

Abstract
Ribosomally-synthesized and Post-translationally-modified Peptides (RiPPs) take advantage of the ribosomal translation machinery to generate linear peptides that are subsequently modified with heterocycles and/or macrocycles to impose three-dimensional structure and thwart degradation by proteases. Although RiPPs are limited to proteinogenic amino acids, post-translational modifications (PTMs) can alter the structure of individual amino acids and thereby improve stability and biological activity of the molecule. These “tailoring modifications” often occur on amino acid side chains—for example, hydroxylation, methylation, halogenation, prenylation, and acylation—but can also take place within the back bone, as in epimerization, or can result in capping of the N or C termini. At one extreme, these modifications can be essential to the activity of the RiPP, either as a compulsory step in reaching the final molecule or by imparting chemical functionality required for biological activity. At the other extreme, tailoring PTMs may have little effect on activity in an in vitro setting—possibly because of test conditions that do not match the biological context in which the PTMs evolved.
Establishing the molecular basis for the function of tailoring PTMs often requires a three-dimensional structure of the RiPP bound to its biological target. These structures have revealed roles for tailoring PTMs that include providing additional hydrogen bonds to targets, rigidifying the RiPP structure to reduce the entropic cost to binding, or altering the secondary structure of the peptide backbone. Bacterial RiPPs are particularly suited to structural characterization as they are relatively easy to isolate from laboratory cultures or to produce in a heterologous host. Identifying new tailoring PTMs within bacteria is also facilitated by clustering of the genes encoding tailoring enzymes with those of the RiPP precursor and primary modification enzymes. In this Account, we describe the effects of tailoring PTMs on RiPP structure, their interactions with biological targets, and their influence on RiPP stability, with a focus on bacterial RiPP classes. We also discuss the enzymes that generate tailoring PTMs and highlight examples of and prospects for engineering of RiPPs.
Graphical abstract
Introduction
Ribosomally-synthesized and Post-translationally-modified Peptides (RiPPs) are subdivided based on class-defining (primary) post-translational modifications (PTMs).1 These primary PTMs, which typically result in macrocyclization,2 generate scaffolds that endow biological activity, often as antimicrobial or cytotoxic agents. After cyclization, many RiPPs are further modified by enzymes that introduce compound-specific functional groups that are important for improving activity or stability or essential for RiPP maturation. These alterations have been termed tailoring modifications.1
Class-defining PTMs have been extensively reviewed,1–4 but tailoring modifications have received less attention. This Account provides a summary of the physiological functions of some tailoring modifications and the promise of tailoring enzymes for bioengineering. Because of limitations in space, our discussion will be limited to modifications of bacterial RiPPs with established biological activity and/or biochemical characterization, but there exist many examples of fungal and plant RiPPs with tailoring modifications.1 This review highlights Nature’s skill as medicinal chemist in taking conformationally-constrained scaffolds and appending substructure to enhance biological activity.
Hydroxylation and epoxidation
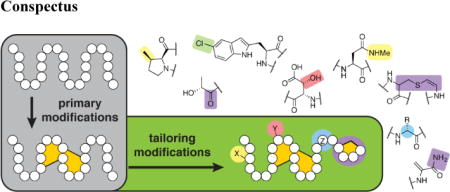
The best-understood examples of oxidative modification of RiPPs are found in thiopeptides, compounds unified by the presence of azolines/azoles and a six-membered nitrogen-containing heterocycle.5–7 In thiostrepton-like peptides, epoxidation of a quinaldic acid moiety and attack by the N-terminal amine on the epoxide generates a second macrocyclic ring (Figure 1A) that may provide rigidity and improve binding to the 50S ribosome.8,9 Ile10 is also hydroxylated at the β and γ1 positions.8 The β-hydroxylation may contribute to the stability of the second macrocycle by providing an intramolecular hydrogen bond to the hydroxyl formed by epoxide ring opening (Figure 1C). Nosiheptide and nocathiacin contain tailoring modifications at the same site as thiostrepton, but utilize a different set of PTMs to generate a second macrocycle (Figure 1B and 1D). A 3,5-dimethylindolic acid moiety is attached to a Glu residue to close the macrocycle through an ester linkage.10
Figure 1.
Thiopeptides targeting the 50S ribosome. Structures of A) thiostrepton, and B) nosiheptide with tailoring modifications highlighted. Also shown are crystal structures of C) thiostrepton (PDB ID 3CF5)47 and D) nosiheptide (PDB ID 2ZJP)47 bound to the 50S ribosome. The 23S RNA binding site is shown as a surface; the L11 protein is not shown in the foreground but binds to the near face of the peptides. E) Structure of thiocillin.
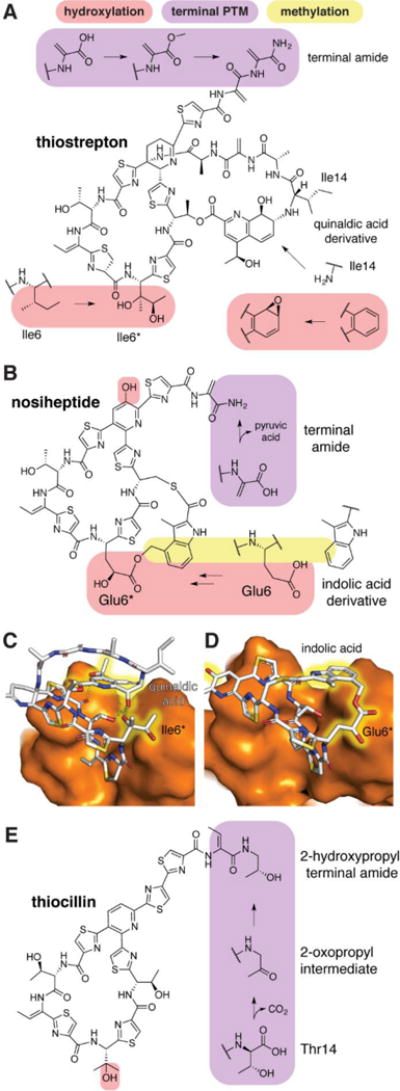
Many thiopeptides contain hydroxylations not involved in macrocycle formation that impact potency by interactions with target molecules. For example, β-hydroxylation of Phe8 of GE2270A (Figure 2A) creates a hydrogen bond donor (Figure 2B) at the surface of the target, elongation factor Tu (EF-Tu).11 As a result, GE2270A in which Phe8 is not hydroxylated displays a 30-fold decrease in activity.12
Figure 2.
Thiopeptides targeting EF-Tu. (A) Structure of GE2270A with tailoring modifications highlighted; Thz, thiazole. (B-D) Different views of GE2270A (PDB ID 2C77)11 bound to EF-Tu. E) Structure of thiomuracin I, and F) X-ray structure bound to EF-TU (PDB ID 4G5G).78
Partial in vitro biosynthesis of thiomuracin (Figure 2E) was recently reported.13 The simplified product, lacking oxidative modifications at Phe5 and Ile8, retained similar activity against test strains as authentic thiomuracin A. Likewise, thiomuracin congeners isolated from the native producer that lack Phe5 hydroxylation have only slightly diminished potency against test strains in vitro.14 Consistent with these observation, Phe5 does not make direct contacts with the target (Figure 2F). In contrast, the oxidized Ile8 is engaged in hydrogen bonds with EF-Tu and congeners with different oxidation products accordingly show more variation in activity.14
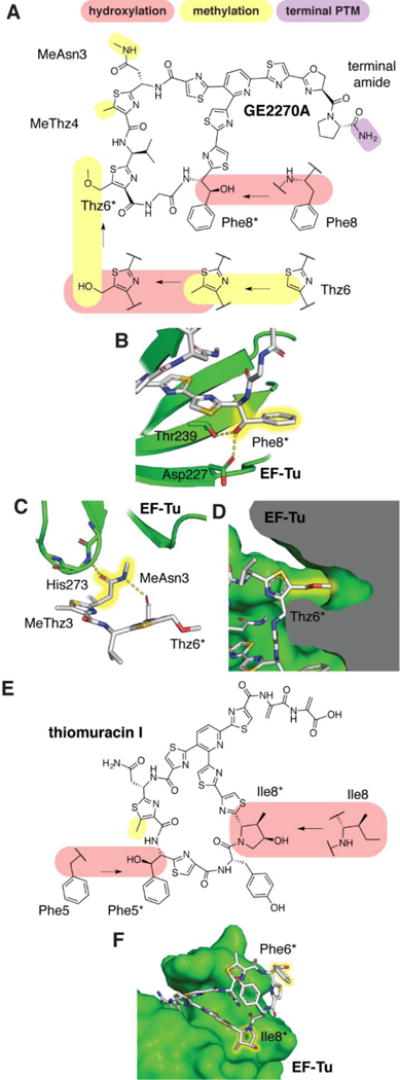
Hydroxylation in other bacterial RiPP classes is rare, but two lanthipeptides bearing hydroxylations have been studied. For the closely related duramycin and cinnamycin, hydroxylation of Asp15 is essential for antimicrobial activity.15,16 The NMR structure of cinnamycin bound to its target lysophosphatidylethanolamine places the head group of ethanolamine in a pocket containing the β-hydroxylated Asp15 (Figure 3),17 with the hydroxyl accepting a hydrogen bond from the ammonium group of ethanolamine. The α-ketoglutarate-dependent oxygenases that hydroxylate the Asp have been characterized and although they are specific for Asp15, they do show some tolerance with respect to surrounding residues.15 The hydroxylation of Pro14 in the C-ring of microbisporicin (also called NAI-107) is non-essential, but generally increases the potency against test strains by 2–4 fold (see the section on halogenation for the microbisporicin structure).18
Figure 3.
Structure of cinnamycin and solution structure (PDB ID 2DDE)17 bound to phosphatidylethanolamine (PE). The β-hydroxylated Asp residue makes a hydrogen bond (black dash) with the ammonium group of PE.
Epimerization
Within RiPPs, two mechanisms have been described for enzymatic conversion of an L-configured amino acid to the D-configuration. This PTM was first observed in lactocin S19, for which inspection of the gene sequence encoding the precursor peptide suggested conversion of a genetically encoded L-Ser to D-Ala by stereospecific reduction of dehydrated L-Ser (dehydroalanine, Dha, Figure 4).20 Alternatively, radical-S-adenosylmethionine (SAM) enzymes can convert L-amino acids into D-amino acids by epimerization at the α-carbon (Figure 5A).21 This mechanism of epimerization is more general than the dehydration-hydrogenation sequence in lanthipeptides as all 19 chiral, proteinogenic amino acids can epimerized. Other epimerizations found in RiPPs include allo-Ile, in cypemycin (Figure 4),22 and the non-enzymatic epimerization of amino acids upstream of a thiazoline residue23 such as in bottromycin24 (Figure 5B).
Figure 4.
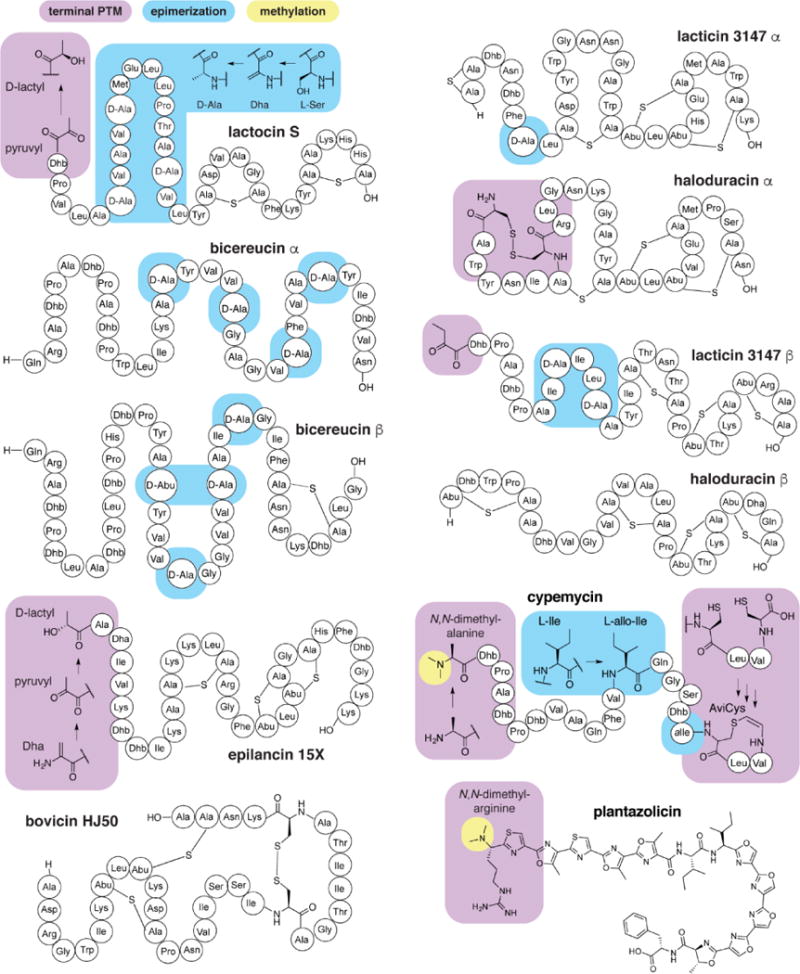
Selection of RiPPs with tailoring modifications as described in the text. Abu, 2-aminobutyric acid.
Figure 5.

Radical-SAM-enzymes catalyze Cα-epimerization and C-methylation. A) The proteusins are a family of Cα-epimerized peptides.21 B) Bottromycin A2 contains multiple C-methylations, an O-methylation, and a nonenzymatic Cα-epimerization.24 C) Chemical38 and solution structure (PDB ID 2RQO)30 of polytheonamide B.
Epimerization at the α-carbon is likely to result in local secondary structures that is not available to only L-configured peptides or to provide protection from proteolysis. Lacticin 3147 displays homology to lanthipeptides that do not contain D-amino acids and yet still have potent activity against susceptible test strains (Figure 4). The NMR structure of the β peptide of lacticin 3147 indicates an α-helical conformation for residues 7–15, including the two D-amino acids.25 In contrast, the corresponding region of related peptides, haloduracin β for example, contain a macrocycle in this region. D-Amino acid-stabilized secondary structures may thus serve as a replacement for thioether or disulfide rings. Accordingly, mutants of lacticin 3147 β with L-Ala at the positions of D-Ala retain activity, although it is diminished relative to wild type.26 In the two-component lanthipeptide bicereucin (Figure 4), in which only a single lanthionine ring is present, epimerization and dehydrobutyrine (Dhb) residues may play a more central role in determining the structure and biological activity.27 Dha/Dhb dehydrogenases appear to be substrate tolerant and have been used in bioengineering studies to modify non-native peptides.15,28
Proteusins are linear peptides that contain D-amino acids introduced by radical-SAM epimerases,21,29 but it remains to be determined what role these PTMs play in biological activity. A survey of three proteusin gene clusters revealed that radical-SAM epimerases are capable of acting on a range of amino acids (Figure 5A). Although the exact substrate specificity rules remain to be determined for radical-SAM epimerases, their ability to act on many residue types suggests utility in engineering applications.21 Epimerization by a radical-SAM enzyme could be considered a primary modification in polytheonamides, where alternating L- and D-amino acids (or achiral glycine) induce formation of a β-helical structure in a membrane-mimicking solvent (Figure 5C).30,31
N- and O-Methylation
Methylations on heteroatoms are prevalent in bacterial RiPPs and are generally installed after the primary modifications. Two peptides contain an N-terminal N,N-dimethylated amino acid: Arg in plantazolicin32 and Ala in cypemycin22 (Figure 4). Dimethylation of plantazolicin is essential to the action of this peptide,33 and the methyltransferase is highly specific for desmethylplantazolicin.34,35 Cypemycin also contains an essential N-terminal N,N-dimethylation (Figure 4), installed by the methyltransferase CypM.36 CypM is substrate tolerant and has been used to methylate a range of unrelated peptides.37
The aforementioned polytheonamides contain both N- and C-methylations in abundance (Figure 5C).38 Particularly intriguing for this class of peptides is the apparent iterative nature of the modifications, with relatively few enzymes acting multiple times on the same peptide.31 Eight Asn residues are N-methylated at the side chain throughout the peptide, likely in conjunction with epimerization of these residues.31 The solution structure of polytheonamide B suggests that these methylations increase the lipophilicity of the intramembrane amide groups and stabilize the β-helical structure of the peptide pore.30 Biological activity has not been reported for peptides lacking Asn methylation, but molecular dynamics simulations suggest that such peptides are more prone to unfolding.39 The N-methylations all result from the action of a single SAM-dependent methyltransferase PoyE.40
The thiopeptide GE2270A (Figure 2A) is also N-methylated at an Asn sidechain.12 The structure of GE2270 bound to EF-Tu reveals this methylation may help reinforce intramolecular hydrogen bonds and assist in desolvation during complex formation by eliminating hydrogen bonding with solvent (Figure 2C).11 Asn methylation at this position is common but not required for activity as a number of non-methylated peptides have been isolated.5 However, mutation of this Asn in a closely related thiopeptide abolishes activity, confirming that this site is crucial for binding.41 Asn side chain methylation has not yet been used in engineering studies with non-native peptides.
The penultimate D-Asp residue of bottromycin A2 is O-methylated in what is believed to be the final step of the biosynthesis of this antibiotic (Figure 5B).24 Removal of this modification diminishes the biological activity substantially.
C-methylation
Methylation at non-nucleophilic carbon centers requires the action of a radical-SAM methyltransferase.42,43 β-Methylation of amino acids is observed in several RiPP classes. For instance, during bottromycin A2 biosynthesis, four β-C-methylations are accomplished by three enzymes (Figure 5B).44 Congeners lacking various methylations were isolated from the native producer and vary in potency by 2–4 fold.45 Two enzymes catalyze at least fourteen β-C-methylations in polytheonamide B (Figure 5C),31,46 including repeated methylation of a Dhb residue, which is hydrolyzed following removal of the leader peptide to form a 2-oxo-5,5-dimethyloctyl group at the N terminus of the mature peptide. A similarly complex transformation is required for the formation of the methoxymethylthiazole of GE2270 (Figure 2A). A C-methyltransferase possibly first generates a methylthiazole, which may be hydroxylated and methylated again by a SAM-dependent O-methyltransferase.12 The effect of these modifications on antibiotic potency has not been reported; however, inspection of the structure of GE2270 in complex with EF-Tu47 reveals a tight binding pocket for this moiety (Figure 2D). The C-methyltransferase that generates a methylthiazole in thiomuracin has been investigated in vitro and has a very high substrate specificity suggesting limited opportunities for engineering.48
Halogenation
The lanthipeptide microbisporicin (Figure 6A) contains a 5-chlorotryptophan (5-Cl-Trp) that is 2–32 fold more potent than deschloromicrobisporicin, depending on the bacterial test strain.49 Overlay of the structure of microbisporicin with the lanthipeptide nisin bound to a lipid II analog suggests that the increase in potency may derive from direct interactions with lipid II as the 5-Cl-Trp residue is directly next to the presumed binding site of the N-acetylmuramic acid moiety (Figure 6B). The halogenase MibH accepts only deschloromicrobisporicin,50 but can perform Trp bromination, resulting in a ~2-fold increase in potency.51 The less potent deschloro-deshydroxy analog serves an unusual dual role as a signaling molecule: accumulation induces expression of the tailoring enzymes and transporters allowing for generation and export of the more potent antimicrobial compound.52
Figure 6.
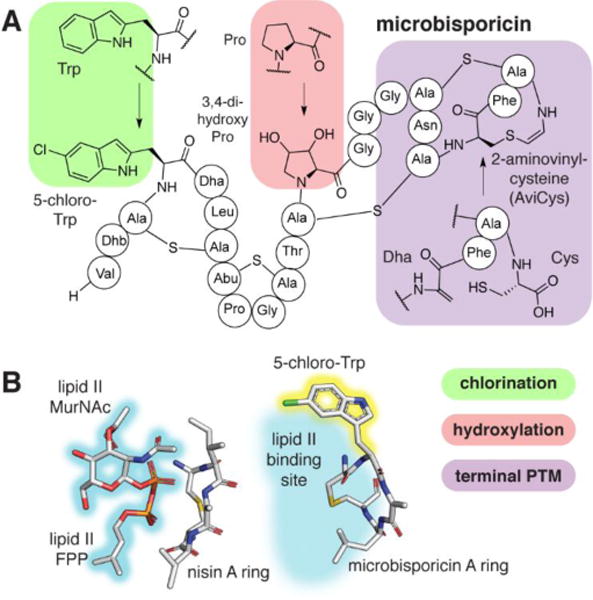
A) Structure of microbisporicin. B) Comparison of solution structures of nisin bound to a lipid II analog (PDB ID 1WCO)79 and microbisporicin in complex with a dodecylphosphocholine micelle (not shown) (PDB ID 2MH5).80 Nisin binds to the farnesylpyrophosphate (FPP) moiety with backbone amide groups. MurNAc, N-acetylmuramic acid.
Disulfides
Disulfides are common in RiPPs from higher organisms1 but infrequent in bacterial RiPPs. One example is bovicin HJ50 and related peptides (Figure 4).53 This class of lanthipeptides, which resemble lacticin 481 but with a disulfide in place of a lanthionine ring, loses antimicrobial activity when reduced and/or alkylated.54,55 Mutagenesis of the cysteines involved in disulfide formation suggests a role for the disulfide linkage in stabilizing the hydrophobic core of the peptide.54 A similar replacement of a primary PTM is observed by comparing haloduracin α with lacticin 3147 α, where a disulfide replaces a lanthionine ring and a D-Ala residue (Figure 4).
Prenylation
Prenylation has been observed in some bacterial RiPPs, but little is known about the function of these PTMs or their importance in target recognition. An example is the competence hormone ComXRO-E-2 which contains a C-geranylated Trp residue (Figure 7A); alteration or removal of the prenyl group reduces the activity of this RiPP.56 O-Prenylation of cyanobactins such as trunkamide (Figure 7A) is carried out by prenyltransferases that may be useful for combinatorial biosynthesis of prenylated RiPPs.57 These enzymes are also capable of generating an ortho-C-prenyl-Tyr through a spontaneous Claisen rearrangement from from O-prenyl-Tyr (Figure 7A).58
Figure 7.
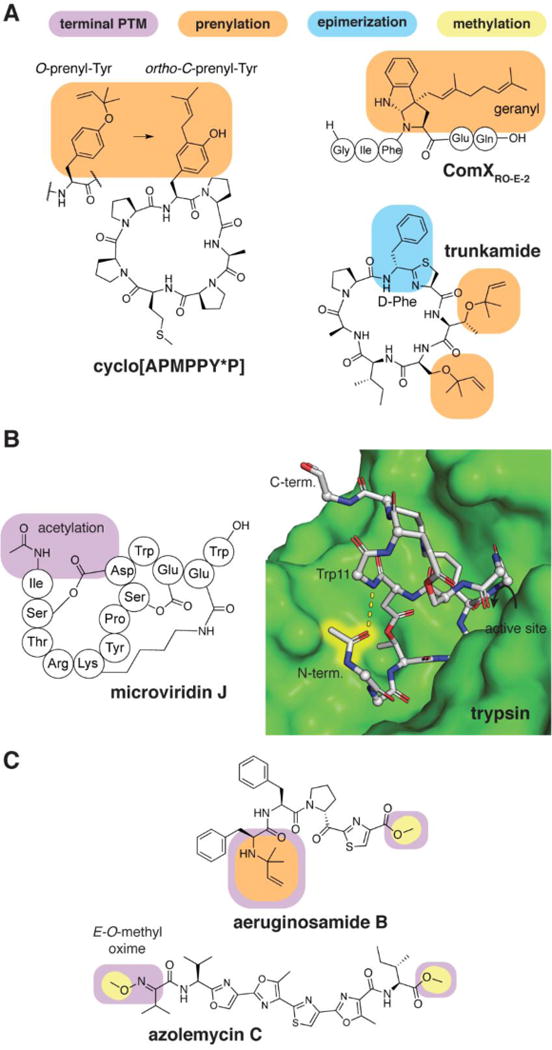
A) Examples of O- and C-prenylated RiPPs. B) Microviridin J bound to bovine trypsin (PDB ID 4KTS).64 A hydrogen bond (black dash) is present between the acetyl group (highlighted) and a backbone amide. C) Examples of short linear peptides with N- and C-terminal PTMs.
N- and C-terminal modifications
Peptidases are ubiquitous in microbe-colonized environments,59 leading to rapid degradation of unstructured peptides. In many cases, primary PTMs provide protection from proteases,60 but tailoring PTMs found at the termini of RiPPs may further enhance their stability. A common theme in lanthipeptides containing N-terminal PTMs is the presence of multiple residues at the N terminus before the first lanthionine ring,61,62 consistent with the modified N-terminus protecting their linear N-terminal segment. Such modification is seen for epilancin 15X (Figure 4), which contains an N-terminal lactyl group that protects a linear model peptide from aminopeptidase hydrolysis.63 The lactyl group is produced by reduction of a pyruvyl group that results from hydrolysis of an N-terminal Dha residue following proteolytic removal of a leader peptide. Non-reduced pyruvyl and 2-oxobutyryl (Obu) groups may serve a similar role. Mutation of the N-terminal Thr (precursor to Obu) of lacticin 3147 β to Ala results in peptides that show a modest decrease in potency.26
Several other means of protecting of the N-terminus of RiPPs have been reported. The N-acetyl group of the protease inhibitor microviridin participates in intramolecular hydrogen bonds (Figure 7B)64 and may account for a slight improvement in trypsin inhibition.65 N- to C-terminal macrocyclization is another common strategy to avoid aminopeptidase sensitivity and is found in cyanobactins and some sactipeptides such as subtilosin.66 Recent work has demonstrated that many cyanobactins are not cyclized, but instead O-methylated at the C terminus and N-prenylated at the N terminus (Figure 7C).67 The role of these modifications in biological activity is not yet known, but it is probable that they influence the stability and three-dimensional structure of the linear peptides.68 Another example is azolemycin C, a cytotoxic, azole-rich peptide that contains modified N- and C-terminal residues which are important for activity (Figure 7C).69
C-terminal modifications are especially prevalent in thiopeptides, with amidation the most common. At least two distinct mechanisms have evolved for generation of this modification in different thiopeptide classes.70 C-terminal O-methylation is a cryptic intermediate in the biosynthesis of the terminal amide in thiostrepton71 and produces a dramatic increase in potency over the free carboxylate. The final amide product is less potent than the O-methylated precursor, but much more soluble. The C-terminus of thiostrepton does not interact directly with the ribosome in the inhibitory complex,47 so these modifications may exert their effects by altering cell permeability or stability rather than mediating direct interactions with the ribosome.
Aminovinyl(methyl)cysteine is a C-terminal modification formed by Michael-type addition of an oxidatively decarboxylated Cys to an internal Dha or Dhb (Figure 4 and 6A).72 At present, little is known about the contribution of AviCys to biological activity, but several cases have demonstrated the importance of this modification in proper maturation and activity of the peptides.73,74 Recent in vitro characterization of MibD, which catalyzes AviCys formation in microbisporicin, revealed that Cys decarboxylation may occur alongside or be required for complete modification by the lanthipeptide synthase.50 Cys oxidative decarboxylases have been shown to tolerate many non-native sequences ending in Cys,28,75 suggesting their possible use for protecting the C termini of RiPPs or other peptides.
AviCys is also present in thioviramides76 and linaridins such as cypemycin (Figure 4).36 These non-lanthipeptides utilize the same oxidative decarboxylase as in AviCys-containing lanthipeptides but presumably require an as-yet uncharacterized enzyme for generation of the Dha residues to form the ring. No specific function for AviCys in these molecules has been assigned, but for cypemycin—similar to microbisporicin—no antibiotic activity is observed when biosynthesis is blocked by knock-out of the decarboxylase, suggesting an important role in biosynthesis or activity.36
Conclusion
Primary modifications of RiPPs are often iterative and convert particular amino acids into conformationally constrained heterocycles or macrocycles. Although further modification of these peptides is not necessary for creation of biologically active molecules, tailoring modifications provide a means to add to these privileged scaffolds, often enhancing stability or binding affinity—and thus efficacy of biological activity. Space considerations did not allow a comprehensive cataloguing of all tailoring reactions in RiPPs in this Account, leading us to focus on those examples for which the molecular details of their interactions with targets are known or that have particular promise for bioengineering purposes. Characterization of the enzymes that produce them has already lead to several examples of engineered RiPPs and peptides with improved properties.
Future opportunities in this field include biochemical study of the numerous uncharacterized enzymes that have been identified from genomic data. Ribosomal peptide scaffolds are attractive targets for genome-based discovery of new compounds and new tailoring enzymes as the molecular nature of the final molecule and the types of likely modifications are often apparent in the DNA sequence. The tailoring enzymes, especially substrate tolerant examples that do not require a leader peptide,1,77 may provide a future toolbox of catalysts that can be used for RiPP engineering, stabilizing therapeutic compounds, or creating novel peptide based materials with desired properties. Better understanding of how primary PTMs determine specificity will be essential to efforts to customize peptides with desirable functions.
Although the chemical structure of RiPPs is often predictable to some extent from genomic data, very little is known about their three-dimensional structures, especially in complex with biological targets. Therefore, increased structural analysis of RiPP-target interactions will be important to provide information about the effects of PTMs on binding affinity, such that tailoring enzymes can be used in rational design of improved compounds, much in the same way as structure-based drug optimization in synthetic molecules.
Acknowledgments
This work was supported by the National Institutes of Health (R37 GM058822 to W.A.V.; F32 GM120868 to M.A.F.)
Biographies
Michael Funk obtained his B.S. in Chemistry and Biology at Vanderbilt University and Ph.D. in Biological Chemistry at MIT with Catherine Drennan. He is a NIH postdoctoral fellow in the van der Donk laboratory.
Wilfred van der Donk obtained his B.S. and M.S. at Leiden University in the Netherlands under the direction of Jan Reedijk. He completed his Ph.D. at Rice University with Kevin Burgess. After postdoctoral studies with JoAnne Stubbe at MIT, he started his independent career at the University of Illinois. He is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Göransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Müller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJ, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Süssmuth RD, Tagg JR, Tang GL, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Truman AW. Cyclisation mechanisms in the biosynthesis of ribosomally synthesised and post-translationally modified peptides. Beilstein J Org Chem. 2016;12:1250–1268. doi: 10.3762/bjoc.12.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ortega MA, van der Donk WA. New insights into the biosynthetic logic of ribosomally synthesized and post-translationally modified peptide natural products. Cell Chem Biol. 2016;23:31–44. doi: 10.1016/j.chembiol.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sardar D, Schmidt EW. Combinatorial biosynthesis of RiPPs: docking with marine life. Curr Opin Chem Biol. 2016;31:15–21. doi: 10.1016/j.cbpa.2015.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Just-Baringo X, Albericio F, Alvarez M. Thiopeptide antibiotics: retrospective and recent advances. Mar Drugs. 2014;12:317–351. doi: 10.3390/md12010317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Q, Liu W. Biosynthesis of thiopeptide antibiotics and their pathway engineering. Nat Prod Rep. 2013;30:218–226. doi: 10.1039/c2np20107k. [DOI] [PubMed] [Google Scholar]
- 7.Zheng Q, Fang H, Liu W. Post-translational modifications involved in the biosynthesis of thiopeptide antibiotics. Org Biomol Chem. 2017;15:3376–3390. doi: 10.1039/c7ob00466d. [DOI] [PubMed] [Google Scholar]
- 8.Zheng Q, Wang S, Liao R, Liu W. Precursor-directed mutational biosynthesis dacilitates the functional assignment of two cytochromes P450 in thiostrepton biosynthesis. ACS Chem Biol. 2016;11:2673–2678. doi: 10.1021/acschembio.6b00419. [DOI] [PubMed] [Google Scholar]
- 9.Zheng Q, Wang S, Duan P, Liao R, Chen D, Liu W. An alpha/beta-hydrolase fold protein in the biosynthesis of thiostrepton exhibits a dual activity for endopeptidyl hydrolysis and epoxide ring opening/macrocyclization. Proc Natl Acad Sci USA. 2016;113:14318–14323. doi: 10.1073/pnas.1612607113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Badding E, Grove TL, Gadsby L, LaMattina J, Boal AK, Booker SJ. Rerouting the pathway for the biosynthesis of the side ring system of nosiheptide: the roles of NosI, NosJ, and NosK. J Am Chem Soc. 2017;139:5896–5905. doi: 10.1021/jacs.7b01497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parmeggiani A, Krab IM, Okamura S, Nielsen RC, Nyborg J, Nissen P. Structural basis of the action of pulvomycin and GE2270 A on elongation factor Tu. Biochemistry. 2006;45:6846–6857. doi: 10.1021/bi0525122. [DOI] [PubMed] [Google Scholar]
- 12.Tocchetti A, Maffioli S, Iorio M, Alt S, Mazzei E, Brunati C, Sosio M, Donadio S. Capturing linear intermediates and C-terminal variants during maturation of the thiopeptide GE2270. Chem Biol. 2013;20:1067–1077. doi: 10.1016/j.chembiol.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 13.Hudson GA, Zhang Z, Tietz JI, Mitchell DA, van der Donk WA. In vitro biosynthesis of the core scaffold of the thiopeptide thiomuracin. J Am Chem Soc. 2015;137:16012–16015. doi: 10.1021/jacs.5b10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris RP, Leeds JA, Naegeli HU, Oberer L, Memmert K, Weber E, LaMarche MJ, Parker CN, Burrer N, Esterow S, Hein AE, Schmitt EK, Krastel P. Ribosomally synthesized thiopeptide antibiotics targeting elongation factor Tu. J Am Chem Soc. 2009;131:5946–5955. doi: 10.1021/ja900488a. [DOI] [PubMed] [Google Scholar]
- 15.Huo L, Ökesli A, Zhao M, van der Donk WA. Insights into the biosynthesis of duramycin. Appl Environ Microbiol. 2017;83:e02698–02616. doi: 10.1128/AEM.02698-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ökesli A, Cooper LE, Fogle EJ, van der Donk WA. Nine post-translational modifications during the biosynthesis of cinnamycin. J Am Chem Soc. 2011;133:13753–13760. doi: 10.1021/ja205783f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hosoda K, Ohya M, Kohno T, Maeda T, Endo S, Wakamatsu K. Structure determination of an immunopotentiator peptide, cinnamycin, complexed with lysophosphatidylethanolamine by 1H-NMR1. J Biochem. 1996;119:226–230. doi: 10.1093/oxfordjournals.jbchem.a021226. [DOI] [PubMed] [Google Scholar]
- 18.Maffioli SI, Iorio M, Sosio M, Monciardini P, Gaspari E, Donadio S. Characterization of the congeners in the lantibiotic NAI-107 complex. J Nat Prod. 2014;77:79–84. doi: 10.1021/np400702t. [DOI] [PubMed] [Google Scholar]
- 19.Skaugen M, Nissen-Meyer J, Jung G, Stevanovic S, Sletten K, Inger C, Abildgaard M, Nes IF. In vivo conversion of L-serine to D-alanine in a ribosomally synthesized polypeptide. J Biol Chem. 1994;269:27183–27185. [PubMed] [Google Scholar]
- 20.Cotter PD, O’Connor PM, Draper LA, Lawton EM, Deegan LH, Hill C, Ross RP. Posttranslational conversion of L-serines to D-alanines is vital for optimal production and activity of the lantibiotic lacticin 3147. Proc Natl Acad Sci USA. 2005;102:18584–18589. doi: 10.1073/pnas.0509371102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morinaka BI, Vagstad AL, Helf MJ, Gugger M, Kegler C, Freeman MF, Bode HB, Piel J. Radical S-adenosyl methionine epimerases: regioselective introduction of diverse D-amino acid patterns into peptide natural products. Angew Chem Int Ed. 2014;53:8503–8507. doi: 10.1002/anie.201400478. [DOI] [PubMed] [Google Scholar]
- 22.Minami Y, Yoshida K-i, Azuma R, Urakawa A, Kawauchi T, Otani T, Komiyama K, Ōmura S. Structure of cypemycin, a new peptide antibiotic. Tetrahedron Lett. 1994;35:8001–8004. [Google Scholar]
- 23.Milne BF, Long PF, Starcevic A, Hranueli D, Jaspars M. Spontaneity in the patellamide biosynthetic pathway. Org Biomol Chem. 2006;4:631–638. doi: 10.1039/b515938e. [DOI] [PubMed] [Google Scholar]
- 24.Crone WJ, Vior NM, Santos-Aberturas J, Schmitz LG, Leeper FJ, Truman AW. Dissecting bottromycin biosynthesis using comparative untargeted metabolomics. Angew Chem Int Ed. 2016;55:9639–9643. doi: 10.1002/anie.201604304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin NI, Sprules T, Carpenter MR, Cotter PD, Hill C, Ross RP, Vederas JC. Structural characterization of lacticin 3147, a two-peptide lantibiotic with synergistic activity. Biochemistry. 2004;43:3049–3056. doi: 10.1021/bi0362065. [DOI] [PubMed] [Google Scholar]
- 26.Cotter PD, Deegan LH, Lawton EM, Draper LA, O’Connor PM, Hill C, Ross RP. Complete alanine scanning of the two-component lantibiotic lacticin 3147: generating a blueprint for rational drug design. Mol Microbiol. 2006;62:735–747. doi: 10.1111/j.1365-2958.2006.05398.x. [DOI] [PubMed] [Google Scholar]
- 27.Huo L, van der Donk WA. Discovery and characterization of bicereucin, an unusual D-amino acid-containing mixed two-component lantibiotic. J Am Chem Soc. 2016;138:5254–5257. doi: 10.1021/jacs.6b02513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Heel AJ, Mu D, Montalban-Lopez M, Hendriks D, Kuipers OP. Designing and producing modified, new-to-nature peptides with antimicrobial activity by use of a combination of various lantibiotic modification enzymes. ACS Synth Biol. 2013;2:397–404. doi: 10.1021/sb3001084. [DOI] [PubMed] [Google Scholar]
- 29.Benjdia A, Guillot A, Ruffié P, Leprince J, Berteau O. Post-translational modification of ribosomally synthesized peptides by a radical SAM epimerase in Bacillus subtilis. Nat Chem. 2017 doi: 10.1038/nchem.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamada T, Matsunaga S, Fujiwara M, Fujita K, Hirota H, Schmucki R, Guntert P, Fusetani N. Solution structure of polytheonamide B, a highly cytotoxic nonribosomal polypeptide from marine sponge. J Am Chem Soc. 2010;132:12941–12945. doi: 10.1021/ja104616z. [DOI] [PubMed] [Google Scholar]
- 31.Freeman MF, Helf MJ, Bhushan A, Morinaka BI, Piel J. Seven enzymes create extraordinary molecular complexity in an uncultivated bacterium. Nat Chem. 2017;9:387–395. doi: 10.1038/nchem.2666. [DOI] [PubMed] [Google Scholar]
- 32.Kalyon B, Helaly SE, Scholz R, Nachtigall J, Vater J, Borriss R, Süssmuth RD. Plantazolicin A and B: structure elucidation of ribosomally synthesized thiazole/oxazole peptides from Bacillus amyloliquefaciens FZB42. Org Lett. 2011;13:2996–2999. doi: 10.1021/ol200809m. [DOI] [PubMed] [Google Scholar]
- 33.Molohon KJ, Melby JO, Lee J, Evans BS, Dunbar KL, Bumpus SB, Kelleher NL, Mitchell DA. Structure determination and interception of biosynthetic intermediates for the plantazolicin class of highly discriminating antibiotics. ACS Chem Biol. 2011;6:1307–1313. doi: 10.1021/cb200339d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee J, Hao Y, Blair PM, Melby JO, Agarwal V, Burkhart BJ, Nair SK, Mitchell DA. Structural and functional insight into an unexpectedly selective N-methyltransferase involved in plantazolicin biosynthesis. Proc Natl Acad Sci USA. 2013;110:12954–12959. doi: 10.1073/pnas.1306101110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piwowarska NA, Banala S, Overkleeft HS, Süssmuth RD. Arg-Thz is a minimal substrate for the N(alpha), N(alpha)-arginyl methyltransferase involved in the biosynthesis of plantazolicin. Chem Commun. 2013;49:10703–10705. doi: 10.1039/c3cc45898a. [DOI] [PubMed] [Google Scholar]
- 36.Claesen J, Bibb M. Genome mining and genetic analysis of cypemycin biosynthesis reveal an unusual class of posttranslationally modified peptides. Proc Natl Acad Sci USA. 2010;107:16297–16302. doi: 10.1073/pnas.1008608107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Q, van der Donk WA. Catalytic promiscuity of a bacterial alpha-N-methyltransferase. FEBS Lett. 2012;586:3391–3397. doi: 10.1016/j.febslet.2012.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamada T, Matsunaga S, Yano G, Fusetani N. Polytheonamides A and B, highly cytotoxic, linear polypeptides with unprecedented structural features, from the marine sponge, Theonella swinhoei. J Am Chem Soc. 2005;127:110–118. doi: 10.1021/ja045749e. [DOI] [PubMed] [Google Scholar]
- 39.Renevey A, Riniker S. The importance of N-methylations for the stability of the β6.3-helical conformation of polytheonamide B. Eur Biophys J. 2017;46:363–374. doi: 10.1007/s00249-016-1179-1. [DOI] [PubMed] [Google Scholar]
- 40.Freeman MF, Gurgui C, Helf MJ, Morinaka BI, Uria AR, Oldham NJ, Sahl HG, Matsunaga S, Piel J. Metagenome mining reveals polytheonamides as posttranslationally modified ribosomal peptides. Science. 2012;338:387–390. doi: 10.1126/science.1226121. [DOI] [PubMed] [Google Scholar]
- 41.Young TS, Dorrestein PC, Walsh CT. Codon randomization for rapid exploration of chemical space in thiopeptide antibiotic variants. Chem Biol. 2012;19:1600–1610. doi: 10.1016/j.chembiol.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bauerle MR, Schwalm EL, Booker SJ. Mechanistic diversity of radical S-adenosylmethionine (SAM)-dependent methylation. J Biol Chem. 2015;290:3995–4002. doi: 10.1074/jbc.R114.607044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding W, Li Q, Jia Y, Ji X, Qianzhu H, Zhang Q. Emerging diversity of the cobalamin-dependent methyltransferases involving radical-based mechanisms. ChemBioChem. 2016;17:1191–1197. doi: 10.1002/cbic.201600107. [DOI] [PubMed] [Google Scholar]
- 44.Huo L, Rachid S, Stadler M, Wenzel SC, Müller R. Synthetic biotechnology to study and engineer ribosomal bottromycin biosynthesis. Chem Biol. 2012;19:1278–1287. doi: 10.1016/j.chembiol.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 45.Nakamura S, Yajima T, Lin Y, Umezawa H. Isolation and characterization of bottromycins A2, B2, C2. J Antibiot. 1967;20:1–5. [PubMed] [Google Scholar]
- 46.Parent A, Guillot A, Benjdia A, Chartier G, Leprince J, Berteau O. The B12-radical SAM enzyme PoyC catalyzes valine Cβ-methylation during polytheonamide biosynthesis. J Am Chem Soc. 2016;138:15515–15518. doi: 10.1021/jacs.6b06697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harms JM, Wilson DN, Schluenzen F, Connell SR, Stachelhaus T, Zaborowska Z, Spahn CM, Fucini P. Translational regulation via L11: molecular switches on the ribosome turned on and off by thiostrepton and micrococcin. Mol Cell. 2008;30:26–38. doi: 10.1016/j.molcel.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 48.Mahanta N, Zhang Z, Hudson GA, van der Donk WA, Mitchell DA. Reconstitution and substrate specificity of the radical S-adenosyl-methionine thiazole C-methyltransferase in thiomuracin biosynthesis. J Am Chem Soc. 2017;139:4310–4313. doi: 10.1021/jacs.7b00693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maffioli SI, Cruz JC, Monciardini P, Sosio M, Donadio S. Advancing cell wall inhibitors towards clinical applications. J Ind Microbiol Biotechnol. 2016;43:177–184. doi: 10.1007/s10295-015-1703-9. [DOI] [PubMed] [Google Scholar]
- 50.Ortega MA, Cogan DP, Mukherjee S, Garg N, Li B, Thibodeaux GN, Maffioli SI, Donadio S, Sosio M, Escano J, Smith L, Nair SK, van der Donk WA. Two flavoenzymes catalyze the post-translational generation of 5-chlorotryptophan and 2-aminovinyl-cysteine during NAI-107 Biosynthesis. ACS Chem Biol. 2017;12:548–557. doi: 10.1021/acschembio.6b01031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cruz JC, Iorio M, Monciardini P, Simone M, Brunati C, Gaspari E, Maffioli SI, Wellington E, Sosio M, Donadio S. Brominated variant of the lantibiotic NAI-107 with enhanced antibacterial potency. J Nat Prod. 2015;78:2642–2647. doi: 10.1021/acs.jnatprod.5b00576. [DOI] [PubMed] [Google Scholar]
- 52.Foulston L, Bibb M. Feed-forward regulation of microbisporicin biosynthesis in Microbispora corallina. J Bacteriol. 2011;193:3064–3071. doi: 10.1128/JB.00250-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiao H, Chen X, Chen M, Tang S, Zhao X, Huan L. Bovicin HJ50, a novel lantibiotic produced by Streptococcus bovis HJ50. Microbiology. 2004;150:103–108. doi: 10.1099/mic.0.26437-0. [DOI] [PubMed] [Google Scholar]
- 54.Wang J, Ma H, Ge X, Zhang J, Teng K, Sun Z, Zhong J. Bovicin HJ50-like lantibiotics, a novel subgroup of lantibiotics featured by an indispensable disulfide bridge. PLoS One. 2014;9:e97121. doi: 10.1371/journal.pone.0097121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kabuki T, Uenishi H, Seto Y, Yoshioka T, Nakajima H. A unique lantibiotic, thermophilin 1277, containing a disulfide bridge and two thioether bridges. J Appl Microbiol. 2009;106:853–862. doi: 10.1111/j.1365-2672.2008.04059.x. [DOI] [PubMed] [Google Scholar]
- 56.Tsuji F, Kobayashi K, Okada M, Yamaguchi H, Ojika M, Sakagami Y. The geranyl-modified tryptophan residue is crucial for ComXRO-E-2 pheromone biological activity. Bioorg Med Chem Lett. 2011;21:4041–4044. doi: 10.1016/j.bmcl.2011.04.123. [DOI] [PubMed] [Google Scholar]
- 57.Ruffner DE, Schmidt EW, Heemstra JR. Assessing the combinatorial potential of the RiPP cyanobactin tru pathway. ACS Synth Biol. 2015;4:482–492. doi: 10.1021/sb500267d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McIntosh JA, Donia MS, Nair SK, Schmidt EW. Enzymatic basis of ribosomal peptide prenylation in cyanobacteria. J Am Chem Soc. 2011;133:13698–13705. doi: 10.1021/ja205458h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bach H-J, Munch JC. Identification of bacterial sources of soil peptidases. Biol Fertility Soils. 2000;31:219–224. [Google Scholar]
- 60.Cooper LE, McClerren AL, Chary A, van der Donk WA. Structure-activity relationship studies of the two-component lantibiotic haloduracin. Chem Biol. 2008;15:1035–1045. doi: 10.1016/j.chembiol.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Repka LM, Chekan JR, Nair SK, van der Donk WA. Mechanistic understanding of lanthipeptide biosynthetic enzymes. Chem Rev. 2017;117:5457–5520. doi: 10.1021/acs.chemrev.6b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Caetano T, Krawczyk JM, Mosker E, Süssmuth RD, Mendo S. Heterologous expression, biosynthesis, and mutagenesis of type II lantibiotics from Bacillus licheniformis in Escherichia coli. Chem Biol. 2011;18:90–100. doi: 10.1016/j.chembiol.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 63.Velásquez JE, Zhang X, van der Donk WA. Biosynthesis of the antimicrobial peptide epilancin 15X and its N-terminal lactate. Chem Biol. 2011;18:857–867. doi: 10.1016/j.chembiol.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weiz AR, Ishida K, Quitterer F, Meyer S, Kehr JC, Muller KM, Groll M, Hertweck C, Dittmann E. Harnessing the evolvability of tricyclic microviridins to dissect protease-inhibitor interactions. Angew Chem Int Ed. 2014;53:3735–3738. doi: 10.1002/anie.201309721. [DOI] [PubMed] [Google Scholar]
- 65.Reyna-Gonzalez E, Schmid B, Petras D, Süssmuth RD, Dittmann E. Leader peptide-free in vitro reconstitution of microviridin niosynthesis enables design of synthetic protease-targeted libraries. Angew Chem Int Ed. 2016;55:9398–9401. doi: 10.1002/anie.201604345. [DOI] [PubMed] [Google Scholar]
- 66.Kawulka K, Sprules T, McKay RT, Mercier P, Diaper CM, Zuber P, Vederas JC. Structure of subtilosin A, an antimicrobial peptide from Bacillus subtilis with unusual posttranslational modifications linking cysteine sulfurs to alpha-carbons of phenylalanine and threonine. J Am Chem Soc. 2003;125:4726–4727. doi: 10.1021/ja029654t. [DOI] [PubMed] [Google Scholar]
- 67.Leikoski N, Liu L, Jokela J, Wahlsten M, Gugger M, Calteau A, Permi P, Kerfeld CA, Sivonen K, Fewer DP. Genome mining expands the chemical diversity of the cyanobactin family to include highly modified linear peptides. Chem Biol. 2013;20:1033–1043. doi: 10.1016/j.chembiol.2013.06.015. [DOI] [PubMed] [Google Scholar]
- 68.Sardar D, Hao Y, Lin Z, Morita M, Nair SK, Schmidt EW. Enzymatic N- and C-protection in cyanobactin RiPP natural products. J Am Chem Soc. 2017;139:2884–2887. doi: 10.1021/jacs.6b12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu N, Song L, Liu M, Shang F, Anderson Z, Fox DJ, Challis GL, Huang Y. Unique post-translational oxime formation in the biosynthesis of the azolemycin complex of novel ribosomal peptides from Streptomyces sp. FXJ1.264. Chem Sci. 2016;7:482–488. doi: 10.1039/c5sc03021h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu Y, Guo H, Zhang Q, Duan L, Ding Y, Liao R, Lei C, Shen B, Liu W. NosA catalyzing carboxyl-terminal amide formation in nosiheptide maturation via an enamine dealkylation on the serine-extended precursor peptide. J Am Chem Soc. 2010;132:16324–16326. doi: 10.1021/ja106571g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liao R, Liu W. Thiostrepton maturation involving a deesterification-amidation way to process the C-terminally methylated peptide backbone. J Am Chem Soc. 2011;133:2852–2855. doi: 10.1021/ja1111173. [DOI] [PubMed] [Google Scholar]
- 72.Sit CS, Yoganathan S, Vederas JC. Biosynthesis of aminovinyl-cysteine-containing peptides and its application in the production of potential drug candidates. Acc Chem Res. 2011;44:261–268. doi: 10.1021/ar1001395. [DOI] [PubMed] [Google Scholar]
- 73.Ottenwälder B, Kupke T, Brecht S, Gnau V, Metzger J, Jung G, Götz F. Isolation and characterization of genetically engineered gallidermin and epidermin analogs. Appl Environ Microbiol. 1995;61:3894–3903. doi: 10.1128/aem.61.11.3894-3903.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Foulston LC, Bibb MJ. Microbisporicin gene cluster reveals unusual features of lantibiotic biosynthesis in actinomycetes. Proc Natl Acad Sci USA. 2010;107:13461–13466. doi: 10.1073/pnas.1008285107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kupke T, Kempter C, Jung G, Götz F. Oxidative decarboxylation of peptides catalyzed by flavoprotein EpiD. Determination of substrate specificity using peptide libraries and neutral loss mass spectrometry. J Biol Chem. 1995;270:11282–11289. doi: 10.1074/jbc.270.19.11282. [DOI] [PubMed] [Google Scholar]
- 76.Izawa M, Kawasaki T, Hayakawa Y. Cloning and heterologous expression of the thioviridamide biosynthesis gene cluster from Streptomyces olivoviridis. Appl Environ Microbiol. 2013;79:7110–7113. doi: 10.1128/AEM.01978-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang X, van der Donk WA. Ribosomally synthesized and post-translationally modified peptide natural products: new insights into the role of leader and core peptides during biosynthesis. Chem Eur J. 2013;19:7662–7677. doi: 10.1002/chem.201300401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.LaMarche MJ, Leeds JA, Dzink-Fox J, Gangl E, Krastel P, Neckermann G, Palestrant D, Patane MA, Rann EM, Tiamfook S, Yu D. Antibiotic optimization and chemical structure stabilization of thiomuracin A. J Med Chem. 2012;55:6934–6941. doi: 10.1021/jm300783c. [DOI] [PubMed] [Google Scholar]
- 79.Hsu ST, Breukink E, Tischenko E, Lutters MA, de Kruijff B, Kaptein R, Bonvin AM, van Nuland NA. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat Struct Mol Biol. 2004;11:963–967. doi: 10.1038/nsmb830. [DOI] [PubMed] [Google Scholar]
- 80.Munch D, Muller A, Schneider T, Kohl B, Wenzel M, Bandow JE, Maffioli S, Sosio M, Donadio S, Wimmer R, Sahl HG. The lantibiotic NAI-107 binds to bactoprenol-bound cell wall precursors and impairs membrane functions. J Biol Chem. 2014;289:12063–12076. doi: 10.1074/jbc.M113.537449. [DOI] [PMC free article] [PubMed] [Google Scholar]