

Abstract
Leptin is an adipocyte-secreted hormone that is delivered via a specific transport system across the blood-brain barrier (BBB) to the brain where it acts on the hypothalamus receptors to control appetite and thermogenesis. Peripheral resistance to leptin due to its impaired brain delivery prevents therapeutic use of leptin in overweight and moderately obese patients. To address this problem, we modified the N-terminal amine of leptin with Pluronic P85 (LepNP85) and administered this conjugate intranasally using the nose-to-brain (INB) route to bypass the BBB. We compared this conjugate with the native leptin, the N-terminal leptin conjugate with poly(ethylene glycol) (LepNPEG5K), and two conjugates of leptin with Pluronic P85 attached randomly to the lysine amino groups of the hormone. Compared to the random conjugates of leptin with P85, LepNP85 has shown higher affinity upon binding with the leptin receptor, and similarly to native hormone activated hypothalamus receptors after direct injection into brain. After INB delivery, LepNP85 conjugate was transported to the brain and accumulated in the hypothalamus and hippocampus to a greater extent than the native leptin and LepNPEG5K and activated leptin receptors in hypothalamus at lower dose than native leptin. Our work suggests that LepNP85 can access the brain directly after INB delivery and confirms our hypothesis that the improvement in brain accumulation of this conjugate is due to its enhanced brain absorption. In conclusion, the LepNP85 with optimized conjugation chemistry is a promising candidate for treatment of obesity.
Keywords: leptin, Pluronic block copolymer, protein-polymer conjugation, nose-to-brain delivery, pharmacokinetics, obesity
Graphical abstract
1. Introduction
According to World Health Organization, 39% of worldwide adults were overweight with body mass index (BMIs) 25.0–29.9 kg/m2 and 13% were obese with BMIs at or above 30 kg/m2. These subjects have higher risk of developing type 2 diabetes, cardiovascular diseases, musculoskeletal disorders, and cancers [1]. Dietary restriction and physical exercise are rarely effective in lasting weight loss [1]. Yet, there are very limited obesity pharmacotherapies available. Safety and toxicity issues have plagued development of small-molecule obesity drugs. Several weight-loss drugs (e.g., sibutramine, rimonabant) were approved but later withdrawn from the market due to unacceptable safety profiles. Currently, Contrav® (naltrexone / bupropion), Xenical® (orlistat), Qsymia® (phentermine/topiramate), and Belviq® (lorcaserin) are the only US food and drug administration (FDA)-approved drugs for long-term treatment of obesity [2]. However, these drugs have low compliance and limited weight-loss efficacy due to significant toxicity issues such as suicide risk (buproprion), unpleasant gastrointestinal (GI) side effects (Xenical), birth defect (topiramate), and heart valvulopathy (lorcaserin) [1–3]. In view of the safety setbacks associated with small-molecule obesity drugs, hormonal therapy has attracted significant attention. Specifically, GI hormones (e.g., leptin, glucagon-like peptide 1 (GLP-1), oxyntomodulin (OXM), peptide YY3-36 (PYY), ghrelin, pancreatic polypeptide) have been tested in clinics as treatments for obesity. Although the clinical trials of these hormones have generally shown satisfactory toxicity profiles, they each have drug delivery issues including short blood half-lives, and their efficacy performance so far has been disappointing [4–6].
Leptin (16 kDa) is secreted by fat cells and then acts in the brain hypothalamus to regulate appetite and thermogenesis [7–9]. It reaches the brain via a saturable transporter system located at the blood-brain barrier (BBB) and choroid plexus [8, 10]. Although Amgen terminated efforts to develop leptin as a treatment for obesity [11, 12], their studies and those of many others have yielded a wealth of information regarding both the biology and pharmacodynamics of the hormone. For example, it is now appreciated that earlier attempts of leptin monotherapy were not successful due to the phenomenon of leptin resistance in obese patients [10]. Nonetheless, leptin was well tolerated and effective in leptin-deficient obese patients [13]. Recent studies have shown that leptin resistance can occur via three mechanisms depending in large part to the degree of obesity present. These mechanisms include: 1) defective transport at the BBB, 2) impaired leptin receptor function in the hypothalamus, and 3) disruption of anorectic downstream neuronal circuitries [14–16]. The latter mechanism of resistance appears to be less important in humans than the first two. In contrast, the ability of clinically relevant amounts of the hormone to cross the BBB (peripheral resistance) and receptor dysfunction (central resistance) are critical considerations in any attempt to develop a leptin-based drug. It is well known that peripheral resistance due to leptin transporter defects predominate over brain receptor defects early on in outbred diet-induced obesity (DIO) rodent models with milder forms of obesity; whereas, both the BBB and receptor are largely dysfunctional in severe obesity [15]. Modeling based on cerebrospinal fluid (CSF) and serum levels of leptin indicates that in advanced obesity in humans (BMI>30 kg/m2, leptin serum levels about 40 ng/ml), transporter defects account for almost two-thirds of resistance to peripherally administered leptin [17]. Thus, just as in the rodent and canine models [18–20], it is expected that moderately obese humans who show a poor response to leptin would respond better to a form of leptin that could reach brain receptors [21]. Indeed, leptin entry to the brain is greatly complicated by obesity, triglycerides, fasting, hyperglycemia, insulin, and inflammation [22–24]. An impaired BBB transport is important in the maintenance and probably in the progression of obesity [15, 18, 25]. Therefore, we believe that improved delivery of leptin to the brain could significantly improve its therapeutic effect when administered to subjects that are at increased risk for disease [26]. Specifically, the target patient population is overweight or mildly obese as it is in these individuals where one would expect peripheral resistance to be the most critical determinant of efficacy (as opposed to severe obese individuals in which central resistance predominates).
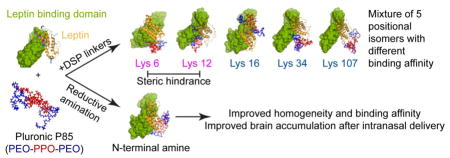
To improve the delivery of leptin to the brain, we previously modified leptin randomly at five lysine amines via a degradable crosslinker with Pluronic P85 (P85), an amphiphilic triblock copolymer of poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO), PEO-PPO-PEO [27, 28]. Modification with a single chain of P85 (1:1 conjugate) or two chains of P85 (1:2 conjugate) prolonged the circulation of leptin in the blood and resulted in increased brain accumulation of leptin after intravenous (IV) administration. While 1:1 conjugate relied on the leptin transporter to enter the brain, the 1:2 conjugate was independent of the transporter [27]. Both random conjugates, however, represented a heterogeneous mixture consisting of several positional isomers and displayed a significant loss of activity [27]. Here, we propose to selectively modify the N-terminal amine of leptin with P85 (LepNP85) to improve the homogeneity of the conjugate and minimize the steric hindrance of the polymer chain upon binding of leptin to its receptor. Additionally, we evaluate the nose-to-brain (INB) administration route to deliver the conjugates directly to the brain, thereby minimizing their exposure to peripheral clearance and bypassing the BBB. We determined here whether the P85 modification of leptin facilitates INB delivery of the hormone by improving its transport across the epithelial barriers.
2. Materials and methods
2.1. Materials
Pluronic P85 (P85, lot no. WPYE537B, average M.W. 4600) was a gift from BASF Corp. (Parspany, NJ). Disuccinimidyl suberate (DSS) linker, dithiobissuccinimidyl propionate (DSP), radioimmunoprecipitation assay buffer (RIPA buffer), 5% 2,4,6-trinitrobenzene sulfonic acid (TNBSA) solution, Corning™ high binding polystyrene 96-well plate, SureBlue Reserve™ tetramethylbenzidine (TMB) peroxidase substrate, and 100 x protease and phosphatase inhibitor cocktail were all purchased from ThermoFisher Scientific (Rockford, IL). Sephadex LH-20, PD-10, and Illustra Nap columns were purchased from GE Healthcare Life Sciences (Pittsburgh, PA). 4–15% Mini-PROTEAN® TGX™Precast Gel, ProteOn™ GLC sensor chip, 1 M ethanolamine·HCl, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), and N-hydroxysulfosuccinimide sodium salt (sulfo-NHS) were purchased from Bio-Rad Laboratories (Hercules, CA). Recombinant mouse leptin and recombinant human leptin receptor-Fc chimeras were purchased from R&D Systems (Minneapolis, MN). Na125I and Na131I were purchased from PerkinElmer (Waltham, MA). Signal transducer and activation of transcription 3 (STAT3) rabbit monoclonal antibodies and phospho-STAT3 (Tyr705) rabbit monoclonal antibodies were purchased from cell signaling (Danvers, MA). O-[2-(6-Oxocaproylamino)ethyl]-O′-methylpolyethylene glycol 5000 (PEG5K-CHO, lot no.BCBM0003V, average molecular mass 5000 Da), 4-methoxytriphenylmethyl chloride (MTr-Cl), 1,1′-carbonyldiimidazole (CDI), sinapinic acid, bovine serum albumin (BSA), protein G, Amicon ultra centrifugal filter units (molecular weight cut-off (MWCO), 10 kDa), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and all other chemicals were purchased from Sigma-Aldrich Co.(St-Louis, MO).
2.2. Synthesis of leptin conjugates
2.2.1. Random lysine conjugate
Leptin random lysine conjugates were synthesized as previously reported (Scheme S1a) [29]. Briefly, mono-amine-P85 was prepared from P85 in three steps (Scheme S1b). First, P85 (1) was reacted with MTr-Cl (1.1 molar equivalent) in anhydrous pyridine for 5 days at 23 °C. Unreacted MTr-Cl, bis-MTr-P85, mono-MTr-P85 (2), and unreacted P85 were then eluted sequentially from silica gel column in dichloromethane with stepwise increase of methanol (2%, 5%, and 10%). Second, 2 was reacted with CDI (5 molar equivalents) in anhydrous acetonitrile for 2 h at 23 °C, quenched with 0.2 ml of water for 20 min, and then mixed with ethylenediamine (20 molar equivalents) to obtain MTr-P85-amine (3). 3 was purified on a Sephadex LH-20 column in methanol and dried in vacuo. Third, 3 was dissolved in dichloromethane and mixed with 2 ml of trifluoroacetic acid (TFA) for 1 h at 23 °C to remove the MTr group and obtain mono-amine-P85 (4). The reaction mixture was dried in vacuo, dissolved in methanol and then neutralized by triethylamine. The product 4 was also purified on a Sephadex LH-20 column in methanol (37% wt. yield).
To attach mono-amine-P85 to leptin, mono-amine-P85 was activated with 20 molar equivalents of DSS or DSP (Supplementary Scheme S1c) in dimethylformamide / 0.1 M sodium borate, pH 8.0 for 30 min, and then purified on an Illustra Nap-25 column in 20% ethanol. Five molar equivalents of P85-DSP or P85-DSS polymers were immediately mixed with leptin in 20% ethanol/0.1 M sodium borate, pH 8.0 and incubated overnight at 4°C.
2.2.2. N-terminal conjugate
Leptin N-terminal amine was selectively conjugated with in house synthesized mono-aldehyde-P85 (CHO-P85-OH) via reductive amination (Supplementary Scheme S2a). Mono-propanediol-P85 was first synthesized and purified similarly to mono-amine-P85 but using 3-amino-1,2-propanediol instead of ethylenediamine (Supplementary Scheme S2b, 46% wt. yield). CHO-P85-OH was synthesized from mono-propanediol-P85 by oxidation with sodium periodate in methanol / 50 mM sodium acetate, pH 5.5 in the dark for 0.5 h at 4°C, and then purified using Illustra Nap-25 column in 20% ethanol. To synthesize LepNP85, leptin was mixed with CHO-P85-OH (20 molar equivalents) in 20% ethanol / 50 mM sodium acetate, pH 5.5 at 4°C for 3 h, and then reduced by NaBH3CN (final concentration 20 mM) at 4°C overnight. The PEGylated analog of leptin N-terminal conjugate (LepNPEG5K) was synthesized similarly using commercial PEG5K-CHO at 5 molar equivalents.
2.3. Purification of leptin conjugates
After the conjugation, leptin conjugates were precipitated in cold acetone to remove excess of polymers. Singly modified leptin conjugate was then purified by size exclusion chromatography (SEC) using ÄKTA FPLC system equipped with a ultraviolet (UV) detector at 254 nm (GE Healthcare Life Sciences). Large hydrodynamic size-protein aggregates, leptin conjugates, and free leptin were eluted sequentially from Superdex 75 100/300 GL column in 10% methanol / 0.25 M sodium phosphate, pH 7.5 at 0.5 ml/min. Peaks were collected and characterized by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF). The pooled fractions were buffer exchanged into 10 mM sodium phosphate buffer, pH 7.5 using Amicon ultra-15 centrifugal filter units (MWCO 10 kDa) and stored at −80 °C for further analysis. The yield of 1:1 modified leptin conjugates after FPLC purification was about 10% based on protein amount.
2.4. N-terminal amino group protection assay
The N-terminal amine of leptin was protected with 2-pyridinecarboxaldehyde (2PCA) as reported [30]. In brief, leptin was mixed with 400 molar equivalents of 2PCA in 0.1 M sodium borate buffer, pH 8 at 37 °C for 24 h, and then purified by PD-10 columns in 10 mM sodium phosphate, pH 7.5. The modification degree was determined by TNBSA assay following product protocol. Briefly, 50 μl of leptin or 2PCA protected leptin (Leptin-2PCA) (100–300 μg/ml) was mixed with 25 μl of 0.01% TNBSA in 0.1 M sodium bicarbonate, pH 8.5 at 37 °C for 2 h. Absorbance at 335 nm was read on a microplate reader SpectraMax M5 (Molecular devices). The modification degree (S) was calculated according to the following equation [31]: where A and C are the absorbance and concentration of native leptin (native;) or Leptin-2PCA (modified), and 8 is the total number of primary amines in leptin monomer. Leptin and Leptin-2PCA was then mixed with CHO-P85-OH or P85-DSP as described in 2.2. The reaction mixtures were characterized by SDS-PAGE and MALDI-TOF.
2.5. Characterization of leptin conjugates
2.5.1. MALDI-TOF
Protein samples were prepared using the sandwich method and matrix of saturated sinapinic acid in acetonitrile/0.1% TFA (50/50, v/v). Briefly, 0.5 μl of matrix was deposited on an Opti-TOF™ 384-well target, air dried, followed by 0.5 μl of protein sample and another 0.5 μl of matrix. The target was then air dried. Mass spectra were collected on a 5800 MALDI-TOF/TOF system (Applied Biosystems/MDS SCIEX) in positive linear mode as previously reported [27].
2.5.2. SDS-PAGE
5 μg of protein samples (as determined by microBCA) were mixed with Laemmli sample buffer without reducing agents, denatured at 95 °C for 5 min, and then resolved on 4–15% Mini-PROTEAN® TGX™ Precast Gel at 120 v for 50 min. The gel was stained with coomassie blue G250 and imaged by FluorChem E System (ProteinSimple).
2.5.3. Far UV-circular dichroism (Far UV-CD)
The secondary structures of leptin and leptin conjugates were characterized by far UV-CD. The proteins were diluted with 10 mM sodium phosphate, pH 7.5 to 0.1 mg/ml. CD spectra from 190 to 260 nm were recorded using Chirascan™-Plus CD Spectrometer (AppliedPhotophysics, Beverly, MA) at the following settings: cell pathlength 10 mm, bandwidth 1.0 nm, step 0.5 nm, time-per-point of 1.25 sec (50000 repeats per point), and temperature 20 °C. For each sample, three repeated scans were recorded, averaged, subtracted the buffer background, smoothed, and then normalized by ellipticity at 222 nm to adjust for minor difference in protein concentrations. Mean residue molar ellipticity (degrees·M−1·m−1) was calculated using equation where θ is the recorded ellipticity in degrees, M the molecular mass (16140 g/mol), C the protein concentration in mg/ml (0.1 mg/ml), l the cell pathlength in cm (0.1 cm), and n the number of amino acid residues in leptin (147) [32]. The composition of secondary structures and the goodness-of-fit parameter normalized root mean square deviation (NRMSD) were obtained using online DichroWeb program CDSSTR and protein reference set 7 [33, 34].
2.5.4. Reverse phase-high performance liquid chromatography (RP-HPLC)
The relative hydrophobicity of leptin and leptin conjugates were compared by RP-HPLC: the hydrophobic proteins were eluted later than the hydrophilic proteins. The proteins were eluted from Jupiter C4 column (particle diameter 5 μm, pore diameter 300 Å, 4.6 × 100 mm) by gradient elution at 1 ml/min and 25 °C, and monitored by absorption at 220 nm. Mobile phase A: water + 0.1% TFA; mobile phase B: acetonitrile + 0.1% TFA: isopropanol 50:50 (v/v). The elution started from 5% B for 5 min, then linearly increased to 95% B at 1%/min, and stayed at 95% B till 100 min.
2.5.5. Surface plasmon resonance (SPR)
The binding affinity of leptin and leptin conjugates to leptin receptor was determined by ProteOn™ XPR protein interaction array system (BioRad Laboratories) similarly as previous reports using BiaCore 2000 system [35] or BiaCore 3000 system [27]. Protein G was covalently immobilized on GLC sensor chip to allow capture of leptin receptor-Fc chimeras. In detail, 1600 resonance units (RU) of leptin receptor was captured by sequential injection of the following compounds to the vertical ligand channels at 30 μl/min: 1) 1:1 (v/v) mixture of 40 mM EDC/10 mM sulfo-NHS for 300 s, 2) 75 μg/ml of protein G in 10 mM sodium acetate, pH 4 for 120s, 3) 1 M ethanolamine for 300 s, and 4) 10 μg/ml of leptin receptor in running buffer (10 mM HEPES, pH 7.4/150 mM NaCl/0.005% Tween 20) for 60 s. After 180 s equilibration at 50 μl/min, a serial dilution of leptin or leptin conjugates in running buffer was simultaneously injected to the horizontal analyte rows. A 150 s binding phase and a 1200 s dissociation phase were recorded at 100 μl/min. The protein G surface was then regenerated by injection of 10 mM glycine buffer, pH 2 at 100 μl/min for 18s. The sensorgrams were auto-processed (injection alignment, baseline alignment and artifact removal), referenced to a ligand channel that had no leptin receptor for non-specific binding, and double referenced to a buffer analyte row for baseline shift (resulted from dissociation of leptin receptors). The equilibrium dissociation constants KD (nM) were obtained by fitting the sensorgrams to a Langmuir 1:1 binding model using the instrument software.
2.6. Animal studies
All animal experiments were approved by the University of North Carolina Institutional Animal Care and Use Committee. 6–8 weeks male CD-1 mice were purchased from Charles River Laboratories (Wilmington, MA).
2.6.1. Leptin receptor activation in hypothalamus after intracerebroventricular (ICV) injection and INB delivery
The mice were anesthetized by intraperitoneal injection of 40% urethane (4 mg/kg), and then injected with leptin or leptin conjugates in 4 μl of phosphate buffered saline (PBS) by ICV administration (0.2 mm caudal to bregma, 1 mm lateral to sagittal suture, and 3.5 mm in depth). Alternatively, the mice were injected intranasally with 8 μl of protein samples in PBS using a gel loading tip attached to a 10 μl pipette. The tip was inserted 4 mm deep to the depth of the cribriform plate [36] (2 μl per injection, alternate nostril between injections with 1 min apart). 30 min after ICV injection or 1 h after INB delivery, the mice were sacrificed. The brains were removed and dissected on ice to sample hypothalamus and hippocampus according to published procedure [37]. The tissue was homogenized in 100 μl of RIPA buffer supplemented with 1 x protease and phosphatase inhibitor cocktail. For ICV samples, 50 μg of homogenate proteins were analyzed by western blotting for phosphorylation of STAT3 at Tyr 705 (pSTAT3) and then total STAT3 after stripping. The band densitometry was quantified using Image J. For intranasal samples, 1 mg/ml of the hypothalamus lysates were analyzed using Wes instrument (ProteinSimple). The activity of leptin and leptin conjugate was reported as densitometric ratio of pSTAT3 to STAT3.
2.6.2. Pharmacokinetic study
2.6.2.1. Radioactive labeling
Leptin and leptin conjugates were labeled with iodine by chloramine T method [27]. Briefly, 10 μg of proteins were mixed with 1 mCi of Na125I or Na131I and 10 μg of chloramine T in 100 μl of 0.25 M sodium phosphate, pH 7.5 for 60 s. The proteins were then eluted from Illustra Nap-5 columns in PBS, collected in tubes pretreated with 1% BSA to prevent nonspecific adsorption, and concentrated to around 80 μl using Amicon ultra-0.5 ml centrifugal filter units (MWCO 10 kDa). The percentage of iodine associated with proteins in the final concentrated samples was determined by trichloroacetic acid (TCA) precipitation [38]. Briefly, 1 μl of the concentrated samples was mixed with 0.5 ml of 1% BSA in PBS and 0.5 ml of 30% TCA by brief vortex followed by centrifugation at 5400 g for 10 min. The radioactivity of the resulting protein pellet and supernatant was counted on r-counter (PerkinElmer). The percentage of iodine associated with protein was calculated as the percentage of pellet radioactivity in total radioactivity. Protein samples with percentage of iodine association higher than 95% were used.
2.6.2.2. Serum clearance and brain influx rate after IV injection
The mice were anesthetized by intraperitoneal injection of 40% urethane (4 mg/kg). 4 × 105 counts per minute (cpm) of 125I- and 131I- labeled compounds in 0.2 ml of lactated ringer’s buffer supplemented with 1% BSA were co-injected to the left jugular vein. At defined time points (1 to 180 min), blood was sampled from the right carotid artery, allowed to clot at 23 °C, and then centrifuged at 5400 g for 10 min to collect serum. Immediately after blood sampling, the mice were sacrificed. The whole brains were removed and weighed. The brain/serum samples and four injection checks representing injected dose (ID, cpm per mouse) were counted on r-counter simultaneously.
The cpm in brain/serum samples was divided by ID and multiplied by 100 to get %ID, and then normalized by weight for brain samples (Am, %ID/g) or by volume for serum samples (Cpt, %ID/ml). Ln(Cpt) was plotted against time (t, min) using Prism 5.0 software (GraphPad, San Diego, CA). The serum clearance rate (k, min−1), half-life (t1/2, min), and volume of distribution (V, ml) were calculated from the slope and y-intercept of the linear regression using the following equations [27].
The unidirectional brain influx rate (Ki, ml/g·min) and initial volume of brain distribution (Vi, ml/g) were determined by multiple-time regression analysis using the following equation [39, 40]
where Am/Cpt is the brain/serum ratios (ml/g), and ∫ Cpt·dt/Cpt the exposure time, which is the integral area under the curve (AUC) of serum concentration from time 0 to time t divided by Cpt at time t.
2.6.2.3. Serum absorption and brain regional distribution after intranasal administration
The mice were anesthetized by intraperitoneal injection of 40% urethane (4 mg/kg). 2 × 106 cpm of 125I- and 131I-labeled samples in 2 μl of PBS were co-injected intranasally into the upper nasal area near cribriform plate by inserting a gel loading pipette tip 4 mm into the left nostril of each mouse [36]. The mice were laid naturally on their backs on a heating pad after injection. 5, 10, 20, 30 and 60 min later, serum was collected as mentioned above. The olfactory bulb, hypothalamus, hippocampus, and the rest of brain were sampled on ice following published procedure [37] and weighed. Radioactivity in serum and brain samples was counted. Six injection checks representing ID were counted simultaneously. Radioactivity in serum and brain samples were presented as %ID/ml and %ID/g, respectively as mentioned above. Note that the mice were not perfused. It is reported that the radioactivity in perfused brain was comparable to non-perfused brain after intranasal delivery of leptin, due to negligible material entering the blood [41].
2.6.2.4. Stability of iodinated proteins in serum and brain samples
The mice were dosed intranasally with 2 × 106 cpm of 125I- and 131I-labeled samples in 2 μl of PBS as mentioned above. 10 and 60 min later, blood was sampled by cardiac puncture and allowed to clot at 23 °C to collect serum. The mice were immediately perfused with ice-cold PBS. The brain was removed, homogenized in 2 ml of ice-cold PBS, and then centrifuged at 5400 g for 10 min. 50 μl of serum and 1 ml of brain homogenate supernatant were precipitated with 1 ml of 30% TCA as mentioned above. Process controls that evaluate process-induced degradation were made by spiking untreated mice serum and brain samples with iodinated proteins and then being treated similarly as dosed animal samples. The stability of iodinated samples in serum and brain samples was evaluated as the percentage of radioactivity in the protein pellet [27].
2.6.2.5. Blood absorption and brain clearance after ICV injection
To study the clearance of leptin and LepNP85 from the brain, leptin and LepNP85 were labeled with biotin to distinguish from endogenous leptin. Briefly, the proteins were mixed with 40 molar equivalents of Sulfo-NHS-biotin in 10 mM sodium phosphate, pH 7.5 on ice for 2 h, and then purified by PD-10 column. 1 μg of biotin-labeled leptin or LepNP85 in 1 μl of PBS were injected locally into brain by ICV injection. At each time point, blood was sampled from carotid artery into heparinized tubes and centrifuged at 2000 g for 15 min to collect plasma. The whole brain was removed and homogenized in 1 ml of RIPA buffer supplemented with 1 x protease inhibitor cocktail. The concentration of biotin-labeled protein in brain lysate and plasma were analyzed by enzyme-linked immunosorbent assay (ELISA). Briefly, a high binding polystyrene 96-well plate was coated with 1 μg/ml of recombinant leptin receptor-Fc chimera in PBS overnight at 4 °C (0.1 ml per well). After trice wash with PBS, each well was blocked with 0.2 ml of 3% BSA/PBS at 23 °C for 1 h, and then incubated with 0.1 ml of biotin-labeled protein standards or brain/plasma samples diluted with 3% BSA/PBS for 2 h. After another trice wash with 0.2 ml of 0.05% Tween-20/PBS, each well was incubated with 0.1 ml of high sensitivity streptavidin-horseradish peroxidase (Pierce, 1:250 dilution) in 3% BSA/PBS for another 1.5 h followed by four washes with 0.05% Tween-20/PBS. Each well was then incubated with 0.1 ml of TMB peroxidase substrate for 15 min. The reaction was stopped by adding 0.1 ml of 2 M H2SO4 and detected at 450 nm.
3. Results
3.1. Synthesis and purification of leptin conjugates
The two leptin-P85 conjugates with the block copolymer attached to the protein’s lysine amino groups via DSP linker (LepDSPP85) or DSS linker (LepDSSP85) were synthesized and purified as previously reported [27] (Supplementary Scheme S1 and Fig. S1).
Leptin N-terminal conjugation was accomplished via reductive amination based on the pKa difference between N-terminal amine (7.6–8.0) and lysine amine (9.3–9.5) [42]. Under acidic conditions, the lysine amines were preferentially protonated, leaving the unprotonated N-terminal amine reacting with aldehyde to form Schiff base, which was then reduced to a stable secondary amine by sodium cyanoborohydride (Supplementary Scheme S2a). By this method, we synthesized leptin N-terminal conjugate modified with P85-aldehyde (CHO-P85-OH, prepared as shown in Supplementary Scheme S2b) (LepNP85) or with commercial PEG5K-CHO of similar molecular mass (LepNPEG5K) at pH 5.5. The appearance in the MALDI-TOF spectra of a 21 kDa peak representing leptin monomer modified with a polymer chain (molar ratio of leptin monomer: polymer 1:1) and a 37 kDa peak representing leptin dimer modified with a polymer chain (2:1) in the reaction mixture (Fig. 1a) indicated successful conjugation of leptin with CHO-P85-OH and PEG5K-CHO.
Figure 1. Synthesis, purification, and composition characterization of LepNP85 and LepNPEG5K.

(a) MALDI-TOF spectra of reaction mixture. The ratio denotes the molar ratio of leptin monomer to polymer. (b) SEC of leptin conjugates eluted from Superdex75 100/300 GL column in 10% MeOH/0.25 M sodium phosphate, pH 7.5 at 0.5 ml/min. (c) SDS-PAGE of pooled fractions in SEC denoted by elution time. Fraction of 20.3–21.9 min in LepNP85 and fraction of 18.5–21 min in LepNPEG5K were used.
LepNP85 and LepNPEG5K were purified by SEC (Fig. 1b). The fractions were characterized by MALDI-TOF (Supplementary Fig. S2) and SDS-PAGE (Fig. 1c). Leptin conjugates and unmodified leptin were eluted sequentially based on their hydrodynamic size. The aggregates of large hydrodynamic size eluted in 14 to 17 min consisted of unmodified leptin (1:0, 16 kDa), singly modified leptin conjugate (1:1, 21 kDa), doubly modified leptin conjugate (1:2, 26 kDa), and heavily modified leptin conjugates (as indicated by the smear in SDS-PAGE [43]). Mixture of 1:2 and 1:1 conjugates was eluted in 18.2 to 20.3 min. A relatively pure fraction of 1:1 conjugate was eluted in 20.3 to 21.9 min. Mixture of 1:1 conjugate, leptin monomer modified by the diblock PEO-PPO (1:1*, 18 kDa), and unmodified leptin was eluted in 21.9 to 24 min. Lastly, unmodified leptin was eluted in 25 to 27 min. Commercial Pluronics including P85 from BASF typically contain about 10–25 wt% diblock PEO-PPO impurities [44]. We detected in mono-amine-P85 synthesized from commercial P85 the presence of low-molecular mass impurities (Supplementary Fig. S3), which should be separated prior to conjugation in subsequent improved procedures. Compared to LepNP85, LepNPEG5K 1:1 conjugate had similar molecular mass (21 kDa) but was eluted earlier (18.5 vs. 21 min) in SEC, indicating their larger hydrodynamic size. This is possibly due to greater swelling of the PEO chain attached to leptin in LepNPEG5K compared to the P85 in LepNP85 in which a hydrophobic PPO block should form a more condense structure. We carried out a molecular dynamics simulation and showed that the PEG chain (5 kDa) in LepNPEG5K is loosely wrapped around leptin whereas P85 in LepNP85 is compressed on the surface of leptin (Supplementary Fig. S4). In the following studies, we used purified 1:1 conjugates of LepDSPP85, LepDSSP85, LepNP85, and LepNPEG5K (Supplementary Fig. S5, chemical structures are shown in Supplementary Scheme. S3).
3.2. N-terminal amino group protection analysis
To confirm that the CHO-P85-OH was attached at the N-terminal amino group of leptin, we protected this group with 2PCA [30] and then examined if the conjugation of leptin with CHO-P85-OH is affected. On average, each leptin molecule was modified with 0.76 ± 0.06 2PCA as determined by TNBSA assay. The modification resulted in an incremental increase of the molecular mass of the leptin monomer, dimer and trimer by 129, 232, and 311 Da respectively, as determined by MALDI-TOF (Supplementary Fig. S6). This increase appears to be higher than expected (89, 178, and 267 Da respectively), which is due to the low accuracy of our MALDI-TOF analysis for the species with the mass-to-charge ratio exceeding 4000. Based on MALDI-TOF (Supplementary Fig. S7), the protection of the N-terminal amine nearly abolished the formation of LepNP85 1:1 conjugate, but did not impede the reaction of the protein with P85-DSP, which modifies leptin at multiple lysine amines [27]. This conclusion was reinforced by the SDS-PAGE (Fig. 2), suggesting that the protection of the N-terminal amine greatly diminished the formation of 1:1 conjugate in the reaction of leptin with CHO-P85-OH, but did not affect the yield of such conjugate in the reaction with P85-DSP. This provides strong evidence of modification of N-terminal amine of leptin with CHO-P85-OH in LepNP85.
Figure 2.

SDS-PAGE of the reaction mixture of leptin and Leptin-2PCA with CHO-P85-OH or with P85-DSP.
3.3. Secondary structures of the conjugates by CD
The secondary structures of leptin and leptin conjugates were characterized by far UV-CD (Fig. 3). We also examined the CD spectra of the physical mixtures of native leptin with P85 or PEG5K to see if these polymers can have any effect on the conformation of the hormone via non-covalent interaction. The native leptin displayed a dominant α-helical structure (63%) as reported elsewhere [27, 45, 46]: two minima at 208 and 222 nm, and one maximum around 190 nm. The CD spectra of leptin in 1:1 physical mixture of leptin with P85 or PEG5K or covalent conjugates of leptin with these polymers were superimposed to that of native leptin. The results suggest that neither the physical mixture nor the covalent conjugation of the polymers perturb the secondary structure of leptin (see Supplementary Table S1 for compositions of secondary structure).
Figure 3. Far UV-CD spectra of leptin and leptin conjugates.

(a) Leptin 1:1 physical mixture with P85 or PEG5K. (b) Leptin 1:1 conjugates.
3.4. Relative hydrophobicity of the conjugates by RP-HPLC
The relative hydrophobicity of leptin and leptin conjugates was compared by RP-HPLC using a gradient of acetonitrile + 0.1% TFA: isopropanol 50:50 (v/v) (Fig. 4). As seen in the figure, leptin modified with a single chain of PEG5K (LepNPEG5K) was eluted at nearly the same concentration of organic solvents as the unmodified leptin, suggesting that this conjugate had similar hydrophobicity to the native protein. As expected, leptin modified with a single chain of P85 (both LepNP85 and LepDSPP85) were eluted at higher concentration of organic solvents than the unmodified leptin and PEGylated analog, which was consistent with their increased hydrophobicity due to incorporation of the hydrophobic PPO block. Notably, despite the increased hydrophobicity of the leptin-P85 conjugates we did not observe aggregation or formation of micelle-like structures of the conjugates in aqueous solution. We characterized the intensity-based size distribution of leptin and LepNP85 at protein concentration of 1 mg/ml (Supplementary Fig. S8). Interestingly, LepNP85 had lower polydispersity and less aggregation than native leptin. Each sample exhibited a main fraction of small particles with an effective diameter of 5.3 nm for leptin and 8.2 nm for LepNP85, which is much smaller than the typical diameter of P85 micelles (around 16 nm [47]).
Figure 4. RP-HPLC analysis of leptin and leptin conjugates.

Leptin and leptin 1:1 conjugates were eluted from Jupiter C4 column (particle diameter 5 μm, pore diameter 300 Å, 4.6 × 100 mm) by gradient elution at 1 ml/min and 25 °C, and monitored by absorption at 220 nm. Mobile phase A: water + 0.1% TFA; mobile phase B: acetonitrile + 0.1% TFA: isopropanol 50:50 (v/v). The elution started from 5% B for 5 min, then linearly increased to 95% B at 1%/min, and stayed at 95% B till 100 min.
3.5. Binding affinity of the conjugates by SPR
Next, we characterized the binding of leptin and leptin conjugates to leptin receptor by SPR (Fig. 5). Native leptin displayed the fast binding and slow dissociation and had a dissociation constant (KD) of 0.089 ± 0.046 nM, which was similar to the previously reported value (0.12 ± 0.08 nM) [35]. Leptin N-terminal conjugates LepNP85 and LepNPEG5K had similar sensorgrams and dissociation constants (0.20 ± 0.13 nM for LepNP85 and 0.28 ± 0.15 nM for LepNPEG5K). These KD values were numerically (~2 to 3 times) higher but not statistically different compared to that of the native leptin. In contrast, the random lysine conjugates LepDSPP85 and LepDSSP85 displayed much faster dissociation rates and over ten times higher dissociation constants (1.09 ± 0.23 nM for LepDSPP85 and 1.08 ± 0.24 nM for LepDSSP85). These KD values were significantly different compared to that of the native leptin. Therefore, consistent with the molecular dynamics prediction [27], the binding affinity (inverse to KD) determined by SPR was only marginally affected in leptin N-terminal conjugates, but greatly decreased in the random lysine conjugates. Since we determined that the conjugation did not affect the conformation of leptin, the observed changes in binding of the leptin conjugates to the receptor are most likely due to steric hindrance of such binding by the attached polymer chains.
Figure 5. Binding affinity of leptin and leptin conjugates to leptin receptor by SPR.

(a) Representative sensorgram for native leptin. (b) Representative sensorgram for LepNP85 1:1 conjugates. (c) Representative sensorgram for LepDSSP85 1:1 conjugates. (d) Dissociation constants (KD) of leptin and leptin 1:1 conjugates. Data are mean ± SD, n= 5~12. *** p < 0.001 and n.s. not significant by One-way ANOVA and post Newman-Keuls Multiple Comparison Test.
3.6. Leptin receptor activation in hypothalamus after ICV injection
Leptin regulates appetite mainly by binding to its receptor in the hypothalamus and activating the downstream janus kinases-STAT3 (JAK-STAT3) pathway [8, 48]. To determine if leptin conjugates are active in vivo, we injected the leptin and leptin conjugates to the brain by ICV administration, and then sampled hypothalamus for western blotting (Fig. 6). The pSTAT3 to total STAT3 ratio (pSTAT3/STAT3) served as a measure of leptin receptor activation [49, 50]. 30 minutes after ICV administration of native leptin at a dose of 100 ng/mouse, this ratio was significantly higher than the one observed after the injection of the vehicle (Fig. 6a). Further increase of leptin dose to 500 and 1000 ng/mouse did not produce any significant change in pSTAT3/STAT3 ratio. LepNP85 displayed essentially the same activity as native leptin, producing nearly the same pSTAT3/STAT3 ratios at the same doses of 100, 500, and 1000 ng/mouse (Fig. 6a), suggesting signal saturation at the dose of at least 100 ng/mouse. Next, we compared all leptin conjugates to native leptin at a dose of 100 ng/mouse and concluded that they all have similar activities as the native leptin (Fig. 6b). It is likely that we cannot detect the differences between these forms as seen in SPR due to signal saturation at this dose and the large animal variability such as receptor expression on plasma membrane, downstream signaling, and diffusion of proteins from injection sites to hypothalamus [51], which reduce the experimental space for us to detect the difference at lower doses. Nonetheless, these data suggest that all leptin conjugations produced in this study were active in live brain.
Figure 6. Phosphorylation of STAT3 in hypothalamus 30 min after ICV injection of leptin and leptin conjugates.

(a) Dose response of leptin and LepNP85 1:1 conjugates. Data are mean ± SEM, n=3. (b) Comparison of leptin and leptin 1:1 conjugates at 100 ng/mouse. Data are mean ± SEM, n= 5~7. ** p < 0.01 and *** p < 0.001 by One-way ANOVA and post Newman-Keuls Multiple Comparison Test.
3.7. Serum clearance and brain influx rates after IV injection
To characterize serum clearance and brain influx rate following IV administration, we labeled leptin and leptin conjugates with 125I and 131I respectively and co-injected them into the left jugular veins of CD1 mice. The half-life of serum clearance was calculated from the linear portion of the regression of Ln serum concentration against time (Fig. 7 and Table 1). Similar to previously reported results for LepDSPP85 [27], LepNP85, LepDSSP85 and LepNPEG5K showed two-phase decay of serum clearance and prolonged half-life of 50.58 ~57.04 min compared to leptin (~11.41 min). The unidirectional brain influx rate (Ki) and the initial volume of brain distribution (Vi) were calculated using multiple-time regression analysis (Table 1). Vi included the vascular space and steady-state exchangeable space that was quickly equilibrated with the blood, such as rapid reversible transporter binding at the brain endothelial cells [39, 40, 52]. The brain influx rate of native leptin was similar to the value reported previously (0.236 μl/g·min) [27]. LepNP85 displayed ~3.5-times lower brain influx rate and slightly lower initial volume of brain distribution compared to co-injected native leptin. We further compared LepNP85 to non-cleavable random lysine-modified conjugate, LepDSSP85. (We did not include in this study a cleavable analog, LepDSPP85 that was evaluated previously [27] to avoid any interference of the disulfide bond reduction and decrease the animal use.) Compared to LepDSSP85, LepNP85 displayed a trend of having ~1.5-time faster brain influx rate (p=0.05718) and slightly albeit significantly higher initial volume of brain distribution. We also compared LepNP85 to the N-terminal PEGylated analog LepNPEG5K and revealed great similarity between these analogs. LepNPEG5K and LepNP85 had similar brain influx rates (p=0.8965) and volumes of brain distribution (p=0.4086). Taken together, these results suggest that the N-terminal leptin conjugates with P85 or PEG5K are slightly superior in terms of brain delivery to the random lysine conjugate by IV injection. However, brain entry of LepNP85 was inhibited by co-injection of cold leptin (Supplementary Fig. S9), suggesting it cannot overcome the leptin peripheral resistance in obesity. Therefore, we administered LepNP85 intranasally using the INB route to bypass the peripheral resistance of leptin transporter.
Figure 7. Serum clearance and unidirectional brain influx rate of leptin and leptin conjugates.

In each row, the two compounds were labeled with 125I and 131I respectively and co-injected into CD-1 mice by IV injection. (a, d, g) Serum concentration over time. (b, e, h) Linear regression of serum concentration. (c, f, i) Multiple-time regression analysis for unidirectional brain influx rate. n=1~2/time point, **p<0.01 and ***p<0.001 by two-way ANOVA.
Table 1.
Serum clearance, unidirectional brain influx rates and initial volumes of brain distribution for leptin and leptin 1:1 conjugates.
| Leptin VS LepNP85# | LepNP85 VS LepDSSP85# | LepNP85 VS LepNPEG5K# | ||||
|---|---|---|---|---|---|---|
| Leptin | LepNP85 | LepNP85 | LepDSSP85 | LepNP85 | LepNPEG5K | |
| t1/2, min | 11.42 | 51.52 | 53.38 | 54.52 | 57.04 | 50.58 |
| Ki, ml/(min · g) | 0.270±0.004 | 0.078±0.001*** | 0.071±0.009 | 0.048±0.007 | 0.074±0.015 | 0.072±0.015 |
| Vi, ml/g | 25.18±0.35 | 20.17±0.19 | 14.31±0.53 | 12.94±0.43** | 16.11±0.94 | 15.29±1.03 |
Pairs were labeled respectively with 125I and 131I and co-injected into mice by IV administration. t1/2, Ki, and Vi were calculated from Fig. 7,
p<0.01 and
p<0.001 by two-way ANOVA.
3.8. Serum absorption and brain regional distribution after intranasal administration
3.8.1. Comparison of leptin and LepNP85
We examined whether Pluronic modification could improve INB delivery of leptin to the brain. Towards this goal, we co-administered 125I-LepNP85 and 131I-leptin into the upper nasal area near cribriform plate [36]. INB delivery of leptin in a rat model follows the traditional pattern of uptake by the olfactory bulb with less amounts of uptake by other brain regions and with little material entering blood [41]. Consistent to this report, the serum absorption of both leptin and LepNP85 increased over time (Fig. 8). The AUC of serum concentration (%ID/ml) over the first 60 min for LepNP85 was ~1.5-times of that for co-injected leptin (Supplementary Table S2), suggesting that the copolymer modification enhanced the serum absorption of LepNP85. However, the serum concentration of LepNP85 was still quite low (1.7 %ID/ml at 60 min). Accordingly, the distribution of leptin and LepNP85 in peripheral organs was minimal and increased over time as the serum concentration increased ( Supplementary Fig. S10 ). Notably, the lungs, which were supplied by iodinated proteins from blood and nose, had significantly higher accumulation compared to the other peripheral organs that had blood as the only source for iodinated proteins.
Figure 8. Serum absorption and brain regional distribution of intranasally delivered leptin and LepNP85 1:1 conjugate.

Male CD-1 mice were co-injected with 125I-LepNP85 and 131I-leptin. Data are mean ± SEM, n=7/time point, * p < 0.05, ** p < 0.01, and *** p < 0.001 by Two-way ANOVA.
The brain distribution pattern of both LepNP85 and native leptin was also similar to that reported for intranasal leptin in rat [41]: the highest concentration was observed in olfactory bulb, followed by hypothalamus and then hippocampus, which was comparable to the rest of brain (Fig. 8). Compared to co-injected leptin, LepNP85 had similar accumulation in olfactory bulb, but 1.9-times higher AUC in hypothalamus and 3.2-times higher AUC in hippocampus over the first 60 min. This result was further reinforced using a DIO mice model suggesting that LepNP85 accumulated in the brain hypothalamus and hippocampus better than the unmodified leptin ( Supplementary Fig. S11a ). Brain distribution of intranasal LepNP85 did not differ between healthy mice and DIO mice ( Supplementary Fig. S11b). These data support our hypothesis that Pluronic modification enhanced INB delivery of leptin.
3.8.2. Comparison of LepNP85 and LepNPEG5K
To explore the function of hydrophobic PPO block in P85, we compared LepNP85 to LepNPEG5K. LepNP85 accumulated 2.4-times more in serum, 1.7-times more in olfactory bulb, 2.5-times more in hypothalamus, and 3.2-times more in hippocampus than co-injected LepNPEG5K (Fig. 9), suggesting that the PPO block in LepNP85 facilitates serum absorption and intranasal delivery to the brain. Note that leptin, LepNP85 and LepNPEG5K demonstrated similar integrity in serum and brain samples after intranasal administration as determined by TCA precipitation ( Supplementary Table S3), suggesting the above comparisons of total radioactivity represent the comparisons of intact proteins.
Figure 9. Serum absorption and brain distribution of intranasally delivered 1:1 conjugates of LepNP85 and LepNPEG5K.

Male CD-1 mice were co-injected with 125I-labeled LepNP85 and 131I-labeled LepNPEG5K. Data are mean ± SEM, n=7/ time point, * p < 0.05 and *** p < 0.001 by Two-way ANOVA.
3.8.3. Comparison of LepNP85, LepDSP85, and LepDSSP85
Next, we compared LepNP85 to the previously developed LepDSPP85. Interestingly, LepNP85 had 0.7-time lower serum absorption, similar accumulation in OB, 3.5-times higher accumulation in hypothalamus, and 5.3-times higher accumulation in hippocampus (Fig. 10). The reduced serum absorption and increased brain accumulation indicated that LepNP85 was superior to LepDSPP85. The difference between LepNP85 and LepDSPP85 in INB delivery cannot be explained by the difference in modification sites, because LepNP85 and LepDSSP85 had similar serum absorption and brain distribution after INB delivery (Supplementary Fig. S12). These suggest that the difference between LepNP85 and LepDSPP85 was mainly due to the reduction of disulfide bond in LepDSPP85. Taken together, LepNP85 is the best conjugate that accumulated most in brain hypothalamus and hippocampus by intranasal delivery.
Figure 10. Serum absorption and brain regional distribution of intranasally delivered 1:1 conjugates of LepNP85 and LepDSPP85.

Male CD-1 mice were co-injected with 125I-labeled LepNP85 1:1 conjugates and 131I-labeled LepDSPP85 1:1 conjugates. Data are mean ± SEM, n=7/time point, ** p < 0.01 and *** p < 0.001 by Two-way ANOVA.
3.8.4. Brain / Serum ratios of leptin and leptin conjugates
The brain/serum ratios for leptin and leptin conjugates after INB delivery were calculated by summing radioactivity of hypothalamus, hippocampus, and the rest of brain (Supplementary Fig. S13). These values for all the conjugates were generally one order higher than the respective brain/serum ratios observed after IV administration. For the native leptin, the difference was somewhat less suggesting possibility of at least partial reabsorption of the hormone from the blood to the brain.
3.8.5. Competitive effect of leptin on distribution of the conjugate
Fliedner, et al reported that in a rat model the intranasal delivery of leptin to the brain is non-saturable although the serum absorption of the protein is saturable [41]. Specifically, co-injection of iodinated leptin with unlabeled leptin (0.2 mg/kg in ~600 g rats, which according to another paper is a therapeutic dose [53]) did not affect the amount of the labeled protein distributed in the brain (including olfactory bulb, hypothalamus and hippocampus) but distribution of this protein in the serum. We obtained similar result in our mouse study for LepNP85 conjugate. In this case, co-administration of the conjugate with unlabeled leptin (10 μg/mouse or 0.4 mg/kg in ~25 g mice) significantly decreased serum absorption of the conjugate, and did not inhibit its uptake in olfactory bulb, hypothalamus, and hippocampus over the first 30 min after INB delivery (Fig. 11). This suggests that although the brain distribution of the LepNP85 appears to be receptor-independent, the serum distribution is still mediated by a saturable system that recognizes both the native leptin and this conjugate.
Figure 11. Serum absorption and brain regional distribution of intranasally delivered 1:1 conjugates of LepNP85 with or without cold leptin.

Each male CD-1 mouse was injected with 125I-labeled LepNP85 with or without 10 μg of cold leptin. Data are mean ± SEM, n=7/time point, * p < 0.05 by Two-way ANOVA.
3.9. Blood absorption and brain clearance of leptin conjugate after ICV injection
We also observed that leptin and LepNP85 can be cleared back into blood when administered locally into the brain by ICV injection (Fig. 12 and Supplementary Fig. S14). Compared to leptin, plasma concentrations of LepNP85 were higher (p<0.001 by Two-way ANOVA), suggesting that P85 modification enhanced the brain to blood clearance of leptin (Fig. 12a). Compared to the initial time points, the plasma concentration of leptin dropped significantly at 4 h while the concentration of LepNP85 still remained high and decreased only at 8 h. The more sustained amount of the conjugate found in the blood may be due to both its greater brain to blood clearance as well as its longer circulation half-life compared to the native leptin (Fig. 7). The brain concentrations of leptin were comparable to those of LepNP85 at all time points (Fig. 12b). Compared to the initial time points, the brain concentrations of leptin and LepNP85 only dropped significantly after 4 h.
Figure 12. Brain clearance and serum absorption of leptin and LepNP85 by ICV injection.

1 μg of biotin-labeled leptin and LepNP85 in 1 μl of PBS were injected locally into brain by ICV injection. The concentrations of biotin-labeled proteins in brain lysate and plasma were analyzed by ELISA. (a) Blood absorption of biotin-leptin and biotin-LepNP85 after ICV injection. (b) Brain retention of biotin-leptin and biotin-LepNP85 after ICV injection. Data are mean ± SEM, n=4/time point, * p < 0.05 by One-way ANOVA and post Dunnett’s Multiple Comparison Test, and *** p < 0.001 by Two-way ANOVA.
3.10. Leptin receptor activation in hypothalamus after intranasal administration
One hour following intranasal administration, LepNP85 activated the downstream STAT3 signaling pathway of leptin receptor in brain hypothalamus at a dose of 5 μg/mouse (Fig. 13). At this dose, unmodified leptin was not active. Leptin was found to be inactive until at dose of 12 μg/mouse. At this higher dose, LepNP85 was not active, which is consistent with the U shape response of leptin [54]. Note that the intranasal route is variable especially at low dose. At the dose of 5 μg/mouse, one out of the nine mice in the leptin group and four out of the eleven mice in the LepNP85 group responded to the treatment.
Figure 13. Phosphorylation of STAT3 in hypothalamus 1 h after intranasal injection of leptin and LepNP85.

(a) At dose of 5 μg/mouse. n=4–11. The western blotting was repeated twice. (b) At dose of 12 μg/mouse. n= 4~5. Data are mean ± SEM, * p < 0.05 by One-way ANOVA and post Newman-Keuls Multiple Comparison Test.
4. Discussion
To improve the delivery of leptin to the brain, we previously modified leptin with P85 via a cleavable disulfide bond linker (LepDSPP85) [27]. In this approach, the polymer was attached to the protein randomly at five lysine amines K6, K12, K16, K34, and K107. The resulting conjugates displayed significantly increased serum stability and circulation time. Their brain accumulation was increased compared to native leptin probably due to the increased exposure to the BBB. Notably, while the 1:1 conjugate relied on the leptin transporter to enter the brain, the 1:2 conjugate was independent of the transporter [27]. However, the random modification also resulted in heterogeneity of the conjugates and considerable loss of their activity, especially in the case of the protein modified with two P85 chains. As previously reported based on the molecular dynamics simulation, the K6 and K12 positional isomers of these conjugates are likely to be inactive since the point modifications of these amino groups should sterically hinder the binding of leptin to its receptor [27]. This conclusion is also reinforced by other modeling studies indicating that K6 and K12 of leptin contribute to the binding specificity by forming salt bridges and hydrogen bonds with D615, E563, N564, and N565 in the leptin-binding domain (LBD) of the leptin receptor [55]. At the same time, based on our modeling study [27], the modification of leptin at the N-terminal amine should not affect the hormone binding to the receptor. Experimentally, leptin has been fused with Fc [56] or Pro/Ala/Ser polypeptides [57] at the N-termini, and the resulting fusion proteins were still active in reducing the body weight in both leptin-deficient ob/ob mice and normal lean mice. Therefore, in the present study, we selectively conjugated P85 to leptin N-terminal amine and showed that this conjugate has improved binding affinity compared to randomly modified forms.
Another limitation of the previously developed random lysine conjugate modified with one P85 polymer chain is that its entry to the brain appeared to be dependent on the BBB transporter, which is impaired in obese patients. We showed here that LepNP85 entered the brain slightly faster than the random conjugate. However, its brain delivery still relied on the BBB transporter as it was inhibited by the unmodified leptin. Hence we explored the INB route to bypass the BBB altogether. Several studies have reported physiological effects of native leptin administered through this route. For example, in a rat seizure model, nasal leptin showed anticonvulsant action via direct effects on glutamate neurotransmission in the hippocampus [58]. More relevantly, food intake was reduced in normal body weight rats and in DIO rats receiving intranasal leptin [49, 53, 59]. These results demonstrate that nasal leptin can access the brain in amounts large enough to produce therapeutic effects. Toxicity was not reported in animals receiving nasal leptin at therapeutic doses [49, 53, 59]. Furthermore, whether used as a weight loss measure or an approach to healthy weight maintenance, an easily and non-invasively administered product should have wide acceptance by the public and the greatest positive impact on our healthcare system [60].
The concept of INB delivery is that compounds administered intranasally gain direct access to the central nervous system (CNS) [61]. Three pathways are believed to be involved in ferrying substances across the epithelial barriers and into the CNS: 1) transcellular transport across the sustencular cells in respiratory or olfactory epithelia, and then diffusion via the CSF and brain interstitial fluid; 2) intraneuronal or paraneuronal retrograde transmission via the olfactory nerves across the cribriform plate to olfactory bulb; 3) intraneuronal or paraneuronal retrograde transmission via the trigeminal nerve to caudal brainstem. Modification of proteins with amphiphilic Pluronic block copolymers such as P85 has been shown to enhance uptake of these proteins in neurons, epithelial cells, and endothelial cells [29, 62, 63]. Therefore, we hypothesized that such modification can also enhance the transmembrane penetration of the polypeptide across these epithelial barriers. Towards this goal, we tested if P85 modification improves INB delivery.
We compared LepNP85 to native leptin and random leptin conjugates (LepDSPP85 and LepDSSP85). We also included in this study a comparison of LepNP85 with N-terminal conjugate with PEG having approximately same molecular mass as P85 (LepNPEG5K). We examined the serum absorption and distribution of these compounds in hypothalamus where leptin regulates feeding behavior and in hippocampus where leptin can mitigate the cognitive dysfunction associated with obesity [64, 65] and Alzheimer’s disease [66]. Based on this study LepNP85 was superior in terms of INB delivery to the native leptin as well as each other conjugate except for the non-cleavable random lysine conjugate LepDSSP85. Several conclusions can be made based on this. First, the P85 modification of leptin improved the intranasal delivery of this protein to the relevant regions of the brain. Second, the hydrophobic PPO block of P85 was important in the INB delivery process of the conjugate. (This conclusion was made by comparing the N-terminal conjugates of leptin with PEG (same as PEO) and P85 having a triblock structure PEO-PPO-PEO). Third, the linker between the protein and the copolymer needs to be stable as the cleavable LepDSPP85 conjugate was less successful in terms of INB delivery than the two non-cleavable conjugates LepNP85 and LepDSSP85. These two latter conjugates displayed virtually same brain distribution after intranasal administration, which means that the site of the P85 attachment to the protein is not as important for this delivery pathway. However, the N-terminal conjugate, LepNP85 is still preferable due to its greater binding affinity compared to any of the randomly modified conjugates, LepDSPP85 or LepDSSP85. Although we showed that reduction of disulfide bond partially recovered the binding affinity of leptin released from LepDSPP85 [27], LepNP85 had comparable binding affinity and central activity to that of native leptin, suggesting that it is unnecessary to release leptin from LepNP85. Taken together, LepNP85 was the best of the studied conjugates for INB delivery of leptin to brain hypothalamus and hippocampus.
This modified form of leptin has displayed the central activity of the hormone after intranasal administration. Specifically, LepNP85 activated the leptin receptor in hypothalamus at 2.4-fold lower dose than the unmodified leptin. This was consistent with the 1.9-fold higher AUC of LepNP85 in hypothalamus compared to the native leptin after INB delivery. Due to the low expression level of leptin receptor in hippocampus, we were not able to detect enhanced phosphorylation of STAT3 in hippocampus following ICV and INB administration of leptin and LepNP85.
Interestingly, P85 modification increased the serum absorption of the LepNP85 conjugate after the intranasal administration compared to the native leptin. The serum distribution of the conjugates appeared to correlate with their hydrophobicity as LepNP85 displayed higher serum levels than LepNPEG5K, but these levels were approximately equal to those of the uncleavable conjugate LepDSSP85. This suggests that the hydrophobic PPO block in P85 facilitates leptin transport across the epithelial barriers into the underneath vasculature. Similar to the native protein, the LepNP85 conjugate at least in part used a saturable pathway for the distribution to the serum that was inhibited by co-administration of the free hormone. It is known that leptin receptors are expressed in human nasal mucosa [67–70] and serum levels of leptin were shown to positively correlate with that of nasal leptin in human [67]. Based on this, we believe that both the membrane binding of the PPO block and the receptor/transporter binding of the leptin might be responsible for the serum penetration of this conjugate after intranasal delivery. We also observed that leptin and LepNP85 were cleared into blood when administered locally into the brain by ICV injection. Compared to native leptin, blood levels of LepNP85 were higher, suggesting that the P85 modification enhanced the clearance pathway from brain to blood. Very interestingly, the cleavable LepDSPP85 conjugate after intranasal delivery displayed even higher serum levels than LepNP85. This could not be explained by the rapid reduction of the disulfide bond and transformation of LepDSPP85 into the free leptin in the nasal cavity, since the free leptin has even lower rate of serum absorption than LepNP85. Perhaps the disulfide bond reduction during the serum absorption process and the resulting change of protein size and hydrophobicity, recovery of receptor binding affinity, and/or disulfide exchange with endogenous proteins may contribute to the higher serum absorption of LepDSPP85 compared to LepNP85.
We believe that one should not be concerned about the increased serum absorption of leptin as a result of P85 modification, because 1) the overall serum absorption of LepNP85 after intranasal delivery is minimal (less than 2% ID/ml at 1 h) and 2) the obese patients with peripheral resistance have compensatory high circulating levels of leptin. However, having in mind that the serum pool of the material can re-enter the brain after BBB penetration, it is important to analyze whether the conjugate has a direct access to the brain from the nose or it enters brain due to reabsorption. Comparing the LepNP85 total brain/serum ratios for IV (~0.02 to 0.04 ml/g) and INB (~0.14 to 0.45 ml/g) suggests that the blood to brain route could explain at the most only ~9 to 14% of the whole brain uptake of this conjugate after intranasal administration. Therefore, we believe that the INB pathway was actually responsible for the increased distribution of the conjugate to the site of desired activity. This conclusion may be less obvious for the native leptin. Indeed, the leptin IV brain/serum ratios (~0.03 to 0.07 ml/g) are relatively closer to the INB ratios (~0.12 to 0.36 ml/g). Therefore, the blood to brain reabsorption of the native hormone at least in part could contribute to its delivery to the brain after intranasal administration in the healthy animals. However, there are previous indications that native leptin can undergo the direct transport from nose to brain as INB delivery of leptin successfully reduced appetite and body weight in DIO models (where impaired transporter would have prevented the blood-to-brain reabsorption) [49, 53, 59]. Besides, Fliedner, et al demonstrated that IV injection of unlabeled leptin cannot inhibit brain accumulation of radiolabeled leptin administered by INB route, suggesting that leptin takes a direct pathway from the nose to the brain [41]. We have also demonstrated that in a DIO mice model INB LepNP85 accumulated in the brain hypothalamus and hippocampus better than the unmodified leptin. Brain distribution of intranasal LepNP85 in healthy mice and DIO mice was essentially same. Taken together, the increased brain accumulation of LepNP85 is not due to enhanced blood absorption and subsequent brain reabsorption, but due to enhanced direct nose-to-brain delivery of the hormone as a result of its modification with P85.
We did not observe aggregation of LepNP85 after the modification of leptin with the block copolymer. Moreover, at the active dose of LepNP85 (5 μg/mouse), the concentration of the injected solution was 0.625 mg/ml (5 μg in 8 μl PBS) or 29.8 μM, which is lower than the critical micelle concentration (CMC) of P85 (0.03% wt. [71] or 65.2 μM). The pharmacokinetic studies used the trace amounts of radiolabeled leptin. This suggests that LepNP85 was unlikely to form micelles under the conditions used in the animal studies. Taken together, we believe that the improvement in the absorption of leptin as a result of P85 modification is likely due to the increased hydrophobicity and transmembrane penetration of the conjugate across the nasal epithelial barrier [29, 62, 63].
To the best of our knowledge, this is the first study showing that P85 modification of leptin can enhance its direct nose-to-brain transport of the hormone and its activity with respect to the receptor. In general, formulation strategies used to enhance direct nose-to-brain delivery of therapeutic proteins remain largely unexplored [72, 73]. Absorption enhancers/tight-junction modulators, bioadhesive polymers, enzyme inhibitors, and nanoparticles have been studied [74–78], aiming to improve the stability, prolong the residence time, create a local drug depot or enhance absorption across the mucosa epithelium. However, many of these strategies enhance systemic absorption following intranasal administration [79, 80]. Specifically for leptin, using an absorption enhancer lysophosphatidylcholine only enhanced systemic absorption but not the brain delivery [49]. In addition, the INB delivery of LepNP85 is independent of its receptors/transporters expressed in the nasal route, likely suggesting that our approach may be generalized for other proteins.
Of the challenges remaining for the development of an efficient treatment for obesity we would like to point out significant variability in the delivery and activity of the INB administered hormone. Limited information is available on the influence of dosing volume, tube insertion depth, head position and anesthesia on the nasal distribution pattern and efficiency of INB delivery [81–84]. Targeting the olfactory epithelium is likely to be more challenging in human than the experiments on rodents which have over 50% nasal cavity surface covered by olfactory epithelium [85]. The drug should pass through the narrow triangular shaped nasal valve to reach the posterior olfactory epithelium [85]. To decrease variability and improve INB delivery one may need to use nasal devices such as one of those reported by Djupesland et al. [85]. Moreover, since obesity is a chronic disease and P85 modification did not enhance the retention of leptin in the brain, the use of a depot system for LepNP85 may further be beneficial for the treatment of obesity.
5. Conclusion
In summary, we improved the brain delivery of leptin for treatment of obesity by conjugating this protein with Pluronic P85 and utilizing the INB route to bypass BBB. The conjugation was carried out by selectively modifying the N-terminal amine of leptin to preserve the protein binding affinity and maximize homogeneity of the conjugate. We showed that this modification enhanced the direct nose-to-brain transport of leptin.
Supplementary Material
Acknowledgments
The National Institutes of Health (RO1 NS051334; 1R41DK108466) and the Carolina Partnership between the UNC Eshelman School of Pharmacy and the University Cancer Research Fund in parts supported this work. We gratefully acknowledge Dr. Ashutosh Tripathy (UNC macromolecular interaction facility) for his expertise in protein characterization and Dr. Tim Bradshaw and Dr. Diane Ignar (Neuronano Pharma, Inc., Research Triangle Park, NC) for the discussion of the results. Dr. A. V. Kabanov is the founder, shareholder and director of Neuronano Pharma, Inc. that develops protein formulations for CNS delivery and thereby declares possible competing interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wharton S. Current Perspectives on Long-term Obesity Pharmacotherapy. Can J Diabetes. 2016;40:184–191. doi: 10.1016/j.jcjd.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 2.Wong D, Sullivan K, Heap G. The pharmaceutical market for obesity therapies. Nat Rev Drug Discov. 2012;11:669–670. doi: 10.1038/nrd3830. [DOI] [PubMed] [Google Scholar]
- 3.Rankin W, Wittert G. Anti-obesity drugs. Curr Opin Lipidol. 2015;26:536–543. doi: 10.1097/MOL.0000000000000232. [DOI] [PubMed] [Google Scholar]
- 4.Colon-Gonzalez F, Kim GW, Lin JE, Valentino MA, Waldman SA. Obesity pharmacotherapy: what is next? Molecular aspects of medicine. 2013;34:71–83. doi: 10.1016/j.mam.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravussin E, Smith SR, Mitchell JA, Shringarpure R, Shan K, Maier H, Koda JE, Weyer C. Enhanced weight loss with pramlintide/metreleptin: an integrated neurohormonal approach to obesity pharmacotherapy. Obesity (Silver Spring) 2009;17:1736–1743. doi: 10.1038/oby.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, Anderson CM, Parkes DG, Baron AD. Leptin responsiveness restored by amylin agonism in diet-induced obesity: evidence from nonclinical and clinical studies. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7257–7262. doi: 10.1073/pnas.0706473105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sainz N, Barrenetxe J, Moreno-Aliaga MJ, Martinez JA. Leptin resistance and diet-induced obesity: central and peripheral actions of leptin. Metabolism. 2015;64:35–46. doi: 10.1016/j.metabol.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 8.Ahima RS, Osei SY. Leptin signaling. Physiology & behavior. 2004;81:223–241. doi: 10.1016/j.physbeh.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 9.Munzberg H, Morrison CD. Structure, production and signaling of leptin. Metabolism. 2015;64:13–23. doi: 10.1016/j.metabol.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banks WA, Lebel CR. Strategies for the delivery of leptin to the CNS. J Drug Target. 2002;10:297–308. doi: 10.1080/10611860290031895. [DOI] [PubMed] [Google Scholar]
- 11.DePaoli AM. 20 years of leptin: leptin in common obesity and associated disorders of metabolism. J Endocrinol. 2014;223:T71–81. doi: 10.1530/JOE-14-0258. [DOI] [PubMed] [Google Scholar]
- 12.Mantzoros CS, Flier JS. Editorial: leptin as a therapeutic agent--trials and tribulations. The Journal of clinical endocrinology and metabolism. 2000;85:4000–4002. doi: 10.1210/jcem.85.11.7062. [DOI] [PubMed] [Google Scholar]
- 13.Mantzoros CS, Flier JS. Leptin as a Therapeutic Agent—Trials and Tribulations. The Journal of Clinical Endocrinology & Metabolism. 2000;85:4000–4002. doi: 10.1210/jcem.85.11.7062. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 15.Levin BE, Dunn-Meynell AA, Banks WA. Obesity-prone rats have normal blood-brain barrier transport but defective central leptin signaling before obesity onset. American journal of physiology. Regulatory integrative and comparative physiology. 2004;286:R143–150. doi: 10.1152/ajpregu.00393.2003. [DOI] [PubMed] [Google Scholar]
- 16.Wu-Peng XS, Chua SC, Jr, Okada N, Liu SM, Nicolson M, Leibel RL. Phenotype of the obese Koletsky (f) rat due to Tyr763Stop mutation in the extracellular domain of the leptin receptor (Lepr): evidence for deficient plasma-to-CSF transport of leptin in both the Zucker and Koletsky obese rat. Diabetes. 1997;46:513–518. doi: 10.2337/diab.46.3.513. [DOI] [PubMed] [Google Scholar]
- 17.Banks WA. Is obesity a disease of the blood-brain barrier? Physiological, pathological, and evolutionary considerations. Current pharmaceutical design. 2003;9:801–809. doi: 10.2174/1381612033455350. [DOI] [PubMed] [Google Scholar]
- 18.Van Heek M, Compton DS, France CF, Tedesco RP, Fawzi AB, Graziano MP, Sybertz EJ, Strader CD, Davis HR., Jr Diet-induced obese mice develop peripheral, but not central, resistance to leptin. The Journal of clinical investigation. 1997;99:385–390. doi: 10.1172/JCI119171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8878–8883. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LeBel C, Bourdeau A, Lau D, Hunt P. Biologic response to peripheral and central administration of recombinant human leptin in dogs. Obes Res. 1999;7:577–585. doi: 10.1002/j.1550-8528.1999.tb00717.x. [DOI] [PubMed] [Google Scholar]
- 21.Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, Lubina JA, Patane J, Self B, Hunt P, McCamish M. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA : the journal of the American Medical Association. 1999;282:1568–1575. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- 22.Kahler A, Geary N, Eckel LA, Campfield LA, Smith FJ, Langhans W. Chronic administration of OB protein decreases food intake by selectively reducing meal size in male rats. The American journal of physiology. 1998;275:R180–185. doi: 10.1152/ajpregu.1998.275.1.R180. [DOI] [PubMed] [Google Scholar]
- 23.Elinav E, Niv-Spector L, Katz M, Price TO, Ali M, Yacobovitz M, Solomon G, Reicher S, Lynch JL, Halpern Z, Banks WA, Gertler A. Pegylated leptin antagonist is a potent orexigenic agent: preparation and mechanism of activity. Endocrinology. 2009;150:3083–3091. doi: 10.1210/en.2008-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kovalszky I, Surmacz E, Scolaro L, Cassone M, Ferla R, Sztodola A, Olah J, Hatfield MP, Lovas S, Otvos L., Jr Leptin-based glycopeptide induces weight loss and simultaneously restores fertility in animal models. Diabetes, obesity & metabolism. 2010;12:393–402. doi: 10.1111/j.1463-1326.2009.01170.x. [DOI] [PubMed] [Google Scholar]
- 25.Banks WA, Farrell CL. Impaired transport of leptin across the blood-brain barrier in obesity is acquired and reversible. American journal of physiology Endocrinology and metabolism. 2003;285:E10–15. doi: 10.1152/ajpendo.00468.2002. [DOI] [PubMed] [Google Scholar]
- 26.L. American Association for the Study of Obesity (NAASO) and the National Heart, and Blood Institute (NHLBI), editor The Practical Guide: Identification, Evaluation, and Treatment of Overweight and Obesity in Adults. Oct, 2000. 00-4084. [Google Scholar]
- 27.Yi X, Yuan D, Farr SA, Banks WA, Poon CD, Kabanov AV. Pluronic modified leptin with increased systemic circulation, brain uptake and efficacy for treatment of obesity. J Control Release. 2014;191:34–46. doi: 10.1016/j.jconrel.2014.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price TO, Farr SA, Yi X, Vinogradov S, Batrakova E, Banks WA, Kabanov AV. Transport across the blood-brain barrier of pluronic leptin. The Journal of pharmacology and experimental therapeutics. 2010;333:253–263. doi: 10.1124/jpet.109.158147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yi X, Batrakova E, Banks WA, Vinogradov S, Kabanov AV. Protein conjugation with amphiphilic block copolymers for enhanced cellular delivery. Bioconjug Chem. 2008;19:1071–1077. doi: 10.1021/bc700443k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacDonald JI, Munch HK, Moore T, Francis MB. One-step site-specific modification of native proteins with 2-pyridinecarboxyaldehydes. Nat Chem Biol. 2015;11:326–331. doi: 10.1038/nchembio.1792. [DOI] [PubMed] [Google Scholar]
- 31.Tong J, Yi X, Luxenhofer R, Banks WA, Jordan R, Zimmerman MC, Kabanov AV. Conjugates of superoxide dismutase 1 with amphiphilic poly(2-oxazoline) block copolymers for enhanced brain delivery: synthesis, characterization and evaluation in vitro and in vivo. Mol Pharm. 2013;10:360–377. doi: 10.1021/mp300496x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corrêa DHA, Ramos CHI. The use of circular dichroism spectroscopy to study protein folding, form and function. African Journal of Biochemistry Research. 2009;3:164–173. [Google Scholar]
- 33.Whitmore L, Wallace BA. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 34.Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic acids research. 2004;32:W668–673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mistrik P, Moreau F, Allen JM. BiaCore analysis of leptin-leptin receptor interaction: evidence for 1:1 stoichiometry. Anal Biochem. 2004;327:271–277. doi: 10.1016/j.ab.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 36.Falcone JA, Salameh TS, Yi X, Cordy BJ, Mortell WG, Kabanov AV, Banks WA. Intranasal Administration as a Route for Drug Delivery to the Brain: Evidence for a Unique Pathway for Albumin. The Journal of pharmacology and experimental therapeutics. 2014;351:54–60. doi: 10.1124/jpet.114.216705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glowinski J, Iversen LL. Regional studies of catecholamines in the rat brain. I. The disposition of [3H]norepinephrine, [3H]dopamine and [3H]dopa in various regions of the brain. Journal of neurochemistry. 1966;13:655–669. doi: 10.1111/j.1471-4159.1966.tb09873.x. [DOI] [PubMed] [Google Scholar]
- 38.Banks WA, Kastin AJ, Huang W, Jaspan JB, Maness LM. Leptin enters the brain by a saturable system independent of insulin. Peptides. 1996;17:305–311. doi: 10.1016/0196-9781(96)00025-3. [DOI] [PubMed] [Google Scholar]
- 39.Patlak CS, Blasberg RG. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. Generalizations. J Cereb Blood Flow Metab. 1985;5:584–590. doi: 10.1038/jcbfm.1985.87. [DOI] [PubMed] [Google Scholar]
- 40.Patlak CS, Blasberg RG, Fenstermacher JD. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab. 1983;3:1–7. doi: 10.1038/jcbfm.1983.1. [DOI] [PubMed] [Google Scholar]
- 41.Fliedner S, Schulz C, Lehnert H. Brain uptake of intranasally applied radioiodinated leptin in Wistar rats. Endocrinology. 2006;147:2088–2094. doi: 10.1210/en.2005-1016. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Z, Zhang J, Sun L, Ma G, Su Z. Comparison of site-specific PEGylations of the N-terminus of interferon beta-1b: selectivity, efficiency, and in vivo/vitro activity. Bioconjug Chem. 2014;25:138–146. doi: 10.1021/bc400435u. [DOI] [PubMed] [Google Scholar]
- 43.Zheng C, Ma G, Su Z. Native PAGE eliminates the problem of PEG-SDS interaction in SDS-PAGE and provides an alternative to HPLC in characterization of protein PEGylation. Electrophoresis. 2007;28:2801–2807. doi: 10.1002/elps.200600807. [DOI] [PubMed] [Google Scholar]
- 44.Mortensen K, Batsberg W, Hvidt S. Effects of PEO–PPO Diblock Impurities on the Cubic Structure of Aqueous PEO–PPO–PEO Pluronics Micelles: fcc and bcc Ordered Structures in F127. Macromolecules. 2008;41:1720–1727. [Google Scholar]
- 45.Churgay LM, Kovacevic S, Tinsley FC, Kussow CM, Millican RL, Miller JR, Hale JE. Purification and characterization of secreted human leptin produced in baculovirus-infected insect cells. Gene. 1997;190:131–137. doi: 10.1016/s0378-1119(97)00041-3. [DOI] [PubMed] [Google Scholar]
- 46.Niv-Spector L, Gonen-Berger D, Gourdou I, Biener E, Gussakovsky EE, Benomar Y, Ramanujan KV, Taouis M, Herman B, Callebaut I, Djiane J, Gertler A. Identification of the hydrophobic strand in the A–B loop of leptin as major binding site III: implications for large-scale preparation of potent recombinant human and ovine leptin antagonists. Biochem J. 2005;391:221–230. doi: 10.1042/BJ20050457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bharatiya B, Aswal VK, Hassan PA, Bahadur P. Influence of a hydrophobic diol on the micellar transitions of Pluronic P85 in aqueous solution. Journal of colloid and interface science. 2008;320:452–459. doi: 10.1016/j.jcis.2008.01.050. [DOI] [PubMed] [Google Scholar]
- 48.Morris DL, Rui L. Recent advances in understanding leptin signaling and leptin resistance. Am J Physiol Endocrinol Metab. 2009;297:E1247–1259. doi: 10.1152/ajpendo.00274.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimizu H, Oh IS, Okada S, Mori M. Inhibition of appetite by nasal leptin administration in rats. Int J Obes (Lond) 2005;29:858–863. doi: 10.1038/sj.ijo.0802951. [DOI] [PubMed] [Google Scholar]
- 50.Faouzi M, Leshan R, Bjornholm M, Hennessey T, Jones J, Munzberg H. Differential accessibility of circulating leptin to individual hypothalamic sites. Endocrinology. 2007;148:5414–5423. doi: 10.1210/en.2007-0655. [DOI] [PubMed] [Google Scholar]
- 51.Maness LM, Kastin AJ, Farrell CL, Banks WA. Fate of leptin after intracerebroventricular injection into the mouse brain. Endocrinology. 1998;139:4556–4562. doi: 10.1210/endo.139.11.6319. [DOI] [PubMed] [Google Scholar]
- 52.Banks WA, Niehoff ML, Martin D, Farrell CL. Leptin transport across the blood-brain barrier of the Koletsky rat is not mediated by a product of the leptin receptor gene. Brain research. 2002;950:130–136. doi: 10.1016/s0006-8993(02)03013-5. [DOI] [PubMed] [Google Scholar]
- 53.Schulz C, Paulus K, Johren O, Lehnert H. Intranasal leptin reduces appetite and induces weight loss in rats with diet-induced obesity (DIO) Endocrinology. 2012;153:143–153. doi: 10.1210/en.2011-1586. [DOI] [PubMed] [Google Scholar]
- 54.Zhang HH, Kumar S, Barnett AH, Eggo MC. Intrinsic site-specific differences in the expression of leptin in human adipocytes and its autocrine effects on glucose uptake. The Journal of clinical endocrinology and metabolism. 1999;84:2550–2556. doi: 10.1210/jcem.84.7.5846. [DOI] [PubMed] [Google Scholar]
- 55.Peelman F, Van Beneden K, Zabeau L, Iserentant H, Ulrichts P, Defeau D, Verhee A, Catteeuw D, Elewaut D, Tavernier J. Mapping of the leptin binding sites and design of a leptin antagonist. The Journal of biological chemistry. 2004;279:41038–41046. doi: 10.1074/jbc.M404962200. [DOI] [PubMed] [Google Scholar]
- 56.Hukshorn CJ, Saris WH, Westerterp-Plantenga MS, Farid AR, Smith FJ, Campfield LA. Weekly subcutaneous pegylated recombinant native human leptin (PEG-OB) administration in obese men. J Clin Endocrinol Metab. 2000;85:4003–4009. doi: 10.1210/jcem.85.11.6955. [DOI] [PubMed] [Google Scholar]
- 57.Morath V, Bolze F, Schlapschy M, Schneider S, Sedlmayer F, Seyfarth K, Klingenspor M, Skerra A. PASylation of Murine Leptin Leads to Extended Plasma Half-Life and Enhanced in Vivo Efficacy. Mol Pharm. 2015;12:1431–1442. doi: 10.1021/mp5007147. [DOI] [PubMed] [Google Scholar]
- 58.Xu L, Rensing N, Yang XF, Zhang HX, Thio LL, Rothman SM, Weisenfeld AE, Wong M, Yamada KA. Leptin inhibits 4-aminopyridine- and pentylenetetrazole-induced seizures and AMPAR-mediated synaptic transmission in rodents. J Clin Invest. 2008;118:272–280. doi: 10.1172/JCI33009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schulz C, Paulus K, Lehnert H. Central nervous and metabolic effects of intranasally applied leptin. Endocrinology. 2004;145:2696–2701. doi: 10.1210/en.2003-1431. [DOI] [PubMed] [Google Scholar]
- 60.Kissileff HR, Thornton JC, Torres MI, Pavlovich K, Mayer LS, Kalari V, Leibel RL, Rosenbaum M. Leptin reverses declines in satiation in weight-reduced obese humans. The American journal of clinical nutrition. 2012;95:309–317. doi: 10.3945/ajcn.111.012385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lochhead JJ, Thorne RG. Intranasal delivery of biologics to the central nervous system. Advanced drug delivery reviews. 2012;64:614–628. doi: 10.1016/j.addr.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 62.Yi X, Kabanov AV. Brain delivery of proteins via their fatty acid and block copolymer modifications. J Drug Target. 2013;21:940–955. doi: 10.3109/1061186X.2013.847098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yi X, Zimmerman MC, Yang R, Tong J, Vinogradov S, Kabanov AV. Pluronic-modified superoxide dismutase 1 attenuates angiotensin II-induced increase in intracellular superoxide in neurons. Free radical biology & medicine. 2010;49:548–558. doi: 10.1016/j.freeradbiomed.2010.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harvey J. Leptin regulation of neuronal morphology and hippocampal synaptic function. Front Synaptic Neurosci. 2013;5:3. doi: 10.3389/fnsyn.2013.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller AA, Spencer SJ. Obesity and neuroinflammation: a pathway to cognitive impairment. Brain Behav Immun. 2014;42:10–21. doi: 10.1016/j.bbi.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 66.Marwarha G, Ghribi O. Leptin signaling and Alzheimer’s disease. Am J Neurodegener Dis. 2012;1:245–265. [PMC free article] [PubMed] [Google Scholar]
- 67.Taildeman J, Demetter P, Rottiers I, Holtappels G, Bachert C, Cuvelier CA, Perez-Novo CA. Identification of the nasal mucosa as a new target for leptin action. Histopathology. 2010;56:789–798. doi: 10.1111/j.1365-2559.2010.03552.x. [DOI] [PubMed] [Google Scholar]
- 68.Song SY, Woo HJ, Bae CH, Kim YW, Kim YD. Expression of leptin receptor in nasal polyps: leptin as a mucosecretagogue. Laryngoscope. 2010;120:1046–1050. doi: 10.1002/lary.20844. [DOI] [PubMed] [Google Scholar]
- 69.Bruno A, Gerbino S, Ferraro M, Siena L, Bonura A, Colombo P, La Grutta S, Gallina S, Ballacchino A, Giammanco M, Gjomarkaj M, Pace E. Fluticasone furoate maintains epithelial homeostasis via leptin/leptin receptor pathway in nasal cells. Mol Cell Biochem. 2014;396:55–65. doi: 10.1007/s11010-014-2142-z. [DOI] [PubMed] [Google Scholar]
- 70.Baly C, Aioun J, Badonnel K, Lacroix MC, Durieux D, Schlegel C, Salesse R, Caillol M. Leptin and its receptors are present in the rat olfactory mucosa and modulated by the nutritional status. Brain Res. 2007;1129:130–141. doi: 10.1016/j.brainres.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 71.Batrakova EV, Kabanov AV. Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J Control Release. 2008;130:98–106. doi: 10.1016/j.jconrel.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de la Monte SM. Intranasal insulin therapy for cognitive impairment and neurodegeneration: current state of the art. Expert Opin Drug Deliv. 2013;10:1699–1709. doi: 10.1517/17425247.2013.856877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ruigrok MJ, de Lange EC. Emerging Insights for Translational Pharmacokinetic and Pharmacokinetic-Pharmacodynamic Studies: Towards Prediction of Nose-to-Brain Transport in Humans. AAPS J. 2015;17:493–505. doi: 10.1208/s12248-015-9724-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kubek MJ, Domb AJ, Veronesi MC. Attenuation of kindled seizures by intranasal delivery of neuropeptide-loaded nanoparticles. Neurotherapeutics. 2009;6:359–371. doi: 10.1016/j.nurt.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang X, Liu L, Chai G, Zhang X, Li F. Brain pharmacokinetics of neurotoxin-loaded PLA nanoparticles modified with chitosan after intranasal administration in awake rats. Drug Dev Ind Pharm. 2013;39:1618–1624. doi: 10.3109/03639045.2012.727828. [DOI] [PubMed] [Google Scholar]
- 76.Gao X, Wu B, Zhang Q, Chen J, Zhu J, Zhang W, Rong Z, Chen H, Jiang X. Brain delivery of vasoactive intestinal peptide enhanced with the nanoparticles conjugated with wheat germ agglutinin following intranasal administration. J Control Release. 2007;121:156–167. doi: 10.1016/j.jconrel.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 77.Appu AP, Arun P, Krishnan JK, Moffett JR, Namboodiri AM. Rapid intranasal delivery of chloramphenicol acetyltransferase in the active form to different brain regions as a model for enzyme therapy in the CNS. J Neurosci Methods. 2016;259:129–134. doi: 10.1016/j.jneumeth.2015.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hackel D, Krug SM, Sauer RS, Mousa SA, Bocker A, Pflucke D, Wrede EJ, Kistner K, Hoffmann T, Niedermirtl B, Sommer C, Bloch L, Huber O, Blasig IE, Amasheh S, Reeh PW, Fromm M, Brack A, Rittner HL. Transient opening of the perineurial barrier for analgesic drug delivery. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2018–2027. doi: 10.1073/pnas.1120800109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Costantino HR, Illum L, Brandt G, Johnson PH, Quay SC. Intranasal delivery: physicochemical and therapeutic aspects. Int J Pharm. 2007;337:1–24. doi: 10.1016/j.ijpharm.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 80.Romeo VD, deMeireles JC, Gries WJ, Xia WJ, Sileno AP, Pimplaskar HK, Behl CR. Optimization of systemic nasal drug delivery with pharmaceutical excipients. Advanced drug delivery reviews. 1998;29:117–133. doi: 10.1016/s0169-409x(97)00064-1. [DOI] [PubMed] [Google Scholar]
- 81.Charlton ST, Davis SS, Illum L. Nasal administration of an angiotensin antagonist in the rat model: effect of bioadhesive formulations on the distribution of drugs to the systemic and central nervous systems. Int J Pharm. 2007;338:94–103. doi: 10.1016/j.ijpharm.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 82.van den Berg MP, Romeijn SG, Verhoef JC, Merkus FW. Serial cerebrospinal fluid sampling in a rat model to study drug uptake from the nasal cavity. J Neurosci Methods. 2002;116:99–107. doi: 10.1016/s0165-0270(02)00033-x. [DOI] [PubMed] [Google Scholar]
- 83.Wu H, Hu K, Jiang X. From nose to brain: understanding transport capacity and transport rate of drugs. Expert Opin Drug Deliv. 2008;5:1159–1168. doi: 10.1517/17425247.5.10.1159. [DOI] [PubMed] [Google Scholar]
- 84.Mayor SH, Illum L. Investigation of the effect of anaesthesia on nasal absorption of insulin in rats. International Journal of Pharmaceutics. 1997;149:123–129. [Google Scholar]
- 85.Djupesland PG, Messina JC, Mahmoud RA. The nasal approach to delivering treatment for brain diseases: an anatomic, physiologic, and delivery technology overview. Ther Deliv. 2014;5:709–733. doi: 10.4155/tde.14.41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.