

Summary
MicroRNA-122, an abundant and conserved liver-specific miRNA, regulates hepatic metabolism and functions as a tumor suppressor, yet systematic and direct biochemical elucidation of the miR-122 target network remains incomplete. To this end, we performed Argonaute crosslinking immunoprecipitation (Ago-CLIP) sequencing in miR-122 knockout and control mouse livers, as well as in matched human hepatocellular carcinoma (HCC) and benign liver tissue to identify miRNA target sites transcriptome-wide in two species. We observed a majority of miR-122 binding on 3’-UTRs and coding exons followed by extensive binding to other genic and non-genic sites. Motif analysis of miR-122 dependent binding revealed a G-bulged motif in addition to canonical motifs. A large number of miR-122 targets were found to be species-specific. Upregulation of several common mouse and human targets, most notably BCL9, predicted survival in HCC patients. These results broadly define the molecular consequences of miR-122 downregulation in hepatocellular carcinoma.
Graphical Abstract
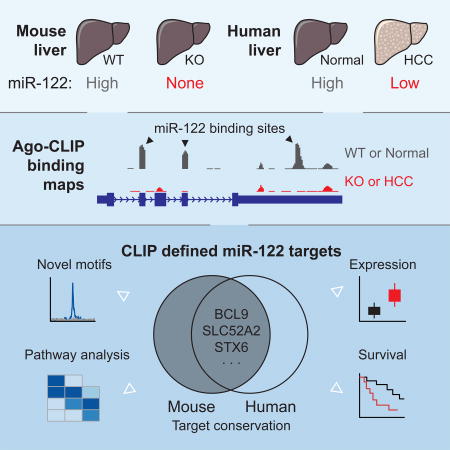
Lack of the liver specific microRNA-122 spontaneously results in hepatocellular carcinoma in mice. Luna et al. provide a comprehensive and biochemically defined catalog of miR-122 targets in mice and humans to uncover novel features of miR-122 targeting that may offer predictive value for human HCC survival.
Introduction
An invariant feature of vertebrate livers is the presence of microRNA (miR)-122. miR-122 is a small (21–22nt) non-coding RNA that post-transcriptionally regulates gene expression by guiding Argonaute (Ago) proteins to engage and repress hundreds of target mRNAs typically via 3’UTR binding (Bartel, 2009). As a predominant miRNA in the liver, miR-122 has been extensively studied and shown to be involved in the regulation of numerous transcripts encoding a variety of hepatic processes, among them, cholesterol and lipid metabolism (Esau et al., 2006; Krützfeldt et al., 2005), mitochondrial function (Burchard et al., 2010), circadian rhythm control (Gatfield et al., 2009; Kojima et al., 2010), polyploidy regulation (Hsu et al., 2016), hepatitis C virus replication (Jopling et al., 2005), and liver tumor suppression (Bai et al., 2009; Gramantieri et al., 2007; Tsai et al., 2009).
The central role of miR-122 as a regulator of hepatic homeostasis is perhaps best demonstrated by in vivo knockout studies (Hsu et al., 2012; Tsai et al., 2012). Genetic ablation of miR-122 in mice does not grossly impair liver development or function, but results in liver inflammation and steatosis in early adult life that proceeds to steatohepatitis, fibrosis, and high-penetrance spontaneous tumors resembling hepatocellular carcinoma (HCC) in late life (Hsu et al., 2012; Tsai et al., 2012). Functionally, growth inhibition of HCC xenografts in nude mice is observed upon delivery of miR-122 mimic (Bai et al., 2009; Hsu et al., 2013; Tsai et al., 2009); similarly, miR-122 precursor gene expression via adeno-associated virus (AAV) vectors is sufficient to rescue MYC-induced liver tumorigenesis (Hsu et al., 2012). Combined with clinical observations that downregulation of miR-122 in HCC patients correlates with poor prognosis (Budhu et al., 2008; Burchard et al., 2010; Coulouarn et al., 2009; Tsai et al., 2009), miR-122 has been recognized as a formidable tumor suppressor, and a potential focus for therapeutic strategies aimed at HCC (Thakral and Ghoshal, 2015). Yet, despite a large set of data on miR-122 target genes, a complete description of the miR-122 regulated target network in liver remains incomplete.
Studies of general miRNA action have benefitted from the ability to biochemically identify microRNA targets in vivo using unbiased, high throughput sequencing methods, broadly known as crosslinking immunopreciptation (CLIP) (Chi et al., 2009; Hafner et al., 2010; Ule et al., 2003). Although computational algorithms can successfully predict miRNA targets, they typically ignore transcriptome information of cell types under study, miss non-canonical or non-conserved binding sites, and are usually restricted to 3’UTRs. CLIP is uniquely suited to overcome these limits, and when combined with miRNA knockout mouse models, provides a framework to empirically define a global miRNA target network. Such a rationale has been fruitful in studies of miR-155 in T cells (Loeb et al., 2012).
Here we present a comprehensive miRNA:target interaction map using Ago-CLIP in miR-122 knockout mice to exhaustively define the miR-122 regulated transcriptome in the liver. We observe widespread canonical and non-canonical binding for miR-122, finding greater than 104 distinct target loci across genic and non-genic regions; however, changes in gene expression are linked only to 3’UTR and coding exon targeting. Intriguingly, the majority of miR-122 targeting was species-specific, a finding confirmed by comparison of AGO-CLIP targets between mouse and human liver, and between human HCC and matched normal liver tissue obtained from the same individuals. We focused on 965 targets shared between humans and mice, many of which were found to be upregulated in HCC and predictive of overall survival. We propose that one such conserved CDS target, BCL9, a component of WNT/β-catenin signaling, is a critical factor for HCC pathogenesis as a consequence miR-122 down regulation.
Results
miR-122 exhibits widespread and non-canonical binding in the mouse liver
To uncover hepatic miRNA:target interactions globally, we performed Ago CLIP and RNA-seq analyses in six-week old 122KO or floxed control mice (Figure S1A). Measuring Ago associated miRNA abundance confirmed specific miR-122 loss in KO livers (Figure S1B). Principal component analysis of all Ago:target RNA peaks separated all WT and KO samples along a single axis, and confirmed that most of the variance (93%) in Ago binding was due to the absence of miR-122 (Figure S1C).
To generate a comprehensive landscape of miR-122-5p (hereafter referred to as miR-122) binding in the mouse liver, we focused on Ago binding peaks that were specifically absent in KO samples (Figure 1A). Motif enrichment analysis of all peaks exhibiting this pattern revealed strong enrichment for canonical miR-122 target sequences as well as a miR-122-related but non-canonical G-bulged motif (Figure 1B). The location of target sequences was centrally distributed around Ago binding sites for either canonical or G-bulged sites (Figure 1C–D), further indicative of specific and miR-122 dependent Ago binding. Of the top 10 miRNA families in mouse liver, capable of engaging 44% of all target loci (Figure 1E), we found that miR-122 targets were the most frequent (n=9280) with the bulged motif adding 1923 additional target sites, in total comprising 18.7% of binding events for the top ten miRNAs. The average transcript 3’UTR contained binding sites for 2.2 distinct miRNAs and 0.4 sites for miR-122 specifically. Genomic annotation of all target loci revealed widespread miR-122 dependent engagement beyond 3’UTRs with the largest set occurring on coding exons and introns (Figure 1F). Coding exon binding predominated among G-bulged targeted sequences as well as “orphan” peaks with no obvious miRNA binding site (Figure 1F). Taken together, these data suggest that miR-122 engages a larger fraction of the transcriptome than previously assumed using both canonical and non-canonical binding modes.
Figure 1.

Non-canonical and widespread binding of miR-122 targets in mouse liver. (A) Example miR-122 dependent binding peak defined by CLIP signal in WT livers that is absent in KO. (B) Motif enrichment analysis of peaks resembling (A). (C) Canonical miR-122 motif enrichment by Ago peak position for loci resembling (A) compared to all observed peaks. Inset depicts canonical binding target interactions. (D) Non-canonical bulged miR-122 motif enrichment by Ago peak position for loci resembling (A) compared to all observed peaks. Inset depicts proposed non-canonical binding target interactions. (E) Summary of mRNA target loci distributed among the top 30 (lower pie) or the top 10 miRNA families in mouse liver (upper pie). (F) Global annotation of miRNA target loci as defined in (E). Int: intergenic. Number of loci per category indicated in parentheses. L2FC, Log-2 fold change. See also Figure S1.
Functional characterization of the miR-122 bound transcriptome
We next sought to address the functional impact of global miR-122 loss observed with CLIP by using RNAseq. Comparing 3’UTR target loci, we observed that both canonical and bulged miR-122 targets displayed dramatically reduced Ago binding in KO livers, yet only canonical miR-122 targets were de-repressed (Figure 2A). Extending these observations, we observed statistically significant loss of binding to miR-122 target loci regardless of annotation (Figure 2B), but observed mRNA depression only for 3’UTR and coding exon targeting for canonical miR-122 binding only (Figure 2C). In addition, both the amount of Ago binding (approximated by binding site read depth) and the level of target derepression correlated with seed type efficacy (Figure S2A–B). These data are consistent with previous observations that primarily canonical 3’UTR targeting, and to a lesser extent canonical coding exon targeting, affect steady state levels of gene expression (Bartel, 2009). Notably, while we observed reduced Ago binding for miR-122-3p targets in KO mouse livers, we did not observe significant changes in expression for miR-122-3p target genes (Figure S2C).
Figure 2.

Functional characterization of the miR-122 bound transcriptome. (A-B) Scatterplot with marginal histograms comparing log2 fold change in CLIP binding and RNAseq expression between WT and KO livers for all 3’UTR loci containing canonical (red) or non-canonical (blue) miR-122-5p binding events, compared to top 10 miRNA targets exclusive of miR-122 (grey). (B) Boxplots per genomic region depicting log2 fold change in CLIP binding between WT and KO livers. (C) Boxplots per genomic region depicting log2 fold change in RNAseq expression between WT and KO livers. (D) Mean PhyloP conservation scores across the core 6mer for miR-122 seed targets compared to seeds from the top miRNA families by genomic annotation. (E) Mouse to human seed sequence level comparison of unique CLIP peaks per top 10 miRNA families by all targets, 3’UTR targets, or CDS targets. Dashed lines indicate percentage of miR-122 seed sites conserved between mouse and humans in CLIP data, and the white line indicates average for top miRNAs. ****P<0.0001, ns P>0.05, two-sided Mann-Whitney U-test. See also Figures S1–S3.
We next grouped miR-122 target transcripts for pathway analysis. As the general biological consequences of miRNA targeting beyond 3’UTRs and coding exons remains unclear, we binned our RNAseq data among altered 3’UTR and CDS targets (“primary), 5’UTR and intronic targets (“secondary”), and all significantly altered genes in KO mice (“indirect”). Only 29% and 15% of canonical primary and secondary targets were upregulated in KO livers, respectively, despite clear miR-122 dependent Ago binding. For primary targets, we observed greater than 3-fold enrichment for upregulation versus down regulation (Figure S2D), while G-bulged targets did not show significant enrichment. The largest effects on steady-state mRNA levels were downstream of miR-122 activity with roughly 1600 and 1300 transcripts up and downregulated in KO livers respectively (Figure S2D). Signaling pathways surrounding cell-cell junctions and cytoskeletal rearrangement were the most significantly perturbed among de-repressed primary targets, and have been previously implicated in enhanced cell proliferation (Yamazaki et al., 2008). Among indirect targets, pathway analysis revealed signatures of fibrosis and stellate cell activation (Figure S2E) and also confirmed previous findings of mitochondrial dysfunction and fatty acid oxidation as the most affected pathways downstream of miR-122 (Burchard et al., 2010; Esau et al., 2006). These results highlight the direct and indirect global responses to miR-122 loss, and further implicate cell-cell junctions and cytoskeletal rearrangement as major signaling pathways and potential disease drivers targeted directly by miR-122.
As a broadly conserved miRNA from humans to fish (Gerlach et al., 2009), we expected that miR-122 targets would bear site conservation similar to other conserved and expressed miRNAs in the liver. Such a result would be consistent with previous reports based on bioinformatics prediction (Friedman et al., 2009). Surprisingly, we observed that 3’UTR targets of both canonical and G-bulged seeds were substantially less conserved than the targets of the top expressed miRNA families, exclusive of miR-122 (Figure 2D). Such lower conservation was also apparent for canonical miR-122 targeting in coding exons, introns, and 5’UTRs (Figure 2D). We further focused on mouse/human chain alignments to examine site conservation specifically for the top 10 miRNAs in liver. We observed that while greater than 30% CLIP targets for most top miRNAs in mouse were conserved in human, only 16% and 23% of canonical or bulged miR-122 targets were conserved at the site level respectively (Figure 2E). The lack of site-level conservation was most apparent on 3’UTR targets and to a lesser extent CDS targets. Moreover, we did not observe functional differences in Ago binding or RNAseq expression for miR-122 targets when binned by high or low conservation (Figure S2F). Lastly, species-specific and G-bulged targeting was confirmed via luciferase assay (Figure S3). Taken together, these data pinpoint wide and functional species-specific targeting for miR-122 in the mouse liver at sites less conserved than those of other top expressed miRNAs in the same tissue. This observation further underscores the importance of biochemical (CLIP) analysis of targets as an important adjunct to bioinformatically predicted identification of binding sites.
A miR-122 focused AGO binding landscape of human liver and HCC
Given the unique species specific miR-122 targeting observed in mice, we reasoned that obtaining a miRNA binding landscape in human liver would aid in linking the spontaneous HCC phenotype of miR-122 KO mice (Hsu et al., 2012; Tsai et al., 2012), to general observations of reduced miR-122 expression in human HCC (Bai et al., 2009; Burchard et al., 2010; Coulouarn et al., 2009; Gramantieri et al., 2007; Kutay et al., 2006; Tsai et al., 2009). Such a link is likely to rely on a set of shared miR-122 targets between humans and mice, thus the species-specific targeting, if confirmed in humans, could assist in filtering for a core set of expressed and AGO bound targets that may drive liver pathology in both organisms. To this end, we performed AGO-CLIP in human HCC tumors and normal adjacent liver tissue from the same patients to account for expected inter-individual variability (Figure S4A). We analyzed samples from a total of nine patients, most with moderate to poorly differentiated HCC (see Supplementary Table 1 for subject characteristics). We performed CLIP in two batches of five and four HCCs with respective matched histologically normal liver samples in each experiment. Measuring AGO associated miRNA abundance from CLIP data confirmed specific miR-122 reduction in tumor samples (Figure S4B). Likewise, principal component analysis of all AGO:target RNA peaks separated all normal and tumor samples along one axis (16% of variance) despite separation by batch (58% of variance) (Figure S4C).
Consistent with our findings in miR-122 KO mice, we found that both canonical and G-bulged miR-122 3’UTR sites were significantly less bound in tumor samples (Figure 3A). We also noted increased AGO association and target binding in tumors for oncogenic miRNAs such as miR-21 (Figure 3A and S4B) (Jiang et al., 2008; Wang et al., 2014), highlighting the multifaceted nature of miRNA perturbation in this cancer. Global loss of miR-122 or gain of miR-21 binding in HCCs was likewise recapitulated in coding exons, introns, and 5’UTRs (Figure 3B). We further observed that the mean CLIP signal for miR-122 binding was highly correlated with tumor miR-122 levels: those tumors with lowest miR-122 levels displayed the greatest reduction in AGO binding on miR-122 targets in contrast to tumors with slightly reduced miR-122 levels (Figure 3C). These results provide direct biochemical evidence of a deregulated miR-122 bound transcriptome in human HCC.
Figure 3.

AGO-CLIP of human HCC displays features of miR-122 loss. (A) Cumulative density function (CDF) of the log2 fold change in CLIP binding between normal adjacent and matched tumor tissue for all 3’UTR targets containing indicated miRNA seeds by family, from nine donors. “All” refers to the top 50 miRNA families, inclusive of miR-122 and miR-21. (B) Boxplots per genomic region depicting log2 fold change in CLIP binding between normal adjacent and matched tumor tissue. (C) Mean log2 fold change in CLIP binding per patient plotted against miR-122 relative abundance as measured by qPCR. Pearson correlation of best-fit line is shown. (D) Overlap among all primary miR-122 target genes found in mice and humans. Pie charts depict expression level proportion from RNAseq in miR-122 KO mouse livers relative to WT livers. ****P<0.0001, ns P>0.05, two-sided Mann-Whitney U-test. See also Figures S4–S5.
We next compared the overlap between all CLIP supported human and mouse targets at the gene level, with a focus on primary targets. Of all target genes identified (n=4771), 20% (n=965) shared any overlap between species (Figure 3D), and several targets associated with maintaining normal liver homeostasis, e.g. GNPDA2, ZBTB4, GPR107, and ARHGAP1, were validated via luciferase assay and qPCR (Figure S5A–B, Supplementary Table 2 for primer sequences). Proportionally, we observed that 76% (n=3973) of mouse and 45% (n=1764) of human targets were unique to each respective species. Grouping shared or species-specific genes for pathway analysis, we observed that cell cycle and tight junction pathways, as well as cancer signaling pathways such as AMPK, PI3K/AKT, and WNT/β-catenin were shared between humans and mice (Figure S5C). Taken together, these data pinpoint a select minority of miR-122 targets shared at the site, gene and pathway level.
A miR-122 target signature in human HCC that correlates with patient survival
To determine the pathological significance of the 965 shared miR-122 targets, we queried the liver cancer HCC (LIHC) cohort of samples from The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/) reasoning that shared targets might be de-repressed in tumors compared to respective benign livers as controls in this dataset. Indeed, we observed general upregulation for a majority of miR-122 target genes in these data (Figure 4A). We next focused on miR-122 targets, shared between humans and mice, which were upregulated in LIHC data and also impacted patient survival. Expression levels of three such targets, B-Cell Lymphoma protein 9 (BCL9), Solute Carrier Family 52 (riboflavin transporter) Member 2 (SLC52A2), and Syntaxin 6 (STX6) were upregulated in tumors (Figure 4B), negatively correlated with miR-122 expression (Figure 4C), indicating direct miR-122 dependence, and also significantly associated with poor patient survival (Figure 4D). Significant correlation with survival was enhanced upon grouping expression of these three genes (data not shown). We readily observed upregulation of these three genes in HCC within additional RNAseq datasets (Liu et al., 2016; Yang et al., 2017), indicating that that above observations are not likely to be limited to TCGA data (Figure S6A–B). In the LIHC cohort, we noted that the expression of genes most significantly negatively correlated with miR-122 expression were all identified as miR-122 targets in Ago-CLIP data: RAVER1, G6PC3, TBC1D10B and MAPRE1 (Figure S6C). Targets positively correlated with miR-122 expression such as F2, TF, ASGR1, and APOB were mostly hepatocyte specific markers (Figure S6D). Collectively, these data implicate de-repression of specific miR-122 targets in poor HCC outcomes.
Figure 4.

Higher expression of select miR-122 targets in human HCC is associated with poor survival. (A) Consensus-clustered heat map of shared human and mouse miR-122 target genes (n = 965) in LIHC tumors (n = 373 patients) from TCGA reveals general upregulation of miR-122 targets. (B) Expression Z-scores for BCL9, SLC52A2 and STX6 by sample type. (C) RSEM expression for miR-122 and targets BCL9, SLC52A2 and STX6 per tumor sample. Regression coefficients of best-fit line and significance P-values are shown. (D) Kaplan-Meier survival analysis of LIHC patients displaying upregulated versus non-altered levels of genes in (B) with indicated log-rank test P-values, hazard ratios (HR), and confidence intervals. *,P<0.001, K–S test. See also Figure S6.
miR-122 mediates β-catenin dependent transcription by suppressing BCL9
We then validated these survival-associated genes as bona fide conserved miR-122 targets. Both SLC52A2 and STX6 contained single canonical miR-122 seeds in their 3’UTRs, while BCL9, a β-catenin co-factor which mediates transactivation of WNT target genes (Deka et al., 2010), contains six and seven miR-122 sites, all in coding exons in human and mouse, respectively (Figure 5A). Expression levels for all three genes were elevated in 122 KO mice (Figure 5B). Further, expression levels for these three genes were suppressed upon addition of miR-122 mimic in both mouse and human hepatoma cells (Figure 5C). Using luciferase reporters, we confirmed specific miR-122 mediated repression of human and mouse 3’UTRs for SLC52A2 and STX6 (Figure 6A–B, Supplementary Table 2 for primer sequences). BCL9 is a unique miR-122 target that harbors 2 conserved canonical and 4 conserved G-bulged sites, as well as one additional canonical site in mice (Figure 6C and S7A). A large 3’UTR BCL9 peak present in both WT and KO is likely targeted by miR-19 and miR-30, showcasing how multiple miRNAs target this transcript (data not shown). Testing human and mouse orthologues of each BCL9 miR-122 site in a luciferase reporter context demonstrated that five mouse and four human binding sites can mediate repression (Figure 6C). To validate BCL9 as miR-122 CDS target, we overexpressed full-length FLAG-tagged human BCL9 (WT-BCL9, Figure S7B) in H293T cells and treated with miR-122 mimic (Figure 7A). Protein and RNA levels of WT-BCL9 were suppressed with miR-122 mimic, an effect that was abrogated when all 6 miR-122 sites were mutated (MUT-BCL9) to disrupt miR-122 binding while preserving the amino acid sequence (Figure 7A and S7A–B). Considering β-catenin requires BCL9 for transcriptional activation (Deka et al., 2010), we next measured β-catenin activity using the TOP/FOP FLASH assay in MHCC-LM3 cells. Cells overexpressing both WT-BCL9 and MUT-BCL9 increased β-catenin activity; however, only WT-BCL9 expressing cells treated with miR-122 mimic suppressed β-catenin-regulated luciferase activity (Figure 7B). Furthermore, BCL9 knockdown in MHCC-LM3 cells resulted in reduced expression β-catenin targets, CD44 and c-JUN (Figure 7C) (Mann et al., 1999; Wielenga et al., 1999). This data was further corroborated by the rescue of CD44 and c-JUN expression in miR-122 and BCL9 double knockout Huh7.5 cells (Figure 7D, Supplementary Table 2 for primer sequences). Lastly, in both MHCC-LM3 and Huh7.5 cells, we observed enhanced proliferation upon BCL9 overexpression that was capable of rescuing endogenous BCL9 knockdown (Figure 7E) or knockout (Figure 7F). These results establish BCL9 as a conserved miR-122 CDS target that may impact WNT mediated progression of HCC specifically through proliferation. More broadly, these results illustrate the advantage of CLIP in decoding disease associated miRNA target networks and suggests that similar strategies may be applied to identify candidate genes in liver pathology.
Figure 5.

BCL9 is a conserved, non-canonical exonic target of miR-122 and is implicated in human HCC. (A–B) Ago-CLIP binding profile (A) and RNaseq expression (B) of the Bcl9, Slc52A2, or Stx6 genes in WT and KO mice. miR-122 sites labeled in black triangles. (C) qPCR of BCL9 levels from mouse (Hepa1-6) or human (SNU449) hepatoma cells following 25nM control (NC) or miR-122 mimic (122) treatment for 48 hours. *P<0.05, two-tailed t-test. All data are represented as mean ± standard deviation (error bars).
Figure 6.

Luciferase validation of Slc52a2, Stx6, and Bcl9 as mouse and human conserved miR-122 targets. (A–B) Luciferase reporter assay of mouse and human conserved (C) Slc52a2 and (D) Stx6. Renilla luciferase activity was normalized to Firefly luciferase (RLU) after transfection with miR-122 mimic or NC scrambled RNA. (E) Luciferase reporter measurements of human or mouse BCL9 miR-122 sites, tested individually. Dashed line indicates respective signal for mutated sites). Mouse and human alignments are shown. *P<0.05, two-tailed t-test. All data are represented as mean ± standard deviation (error bars). See also Figure S7.
Figure 7.

miR-122 modulates β-catenin dependent transcription by suppressing BCL9. (A) FLAG-tagged human BCL9 expression levels in 293T cells after transfection with 50nM control (NC) or miR-122 mimic (122). WT BCL9 is compared to a mutant where all 6 exonic miR-122 sites were mutated while preserving amino acid sequence. (B) β-catenin dependent activity measured in MHCC-LM3 transfected with 50nM control (NC) or miR-122 mimic (122). TOP/FOP luciferase reporters and WT or MUT BCL9 constructs were transfected 12 hours after miRNA transfection and measured after 48 hours. Firefly luciferase activity was normalized to Renilla luciferase. For immunoblots, fold change (F.C.) represents the protein level normalized to that of NC treated cells. (C) Expression of the downstream targets of Wnt signaling (e.g. CD44 and c-JUN) determined in MHCC-LM3 shBCL9 knockdown cells via qPCR. (D) qPCR expression of targets in (C) in WT, miR-122 knockout (122KO), or miR-122 and BCL9 double knockout (DKO) Huh7.5 cells. (E) Cell viability assay of MHCCC-LM3 cells under BCL9 knockdown or overexpression conditions. (F) Cell viability assay of Huh7.5 cell mutants in (D) upon BCL9 rescue expression (*P<0.05, two-tailed t-test). All data are represented as mean ± standard deviation (error bars). See also Figure S7.
Discussion
In the present study, we biochemically define the endogenous miR-122 target network in the mouse liver and find greater than 11,000 genomic loci displaying clear miR-122 binding dependence. Unambiguous target sites of this broadly conserved miRNA were found throughout all major genomic annotations and encompassed non-canonical G-bulged binding sites, yet only 3’UTR and coding exon binding of canonical miR-122 targets had any significant impact on steady state RNA levels. These results are broadly consistent with previous work (Bartel, 2009; Chi et al., 2009; Loeb et al., 2012). The breadth of miR-122 targeting that had no discernable impact on steady-state RNA levels likely reflects the many inputs beyond miRNAs that influence RNAseq level measurements. Still, the clarity of our Ago-CLIP results expands definitions of miRNA target abundance by including non-3’UTR regions and G-bulged motifs which may portend additional roles for miRNA targeting. While G-bulged motifs between positions 5–6 have been seen previously in Ago-CLIP data (Chi et al., 2012), for miR-122 a single and unique G-bulged motif between positions 6–7 was observed, which suggests that different miRNAs may have unique enthalpy considerations for tolerating mRNA bulges (Moore et al., 2015). Thus, predicting non-canonical binding sites will likely remain a significant challenge in the absence of direct biochemical Ago-CLIP data, where the ideal strategy, we believe as taken here, is to compare miRNA KO to WT Ago-CLIP datasets. Lastly, the degree to which non-repressive miRNA binding may reflect competing endogenous RNA (ceRNA) activity has been recently a much explored and debated topic (Bosson et al., 2014; Denzler et al., 2014; 2016; Jens and Rajewsky, 2014; Thomson and Dinger, 2016). These CLIP data provide an empirical basis for modeling approaches to probe for ceRNA or other as yet unknown functions, especially for miR-122 studies in the liver (Denzler et al., 2014; 2016).
The high species specificity observed for miR-122 targeting at the site and gene levels between mice and humans raises a number of significant questions. Pioneering studies of miRNA target conservation have established clear links between miRNA conservation, target site conservation, and effective target repression for many miRNAs (Bartel, 2009). And yet, our data with the highly liver specific miR-122 appear to contradict this general principle. It’s conceivable that the targets of certain highly conserved miRNAs are under less selective pressure to maintain miRNA binding sites, this may be particularly true for miRNAs with high tissue specificity and/or moderate fitness effects when ablated. Since miR-122 appears mostly dispensable for liver development, it is possible that fitness costs associated with gain or loss of miR-122 binding sites are relatively neutral. Under this framework, many miRNA target sites would be expected to tolerate random genetic drift (Lynch and Hagner, 2015) whereas sites deleteriously sensitive to gain of miR-122 binding would be expected to undergo selective avoidance (Farh et al., 2005). Consequently, miRNA target repertoires should diverge among distantly related species, but should be relatively conserved among closely related species, as has been argued elsewhere (Xu et al., 2013). Supporting this view, miR-122 CLIP target site conservation as measured by PhyloP scores approached neutrality (near zero), suggesting drift. In addition, other RNA centric processes in the liver such as splicing are known to exhibit species specificity over tissue specificity, in contrast to tissues such as the brain and heart where tissue specificity dominates (Merkin et al., 2012). Future work should determine of miRNA target networks behave similarly.
The species specificity and unambiguous target identification afforded by CLIP enables a rigorous definition of a set of conserved targets that may underlie the shared pathology between miR-122 knockout mice and human HCC patients. In humans, HCC typically arises from many specific etiologies that develop through the accumulation of cellular alterations to eventually drive tumor development (Schulze et al., 2015). miR-122 KO mice recapitulate a common and predictable progression of human HCC beginning with steatosis, inflammation, fibrosis and ending in spontaneous tumor development (Hsu et al., 2012; Tsai et al., 2012). Mice in these studies however, did not develop HCC until ∼10–12 months of age. Given that our mouse CLIP data was derived from six-week old knockout animals with liver inflammation and microsteatosis, comparison to human HCC CLIP data suggests that loss of miR-122 may result in continuous and chronic de-repression of key targets and pathways. Such long-term de-repression has been suggested for chronic hepatitis B and C virus infections as both viruses can functionally sequester miR-122 (Liang et al., 2016; Luna et al., 2015). In the data presented here, known pathways that contribute to HCC development such as AMPK and PI3/AKT signaling, hepatic fibrosis, actin cytoskeletal signaling, and WNT signaling were consistently targeted by miR-122 in both mice and humans.
Our focus on CLIP derived miR-122 targets in TCGA data revealed at least three targets significantly correlated with overall survival in human liver cancer patients. BCL9, SLC52A2, STX6 are involved in WNT/β-catenin activation of TCF/LEF transcriptional activity (Deka et al., 2010), riboflavin membrane transport (Haack et al., 2012) and intracellular vesicular transport (Du et al., 2016), respectively. These targets propose a number of hypotheses for miR-122-mediated maintenance of liver homeostasis. For example, dysfunctional STX6 expression leads to cholesterol accumulation in the liver, which has been shown to alter invasive and migratory properties of cells (Du et al., 2016; Reverter et al., 2014). This function of STX6 may also contribute to the altered cholesterol homeostasis observed after depletion of miR-122 in mice (Esau et al., 2006; Hsu et al., 2012; Tsai et al., 2012). Similarly, SLC52A2 may regulate tumor progression as liver cancer cell lines depleted of riboflavin display reduced growth and proliferation as well as increased DNA damage due to oxidative stress (Manthey et al., 2005).
In this study, we focused on BCL9 as a component of the WNT signaling pathway (Deka et al., 2010; Schulze et al., 2015). Previous genomic sequencing studies have established β-catenin as a primary driver of human HCC via mutations found in >37% of patients that are predicted to increase the stability of this key canonical WNT signaling effector (Anastas and Moon, 2013; Schulze et al., 2015). Additional studies have implicated miR-122 in regulating WNT/β-catenin signaling to inhibit downstream proliferation, migration and epithelial-mesenchymal transition (EMT) through 3’UTR targeting of WNT1 (Wang et al., 2016; Xu et al., 2012). BCL9 represents an unexpected addition to previous findings, as a conserved miR-122 target harboring seven miRNA binding sites in coding exons where half contain non-canonical G-bulges. Previous work has shown that BCL9 promotes proliferation and metastases in myeloma and colon cancer cells (Mani et al., 2009), while in HCC, BCL9 inhibition results in reduced proliferation and migration (Xu et al., 2017). We functionally demonstrated that β-catenin transcriptional activity could be repressed by miR-122 through regulation of downstream effector BCL9. Further, knocking out BCL9 in the human hepatoma cells lacking miR-122 suppressed the expression of WNT/β-catenin downstream targets CD44 and c-JUN. These results establish BCL9 as a key miR-122 target in WNT/β-catenin signaling and as a potential therapeutic target in patients with low levels of miR-122. Current drugs in development to specifically target and inhibit BCL9 (Wisniewski et al., 2016) may be useful in future studies of liver pathology.
In summary, we have determined the miR-122 target network using Ago-CLIP in both mice and humans to uncover widespread, non-canonical and species-specific miR-122 target engagement. These data enabled identification of WNT/β-catenin signaling as a major pathway regulated by miR-122 via its interaction with BCL9 mRNA. Overall, these results illustrate but one of many likely molecular consequences of miR-122 downregulation in hepatocellular carcinoma. We propose that the CLIP data presented here may be used to predict patient survival and to identify additional potential drug targets for HCC and other liver pathologies.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Robert B. Darnell (darnelr@rockefeller.edu).
Experimental Model and Subject Details
Mice
miR-122 floxed (WT) and KO mice were generated as described in (Hsu et al., 2012). Animals were housed in temperature-controlled and Helicobacter-free facility under a 12/12 hour light/dark cycle. Five week-old miR-122fl/fl (WT) and miR-122−/− (KO) littermates were sacrificed by CO2 asphyxiation, blood was collected by cardiac puncture and liver tissues were snap-frozen in liquid N2 and stored at −80°C freezer for subsequeny RNA isolation and Ago-CLIP analysis. All animals were handled following the approved by The Ohio State University Institutional Laboratory Animal Care Committee. In total, nine animals (five floxed and four KO litermates) were used in the final analysis and processed for CLIP in a single batch.
Human HCC and matching liver tissues
Primary human benign liver and matching HCC tissues were obtained from the Collaborative Human Tissue Network (CHTN). Surgically removed tissues from patients not infected with HBV, HCV or HIV and that not undergone any treatment were used for this study. Tissue specimens were obtained following The Ohio State University Institutional Review Board approved protocol and stored at −80°C until use. A total of eighteen samples from nine human patients (HCC and matched control from each patient) were used in the final analysis. Given higher expected gene expression variability compared to isogenic mouse lines, we performed CLIP in two batches of five and four patients respectively.
Cell lines and culture conditions
Mouse hepatoma cell line (HEPA1–6) were a gift from Dr. Gretchen Darlington (Baylor College of Medicine). SNU449 cells were obtained from ATCC. HEPA1–6, SNU449 were cultured in Minimum Essential Media (MEM) (Corning) supplemented with 10% fetal bovine serum (FBS, Sigma), 0.1 mM nonessential amino acids (NEAA, Invitrogen, Life Technologies) and Penicillin-Streptomycin (Corning). MHCC-LM3 cells were provided by Dr. Bo Yu (Hangzhou Push-Kang Biotechnology, China). MHCC-LM3 H293-TN, and H293-T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Corning) supplemented with 10% fetal bovine serum (FBS, Sigma) and Penicillin-Streptomycin while maintaining (Corning). Huh7.5 wild type (WT) (Blight et al., 2002), Huh7.5 miR-122 knockout cells (122KO) (Luna et al., 2015), and Huh7.5 miR-122/BCL9 double knockout cells (DKO) were cultured in DMEM (Corning) supplemented with 5% fetal bovine serum (FBS, Sigma), 0.1 mM nonessential amino acids (NEAA, Invitrogen, Life Technologies) and Penicillin-Streptomycin (Corning).
Method Details
RNA Isolation and RT-PCR
Total RNA was isolated using TRIzol (Life Technologies, #15596018) followed by DNase I treatment (NEB, M0303L). DNase treated RNA (1µg) was converted into complementary DNA (cDNA) by High-capacity cDNA reverse transcription kit (Applied Biosystem, # 4368813). Real-time RT-PCR was performed using SYBR Green chemistry. Expression of specific genes was normalized to that of ACTB. Relative expression was calculated by comparative CT method. Primer sequences are provided in the Supplemental Table 2.
Generation of Plasmid Constructs
For validation of mouse and human-specific targets, 3’UTRs or coding exons of genes of interest harboring miR-122 binding site(s), were amplified from mouse or human liver cDNA with primers (sequences provided in the Supplementary Table 2) and cloned into the 3’UTR of Renilla luciferase CDS of psiCHECK2 (Promega) dual luciferase reporter vector at the MCS.
Human BCL9 cDNA was obtained from (Transomic cat# TCHS1003 – MGC) and was subcloned into pCMV-FLAG-expression vector (Transomic cat# TOC1003). Mutation of individual miR-122 binding sites was generated using Gibson Assembly kit (NEB, E2611S) according to the manufacture’s protocol with oligos designed to maintain amino acid sequence integrity (sequences provided in Supplementary Table 2). Mutation of each individual site was verified by Sanger sequencing.
Luciferase reporter assays
Wild-type and mutant 3’UTRs of target genes harboring miR-122 binding sites were cloned into the 3’UTR of Renilla Luciferase of psiCHECK2 (Promega) dual luciferase reporter. psiCHECK2 vectors (50ng) harboring miR-122 target 3’UTRs were co-transfected with of either miR-122 or scrambled (NC) RNA mimics (25 nM) into Hepa or Huh-7 cell lines. Luciferase activity of activity was measured after 48 h per manufacturer’s protocol (Promega) and normalized to Firefly Luciferase activity.
Western blot analysis
Cell lysates were fractionated using SDS-PAGE, transferred onto nitrocellulose membranes, and probed with primary antibodies. Proteins of interests were detected using IRDye® secondary antibodies (LICOR), visualized using Odyssey® CLx Imaging System (LICOR) and signals were quantified using Image Studio Software (Ver. 5.2).
Cell proliferation assays
Cells (MHCC-LM3 + shCTRL/shBCL9, Huh7.5 WT/122KO/122-BCL9DKO) were seeded into a 60mm plate (∼750,000 cells/plate) and allowed to attach overnight (24 h). Cells were then transfected with 1µg of pCMV-Flag Empty Vector control (E.V.) or pCMV-N-terminal FLAG tagged BCL9 overexpression vector (Flag-BCL9) using Lipofectamine™ 3000 following the manufacturer’s protocol (Thermo Fischer Scientific). After 6 h of incubation, media was replaced and cells were allowed to recover for 24 h. Cells were then seeded into a 96-well strip-well plate (8 wells/strip, Corning #9102). Cells were allowed to recover for 4 h, and CellTiter-Glo® was added to first strip-well following the manufacturer’s protocol (Promega) and assigned as time point 0 h. The luminescent supernatant was transferred to an opaque luminometer 96-well plate prior to measuring luminescence in quadruplicates. The same procedure was followed for subsequent time points (12, 24, 36, 48, 72, 96 h) and the data normalized to 0 h to calculate fold change.
Generation of BCL9 knockdown cells
shBCL9 cells were generated by infecting MHCC-LM3 cell with shBCL9 lentivirus pLKO.1 shRNA construct (Sigma cat# SHCLND-NM_004326). Briefly, lentiviral shBCL9 vector and shScrambled (Sigma cat#SHC002, TRCN0000005511) vectors were each individually transfected into H293TN cells. Viral supernatants were collected at 24 hours and 48 hours after transfection. Viral particles were condensed by PEG-it™ (SBI, LV810A-1) and transduced in MHCC-LM3 cells twice. Positive selection was carried out using puromycin treated media. BCL9 shRNA sequence: CCTCTGTGAATATCCCTGGAA.
Generation of miR-122/BCL9 double knockout cells
Huh7.5 miR-122 knockout cells (122KO) were generated as described in Luna et al., 2015. Huh7.5 miR-122/BCL9 double knockout cells (DKO) were generated by CRISPR technology from 122KO cells. Briefly, sgRNA of BCL9 (listed below) was cloned into pSpCas9(BB)-2A–GFP (PX458) (Addgene). sgBCL9-px458 targeting vector was transfected into Huh7.5 122KO cells by Lipofectamine™ 3000 (Thermo Fisher Scientific) following the manufacturer’ protocol. Transfected cells were sorted by GFP at 48hours after transfection. Individual clones were selected and expanded. sgRNA for BCL9: Forward: 5’-CACCgATTCTCCAATCAGGGTAAACAGG-3’ Reverse: 5’-AAACCCTGTTTACCCTGATTGGAGAATc-3’
TOP/FOP FLASH Assay
TOP and FOP plasmids were obtained from Addgene. Briefly, MHCC-LM3 cells (a kind gift from Dr. Bo Yu at Hangzhou Push-Kang Biotechnology) were transfected with scrambled RNA (25nM) or miR-122 mimic RNA (25nM). After 16 hours, cells were co-transfected with TOP or FOP luciferase reporter (80ng) and FLAG-WT-BCL9 or FLAG-MUT-BCL9 (400ng) expression vectors. pRLTK (Renilla luciferase expression plasmid) was co-transfected as a control for transfection efficiency normalization. Cells were harvested after 72 hours and Firefly (driven by TOP/FOP) and Renilla luciferase activities were measured using Dual-Luciferase® Reporter Assay System (Promega). Similarly, methods were repeated for BCL9 knockout/KD cells excluding transfection of RNA. Cells were transfected with TOP or FOP plasmid (100ng) and pRLTK (10ng). These cells were harvested after 48 hours and luciferase activity was measured. All treatments were performed in biological quadruplicates and statistical significance was calculated using Student T-Test.
mRNA-seq library construction
mRNA-seq libraries were prepared from Trizol extracted RNA following Illumina TruSeq protocols for poly-A selection, fragmentation, and adapter ligation. Multiplexed libraries were sequenced as 100nt paired-end runs on HiSeq-2000 or HiSeq 2500 sequencers.
RNA-seq bioinformatics pipeline
Analysis of mRNA-Seq was carried out using kallisto (Bray et al., 2016) and sleuth (Pimentel et al., 2016). Transcript indices for kallisto were created from FASTA files from Ensembl release 85 for mm10 and hg38 for all annotated cDNA and ncRNA transcripts. Quantification was done on paired end RNAseq data with the respective transcript index and 60 bootstraps (−b 60). The kallisto output was next analyzed with sleuth for differential expression testing. Importantly for human data, we leveraged the paired nature of normal and tumor samples per patient in our model matrix (∼patient + condition). Gene names and attributes were queried from Ensembl using biomaRt. For gene containing multiple transcripts, we chose the transcript with the highest mean expression across all samples as the representative example for that gene.
Argonaute CLIP library construction
Multiplexed Argonaute CLIP was performed as previously described (Luna et al., 2015). Sample crosslinking of liver tissue was as follows: mouse and human liver tissue was pulverized in a mortar and pestle cooled with liquid nitrogen. A small portion (20–50mg) of the resulting powder was saved for total RNA isolation and subsequent RNAseq. The remaining powder was crosslinked on a bed of dry ice with three irradations of 254nm UV-light at 400mJ/cm2 in a Stratlinker XL-1500 (Stratagene). All samples were stored at −80°C until lysis.
Protein A Dynabeads (Invitrogen) were washed 3x and resuspended in antibody binding (AB) buffer (AB: PBS, 0.02% Tween-20). Per liver sample, 100µl of beads was used. Beads were rotated with 12.5µl of bridging antibody (rabbit anti-mouse IgG, Jackson Immunoresearch) per 100µl of beads for 30 minutes at room temperature. Beads were then washed 3x with AB before adding 3µl of the pan-AGO antibody (2A8, EMD millipore) per 100µl beads. For IgG only antibody controls, AGO antibody was omitted. Beads were rotated at for 30 min at room temperature or overnight at 4°C. Just prior to IP, beads were washed in 1X PXL lysis buffer (1x PXL: 1X PBS tissue culture grade without magnesium or calcium, 0.1% SDS, 0.5% Sodium-deoxycholate, 0.5% NP-40, with protease inhibitors)
Frozen lysates of powdered liver tissue were prepared by adding 1ml of 1X PXL lysis buffer and triturating to disrupt cells. Lysates treated with 10µl DNAse (RQ1, Promega) for 5 minutes, followed by thermomixing at 37°C at 1100rpm. Samples were placed on ice and treated with RNaseI, first diluted to the indicated concentration by volume (e.g. 1:100 for “High RNase”, or 1:10,000 for “Low RNase) in lysis buffer and then added at 10µl per ml of lysate. Lysates underwent thermomixing again for 5 minutes at 37°C at 1100rpm before being treated with 10µl RNAsin RNase inhibitor (Promega) and spun at 4 °C on max speed of a table-top micro centrifuge for 30 minutes. Supernatants, along with any lipid layer, were harvested and mixed with PXL equilibrated antibody bound beads for immunoprecipitation. Samples were rotated with beads at 4 °C for 2–4 hours. Beads were washed sequentially twice each with 1X PXL, 5x PXL (same as 1X but using 5X PBS), and 1X PNK buffer (50mM Tris-HCl, pH 7.5, 10mM MgCl2, 0.5% NP-40).
To prepare RNA 3’ends for linker ligation, IPs were treated with alkaline phosphatase. Beads were resuspended in 80µl containing 1x dephosphorylation buffer, 3U of CIAP (Roche), RNasin inhibitor (Promega), and thermomixed for 20 minutes at 37°C, shaking at 1100rpm for 15s ever 2 minutes. Samples were washed as above sequentially in 1X PNK, 1X PNK plus 20mM EGTA, and twice with 1X PNK.
Radiolabeled linkers for 3’ linker ligation per sample were set up by preparing a T4 phosphonucleotide kinase (PNK, NED) reaction according to the manufacturers instructions using 2.5µl 32P-γ-ATP and 20pmol of a dephosphorylated and 3’ inverted ddT blocked L32 RNA linker (sequence: GUGUCAGUCACUUCCAGCGG/3InvdT/, IDT) and incubating for 30 minutes at 37°C. To drive the reaction to completion, 0.5µl of 1mM ATP was added and incubated for an additional 5 minutes. Linkers were purified from free nucleotides using G-25 columns following the manufacturers instructions (GE Healthcare). Ligation reactions on bead were prepared using Rnl1 RNA ligase (Fermentas) and 12pmol radiolabeled linker following the manufacturers instructions. Samples were incubated for one hour at 16°C, shakin g at 1100rpm for 15s every 4 minutes. After this hour, 60pmol cold linker and phosphorylated L32 (sequence: /5Phos/GUGUCAGUCACUUCCAGCGG/3InvdT/, IDT) was added and the reaction allowed to go overnight. The next day, beads were washed twice each with 1XPXL, 5X PXL, and 1XPNK. A final PNK reaction was performed on bead to restore the 5’ phosphate. To each sample an 80µl mix containing 1mM ATP, 1X T4 PNK buffer, 4µl T4 PNK and RNasin inhibitor. Samples were incubated for 20 minutes at 37°C, shaking at 1100rpm for 15s ever 2 minutes.
Beads were washed twice each with 1XPXL, 5X PXL, and 1XPNK. Protein was eluted off the beads by incubating with in 30µl of 1X LDS loading buffer (Invitrogen) with reducing agent for 10 minutes at 70°C, shaking at 1100rpm. Supernatants were run on Novex NuPAGE 8% Bis-Tris cels (Invitrogen) in SDS-MOPS buffer at 4°C. Radiolabeled protein RNA complexes were transferred to BA85 nitrocellulose. After transfer, the membrane was rinsed with RNAse-free PBS, and exposed to Biomax MR film (Kodak) at −70°C typically from 3 hours to up to 5 days.
Nitrocellulose membranes were aligned with the exposed film and regions of the membrane from low RNase IP lanes were excised corresponding to signal intensities between 110 and 150kDa. RNA was liberated from membrane fragments using 200µl of a proteinase K solution (Roche) diluted in PK buffer (100mM Tris-HCl, pH 7.5, 50mM NaCl, 10mM EDTA) to 4mg/ml and incubated for 20 minutes at 37°C, shaking at 1000rpm. RNA was further denatured by adding 200µl of PK buffer with 7M urea and incubating for 20 minutes at 37°C, shaking at 1000r pm. RNA fragments underwent acid phenol:chloroform extraction and were precipitated overnight at − 20°C. RNA was pelleted by spinning at max speed (>13,000rpm) in a table top centrifuge at 4°C, and washed twice with 75% ethanol. Following speedvac drying of the RNA, the pellet was dissolved in 6µl RNase-free water. The 5’ linker was ligated using Rnl1 (Fermantas) and 20 pmol of the 5’ linker (sequence: /5InvddT/AGGGAGGACGAUGCGGNNNNG, IDT) following the manufacturers instructions and in a total volume of 10µl. Samples were incubated at 16°C for 5 hours with intermittent shaking. Ligated RNA was DNase digested by adding 30µl RNase-free water, 5µl 10X RQ1 buffer, 2.5µl RQ1 DNase and 2.5µl RNasin inhibitor; and incubating at 37°C for 20 minutes. Samples then underwent phenol chloroform extraction as above.
RT-PCR of adapter ligated CLIPped RNA was carried out using Superscript III (Invitrogen) following the manufacturer’s instructions using the DP3 primer for reverse transctiption (sequence: CCGCTGGAAGTGACTGACAC). PCR was performed immediately after RT using 27µl Accuprime Pfx (Invitrogen), 0.15µl DP5 primer (at 100 pmol/µl, sequence: AGGGAGGACGATGCGG), 0.15µl DP3 primer (at 100 pmol/µl) , and 4µl of the RT reaction, for a total of 4 reactions per sample. PCR conditions were 95°C for 2 minutes, 20–35 cycles of 95°C for 20s de nature, 58°C for 30s anneal, and 68°C for 30s extension. Reactions were taken out of the thermocycler following extension starting at 22 cycles, every two or three cycles. Samples were run on 10% denaturing PAGE gels for size selection from the cycle number displaying the least signal saturation, and DNA fragments were excised between 80–150bp. DNA was extracted by soaking gel slices in DNA diffusion buffer (0.5M ammonium acetate, 10mM Mg-acetate, 1mM EDTA, 0.1% SDS) and incubating at 50°C for 30 minutes, shaking at 1200rpm. DNA was recovered from the filtrate using Qiaquick gel purification (Qiagen) and resuspended in 30µl. A short 2nd PCR (4–7 cycles) was used to append illumina adapters as in (Luna et al., 2015). Samples were sequenced on Hiseq2000 or Hiseq2500 machines as 100nt SE runs.
Ago-CLIP bioinformatics pipeline
Analysis of AGO-CLIP data was carried out similar to previous work (Luna et al., 2015; Moore et al., 2014; Scheel et al., 2016). Data processing was carried out using the Galaxy suite of bioinformatics tools (http://galaxy.psu.edu (Goecks et al., 2010)) and the UCSC genome and table browsers (http://genome.ucsc.edu (Kent et al., 2002)), in addition to in-house tools. FASTQ sequence files were first filtered in Galaxy for quality such that 80% of the read contained a mean score of 20. Reads were then collapsed on sequence to remove sequencing and PCR duplicates. Barcoded samples, if any, were split at this stage using the barcode splitter. 5’-degenerate linker sequences were removed for later estimation of unique binding events. The L32 3’-adapter sequence (GTGTCAGTCACTTCCAGCGG) was also removed if present using the clip adapters tool. Reads shorter than 18nt were discarded. This final set of groomed reads represented the input for subsequent alignment.
Groomed reads were mapped to mm10 for mouse samples and hg38 for human samples using Bowtie2 with default parameters, permitting up to two mismatches. Mapped reads were collapsed on degenerate linker sequence as previously described (Moore et al., 2014). Peak finding to define clusters of reads corresponding to miRNA binding loci was carried out with the HOMER suite of tools (Heinz et al., 2010); specifically, using the findPeaks tool on all mapped reads with the following parameters: -style factor -L 2 -localSize 10000 -strand separate -minDist 50 -size 10 -fragLength 25. The resulting 10nt window-sized “peaks” from this output were extended +/− 32 nucleotides for subsequent seed searching. Any overlapping peaks were combined to generate clusters for seed searching.
Argonaute-CLIP, motif enrichment in miR-122 dependent peaks
Log2 fold changes comparing WT versus KO or Normal versus Tumor conditions were calculated as follows. A pseudo-count of 1 was added to all clusters to allow for incorporation of conditions with zero reads. After binning by biologic complexity, where we stipulate that a cluster must be composed of reads derived form at least 4 libraries, the sum total number of reads per cluster per condition was normalized to the read depth for that condition. Dividing these normalized CLIP abundance measurements per condition and log2 transforming, we arrived at changes in CLIP binding due to KO or Tumor conditions. Peaks displaying WT/KO L2FC values of greater than 5 were then used for motif enrichment. Peak coordinates were extended by 15nt on either side and used for Multiple Em for Motif Elicitation (MEME) analysis (Bailey and Elkan, 1994) of enriched 8mer sequences. Final count numbers per enriched motif category were determined after seed searching.
Argonaute-CLIP, defining miRNA abundance by chimeras
To obtain a ranked list of miRNAs present in liver samples that were engaged with AGO and a target RNA, we turned quantifying miRNAs in miRNA:target RNA chimeras (Moore et al., 2015; Scheel et al., 2016). Sequence reads containing miRNA sequences were identified by reverse mapping mature miRNA sequences against each sample library using Bowtie2 with the following changes to default parameters: 5’ trimming (−5=1), maximum mismatches allowed in the seed (−n=1); seed length (−l=18). This permitted mapping of miRNAs with a maximum of 1 mutation in the first 18 nts and 5’ or 3’ truncation of 1 nt. Downstream sequences from miRNAs within reads were extracted in “R” using the coordinates supplied by Bowtie. Resulting downstream target sequences of >=19 nts were then mapped to mm10 or hg38 reference sequences. PCR duplicates were collapsed on the basis of genomic coordinates and a degenerate barcode present in the 5’ RNA linker as above. Chimeric fragments mapping to miRNA precursor genes were removed and miRNA family nomenclature (miRbase) was added. The resulting list of miRNA chimeras was then summed by miRNA. Importantly, for ranking we used only WT mice or normal human liver tissue to determine proportional abundances of AGO bound and target engaged miRNAs.
Argonaute-CLIP, seed searching across biologic replicates and annotation of sites
Ago cluster regions (10nt peaks +/− 32nts) were then searched using the ranked list of miRNA seeds determined from chimeras where we selected perfect base pairing for miRNA seeds to targets following 8mer, 7mer-A1, 7mer-m8, and 6mer(2–7) pairing rules (Bartel, 2009). G-bulged miR-122 targets were determined by searching for RUGACUCC motifs in clusters. All clusters were annotated against genes from Ensembl release 85 for mouse (mm10) or human (hg38). Clusters were further intersected with RepeatMasker annotations (repeatmasker.org) for each respective genome build for the following repeat or RNA categories: tRNA, rRNA, LINEs, LTRs, low complexity regions, simple repeats, SINEs, and satellite repeats. Additionally, we defined up to 10kb downstream of annotated 3’UTRs as encompassing possible 3’UTR extensions. Clusters overlapping multiple annotations were called randomly for initial analysis. For final analysis we prioritized annotations based on the following heuristic: 3’UTRs > CDS > 5’UTRs > introns > 10kb downstream of 3’UTRs > ncRNA > RepeatMasker regions > deep intergenic regions.
Conservation analysis
Interval coordinates of miRNA seed target sites within CLIP clusters were intersected with PhyloP scores (Pollard et al., 2010) for available mammalian nucleotides from the UCSC table browser. The resulting per-nucleotide scores were averaged across the core 6mer and plotted. For site level comparisons, human:mouse chain alignments were extracted from UCSC. Mouse 6mer coordinates for CLIP sites for the top 10 miRNA familes were then lifted over to human (using LiftOver) where we required all bases to remap. Coordinates that failed to remap due to deletions were labeled as such. Sequences from remapped coordinates were then extracted and compared to original mouse sequences.
Statistical tests
Statistical analysis was carried out in Graphpad Prism or in R. Two-sided KS-Test was employed for CDFs to test both for goodness of fit and a difference in means. Mann Whitney U-test (non parametric t-tests) were used for datasets for which no normality assumption could be made, Student’s t-test were used otherwise.
Analysis of TCGA data
The Cancer Genome Atlas was queried for expression data for liver cancer hepatocellular carcinoma (LIHC) at firebrowse.org. We extracted RSEM calculated Z-scores for shared primary targets, defined as genes with miR-122 binding sites in either 3’UTRs or CDS regions found in both mouse and human CLIP data. Sites with more than one miR-122 site were collapsed such that no gene was represented more than once. RSEM calculated expression values were likewise extracted for shared targets. RPKM calculated miRNA expression was also queried through firebrowse.org. Survival data was queried from LIHC data at the cBioPortal (Cerami et al., 2012; Gao et al., 2013) for all select targets with z-scores greater than 2 (EXP >2). Linear Regression modeling analysis of highly predictive miR-122 targets was calculated using linear modeling function lm() in R version 3.3. Correlation coefficients for all targets were calculated using cor.test() function. The results shown here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Analysis of published RNAseq datasets
Processed count files for datasets GSE77509 and GSE77314 were downloaded from GEO database (https://www.ncbi.nlm.nih.gov/geo/). For GSE77509, we focused on Normal (N) and Tumor (T) samples. Expression counts per sample per gene were log2 transformed and plotted in R. Significance was calculated with Wilcox.test() function.
Quantification and Statistical Analysis
All data are shown in figures are represented as mean ± standard deviation (error bars)
Data and Software Availability
The accession numbers for the sequencing data reported in this paper are GEO: GSE97058, GSE97060, and GSE97056. Other raw data files are available via Mendeley Data: http://dx.doi.org/10.17632/v4wp2mmztd.2.
Supplementary Material
Highlights.
Genome-wide miRNA binding sites were elucidated in miR-122 KO mice and human HCC
Widespread, non-canonical, and species-specific miR-122 binding was observed
miR-122 binding sites in human HCC revealed a core set of conserved targets
Conserved miR-122 targets, most notably BCL9, correlated with human HCC survival
Acknowledgments
We would like to thank Vivek Chowdhary, Huban Kutay, Chelsey Williams, and Maria Guerrero for technical assistance, Lianbo Yu for database access assistance, Troels K.H. Scheel, Chris Y. Park and members of the Darnell and Rice labs for thoughtful discussion. We also acknowledge the New York Genome Center for high throughput sequencing and the OSUCCC sequencing core for Sanger sequencing. We would like to acknowledge the individuals and institutions that have provided data for The Cancer Genome Atlas Liver Hepatocellular Carcinoma (TCGA-LIHC) dataset. This study was supported, in part, by grants R01CA193244, R01CA086978 from NIH and a Pelotonia Idea grant to K.G. Kun-yu Teng is a Pelotonia fellow. This work was also in part supported by grants to R.B.D. from the National Institutes of Health (R01NS034389, R01NS081706, and R35NS097404) and to C.M.R by NIAID (R01AI116943). R.B.D. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
J.M.L. and J.M.B. designed and performed CLIP experiments, analyzed and interpreted data, and drafted the manuscript. K.Y.T. designed and performed validation experiments, analyzed and interpreted data, and drafted the manuscript. H.S. designed experiments. M.J.M. performed initial chimera analyses. C.M.R., R.B.D. and K.G. designed experiments, interpreted data, and revised the manuscript. All authors reviewed the results and contributed to writing the manuscript.
References
- Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- Bai S, Nasser MW, Wang B, Hsu S-H, Datta J, Kutay H, Yadav A, Nuovo G, Kumar P, Ghoshal K. MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. Journal of Biological Chemistry. 2009;284:32015–32027. doi: 10.1074/jbc.M109.016774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosson AD, Zamudio JR, Sharp PA. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Molecular Cell. 2014;56:347–359. doi: 10.1016/j.molcel.2014.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Budhu A, Jia H-L, Forgues M, Liu C-G, Goldstein D, Lam A, Zanetti KA, Ye Q-H, Qin L-X, Croce CM, et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology. 2008;47:897–907. doi: 10.1002/hep.22160. [DOI] [PubMed] [Google Scholar]
- Burchard J, Zhang C, Liu AM, Poon RTP, Lee NPY, Wong K-F, Sham PC, Lam BY, Ferguson MD, Tokiwa G, et al. microRNA-122 as a regulator of mitochondrial metabolic gene network in hepatocellular carcinoma. Molecular Systems Biology. 2010;6:1–12. doi: 10.1038/msb.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Hannon GJ, Darnell RB. An alternative mode of microRNA target recognition. Nat. Struct. Mol. Biol. 2012;19:321–327. doi: 10.1038/nsmb.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulouarn C, Factor VM, Andersen JB, Durkin ME, Thorgeirsson SS. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28:3526–3536. doi: 10.1038/onc.2009.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deka J, Wiedemann N, Anderle P, Murphy-Seiler F, Bultinck J, Eyckerman S, Stehle J-C, André S, Vilain N, Zilian O, et al. Bcl9/Bcl9l are critical for Wnt-mediated regulation of stem cell traits in colon epithelium and adenocarcinomas. Cancer Research. 2010;70:6619–6628. doi: 10.1158/0008-5472.CAN-10-0148. [DOI] [PubMed] [Google Scholar]
- Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Molecular Cell. 2014;54:766–776. doi: 10.1016/j.molcel.2014.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler R, McGeary SE, Title AC, Agarwal V, Bartel DP, Stoffel M. Impact of MicroRNA Levels, Target-Site Complementarity, and Cooperativity on Competing Endogenous RNA-Regulated Gene Expression. Molecular Cell. 2016;64:565–579. doi: 10.1016/j.molcel.2016.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Liu X, Wu Y, Zhu J, Tang Y. Essential role of STX6 in esophageal squamous cell carcinoma growth and migration. Biochem Biophys Res Commun. 2016;472:60–67. doi: 10.1016/j.bbrc.2016.02.061. [DOI] [PubMed] [Google Scholar]
- Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metabolism. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Farh KK-HK, Grimson AA, Jan CC, Lewis BPB, Johnston WKW, Lim LPL, Burge CBC, Bartel DPD. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- Friedman RCR, Farh KK-HK, Burge CBC, Bartel DPD. Most mammalian mRNAs are conserved targets of microRNAs. Genes Dev. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1–pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatfield D, Le Martelot G, Vejnar CE, Gerlach D, Schaad O, Fleury-Olela F, Ruskeepaa A-L, Oresic M, Esau CC, Zdobnov EM, et al. Integration of microRNA miR-122 in hepatic circadian gene expression. Genes Dev. 2009;23:1313–1326. doi: 10.1101/gad.1781009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach D, Kriventseva EV, Rahman N, Vejnar CE, Zdobnov EM. miROrtho: computational survey of microRNA genes. Nucleic Acids Res. 2009;37:D111–D117. doi: 10.1093/nar/gkn707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J Galaxy Team. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramantieri L, Ferracin M, Fornari F, Veronese A, Sabbioni S, Liu C-G, Calin GA, Giovannini C, Ferrazzi E, Grazi GL, et al. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Research. 2007;67:6092–6099. doi: 10.1158/0008-5472.CAN-06-4607. [DOI] [PubMed] [Google Scholar]
- Haack TB, Makowski C, Yao Y, Graf E, Hempel M, Wieland T, Tauer U, Ahting U, Mayr JA, Freisinger P, et al. Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J. Inherit. Metab. Dis. 2012;35:943–948. doi: 10.1007/s10545-012-9513-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano MJ, Jungkamp A-C, Munschauer M, et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Molecular Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu S-H, Delgado ER, Otero PA, Teng K-Y, Kutay H, Meehan KM, Moroney JB, Monga JK, Hand NJ, Friedman JR, et al. MicroRNA-122 Regulates Polyploidization in the Murine Liver. Hepatology. 2016;64:599–615. doi: 10.1002/hep.28573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu S-H, Wang B, Kota J, Yu J, Costinean S, Kutay H, Yu L, Bai S, La Perle K, Chivukula RR, et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Invest. 2012;122:2871–2883. doi: 10.1172/JCI63539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu S-H, Yu B, Wang X, Lu Y, Schmidt CR, Lee RJ, Lee LJ, Jacob ST, Ghoshal K. Cationic lipid nanoparticles for therapeutic delivery of siRNA and miRNA to murine liver tumor. Nanomedicine. 2013;9:1169–1180. doi: 10.1016/j.nano.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jens M, Rajewsky N. Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat Rev Genet. 2014:1–14. doi: 10.1038/nrg3853. [DOI] [PubMed] [Google Scholar]
- Jiang J, Gusev Y, Aderca I, Mettler TA, Nagorney DM, Brackett DJ, Roberts LR, Schmittgen TD. Association of MicroRNA expression in hepatocellular carcinomas with hepatitis infection, cirrhosis, and patient survival. Clinical Cancer Research. 2008;14:419–427. doi: 10.1158/1078-0432.CCR-07-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Research. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima SS, Gatfield DD, Esau CCC, Green CBC. MicroRNA-122 modulates the rhythmic expression profile of the circadian deadenylase Nocturnin in mouse liver. PLoS ONE. 2010;5:e11264–e11264. doi: 10.1371/journal.pone.0011264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- Kutay H, Bai S, Datta J, Motiwala T, Pogribny I, Frankel W, Jacob ST, Ghoshal K. Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J Cell Biochem. 2006;99:671–678. doi: 10.1002/jcb.20982. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Liang H-W, Wang N, Wang Y, Wang F, Fu Z, Yan X, Zhu H, Diao W, Ding Y, Chen X, et al. Hepatitis B virus-human chimeric transcript HBx-LINE1 promotes hepatic injury via sequestering cellular microRNA-122. J Hepatol. 2016;64:278–291. doi: 10.1016/j.jhep.2015.09.013. [DOI] [PubMed] [Google Scholar]
- Liu G, Hou G, Li L, Li Y, Zhou W, Liu L. Potential diagnostic and prognostic marker dimethylglycine dehydrogenase (DMGDH) suppresses hepatocellular carcinoma metastasis in vitro and in vivo. Oncotarget. 2016;7:32607–32616. doi: 10.18632/oncotarget.8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb GB, Khan AA, Canner D, Hiatt JB, Shendure J, Darnell RB, Leslie CS, Rudensky AY. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Molecular Cell. 2012;48:760–770. doi: 10.1016/j.molcel.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna JM, Scheel TKH, Danino T, Shaw KS, Mele A, Fak JJ, Nishiuchi E, Takacs CN, Catanese MT, de Jong YP, et al. Hepatitis C Virus RNA Functionally Sequesters miR-122. Cell. 2015;160:1099–1110. doi: 10.1016/j.cell.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Hagner K. Evolutionary meandering of intermolecular interactions along the drift barrier. Proceedings of the National Academy of Sciences. 2015;112:E30–E38. doi: 10.1073/pnas.1421641112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani M, Carrasco DE, Zhang Y, Takada K, Gatt ME, Dutta-Simmons J, Ikeda H, Diaz-Griffero F, Pena-Cruz V, Bertagnolli M, et al. BCL9 promotes tumor progression by conferring enhanced proliferative, metastatic, and angiogenic properties to cancer cells. Cancer Research. 2009;69:7577–7586. doi: 10.1158/0008-5472.CAN-09-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, et al. Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci USA. 1999;96:1603–1608. doi: 10.1073/pnas.96.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manthey KC, Chew YC, Zempleni J. Riboflavin deficiency impairs oxidative folding and secretion of apolipoprotein B-100 in HepG2 cells, triggering stress response systems. J. Nutr. 2005;135:978–982. doi: 10.1093/jn/135.5.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science. 2012;338:1593–1599. doi: 10.1126/science.1228186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Scheel TKH, Luna JM, Park CY, Fak JJ, Nishiuchi E, Rice CM, Darnell RB. miRNA-target chimeras reveal miRNA 3’-end pairing as a major determinant of Argonaute target specificity. Nature Communications. 2015;6:8864. doi: 10.1038/ncomms9864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Zhang C, Gantman EC, Mele A, Darnell JC, Darnell RB. Mapping Argonaute and conventional RNA-bindingprotein interactions with RNA at single-nucleotideresolution using HITS-CLIP and CIMS analysis. Nat Protoc. 2014;9:263–293. doi: 10.1038/nprot.2014.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel HJ, Bray N, Puente S, Melsted P, Pachter L. Differential analysis of RNA-Seq incorporating quantification uncertainty (Cold Spring Harbor Labs Journals) 2016 doi: 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Research. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reverter M, Rentero C, Garcia-Melero A, Hoque M, Vilà de Muga S, Alvarez-Guaita A, Conway JRW, Wood P, Cairns R, Lykopoulou L, et al. Cholesterol regulates Syntaxin 6 trafficking at trans-Golgi network endosomal boundaries. CellReports. 2014;7:883–897. doi: 10.1016/j.celrep.2014.03.043. [DOI] [PubMed] [Google Scholar]
- Scheel TKH, Luna JM, Liniger M, Nishiuchi E, Rozen-Gagnon K, Shlomai A, Auray G, Gerber M, Fak J, Keller I, et al. A Broad RNA Virus Survey Reveals Both miRNA Dependence and Functional Sequestration. Cell Host Microbe. 2016;19:409–423. doi: 10.1016/j.chom.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genetics. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakral S, Ghoshal K. miR-122 is a unique molecule with great potential in diagnosis, prognosis of liver disease, and therapy both as miRNA mimic and antimir. Curr Gene Ther. 2015;15:142–150. doi: 10.2174/1566523214666141224095610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat. Genetics. 2016;17:272–283. doi: 10.1038/nrg.2016.20. [DOI] [PubMed] [Google Scholar]
- Tsai W-C, Hsu PW-C, Lai T-C, Chau G-Y, Lin C-W, Chen C-M, Lin C-D, Liao Y-L, Wang J-L, Chau Y-P, et al. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 2009;49:1571–1582. doi: 10.1002/hep.22806. [DOI] [PubMed] [Google Scholar]
- Tsai W-C, Hsu S-D, Hsu C-S, Lai T-C, Chen S-J, Shen R, Huang Y, Chen H-C, Lee C-H, Tsai T-F, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Invest. 2012;122:2884–2897. doi: 10.1172/JCI63455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- Wang Q, Wang N, Shen D, Sun X, Cao X, Wu D. Downregulation of microRNA-122 promotes proliferation, migration, and invasion of human hepatocellular carcinoma cells by activating epithelial–mesenchymal transition. Ott. 2016:2035–13. doi: 10.2147/OTT.S92378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W-Y, Zhang H-F, Wang L, Ma Y-P, Gao F, Zhang S-J, Wang L-C. miR-21 expression predicts prognosis in hepatocellular carcinoma. Clin Res Hepatol Gastroenterol. 2014;38:715–719. doi: 10.1016/j.clinre.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Wielenga VJ, Smits R, Korinek V, Smit L, Kielman M, Fodde R, Clevers H, Pals ST. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am. J. Pathol. 1999;154:515–523. doi: 10.1016/S0002-9440(10)65297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski JA, Yin J, Teuscher KB, Zhang M, Ji H. Structure-Based Design of 1,4-Dibenzoylpiperazines as β-Catenin/B-Cell Lymphoma 9 Protein-Protein Interaction Inhibitors. ACS Med Chem Lett. 2016;7:508–513. doi: 10.1021/acsmedchemlett.5b00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhang R, Shen Y, Liu G, Lu X, Wu CI. The evolution of evolvability in microRNA target sites in vertebrates. Genome Research. 2013;23:1810–1816. doi: 10.1101/gr.148916.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhu X, Wu L, Yang R, Yang Z, Wang Q, Wu F. MicroRNA-122 suppresses cell proliferation and induces cell apoptosis in hepatocellular carcinoma by directly targeting Wnt/beta-catenin pathway. Liver Int. 2012;32:752–760. doi: 10.1111/j.1478-3231.2011.02750.x. [DOI] [PubMed] [Google Scholar]
- Xu W, Zhou W, Cheng M, Wang J, Liu Z, He S, Luo X, Huang W, Chen T, Yan W, et al. Hypoxia activates Wnt/β-catenin signaling by regulating the expression of BCL9 in human hepatocellular carcinoma. Sci. Rep. 2017;7:40446. doi: 10.1038/srep40446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Katsuno T, Tamura A, Tsukita S. Tight junction-based epithelial microenvironments and cell proliferation. Oncogene. 2008;27:6930–6938. doi: 10.1038/onc.2008.344. [DOI] [PubMed] [Google Scholar]
- Yang Y, Chen L, Gu J, Zhang H, Yuan J, Lian Q, Lv G, Wang S, Wu Y, Yang Y-CT, et al. Recurrently deregulated lncRNAs in hepatocellular carcinoma. Nature Communications. 2017;8:14421. doi: 10.1038/ncomms14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.