

Editor’s Note
As part of our continuing effort to highlight innovative approaches to improving the health and environment of communities, the Journal is pleased to publish a bimonthly column from the Agency for Toxic Substances and Disease Registry (ATSDR). ATSDR is a federal public health agency of the U.S. Department of Health and Human Services (HHS) and shares a common office of the Director with the National Center for Environmental Health (NCEH) at the Centers for Disease Control and Prevention (CDC). ATSDR serves the public by using the best science, taking responsive public health actions, and providing trusted health information to prevent harmful exposures and diseases related to toxic substances.
The purpose of this column is to inform readers of ATSDR’s activities and initiatives to better understand the relationship between exposure to hazardous substances in the environment and their impact on human health and how to protect public health. We believe that the column will provide a valuable resource to our readership by helping to make known the considerable resources and expertise that ATSDR has available to assist communities, states, and others to assure good environmental health practice for all is served.
The conclusions of this column are those of the author(s) and do not necessarily represent the views of ATSDR, CDC, or HHS.
Kevin Horton is chief of the Environmental Health Surveillance Branch within the Division of Toxicology and Human Health Sciences at ATSDR. Wendy Kaye is a senior epidemiologist at McKing Consulting Corporation. Laurie Wagner is a research associate at McKing Consulting Corporation.
Introduction
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a progressive, fatal neurodegenerative disorder that causes the loss of motor neurons, typically resulting in paralysis, respiratory failure, and death within 3–5 years of symptom onset (Mitsumoto, Chad, & Pioro, 1998). Despite ALS being initially identified in 1869, the actual pathogenesis and cause remain unknown and there is currently no cure. An estimated 5%–10% of cases are attributed to heredity, while the remaining 90%–95% are of unknown etiology (Andersen, 2006). For these latter sporadic cases, many potential risk factors have been explored such as smoking and alcohol consumption; exposures to heavy metals, pesticides, and volatile organic compounds; head trauma; and occupational exposures (Oskarsson, Horton, & Mitsumoto, 2015). The most consistently known risk factors for sporadic cases are being male, Caucasian, and older in age (Chiò et al., 2013; Hirtz et al., 2007; Mehta et al., 2016).
New advances in science, particularly in the area of high-dimensional biology (e.g., genomics, proteomics, metabolomics, transcriptomics), are leading to an improved understanding of diseases like ALS on a molecular level (Horgan & Kenny, 2011). To keep up with “omic” technologies, there is a critical need for high-quality biospecimens for analysis (e.g., blood, serum, tissue). Biospecimens are a vital resource for studying biochemical and genetic differences among diseased and nondiseased individuals (Vaught et al., 2011). Moreover, biospecimens are useful for current and future etiological research studies and for the potential development of new diagnostic markers and therapeutic targets. Biospecimen analysis has already proven useful in the discovery of important genes related to ALS and other motor neuron diseases (Renton, Chiò, & Traynor, 2014).
Because ALS is considered a rare disease, with an estimated U.S. prevalence rate of 5.0 cases per 100,000 population (Mehta et al., 2016) and an incidence rate of 1.5 per 100,000 person-years (Wagner et al., 2015), obtaining biospecimens on an ongoing basis can be challenging for researchers. While there are some local and regional biorepositories (i.e., facilities that collect and store samples of biological material) that include biospecimens from persons with ALS, these biorepositories tend to be limited by specimen type and availability, sample size, geographical coverage, and demographic characteristics. To help address this scientific gap, the Agency for Toxic Substances and Disease Registry (ATSDR) recently conducted a 4-year pilot study to test the practicality, utility, and feasibility of creating a national biorepository of pre and postmortem biospecimens from persons with ALS enrolled in ATSDR’s congressionally-mandated National ALS Registry. Briefly, the purpose of the National ALS Registry (Registry) is to quantify the incidence and prevalence of ALS in the U.S., describe the demographics of persons with ALS, and examine risk factors for the disease (Antao & Horton, 2012; Horton, Mehta, & Antao, 2014; Mehta et al., 2016).
Biorepository Pilot Study
During 2011–2015, ATSDR conducted a study to pilot methods for collecting and banking biospecimens from participants in ATSDR’s Registry. Throughout this pilot study, ATSDR held a series of expert panel meetings to solicit guidance and recommendations on topics such as sample types, storage, and biospecimen governance. The expert panel included prominent neurologists, laboratorians, researchers, and bioethicists from around the country. Once the pilot study protocol was developed and approved by ATSDR and the institutional review board, consent, recruitment, and specimen collection began in 2013 on a nationally representative sample of patients enrolled in the Registry. The pilot study included two specimen collection components: biological specimens from living participants (in-home) and postmortem specimens. The in-home collection aimed to enroll approximately 300 participants, from whom specimens would be collected on two occasions by trained phlebotomists, approximately six months apart. In-home samples collected included blood, urine, hair, and nails (saliva was collected from those who could not provide a blood sample). The postmortem component aimed to enroll 30 participants, who could have also participated in the in-home study. Postmortem collection included brain, spinal cord, cerebrospinal fluid, muscle, bone, and skin specimens for the creation of cell lines.
There were 330 in-home participants (~61% male), from all 50 states, who completed the first specimen collection (Figure 1). Of these, 309 (93.6%) provided at least one blood specimen. Most participants (80.6%) were able to provide specimens at both collection appointments (Table 1). The reasons for not completing the second draw included death (n = 36), too ill or unable to contact (n = 9), and no longer interested or scheduling difficulties (n = 13). DNA and RNA were extracted from blood and other blood specimens were processed into aliquots of plasma, serum, and whole blood. There were 30 postmortem participants with equal numbers of males and females. Eighteen postmortem participants have donated tissues as of May 31, 2016. The length of time in the study for these participants from date of consent to date of death ranged from 1–24 months. The age at death for the deceased participants ranged from 43–76 years of age.
FIGURE 1.
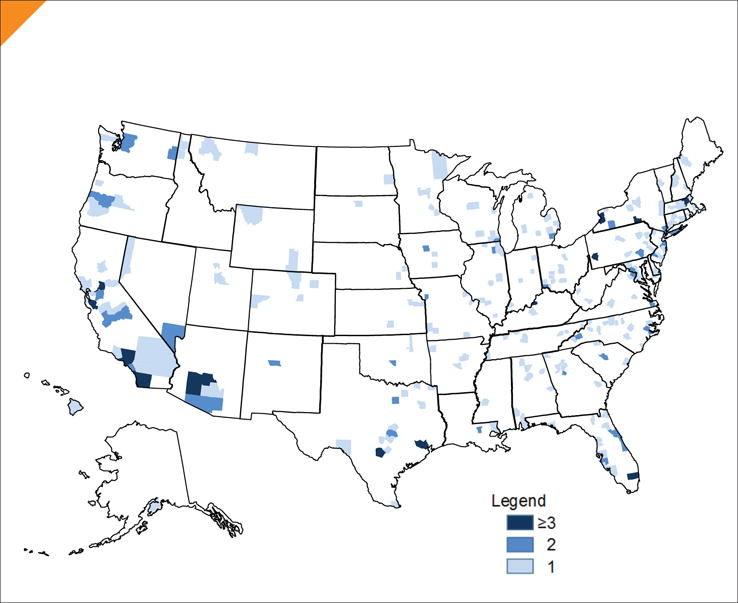
Map of In-Home Participants
TABLE 1.
Number of Specimen Types Collected
| Specimen Type | # of 1st Collection Participants (N = 330) | # of 2nd Collection Participants (N = 266) |
|---|---|---|
| Whole blood (plasma, buffy coat, RBC) | 309 | 255 |
| Whole blood (metals free) | 308 | 248 |
| Plain blood (serum) | 302 | 246 |
| PAXgene1 (RNA) | 303 | 248 |
| PAXgene2 (RNA) | 303 | 247 |
| Urine | 321 | 256 |
| Hair | 310 | 264 |
| Nails | 326 | 271 |
| Saliva | 15 | 0 |
RBC = red blood cells.
Creating a geographically diverse biorepository for this pilot study had unique challenges. Recruitment was slower than expected, finding reliable phlebotomists across the country was difficult, and there were unexpected issues related to shipping specimens, including higher than average temperatures and mechanical failure. In addition, after the first specimens were collected, there was a larger than expected number of individuals who could not participate in the second specimen collection (i.e., were too sick or deceased), thereby decreasing the number of paired specimens. ATSDR was able, however, to recruit the target sample size and process the various specimen types.
The pilot study, which concluded in September 2015, demonstrated that a nationwide collection of pre and postmortem biospecimens from Registry enrollees is feasible, warranted, and can be done in a cost-effective manner. Based on these finding, the expert panel recommended that ATSDR establish a permanent biorepository as part of the Registry (McKing Consulting Corporation, 2015). In the meantime, biospecimens collected through the pilot study are available for researchers to use by contacting the Registry (alsbiorepository@secure.mcking.com).
The National ALS Biorepository
ATSDR is currently moving forward with integrating the National ALS Biorepository (Biorepository) into the Registry. Once the appropriate governmental approvals have been obtained, ATSDR anticipates launching the Biorepository in fall 2016. Although much of the pilot study’s standard operating procedures will be used in the Biorepository, changes based upon lessons learned and recommendations from the expert panel will be implemented, such as increasing/decreasing biospecimen collection type based upon researcher demand, modifying collection frequency, and marketing the Biorepository to ensure maximum use. Once the Biorepository is launched, researchers will be able to complete an online application through the Registry Web site to request samples.
By design, the Biorepository is significantly different from other biorepositories in that it links risk factor data (e.g., occupational, military, and smoking history) with biospecimens, is nationally representative, and uses phlebotomists for in-home collection.
Conclusions
The Registry is the largest cohort of ALS patients in the U.S. Therefore, the Registry is the ideal group in which to collect biologic specimens on a large geographically representative group of persons with ALS. Furthermore, combining biospecimens with risk factor data currently being collected through the Registry is a unique and invaluable resource to help researchers in the U.S. and abroad better understand the etiology of ALS. More information on the Registry and Biorepository can be found at www.cdc.gov/als.
Acknowledgments
The authors thank the many people living with ALS across the U.S. for their generosity in contributing valuable epidemiological data and biospecimens to the Registry.
Biographies

D. Kevin Horton, MSPH, DrPH

Wendy Kaye, PhD
Contributor Information
D. Kevin Horton, Agency for Toxic Substances and Disease Registry.
Wendy Kaye, McKing Consulting Corporation.
Laurie Wagner, McKing Consulting Corporation.
References
- Andersen PM. Amyotrophic lateral sclerosis associated with mutations in the CnZn superoxide dismutase gene. Current Neurology and Neuroscience Reports. 2006;6(1):37–46. doi: 10.1007/s11910-996-0008-9. [DOI] [PubMed] [Google Scholar]
- Antao VC, Horton DK. The National Amyotrophic Lateral Sclerosis (ALS) Registry. Journal of Environmental Health. 2012;75(1):28–30. [PMC free article] [PubMed] [Google Scholar]
- Chiò A, Logroscino G, Traynor BJ, Simeone JC, Goldstein LA, White LA. Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology. 2013;41(2):118–130. doi: 10.1159/000351153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirtz D, Thurman DJ, Gwinn-Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R. How common are the “common” neurologic disorders? Neurology. 2007;68(5):326–337. doi: 10.1212/01.wnl.0000252807.38124.a3. [DOI] [PubMed] [Google Scholar]
- Horgan RP, Kenny LC. ‘Omic’ technologies: Genomics, transcriptomics, proteomics and metabolomics. The Obstetrician & Gynaecologist. 2011;13(3):189–195. [Google Scholar]
- Horton K, Mehta P, Antao VC. Quantifying a nonnotifiable disease in the United States: The National Amyotrophic Lateral Sclerosis Registry model. The Journal of the American Medical Association. 2014;312(11):1097–1098. doi: 10.1001/jama.2014.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKing Consulting Corporation. National ALS Biorepository pilot study: Summary report. Atlanta, GA: Author; 2015. Retrieved from https://wwwn.cdc.gov/als/Download/ALS%20Biorepository%20Pilot%20Study%20Summary%20Report.pdf. [Google Scholar]
- Mehta P, Kaye W, Bryan L, Larson T, Copeland T, Wu J, Horton DK. Prevalence of amyotrophic lateral sclerosis—United States, 2012–2013. Morbidity and Mortality Weekly Report. 2016;65(8):1–12. doi: 10.15585/mmwr.ss6508a1. Retrieved from http://www.cdc.gov/mmwr/volumes/65/ss/ss6508a1.htm. [DOI] [PubMed] [Google Scholar]
- Mitsumoto H, Chad DA, Pioro EP, editors. Differential diagnoses in amyotrophic lateral sclerosis. Philadelphia, PA: F.A. Davis Company; 1998. [Google Scholar]
- Oskarsson B, Horton DK, Mitsumoto H. Potential environmental factors in amyotrophic lateral sclerosis. Neurologic Clinics. 2015;33(4):877–888. doi: 10.1016/j.ncl.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nature Neuroscience. 2014;17(1):17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaught J, Rogers J, Myers K, Lim MD, Lockhart N, Moore H, Compton C. An NCI perspective on creating sustainable biospecimen resources. Journal of the National Cancer Institute Monographs. 2011;42:1–7. doi: 10.1093/jncimonographs/lgr006. [DOI] [PubMed] [Google Scholar]
- Wagner L, Rechtman L, Jordan H, Ritsick M, Sanchez M, Sorenson E, Kaye W. State and metropolitan area-based amyotrophic lateral sclerosis (ALS) surveillance. Amyotrophic Lateral Sclerosis Frontotemporal Degeneration. 2015;17(1–2):128–134. doi: 10.3109/21678421.2015.1074699. [DOI] [PMC free article] [PubMed] [Google Scholar]