Treg [35]. The outcome of FAK inhibitor reprogramming of the TME is increased responsiveness to PD-1 checkpoint antagonists due to increased CD8+ CTL infiltration into PDAC tumors during therapy. These studies have been translated into clinical testing in pancreatic and other cancer patients (NCT02546531 and NCT02758587).
Taken together, these data suggest that targeted agents could be employed to reduce tumor-associated fibrosis to the benefit of immunotherapy. The attraction of this approach is that it could simultaneously reduce fibrosis while targeting tumor survival pathways, thus rendering both malignant and stromal compartments more responsive to immunotherapeutic assault.
Abbreviations
- 4-MU
4-Methylumbelliferone
- A2AR
AMP-elevating A2A adenosine receptors
- ATRA
All-trans retinoic acid
- CAFs
Cancer-associated fibroblasts
- CSF-1
Colony-stimulating factor-1
- CTL
Cytotoxic T lymphocyte
- CXCL1
Chemokine (C-X-C motif) ligand 1
- DDR2
Discoidin domain-containing receptor 2
- ECM
Extracellular matrix
- EMT
Epithelial–mesenchymal transition
- FAK
Focal adhesion kinase
- FAP
Fibroblast activation protein
- FSP1
Fibroblast-specific protein 1
- GM-CSF
Granulocyte-macrophage colony stimulating factor
- HA
Hyaluronic acid
- IDO
Indoleamine 2,3-dioxygenase
- LOX
Lysyl oxidase
- MCP1
Monocyte chemotactic protein-1
- MDSCs
Myeloid derived suppressive cells
- MSCs
Mesenchymal stem cells
- PD-L1
Programmed death-ligand 1
- PDAC
Pancreatic ductal adenocarcinoma
- PDGF
Platelet-derived growth factor
- PEGPH20
PEG-fused hyaluronidase
- PGE2
Prostaglandin E2
- PSCs
Pancreatic stellate cells
- ROCK
Rho-associated protein kinase
- SDF-1
Stromal cell-derived factor 1
- SHH
Sonic hedgehog
- SMA
Smooth muscle actin
- STAT3
Signal transducer and activator of transcription 3
- TAMs
Tumor-associated macrophages
- TGF-β
Transforming growth factor beta
- TME
Tumor microenvironment
- Treg
Regulatory T cells
- TSLP
Thymic stromal lymphopoietin
- TuDCs
Tumor-infiltrating DCs
- VDR
Vitamin D receptor
- YAP1
Yes-associated protein 1
Compliance with ethical standards
Funding
The Authors acknowledges support from the Pancreatic Cancer Action Network and the American Association for Cancer Research (AACR/PANCAN Award), the National Cancer Institute (NCI awards R01-CA177670, R01-CA203890, P50-CA196510, P30-CA91842) and the BJCIH/Siteman Cancer Center Cancer Frontier Fund.
Conflict of interest
The authors declare that there is no conflict of interest.
Footnotes
This paper is a Focussed Research Review based on a presentation given at the conference Regulatory Myeloid Suppressor Cells: From Basic Discovery to Therapeutic Application which was hosted by the Wistar Institute in Philadelphia, PA, USA, 16th – 19th June, 2016. It is part of a Cancer Immunology, Immunotherapy series of Focussed Research Reviews.
References
- 1.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214(2):199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 4.Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. 2006;5(12):1640–1646. doi: 10.4161/cbt.5.12.3354. [DOI] [PubMed] [Google Scholar]
- 5.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 6.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, De Jesus-Acosta A, Sharma P, Heidari P, Mahmood U, Chin L, Moses HL, Weaver VM, Maitra A, Allison JP, LeBleu VS, Kalluri R. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, Fernandez-Zapico ME, Iacobuzio-Donahue C, Olive KP, Stanger BZ. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17(2):135–147. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 9.Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, Martin P, Tseng TW, Dawson DW, Donahue TR, Masamune A, Shimosegawa T, Apte MV, Wilson JS, Ng B, Lau SL, Gunton JE, Wahl GM, Hunter T, Drebin JA, O’Dwyer PJ, Liddle C, Tuveson DA, Downes M, Evans RM. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159(1):80–93. doi: 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, Friess H. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol. 2012;9(8):454–467. doi: 10.1038/nrgastro.2012.115. [DOI] [PubMed] [Google Scholar]
- 11.Davies LC, Heldring N, Kadri N, Le Blanc K. Mesenchymal stromal cell secretion of programmed death-1 ligands regulates T cell mediated immunosuppression. Stem Cells. 2017;35(3):766–776. doi: 10.1002/stem.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Lourdes Mora-Garcia M, Garcia-Rocha R, Morales-Ramirez O, Montesinos JJ, Weiss-Steider B, Hernandez-Montes J, Avila-Ibarra LR, Don-Lopez CA, Velasco-Velazquez MA, Gutierrez-Serrano V, Monroy-Garcia A. Mesenchymal stromal cells derived from cervical cancer produce high amounts of adenosine to suppress cytotoxic T lymphocyte functions. J Transl Med. 2016;14(1):302. doi: 10.1186/s12967-016-1057-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ino Y, Yamazaki-Itoh R, Oguro S, Shimada K, Kosuge T, Zavada J, Kanai Y, Hiraoka N. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PLoS One. 2013;8(2):e55146. doi: 10.1371/journal.pone.0055146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, Teichmann SA, Janowitz T, Jodrell DI, Tuveson DA, Fearon DT. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110(50):20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, Fearon DT. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330(6005):827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 16.Lo A, Wang LC, Scholler J, Monslow J, Avery D, Newick K, O’Brien S, Evans RA, Bajor DJ, Clendenin C, Durham AC, Buza EL, Vonderheide RH, June CH, Albelda SM, Pure E. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. 2015;75(14):2800–2810. doi: 10.1158/0008-5472.CAN-14-3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS One. 2009;4(11):e7965. doi: 10.1371/journal.pone.0007965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simonian PL, Roark CL, Wehrmann F, Lanham AK, Diaz del Valle F, Born WK, O’Brien RL, Fontenot AP. Th17-polarized immune response in a murine model of hypersensitivity pneumonitis and lung fibrosis. J Immunol. 2009;182(1):657–665. doi: 10.4049/jimmunol.182.1.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palucka AK, Coussens LM. The basis of oncoimmunology. Cell. 2016;164(6):1233–1247. doi: 10.1016/j.cell.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su X, Ye J, Hsueh EC, Zhang Y, Hoft DF, Peng G. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol. 2010;184(3):1630–1641. doi: 10.4049/jimmunol.0902813. [DOI] [PubMed] [Google Scholar]
- 21.Barnas JL, Simpson-Abelson MR, Brooks SP, Kelleher RJ, Jr, Bankert RB. Reciprocal functional modulation of the activation of T lymphocytes and fibroblasts derived from human solid tumors. J Immunol. 2010;185(5):2681–2692. doi: 10.4049/jimmunol.1000896. [DOI] [PubMed] [Google Scholar]
- 22.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+ CD25- naive T cells to CD4+ CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198(12):1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, Braga M, Di Carlo V, Doglioni C, Protti MP. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. 2011;208(3):469–478. doi: 10.1084/jem.20101876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41(1):21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chomarat P, Banchereau J, Davoust J, Palucka AK. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol. 2000;1(6):510–514. doi: 10.1038/82763. [DOI] [PubMed] [Google Scholar]
- 26.Wu MH, Hong HC, Hong TM, Chiang WF, Jin YT, Chen YL. Targeting galectin-1 in carcinoma-associated fibroblasts inhibits oral squamous cell carcinoma metastasis by downregulating MCP-1/CCL2 expression. Clin Cancer Res. 2011;17(6):1306–1316. doi: 10.1158/1078-0432.CCR-10-1824. [DOI] [PubMed] [Google Scholar]
- 27.Torres S, Bartolome RA, Mendes M, Barderas R, Fernandez-Acenero MJ, Pelaez-Garcia A, Pena C, Lopez-Lucendo M, Villar-Vazquez R, de Herreros AG, Bonilla F, Casal JI. Proteome profiling of cancer-associated fibroblasts identifies novel proinflammatory signatures and prognostic markers for colorectal cancer. Clin Cancer Res. 2013;19(21):6006–6019. doi: 10.1158/1078-0432.CCR-13-1130. [DOI] [PubMed] [Google Scholar]
- 28.Mathew E, Brannon AL, Del Vecchio A, Garcia PE, Penny MK, Kane KT, Vinta A, Buckanovich RJ, di Magliano MP. Mesenchymal stem cells promote pancreatic tumor growth by inducing alternative polarization of macrophages. Neoplasia. 2016;18(3):142–151. doi: 10.1016/j.neo.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JH, Oh SH, Kim EJ, Park SJ, Hong SP, Cheon JH, Kim TI, Kim WH. The role of myofibroblasts in upregulation of S100A8 and S100A9 and the differentiation of myeloid cells in the colorectal cancer microenvironment. Biochem Biophys Res Commun. 2012;423(1):60–66. doi: 10.1016/j.bbrc.2012.05.081. [DOI] [PubMed] [Google Scholar]
- 30.Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, Fuchs JR, Eubank TD, Frankel WL, Bekaii-Saab T, Bloomston M, Lesinski GB. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013;73(10):3007–3018. doi: 10.1158/0008-5472.CAN-12-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khosravi-Maharlooei M, Pakyari M, Jalili RB, Salimi-Elizei S, Lai JC, Poormasjedi-Meibod M, Kilani RT, Dutz J, Ghahary A. Tolerogenic effect of mouse fibroblasts on dendritic cells. Immunology. 2016;148(1):22–33. doi: 10.1111/imm.12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS, Chen WJ, Liu W, Tai Y, Peng YW, Zhang Q. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis. 2016;5:e198. doi: 10.1038/oncsis.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen HN, Cuevas C, Dotson AM, Tan X, Hotes JL, Greenberg PD, Hingorani SR. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell. 2015;28(5):638–652. doi: 10.1016/j.ccell.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartmann N, Giese NA, Giese T, Poschke I, Offringa R, Werner J, Ryschich E. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin Cancer Res. 2014;20(13):3422–3433. doi: 10.1158/1078-0432.CCR-13-2972. [DOI] [PubMed] [Google Scholar]
- 35.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, Pachter JA, Wang-Gillam A, DeNardo DG. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–860. doi: 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, Mami-Chouaib F, Donnadieu E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122(3):899–910. doi: 10.1172/JCI45817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, Friedl P. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol. 2013;201(7):1069–1084. doi: 10.1083/jcb.201210152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Connor RS, Hao X, Shen K, Bashour K, Akimova T, Hancock WW, Kam LC, Milone MC. Substrate rigidity regulates human T cell activation and proliferation. J Immunol. 2012;189(3):1330–1339. doi: 10.4049/jimmunol.1102757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He J, Baum LG. Presentation of galectin-1 by extracellular matrix triggers T cell death. J Biol Chem. 2004;279(6):4705–4712. doi: 10.1074/jbc.M311183200. [DOI] [PubMed] [Google Scholar]
- 40.Bollyky PL, Wu RP, Falk BA, Lord JD, Long SA, Preisinger A, Teng B, Holt GE, Standifer NE, Braun KR, Xie CF, Samuels PL, Vernon RB, Gebe JA, Wight TN, Nepom GT. ECM components guide IL-10 producing regulatory T-cell (TR1) induction from effector memory T-cell precursors. Proc Natl Acad Sci USA. 2011;108(19):7938–7943. doi: 10.1073/pnas.1017360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McWhorter FY, Davis CT, Liu WF. Physical and mechanical regulation of macrophage phenotype and function. Cell Mol Life Sci. 2015;72(7):1303–1316. doi: 10.1007/s00018-014-1796-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Goethem E, Poincloux R, Gauffre F, Maridonneau-Parini I, Le Cabec V. Matrix architecture dictates three-dimensional migration modes of human macrophages: differential involvement of proteases and podosome-like structures. J Immunol. 2010;184(2):1049–1061. doi: 10.4049/jimmunol.0902223. [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi N, Miyoshi S, Mikami T, Koyama H, Kitazawa M, Takeoka M, Sano K, Amano J, Isogai Z, Niida S, Oguri K, Okayama M, McDonald JA, Kimata K, Taniguchi S, Itano N. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Can Res. 2010;70(18):7073–7083. doi: 10.1158/0008-5472.CAN-09-4687. [DOI] [PubMed] [Google Scholar]
- 44.Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, Mecham RP, Senior RM, Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Investig. 2006;116(3):753–759. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12(3):317–323. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- 46.Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES, Weaver VM. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol (Camb) 2015;7(10):1120–1134. doi: 10.1039/C5IB00040H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, Ireland L, Sakai T, Sakai K, Kim YS, Engle D, Campbell F, Palmer D, Ko JH, Tuveson DA, Hirsch E, Mielgo A, Schmid MC. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol. 2016;18(5):549–560. doi: 10.1038/ncb3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McWhorter FY, Wang T, Nguyen P, Chung T, Liu WF. Modulation of macrophage phenotype by cell shape. Proc Natl Acad Sci USA. 2013;110(43):17253–17258. doi: 10.1073/pnas.1308887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Springer NL, Fischbach C. Biomaterials approaches to modeling macrophage-extracellular matrix interactions in the tumor microenvironment. Curr Opin Biotechnol. 2016;40:16–23. doi: 10.1016/j.copbio.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perri RT, Kay NE, McCarthy J, Vessella RL, Jacob HS, Furcht LT. Fibronectin enhances in vitro monocyte-macrophage-mediated tumoricidal activity. Blood. 1982;60(2):430–435. [PubMed] [Google Scholar]
- 51.Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014;15(12):1243–1253. doi: 10.15252/embr.201439246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stahl M, Schupp J, Jager B, Schmid M, Zissel G, Muller-Quernheim J, Prasse A. Lung collagens perpetuate pulmonary fibrosis via CD204 and M2 macrophage activation. PLoS One. 2013;8(11):e81382. doi: 10.1371/journal.pone.0081382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wesley RB, 2nd, Meng X, Godin D, Galis ZS. Extracellular matrix modulates macrophage functions characteristic to atheroma: collagen type I enhances acquisition of resident macrophage traits by human peripheral blood monocytes in vitro. Arterioscler Thromb Vasc Biol. 1998;18(3):432–440. doi: 10.1161/01.ATV.18.3.432. [DOI] [PubMed] [Google Scholar]
- 54.Meyaard L. The inhibitory collagen receptor LAIR-1 (CD305) J Leukoc Biol. 2008;83(4):799–803. doi: 10.1189/jlb.0907609. [DOI] [PubMed] [Google Scholar]
- 55.Falanga V, Martin TA, Takagi H, Kirsner RS, Helfman T, Pardes J, Ochoa MS. Low oxygen tension increases mRNA levels of alpha 1 (I) procollagen in human dermal fibroblasts. J Cell Physiol. 1993;157(2):408–412. doi: 10.1002/jcp.1041570225. [DOI] [PubMed] [Google Scholar]
- 56.Bentovim L, Amarilio R, Zelzer E. HIF1alpha is a central regulator of collagen hydroxylation and secretion under hypoxia during bone development. Development. 2012;139(23):4473–4483. doi: 10.1242/dev.083881. [DOI] [PubMed] [Google Scholar]
- 57.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. doi: 10.1016/S0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, Deschoemaeker S, Van Ginderachter JA, Tamagnone L, Mazzone M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24(6):695–709. doi: 10.1016/j.ccr.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 59.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211(5):781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang C, Kim JH, Li F, Qu A, Gavrilova O, Shah YM, Gonzalez FJ. Hypoxia-inducible factor 1alpha regulates a SOCS3-STAT3-adiponectin signal transduction pathway in adipocytes. J Biol Chem. 2013;288(6):3844–3857. doi: 10.1074/jbc.M112.426338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res. 2008;14(19):5947–5952. doi: 10.1158/1078-0432.CCR-08-0229. [DOI] [PubMed] [Google Scholar]
- 62.Zion O, Genin O, Kawada N, Yoshizato K, Roffe S, Nagler A, Iovanna JL, Halevy O, Pines M. Inhibition of transforming growth factor beta signaling by halofuginone as a modality for pancreas fibrosis prevention. Pancreas. 2009;38(4):427–435. doi: 10.1097/MPA.0b013e3181967670. [DOI] [PubMed] [Google Scholar]
- 63.Pines M, Knopov V, Genina O, Lavelin I, Nagler A. Halofuginone, a specific inhibitor of collagen type I synthesis, prevents dimethylnitrosamine-induced liver cirrhosis. J Hepatol. 1997;27(2):391–398. doi: 10.1016/S0168-8278(97)80186-9. [DOI] [PubMed] [Google Scholar]
- 64.Juarez P, Mohammad KS, Yin JJ, Fournier PG, McKenna RC, Davis HW, Peng XH, Niewolna M, Javelaud D, Chirgwin JM, Mauviel A, Guise TA. Halofuginone inhibits the establishment and progression of melanoma bone metastases. Cancer Res. 2012;72(23):6247–6256. doi: 10.1158/0008-5472.CAN-12-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, Le QT, Giaccia AJ. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15(1):35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cox TR, Bird D, Baker AM, Barker HE, Ho MW, Lang G, Erler JT. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013;73(6):1721–1732. doi: 10.1158/0008-5472.CAN-12-2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gilkes DM, Chaturvedi P, Bajpai S, Wong CC, Wei H, Pitcairn S, Hubbi ME, Wirtz D, Semenza GL. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res. 2013;73(11):3285–3296. doi: 10.1158/0008-5472.CAN-12-3963. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E, Timpson P, Leach J, McGarry L, Shanks E, Bailey P, Chang D, Oien K, Karim S, Au A, Steele C, Carter CR, McKay C, Anderson K, Evans TR, Marais R, Springer C, Biankin A, Erler JT, Sansom OJ. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med. 2015;7(8):1063–1076. doi: 10.15252/emmm.201404827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chronopoulos A, Robinson B, Sarper M, Cortes E, Auernheimer V, Lachowski D, Attwood S, Garcia R, Ghassemi S, Fabry B, Del Rio Hernandez A. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat Commun. 2016;7:12630. doi: 10.1038/ncomms12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sato N, Cheng XB, Kohi S, Koga A, Hirata K. Targeting hyaluronan for the treatment of pancreatic ductal adenocarcinoma. Acta Pharm Sin B. 2016;6(2):101–105. doi: 10.1016/j.apsb.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kultti A, Pasonen-Seppanen S, Jauhiainen M, Rilla KJ, Karna R, Pyoria E, Tammi RH, Tammi MI. 4-Methylumbelliferone inhibits hyaluronan synthesis by depletion of cellular UDP-glucuronic acid and downregulation of hyaluronan synthase 2 and 3. Exp Cell Res. 2009;315(11):1914–1923. doi: 10.1016/j.yexcr.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 74.Hajime M, Shuichi Y, Makoto N, Masanori Y, Ikuko K, Atsushi K, Mutsuo S, Keiichi T. Inhibitory effect of 4-methylesculetin on hyaluronan synthesis slows the development of human pancreatic cancer in vitro and in nude mice. Int J Cancer. 2007;120(12):2704–2709. doi: 10.1002/ijc.22349. [DOI] [PubMed] [Google Scholar]
- 75.Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, Antzis M, Cotner CE, Johnson LA, Durham AC, Solomides CC, June CH, Pure E, Albelda SM. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2(2):154–166. doi: 10.1158/2326-6066.CIR-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen M, Xiang R, Wen Y, Xu G, Wang C, Luo S, Yin T, Wei X, Shao B, Liu N, Guo F, Li M, Zhang S, Li M, Ren K, Wang Y, Wei Y. A whole-cell tumor vaccine modified to express fibroblast activation protein induces antitumor immunity against both tumor cells and cancer-associated fibroblasts. Sci Rep. 2015;5:14421. doi: 10.1038/srep14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wen Y, Wang CT, Ma TT, Li ZY, Zhou LN, Mu B, Leng F, Shi HS, Li YO, Wei YQ. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. 2010;101(11):2325–2332. doi: 10.1111/j.1349-7006.2010.01695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gottschalk S, Yu F, Ji M, Kakarla S, Song XT. A vaccine that co-targets tumor cells and cancer associated fibroblasts results in enhanced antitumor activity by inducing antigen spreading. PLoS One. 2013;8(12):e82658. doi: 10.1371/journal.pone.0082658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006;116(7):1955–1962. doi: 10.1172/JCI26532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30(3):245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. IFNgamma and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov. 2016;6(4):400–413. doi: 10.1158/2159-8290.CD-15-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zippelius A, Schreiner J, Herzig P, Muller P. Induced PD-L1 expression mediates acquired resistance to agonistic anti-CD40 treatment. Cancer Immunol Res. 2015;3(3):236–244. doi: 10.1158/2326-6066.CIR-14-0226. [DOI] [PubMed] [Google Scholar]
- 83.Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, Clendenin C, Stanger BZ, Furth EE, Wherry EJ, Vonderheide RH. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res. 2015;3(4):399–411. doi: 10.1158/2326-6066.CIR-14-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14(9):598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rustad KC, Wong VW, Gurtner GC. The role of focal adhesion complexes in fibroblast mechanotransduction during scar formation. Differentiation. 2013;86(3):87–91. doi: 10.1016/j.diff.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 87.Lagares D, Kapoor M. Targeting focal adhesion kinase in fibrotic diseases. BioDrugs. 2013;27(1):15–23. doi: 10.1007/s40259-012-0003-4. [DOI] [PubMed] [Google Scholar]
- 88.Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK, LeBleu VS, Roy N, Novitskiy SV, Johansen JS, Poli V, Kalluri R, Iacobuzio-Donahue CA, Wood LD, Hebrok M, Hansen K, Moses HL, Weaver VM. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016;22(5):497–505. doi: 10.1038/nm.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stokes JB, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, Hershey ED, Lowrey B, Thomas KS, Bouton AH, Hwang RF, Stelow EB, Parsons JT, Bauer TW. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther. 2011;10(11):2135–2145. doi: 10.1158/1535-7163.MCT-11-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Serrels A, Lund T, Serrels B, Byron A, McPherson RC, von Kriegsheim A, Gomez-Cuadrado L, Canel M, Muir M, Ring JE, Maniati E, Sims AH, Pachter JA, Brunton VG, Gilbert N, Anderton SM, Nibbs RJ, Frame MC. Nuclear FAK controls chemokine transcription, tregs, and evasion of anti-tumor immunity. Cell. 2015;163(1):160–173. doi: 10.1016/j.cell.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]