

Abstract
Prostate cancer (PCa) is one of the leading causes of cancer-related deaths in men worldwide. Fatty acid synthase (FASN) is reported to be overexpressed in several cancers including PCa, and this has led to clinical cancer treatments that utilize various FASN inhibitors such as the anti-obesity drug, Orlistat. However, pharmacological limitations have impeded the progress in cancer treatments expected thus far with FASN inhibition. In this study, we investigated a novel therapeutic combination to enhance the toxic potential of Orlistat in three different PCa cell-lines (DU145, PC3, and LNCaP). We show that Orlistat and 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) (AMP-activated protein kinase (AMPK) activator) co-treatment induces significant downregulation of two key fatty acid synthesis regulatory proteins (FASN, Sterol regulatory element-binding protein 1 (SREBP-1c)) as compared to control and Orlistat alone. Orlistat and AICAR co-treatment induced a significant decrease in cell viability and proliferation, and a significant increase in apoptosis in all three PCa cell-lines. Apoptosis induction was preceded by a marked increase in reactive oxygen species (ROS) production followed by G0/G1 cell cycle arrest and activation of pro-apoptotic caspases. We also observed a significant decrease in migration potential and VEGF expression in Orlistat and AICAR co-treated samples in all three PCa cell-lines. Compound C (AMPK inhibitor) negatively affected the enhanced effects of Orlistat and AICAR co-treatment. We conclude that AICAR co-treatment potentiates the anti-proliferative effects of Orlistat at a low dose (100 μM), and this combination has the potential to be a viable and effective therapeutic option in PCa treatment.
Keywords: Prostate cancer (PCa), Fatty Acid Synthase, AMPK, Orlistat, AICAR
Prostate cancer (PCa) is the most common type of cancer detected in men in the United States, and is the sixth leading cause of cancer-related deaths in men worldwide [Jemal et al., 2011]. The demographics of PCa detection vary with race epidemiology indicating stark differences: such as being most common in the African-American populous while lowest among Asian men [Carlsson et al., 2012; Jemal et al., 2011]. The variation in PCa rates worldwide and within races and cultures strongly hints at a diet and lifestyle-based component in cancer development. Several studies have directly linked high PCa rates with an enriched fat diet [Kurahashi et al., 2008]. PCa cells typically rely on androgen (testosterone, dihydrotestosterone (DHT), among others) stimulation to maximize proliferation. This dependence allows for androgen deprivation therapy (ADT) treatment which lowers endogenous androgen levels leading to slower cell growth and tumor shrinkage. However, this treatment solely does not cure PCa, and long-term ADT treatment usually results in cells and tumors becoming androgen-independent thereby rendering ADT ineffective [Knudsen and Scher, 2009]. ADT therapy treatment issues combined with other treatment complications related to surgery and radiation therapy have made the development of novel therapeutic options to treat PCa an imminent necessity. To combat the prohibitive treatment issues that androgen-resistant PCa cells present; cell lines that are either androgen-independent (DU145, PC3) or androgen-dependent (LNCaP) have been established, and are used ubiquitously to study the various intracellular mechanisms involved in PCa progression [Ford et al., 2003; Linja et al., 2001].
Fatty acid synthase (FASN) is a key 270 kDa metabolic enzyme that catalyzes the synthesis of fatty acids (FA) in a NADPH-dependent sequential manner, with the terminal product being free palmitate synthesized from acetyl-CoA and malonyl-CoA [Jayakumar et al., 1995; Smith et al., 2003; Wakil, 1989]. Recent data has shown that several common cancers, including PCa, express elevated levels of FASN in comparison to normal tissue [Carvalho et al., 2008; Yoshii et al., 2013]. Upregulated FASN expression is indicative of lipid accumulation which is facilitated by the dysregulation of lipid metabolism in PCa cells. This dysregulation is a result of an increase in malignancy due to the rapid elevation in cell proliferation [Carvalho et al., 2008; Yoshii et al., 2013]. FASN is now considered as a possible oncogenic marker for cancer malignancy, and several FASN inhibitors have been established and used in experimental trials to determine the effect of FASN inhibition on cancer progression and cell death. Orlistat has been established as an effective FASN inhibitor that is marketed as an anti-obesity drug due to its direct inhibitory effect on lipases [Davidson et al., 1999; Zhi et al., 1995]. Lipase inhibition prevents the enzymatic breakdown of diet triglycerides thereby preventing FA absorption from the diet. However, due to its poor bioavailability, there are complications with using Orlistat as an effective anti-cancer agent [Davidson et al., 1999; Menendez et al., 2005; Zhi et al., 1995].
FA synthesis is regulated by Acetyl-CoA carboxylase (ACC), an enzyme responsible for catalyzing the carboxylation of acetyl-CoA to malonyl-CoA; the FA synthesis substrate [Tong, 2005]. ACC regulates lipogenesis by inhibiting malonyl-CoA transfer to Carnitine O-palmitoyltransferase (CPT1), the rate-limiting mitochondrial enzyme for FA oxidation [Thupari et al., 2004]. The transcription factor family; Sterol regulatory element-binding protein 1 (SREBP-1), is responsible for cholesterol synthesis within the cell, and the SREBP precursor; SREBP-1c, is responsible for the transcription of genes involved in FA synthesis [Field et al., 2002]. During lipid synthesis, inactive precursor forms of SREBP-1c leave the endoplasmic reticulum (ER) membrane and move to the Golgi apparatus. Here, the protein is cleaved to the active form which enters the nucleus to trigger FA gene transcription [Field et al., 2002].
AMP-activated protein kinase (AMPK) is a cellular enzyme that plays a ubiquitous role in various cellular processes including apoptosis and autophagy. However, its most important function involves regulating cellular energy homeostasis and metabolism [Hardie, 2004]. AMPK is activated when the cellular AMP/ATP ratio is elevated. AMPK activation results in the inhibition of FA and cholesterol synthesis while simultaneously stimulating FA oxidation. This two-way process releases the ATP needed to restore cellular homeostasis; resulting in AMPK functioning as a regulator of FA metabolism [Hardie, 2004; Zhang et al., 2009]. AMPK is activated through upstream kinase phosphorylation, and once active, phosphorylates ACC rendering it inactive. AMPK can also phosphorylate SREBP-1c thereby inhibiting precursor cleavage, and the subsequent release of active SREBP-1c into the nucleus for FA gene transcription. Due to its ubiquitous role in cellular functions that include a functional role in tumor growth and survival in various cancer forms; AMPK is considered a viable target for cancer treatments [Zhang et al., 2009]. This has led to the establishment of AMPK activators and inhibitors such as 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) and compound C respectively. AICAR is one of the first identified AMPK mimetics that is used clinically to treat cardiac ischemic injury, but also used to activate AMPK in several in vitro and in vivo clinical studies [Galinanes et al., 1992; Sullivan et al., 1994]. In contrast, compound C is used in in vitro and in vivo studies, along with AICAR, to study AMPK-dependent cellular functions because it is a well-defined, potent AMPK inhibitor [Viollet et al., 2010].
The pathways and mechanisms involved in apoptosis induction due to FASN modulation in cancer cells are still ambiguous and require elucidation. Numerous reports have consistently shown that FASN inhibition by different drugs (cerulenin, C75) in various tumor cell lines may not be sufficient for apoptosis induction [Gabrielson et al., 2001; Pizer et al., 2000]. Other publications have indicated that FASN inhibition-related apoptosis occurs through ROS accumulation, and the intrinsic cell death pathway that is characterized by mitochondrial cytochrome c release, and the activation of caspases including caspase 3 and 9 [Heiligtag et al., 2002; Zecchin et al., 2011]. As discussed earlier, poor bioavailability of Orlistat coupled with inconsistent data regarding its efficiency in inducing cancer cell death presents the need to delineate the underlying pathways and explore mechanisms that may enable the use of combination treatments which enhance the potency of Orlistat. In this study, we show that AICAR co-treatment potentiates the anti-proliferative and pro-apoptotic effects of Orlistat in PC3, DU145, and LNCaP PCa cells. These pro-apoptotic effects included increases in ROS production and caspase activity that was coupled with the inhibition of cell cycle progression at the G0/G1 checkpoint. We also observed downregulation in VEGF levels and FA synthesis proteins. Other anti-cancer effects included inhibition of the migration potential in PC3, DU145, and LNCaP cells. The significant effect AMPK activation had on potentiating Orlistat-induced anti-cancer effects was verified by using compound C to inhibit AMPK activation, which negatively affected some of the anti-cancer effects observed with Orlistat.
Materials and methods
Chemicals and Reagents
AICAR and antibodies against Caspase 3 and 9, Poly (ADP-ribose) polymerase (PARP), pACC, ACC, FASN, pAMPK, AMPK, p21, Cyclin-dependent kinase (CDK) 4, CDK 6, and peroxidase-labeled secondary antibodies were obtained from Cell Signaling Technology (Danvers, MA). SREBF1 (SREBP-1c) and β-actin antibodies were obtained from Sigma-Aldrich (St. Louis, MO). The oxidative probes, dichlorofluorescein diacetate (DCF-DA) and dihydroethidium (DHE) were from Molecular Probes (Eugene, OR). Compound C was purchased from Millipore (Billerica, MA). Orlistat was purchased from Adipogen (San Diego, CA).
Cell Culture
The PC3, DU145, and LNCaP cell lines were obtained from American Type Culture Collection (Manassas, VA). PC3 and LNCaP cells were cultured in RPMI (Thermo Scientific) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin. DU145 cells were cultured in Dulbecco’s Modified Eagle medium (DMEM) (Thermo Scientific) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin. All cell lines were grown in a 5% CO2 environment at 37°C.
MTT Assay (Cellular Viability)
The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) colorimetric assay was performed in 96-well plates as previously described [Venkatadri et al., 2016]. Absorbance was measured at 570 nm using a Gen 5 2.0 All-In-One Microplate Reader (BioTek Instruments Inc.).
Western Blotting
After specific treatments, cells were incubated in lysis buffer and prepared for Western blotting as previously described [Venkatadri et al., 2016]. Immune complexes were detected by chemiluminescence (Supersignal West Pico; Pierce Biotechnology) using a MyECL Imager (Thermo Scientific). Quantification was done by imaging densitometry using ImageJ (NIH, Image analysis using Java) digitizing software. Mean densitometry data from independent experiments were normalized to the control.
Cell proliferation assay
Proliferation was analyzed using CyQUANT® dye-binding solution (Invitrogen) according to the manufacturer’s protocol. The fluorescence intensity of each sample was measured at the excitation and emission wavelengths of 485 and 535 nm, respectively using a Gen 5 2.0 All-In-One Microplate Reader (BioTek Instruments Inc.).
Apoptosis assay
Apoptosis was determined by Hoechst 33342 DNA fragmentation assay. Cells were incubated with 10 mg/mL Hoechst 33342 nuclear stain (Life Technologies) for 30 min at 37°C, and apoptosis determined by scoring the percentage of cells having intensely condensed chromatin and/or fragmented nuclei by fluorescence microscopy (EVOS All-in-one digital inverted fluorescence microscope). From random fields, 1,000 nuclei were analyzed for each sample. The apoptotic index was calculated as apoptotic nuclei/total nuclei × 100 (%) using ImageJ software (Java image processing, NIH).
Caspase 8/9 Activity Assay
Caspase 8 and 9 activities were detected using the CaspGLOW™ Fluorescein Active Caspase-8 and -9 Staining Kit respectively (BioVision) according to the manufacturer’s instructions. Briefly, cells were seeded in 96-well plates at a concentration of 6.0 × 103 cells/well, and treated for 6 h. Trypsinized cells were centrifuged at 3000 rpm for 5 min and the supernatant was removed before resuspension in Wash Buffer for additional centrifugation as above. Fluorescence intensity was measured at 485/535 nm using a Gen 5 2.0 All-In-One Microplate Reader (BioTek Instruments Inc.). Wells containing unlabeled cells were used for control. The caspase inhibitor Z-VAD-FMK (1 μl/ml) was used as a negative control by pre-treating cells for 1 h to inhibit caspase activation.
Reactive oxygen species (ROS) detection
Cellular ROS production was determined fluorometrically using DHE and DCF-DA as fluorescent probes for superoxide and hydrogen peroxide, respectively. After 6 h treatments, cells were incubated with the probes (5 μM) for 30 min at 37°C, after which they were washed, resuspended in PBS and analysed for fluorescence intensity using All-In-One Microplate Reader (BioTek Instruments Inc.) at the excitation/emission wavelengths of 485/535 for DHE and 485/610 nm for DCF.
Flow cytometry
Cells were seeded in six-well plates for three days in serum free medium (SFM). Cells were then placed in complete media (CM) before drug treatment for 24 h. After treatment, cells were trypsinized and suspended in 70% ethanol overnight. Cell suspensions were then pelleted, washed in PBS, and stained with a propidium iodide (PI) solution. Samples were then run using a NovoCyte flow cytometer (ACEA Biosciences, Inc., San Diego, CA, USA) and analyzed using NovoExpress 1.0.2 software. Samples were initially screened with SSC-H versus FSC-H density plot in linear scale and the gates were set to exclude debris. Another density plot was generated with the cells in the set gates and analyzed in PE-H versus PE-A for doublet discrimination. Singlet cells were gated and exported as FCS files and analyzed using ModFit 4.0.5 LT cell cycle analysis software (Verity Software House, Topsham, ME, USA).
Scratch wound assay (cell migration analysis)
Cells were seeded overnight in 12-well plates and grown to 80% confluency. A 200 μl pipette tip was used to scratch a line through the center of each well to produce a wound. Loose cells were washed away with PBS. Cells were treated for 24 and 48 h time points and then pictures of wells were taken using a EVOS All-in-one digital inverted fluorescence microscope. Wound healing effects were determined using ImageJ software (Java image processing, NIH) for measuring the wound area as compared with total cell area. Assay was done in triplicate, and data tabulated from 3 independent experiments.
Enzyme-linked Immunosorbent Assay (ELISA)
Cells were plated in a six-well plate at a density of 2 × 105 cells/well in culture medium. After treatment, cell supernatants were collected and analyzed for VEGF protein levels using a Human VEGFA ELISA kit (Thermo Scientific) per the manufacturer’s protocol. After the final wash step, 100 μl of substrate solution was added and the optical density was determined on a Gen 5 2.0 All-In-One Microplate Reader (BioTek Instruments Inc.) at 450 nm.
Bliss Independency Model
MTT data generated as above from all three PCa cell lines (PC3, DU145, LNCaP) with Orlistat and AICAR; Orlistat and compound C drug combinations relative to untreated control cells was used as input. The Bliss model was used to compute the expected combined effects of two drugs as the product of their individual effects based on the assumption that there is no effect from drug-drug interactions, as previously described [Bliss, 1939; Zhao et al., 2014]. The drug combinations are: synergistic if the observed effects of drug combinations are greater than the expected combined effects; antagonistic if the observed effects of drug combinations are lesser than the expected combined effects and additive if the observed effects of drug combinations are equal to the expected effects. For the current study, a bliss value ≥1.1 was considered synergy; 1.0 was considered additive; and ≤ 0.95 was considered antagonism. Data from three independent MTT experiments done in triplicate was used.
Statistical Analysis
The data represent means (±S.E.M) from three or more independent experiments. Statistical analysis was performed by Student’s t test at a significance level of p < 0.05.
Results
Effect of FASN and AMPK modulators on PCa cell viability and FA synthesis proteins
The individual effect of Orlistat, AICAR, and compound C on PCa cell viability was first analyzed by MTT assay. Increasing concentrations of Orlistat led to a dose-dependent decrease of cell viability in all three PCa cell-lines tested (PC3, DU145, LNCaP) (Figure 1A). 24h treatment with AICAR or compound C alone had no significant effect on cell viability in the tested dose range (Figures 1, B and C). However, titration analysis with increasing doses and time points resulted in decreases in cell viability and apoptosis using both AICAR and Compound C (data not shown). Orlistat and AICAR are well established FA inhibitor and AMPK activator, respectively. We confirmed the effect of Orlistat and AICAR on the expression of proteins involved in FA synthesis including FASN, SREBP-1c, ACC, and AMPK by Western blotting. Orlistat treatment alone or in combination with AICAR significantly downregulated the expression of FASN and the mature form of SREBP-1c in all three PCa cell lines (PC3, DU145, LNCaP) (Figure 2). The effect of AICAR treatment was verified by the clear increase in AMPK phosphorylation (pAMPK) and the subsequent phosphorylation and inactivation of ACC (pACC); a vital regulator of FA synthesis. Compound C co-treatment with Orlistat negatively affected AMPK activation and the subsequent inhibition of ACC. Compound C did not inhibit the negative effect of Orlistat on FA protein expression, but clearly did not enhance the downregulation of FASN and SREBP-1c as observed with Orlistat and AICAR co-treatment in all three PCa cell-lines.
Figure 1. Orlistat reduces PCa cell viability in a dose-dependent manner.
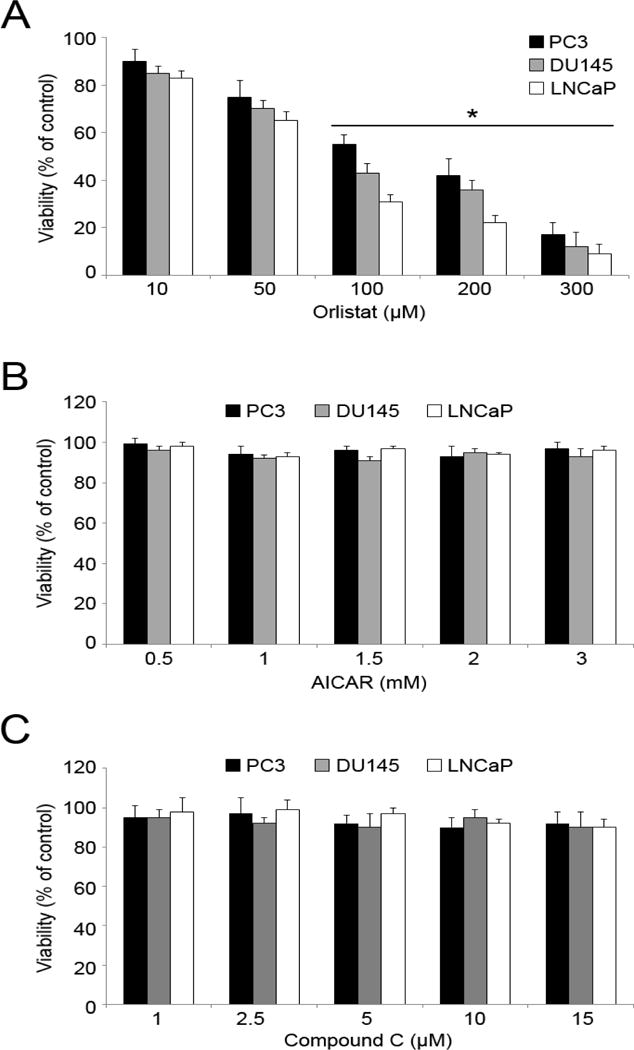
PC3, DU145, and LNCaP cells were treated with varying doses of (A) Orlistat (0–300 uM), (B) AICAR (0.5–3 mM) and (C) compound C (1–15 uM) for 24 h. MTT assay was used to determine cell viability. Values are mean ± S.E.M (n = 3). *, p < 0.05 versus control.
Figure 2. Orlistat and AICAR combination downregulates FA synthesis proteins.
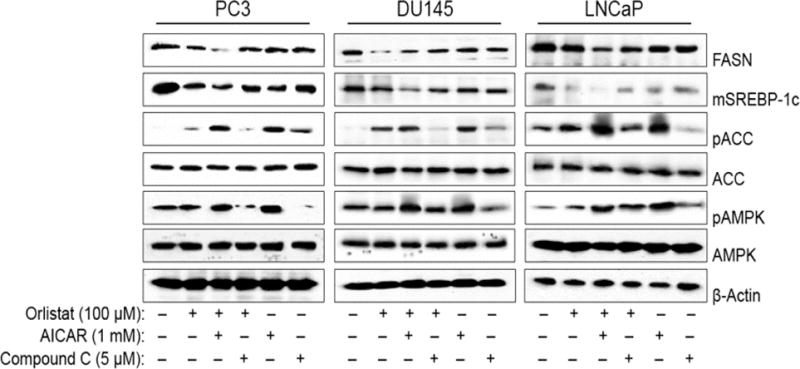
PC3, DU145, and LNCaP cells were pre-treated with AICAR (1 mM) or compound C (5 μM) for 1 h. The cells were then either left untreated or were treated with Orlistat (100 μM) for 24 h. Cell lysates were collected and analyzed for FASN, mature SREBP-1c, pACC, and pAMPK protein expression by Western blotting. Blots were stripped and reprobed for ACC, AMPK, and β-actin protein expression as a loading control. Representative blots from three or more independent experiments are shown.
Effect of Orlistat and AICAR co-treatment on cellular apoptosis
To assess the effect of Orlistat and AICAR together on PCa cell survival, we analyzed treated PC3, DU145, and LNCaP for cell viability. Orlistat and AICAR co-treatment significantly decreased cell viability, both in comparison to control and cells treated with Orlistat alone (Figure 3A). Similarly, a significant reduction in cell proliferation in Orlistat and AICAR co-treated PCa cells was observed, as compared to untreated cells and cells treated with Orlistat alone (Figure 3B). Co-treatment with compound C had negligible effects on both cell viability and proliferation in comparison to treatment with Orlistat alone. We next investigated the effect of Orlistat and AICAR co-treatment on apoptosis using Hoechst assay to analyze cell death. Apoptotic cells demonstrate nuclear condensation and DNA fragmentation which can be detected by Hoechst 33342 staining. Orlistat and AICAR co-treatment resulted in a marked increase in apoptosis in all three PCa cell lines (PC3, DU145, LNCaP) (Figure 3C). Compound C co-treatment was unable to reverse the induction of apoptosis observed with Orlistat treatment alone, however there was clearly no enhanced apoptosis observed as with Orlistat and AICAR co-treatment.
Figure 3. Combination of Orlistat and AICAR treatment induces apoptosis.
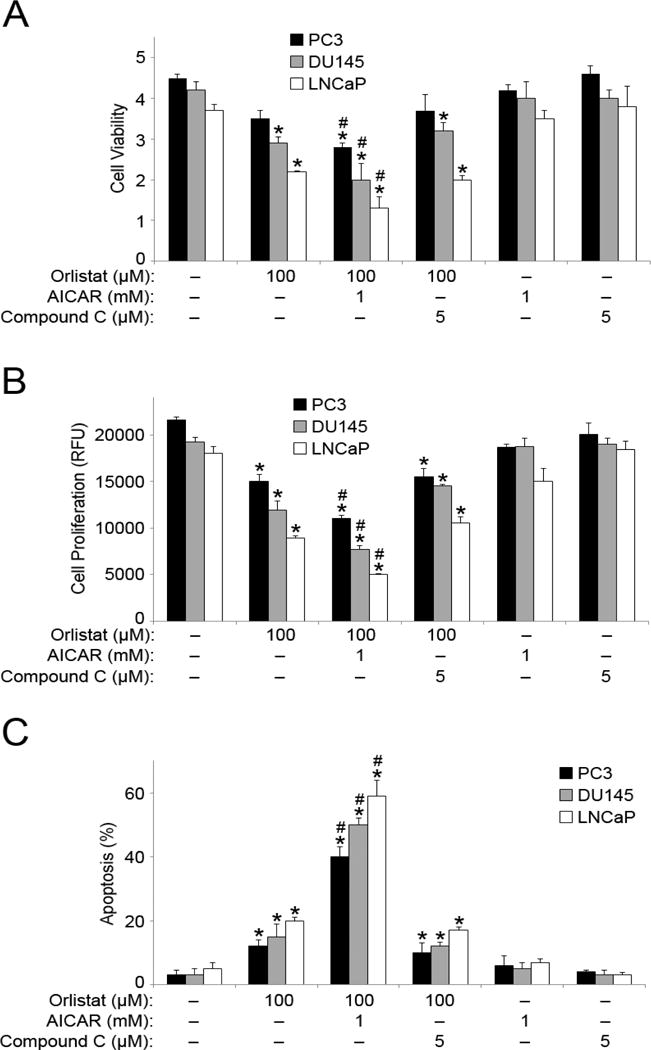
(A) PC3, DU145, and LNCaP cells were pre-treated with AICAR (1 mM) or compound C (5 μM) for 1 h. The cells were then either left untreated or were treated with Orlistat (100 μM) for 24 h. Cell viability was assessed by MTT assay. (B) PC3, DU145, and LNCaP cells were pre-treated with AICAR (1 mM) or compound C (5 μM) for 1 h. The cells were then either left untreated or were treated with Orlistat (100 μM) for 48 h. Cell proliferation was assessed by CyQUANT® assay. (C) PC3, DU145, and LNCaP cells were pre-treated with AICAR (1 mM) or compound C (5 μM) for 1 h. The cells were then either left untreated or were treated with Orlistat (100 μM) for 24 h. Apoptosis was analyzed by Hoechst 33342 assay. Graphs represent relative fluorescence intensity. Values are mean ± S.E.M (n = 3). *, p < 0.05 versus control. #, p < 0.05 versus Orlistat treatment.
Effect of Orlistat and AICAR co-treatment on apoptosis regulatory proteins and ROS production
To identify the major pathway involved in Orlistat-AICAR-mediated apoptosis, Caspase 8 and caspase 9 activities were measured. Minimal changes in caspase 8 activity in response to Orlistat, AICAR and compound C treatments were observed in PC3, DU145 and LNCaP cells (Figure 4A). However, Orlistat and AICAR co-treatment resulted in a two-fold increase in caspase 9 activation as compared to the control in all three PCa cell lines (Figure 4B). Compound C and Orlistat combination resulted in a marked decrease in Orlistat-induced caspase 9 activity. To further characterize the apoptotic effects of Orlistat and AICAR co-treatment; treated PCa cell lysates were assayed for caspase 9, 3, and PARP expression levels by Western blotting. Activation of both caspase 9 and 3, in addition to cleaved PARP was observed in samples co-treated with either Orlistat alone or with Orlistat and AICAR together (Figure 4C). Compound C co-treatment with Orlistat showed the opposite effect as evidenced by the inhibition of caspase 9 and PARP cleavage, and the inhibition of full length caspase 3 downregulation. As stated before, published data has linked ROS accumulation and the intrinsic cell death pathway with FASN inhibition-related apoptosis. To determine if ROS accumulation is involved in the apoptosis observed here, we next analyzed the effect of Orlistat and AMPK modulators on ROS production. Orlistat treatment induced ROS production (hydrogen peroxide and superoxide levels) and the effect was potentiated with AICAR co-treatment as compared to control and Orlistat alone in all three PCa cell lines (Figures 4, D and E). Compound C co-treatment had a negative effect on Orlistat-induced increase in hydrogen peroxide production but not on superoxide production.
Figure 4. Orlistat and AICAR-co-treatment upregulates caspase and ROS activity.
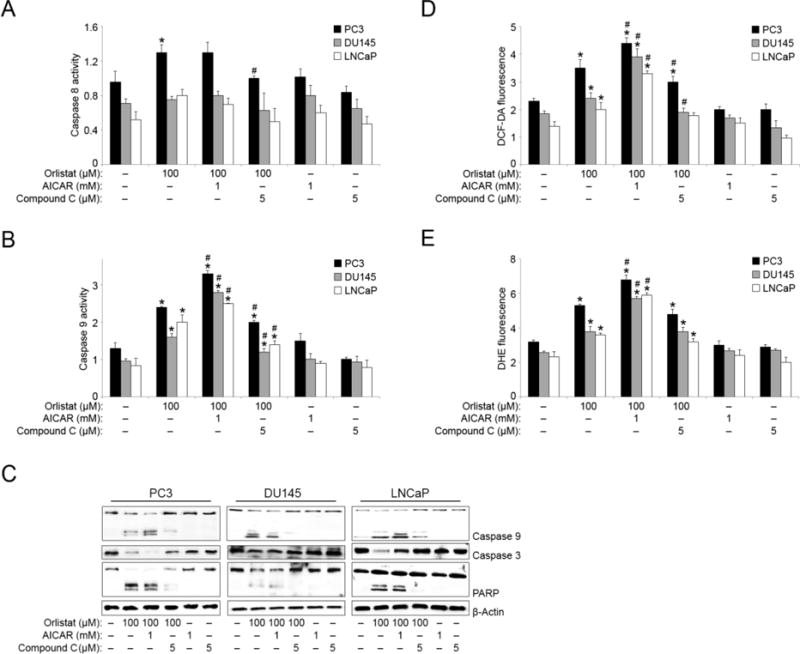
PC3, DU145, and LNCaP cells were pre-treated with AICAR (1 mM) or compound C (5 μM) for 1 h. The cells were then either left untreated or were treated with Orlistat (100 μM) for 6 h. Cell lysates were analyzed for (A) caspase 8 and (B) caspase 9 activity using CaspGLOW activity assay. (C) PC3, DU145, and LNCaP cells were pre-treated with AICAR (1 mM) or compound C (5 μM) for 1 h. The cells were then either left untreated or were treated with Orlistat (100 μM) for 24 h. Cell lysates were collected and analyzed for caspase 9, caspase 3, and PARP protein expression by Western blotting. Blots were reprobed for β-actin protein expression as a loading control. Representative blots from three or more independent experiments are shown. PC3, DU145, and LNCaP cells were pre-treated with AICAR (1 mM) or compound C (5 μM) for 1 h. The cells were then either left untreated or were treated with Orlistat (100 μM) for 6 h. (D) Hydrogen peroxide and (E) superoxide levels were analyzed by spectrofluorometric measurement of DCF and DHE fluorescence, respectively. Values indicate relative fluorescence intensity. Values are mean ± S.E.M (n = 3). *, p < 0.05 versus control. #, p < 0.05 versus Orlistat treatment.
Effect of Orlistat and AICAR co-treatment on cell cycle progression
Orlistat and AICAR co-treatment inhibited cell cycle progression by significantly reducing the number of cells in S phase and increasing the number of cells trapped in G0/G1 phase for all three tested cell-lines (PC3, DU145, LNCaP) (Figures 5, A and B). While there were minimal changes in the ratio of cells in G2/M phase, Orlistat and AICAR co-treatment resulted in a >20% increase in cells for all three PCa cell lines in the G0/G1 phase, with a corresponding decrease in the S phase ratio as compared to control and treatment with Orlistat alone. Interestingly, compound C co-treatment with Orlistat resulted in the ablation of Orlistat-induced G0/G1 arrest; returning G0/G1 cell cycle ratios to or below control levels and increasing the number of cells in S phase for all three PCa cell lines. Furthermore, analysis of three critical cell cycle proteins (p21, CDK 4, CDK 6) by Western blotting corroborated the inhibition of cell cycle progression observed. There was marked upregulation of p21 (cell cycle inhibition marker) in both Orlistat alone and Orlistat and AICAR co-treated samples in all three PCa cell lines (PC3, DU145, LNCaP) (Figure 5C). Analysis of cell cycle progression markers (CDK 4, CDK 6) showed significant downregulation in Orlistat and AICAR co-treated samples in all three PCa cell lines. Compound C co-treatment with Orlistat reversed the effect of Orlistat on these three cell cycle proteins (p21, CDK4, CDK 6).
Figure 5. Orlistat and AICAR co-treatment inhibits cell cycle progression.
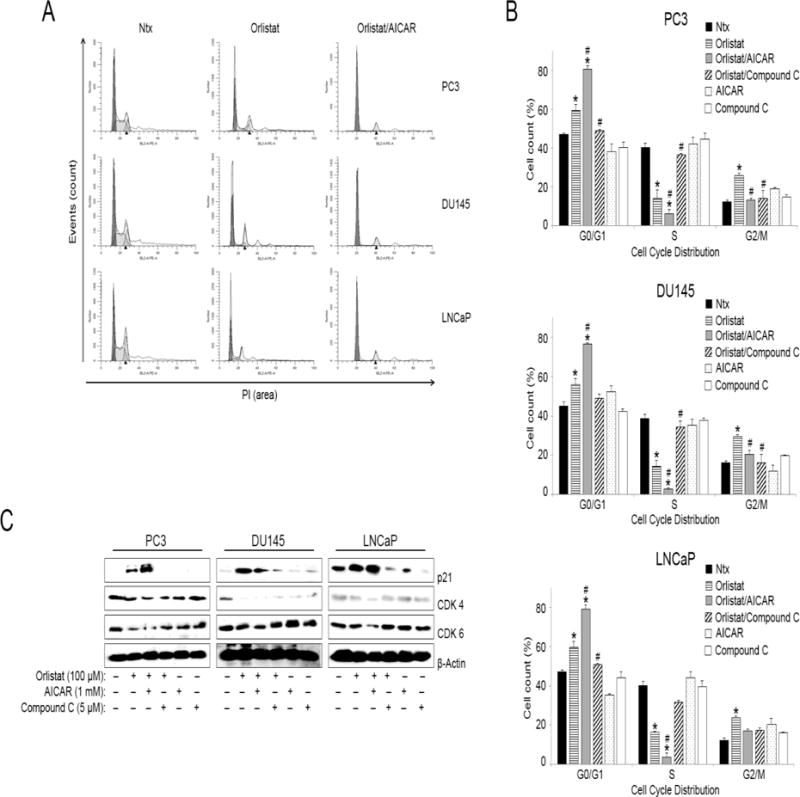
PC3, DU145, and LNCaP cells were pre-treated with AICAR (1 mM) or compound C (5 μM) for 1 h. The cells were then either left untreated or were treated with Orlistat (100 μM) for 24 h. Cell cycle analysis was done using flow cytometry (A) Representative cell cycle plots of PC3, DU145, and LNCaP cells for control (Ntx-no treatment), Orlistat, and Orlistat and AICAR-treated conditions. (B) Cell cycle distribution analysis for PC3, DU145, and LNCaP cells based upon the treatment described above. Values are mean ± S.E.M (n = 3). *, p < 0.05 versus control. #, p < 0.05 versus Orlistat treatment. (C) PC3, DU145, and LNCaP cells were pre-treated with AICAR (1 mM) or compound C (5 μM) for 1 h. The cells were then either left untreated or were treated with Orlistat (100 μM) for 24 h. Cell lysates were collected and analyzed for cell cycle protein (p21, CDK 4, CDK 6) expression by Western blotting. Blots were reprobed for β-actin protein expression as a loading control. Representative blots from three or more independent experiments are shown.
Effect of Orlistat and AICAR co-treatment on cell migration and VEGF levels
We performed wound healing scratch assay to determine the effect of Orlistat and AICAR co-treatment on cellular migration. The migration potential of PC3, DU145, and LNCaP cells was significantly inhibited by Orlistat treatment, and compounded by the additive effect of AICAR co-treatment at both 24 and 48 h time-points (Figures 6, A and B). AMPK inhibition using compound C had no significant effect on the ability of Orlistat to inhibit migration. At the 24 h time-point, there was a marked decrease (50%) in continuous cell movement in samples co-treated with Orlistat and AICAR as compared to control, and a 25% decrease as compared to treatment with Orlistat alone. We also analyzed VEGF levels in PCa cell-lines (PC3, DU145, LNCaP) by ELISA. Orlistat and AICAR co-treatment significantly inhibited VEGF levels 2-fold as compared to control in all three PCa cell lines (Figure 6C). Compound C co-treatment with Orlistat resulted in a slight abrogation of the Orlistat-induced decrease in VEGF production in PCa cells. Lastly, based on the MTT data in Figures 1 and 3, Bliss Independency model formulation system was used to determine the type of drug interaction between Orlistat and AICAR as well as Orlistat and compound C. The effect of Orlistat and AICAR combination treatment was synergism, whereas Orlistat and compound C interaction was antagonism in all three PCa cell lines.
Figure 6. Orlistat and AICAR combination inhibits cell migration and angiogenesis.
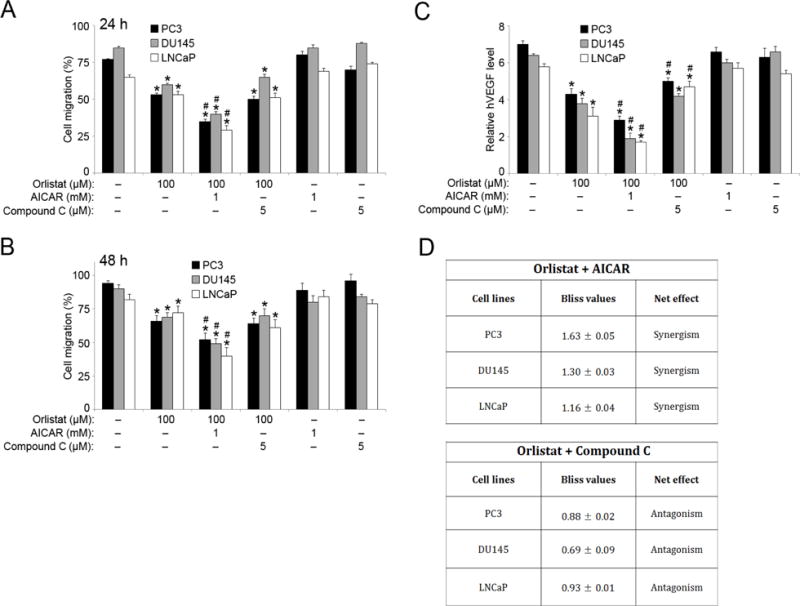
PC3, DU145, and LNCaP cells were seeded in 12-well plates separately and a 1 ml pipette tip was used to scratch a line in the center of each well. Cells were washed with PBS before being pre-treated with AICAR (1 mM) or compound C (5 μM) for 1 h. The cells were then either left untreated or were treated with Orlistat (100 μM) for 24 and 48 h time points. Pictures were taken after (A) 24 h and (B) 48 h. ImageJ software (Java image processing, NIH) was used to measure the distance (%) of migration versus control. (C) PC3, DU145, and LNCaP cells were pre-treated with AICAR (1 mM) or compound C (5 μM) for 1 h. The cells were then either left untreated or were treated with Orlistat (100 μM) for 48 h, and VEGF levels were assessed by ELISA. (D) PC3, DU145, and LNCaP cells were pre-treated with AICAR (1 mM) or compound C (5 μM) for 1 h. The cells were then either left untreated or were treated with Orlistat (100 μM) for 24 h. MTT assay values were then used to determine the combinatorial effects of Orlistat (100 μM)-AICAR (1 mM) and Orlistat (100 μM)-compound C (5 μM) in PC3, DU145, and LNCaP cells predicted by Bliss Independency model. Representative data from three or more independent experiments is shown. Values are mean ± S.E.M (n = 3). *, p < 0.05 versus control. #, p < 0.05 versus Orlistat treatment.
Discussion
FASN has moved from being a potential oncogenic marker to a definite chemotherapeutic target in several cancer types including breast and PCa [Giro-Perafita et al., 2016; Sadowski et al., 2014]. During this time it has become clear that cancer cells synthesize the vast majority of their fatty acids de novo, thereby relying on the cells energy machinery and lipogenic potential to increase their proliferation [Carvalho et al., 2008]. Utilizing FASN inhibitors, such as Orlistat, have shown tremendous potential against multiple cancer forms; resulting in significant inhibition of cell proliferation and cell cycle blockage, and induction of apoptosis [Furuya et al., 1997; Pizer et al., 1998].
In the present study, we demonstrate that utilizing the AMPK activator AICAR potentiates the antiproliferative effects of Orlistat seen in androgen-dependent (LNCaP) as well as androgen-independent (PC3, DU145) PCa cell-lines in a significant and almost identical manner. AICAR treatment induced AMPK activation which in turn phosphorylated and inactivated ACC. Most noticeably however was the significant effect Orlistat and AICAR combination treatment had on FASN and SREBP-1c expression, which are two terminal rate-limiting FA synthesis proteins (Figure 2). There was marked downregulation of FASN in PC3 and LNCaP cells in response to Orlistat and AICAR co-treatment as compared to treatment with Orlistat alone. However, in DU145 cells there was more significant downregulation of FASN in Orlistat alone treatments. It is possible that this discrepancy is due to the variation in FASN expression in the tested PCa cell-lines. LNCaP cells are known to express high levels of FASN as opposed to DU145 cells which express very low levels of FASN protein. SREBP1 has only recently emerged as a therapeutic target for cancer treatment due to its multi-layered involvement in various cancers. As detailed above, SREBP is a transcription factor that regulates target genes of the FA synthesis and cholesterol pathway, most notably in this case, FASN. SREBP1 has also been shown to interact with crucial cell cycle regulatory proteins [Reed et al., 2008]. Overexpression of SREBP1 has been directly linked to the pathogenesis of several cancer types [Furuta et al., 2008; Osada et al., 2006]. Specifically in PCa; SREBP1 upregulation is seen in the progression of androgen-independent cancer [Ettinger et al., 2004]. The significant downregulation of SREBP-1c in Orlistat and AICAR co-treated PCa cells suggests a clear role for SREBP1 in promoting PCa progression. In contrast, AMPK inhibition with compound C had no effect on FASN expression, including when co-treated with Orlistat. While SREBP-1c expression was still downregulated in compound C-treated cells in comparison to control, compound C co-treatment with Orlistat had the opposite effect on SREBP-1c expression in comparison to Orlistat and AICAR co-treatment.
While analyzing the expression levels of FA synthesis proteins is not a direct indicator of FA synthesis, it does serve to identify the mechanistic pathway whereby Orlistat and AICAR negatively affect FA activity. Orlistat clearly has a negative effect on FA synthesis by downregulating FASN and SREBP-1c protein expression. AICAR has a negative effect on FA synthesis by activating AMPK which in turn inactivates ACC. In combination, these two compounds serve to additively inhibit FA activity by downregulating and inhibiting different proteins within the FA synthesis pathway. However, future experiments will focus on FA synthesis activity assays in order to determine if the Orlistat and AICAR drug combination treatment directly inhibits lipid synthesis. Knockdown assays or siRNA targeting FASN, SREBP-1c, and AMPK in future experiments will also serve to corroborate the effects observed here with Orlistat and AICAR in PCa cells. We will also investigate the combinatory effects of different FASN and AMPK inhibitory compounds in order to further establish that the additive potent toxicity we observe in PCa cells is FASN and AMPK-inhibition-specific.
While Figure 1 shows that AICAR and compound C alone have no effect on PCa cell viability at lower doses and a 24h time point; the combination of AICAR treatment with Orlistat has a significant inhibitory effect on cell viability and proliferation accompanied by apoptosis induction possibly through the intrinsic apoptotic pathway (Figure 3). Orlistat induced apoptosis by ~15% as compared to control, whereas Orlistat and AICAR co-treatment led to a ~35% and ~45% increase in apoptosis when compared to treatment with Orlistat alone and control, respectively. Compound C had no inhibitory effect on Orlistat-induced effects on cell viability, proliferation and apoptosis. The significance of Orlistat and AICAR combination leads back to the FA synthesis pathway that can be regulated by AMPK activation. Changes in cellular energy levels can lead to AMPK activation that is then responsible for returning cellular energy homeostasis by manipulating FA synthesis and oxidation as required by the cell through the phosphorylation of multiple FA synthesis pathway proteins [He et al., 2016; Kemmerer et al., 2015]. The apoptosis induced by Orlistat and AICAR co-treatment is preceded by an increase in ROS production and caspase 9 and 3 activation (Figure 4). AICAR has been reported to inhibit cell growth and induce apoptosis in DU145 PCa cells in a ROS and caspase 3-dependent manner [Sauer et al., 2012]. AICAR has also been implicated in the potentiation of ROS production induced by high glucose levels in pancreatic beta cells [Kim et al., 2007]. Fujiwara et al reported that Orlistat induced ROS production in DU145 and PC3 PCa cells [Fujiwara et al., 2012]. We report here that a dose range of 0.5–3 mM of AICAR has non-significant effects on all three PCa cell lines tested (PC3, DU145, LNCaP) for a 24 h time point. However, time dependent analysis (>48h) does indicate cell death in all three PCa cell lines (data not shown). However, combining AICAR with Orlistat potentiates the effects of Orlistat on PCa cell apoptosis induction that is preceded by ROS production and caspase activation (Figure 4). The significant increase in apoptosis that co-treatment with AICAR induces is novel, and establishes a critical role for AMPK activation along with FASN inhibition in the cell death we observe in PCa cell lines.
Future experiments focused on testing this combination drug effect in normal prostate cells is needed to confirm the specificity of the apoptosis observed here. However, since FASN and therefore FA synthesis is elevated in PCa cells, it is a feasible assumption that this combination therapy may be PCa cell-specific. Analysis on other cancer types that exhibit elevated FA synthesis levels would also determine if this Orlistat-AICAR combination treatment has applications in multiple cancer types. While Orlistat and AICAR individually have been shown to have non-specific side effects in multiple in vitro studies, it is possible that at low doses, the independent manner in which both compounds work to negatively affect FA protein expression coupled with their combined effects on pro- and anti-apoptotic proteins ensures an additive effect on cell viability, proliferation, cell cycle progression, and apoptosis.
Caspase 9 is a vital member of the caspase-apoptosis signaling cascade family that is not active until proteolytically cleaved upon apoptotic stimulation [Slee et al., 1999]. Cleaved caspase 9 then participates in the processing and activation of downstream caspases including caspase 3 which is involved in late-stage apoptosis induction [Nicholson et al., 1995; Slee et al., 1999]. In addition to a clear increase in caspase 9 cleavage in Orlistat and AICAR co-treated samples; we show ablation of caspase cleavage in Orlistat and compound C co-treated samples in all three PCa cell lines (PC3, DU145, LNCaP) (Figure 4). Similarly, compound C co-treatment with Orlistat ablated caspase 3 and PARP cleavage. This pattern is significant to establish apoptosis because caspase 9 cleavage and activation facilitates caspase 3 cleavage and activation (as evidenced by the downregulation of full length caspase 3 (Figure 4). Caspase 3 is a critical end-stage apoptosis protein that is also responsible for the cleavage of PARP; a marker for apoptotic cells that triggers cellular disassembly and viability reduction [Oliver et al., 1998]. While Orlistat and AICAR co-treatment does not increase PARP cleavage, it does increase caspase 9 cleavage and the Hoechst assay data in Figure 3 serves to bolster our conclusion that Orlistat and AICAR co-treatment significantly increases apoptosis in PCa cells.
To further understand the combined effect of Orlistat and AICAR on PCa cell lines; we analyzed cell cycle events after treatment. We observed significant G0/G1 cell cycle arrest with Orlistat that is markedly potentiated when AICAR is introduced (Figure 5). This cell cycle arrest is accompanied by increased expression of the cell cycle inhibitor p21 and corresponding decreases in CDK 4 and CDK 6, which are two kinases that function in cell cycle progression. P21 is a potent cyclin dependent inhibitor that regulates G1 to S phase progression, and is tightly regulated by the tumor suppressor p53, but can also function in a p53-independent role [Gartel and Radhakrishnan, 2005; Rodriguez and Meuth, 2006]. P53 analysis in Orlistat and AICAR co-treated samples showed no changes in expression, suggesting a possible p53-independent role here for p21 (data not shown). CDK 4 and CDK 6 are cyclin D-regulated cell cycle proteins that are critical for cell cycle progression through the G1 phase [Diaz-Moralli et al., 2013]. The coordinated downregulation of CDK 4 and CDK 6 with the upregulation in p21 expression indicates clear G0/G1 cell cycle arrest in Orlistat-treated samples that is increased by over 20% when AICAR is introduced. Cell cycle arrest that follows FASN inhibition has been observed in several cancer types including melanoma mouse models [Menendez et al., 2005; Zecchin et al., 2011]. However, the potentiated effect seen with AICAR co-treatment that results in a significant increase in the inhibition of cell cycle progression as compared to treatment with Orlistat alone is novel. The significant role AMPK activation plays here was confirmed by co-treating cells with Orlistat and compound C, which resulted in the inhibition of Orlistat-induced G0/G1 phase arrest and was accompanied by an increase in the number of cells pushed into S phase (Figure 5). The ablation of Orlistat-induced cell cycle arrest seen with compound C treatment was also evident at the protein level. P21 levels were restored to control (baseline), and the same was seen for CDK4 and CDK6 expression. Compound C co-treatment with Orlistat clearly reversed the inhibitory effects on cell cycle progression observed with either Orlistat alone or Orlistat and AICAR co-treatment.
One of the hallmarks of cancer cells is their ability to form tumors. Part of this characteristic involves the ability to migrate and invade surrounding tissue to facilitate growth. We show that Orlistat and AICAR co-treatment significantly suppressed the migration potential of all three PCa cell lines tested (PC3, DU145, LNCaP) as compared to control (Figure 6). This is significant because FASN functions in phospholipid synthesis that can be directly linked to intracellular trafficking and cell migration [Swinnen et al., 2003]. Therefore, FASN inhibition can cause dysfunction in the cellular phospholipid membrane due to impaired lipid production which can directly have a negative effect on cell to cell adhesion, migration, and invasion potential [Menendez and Lupu, 2007]. AMPK is directly involved in cellular lipid synthesis regulation despite the fact that co-treatment with Orlistat and compound C did not ablate Orlistat-induced inhibition of PCa cell migration (Figure 6). However, the significant potentiated effect AMPK activation along with Orlistat treatment has on cell migration was clearly nonexistent when compound C was used to inhibit AMPK.
Angiogenesis is also a major prerequisite for cancer cell progression because it facilitates the increase in blood supply that is required for tumor growth. Multiple growth factors have been identified as regulators of angiogenesis; including transforming growth factor-α (TGF-α), fibroblast growth factor (bFGF), and VEGF-A [Ferrara et al., 1992; Goldfarb, 1990]. VEGF has become a key marker for multiple cancer types (colorectal, lung, breast) due to its high expression that correlates with increased cell proliferation, tumor vascularization and overall cancer progression [Folkman, 1993]. This has led to clinical development and use of drugs that target VEGF in various cancer treatments including colon and lung cancer [Mavrou and Oltean, 2016]. Several reports have shown increased angiogenesis in PCa, and VEGF overexpression has been directly linked to increased invasion and poor differentiation [Grivas et al., 2016]. Elevated VEGF levels have also been found in the urine and plasma of advanced PCa patients [Mavrou and Oltean, 2016]. We demonstrate significant inhibition of VEGF expression with Orlistat treatment that is markedly potentiated with AICAR co-treatment (Figure 6C). Orlistat and compound C co-treatment decreased the inhibitory effect of Orlistat on VEGF expression in all three PCa cell types, confirming the importance of AMPK activation in the potentiated anti-cancer effects observed with Orlistat treatment.
When using multiple drugs in a combination study, it is critical to determine the drug-drug interaction effect of the compounds being administered simultaneously. This information is also crucial from a clinical, pharmacological perspective because it is needed to triage any side effects that may manifest in the host organism being administered the combination of drugs. In Figure 6, using the Bliss independency model, we demonstrate that the drug-drug interaction between Orlistat and AICAR is synergistic that causes an increase in the anti-cancer effects of Orlistat on PCa cells, whereas the drug-drug interaction between Orlistat and compound C is antagonistic. This corroborates the earlier data which demonstrated that the interaction between Orlistat and compound C predominantly led to a decrease in the anti-cancer effects of Orlistat.
PCa therapy is approached by a combination of surgical intervention, radiation and chemotherapy, and anti-androgen drug treatments. However, more than 30,000 men in the USA alone still died from complications surrounding aggressive and advanced metastatic PCa in 2015 [Siegel et al., 2015]. New approaches to PCa treatment are being tested annually, but there is still a significant need for more efficient and effective forms of treatment worldwide. While there is evidence that Orlistat can be used as an effective anti-cancer agent, its poor bioavailability and non-specific side effects are still a major therapeutic issue that needs to be addressed. Here we provide data evaluating the therapeutic value of a novel combination of Orlistat and AICAR co-treatment which results in the potentiation of Orlistat-induced anti-cancer effects on three PCa cell lines (PC3, DU145, LNCaP). This novel combination therapy could possibly provide an opportunity to significantly increase the potency and efficacy of Orlistat against PCa.
Acknowledgments
Contract grant sponsor: National Institutes of Health
Contract grant number: HL112630 and CA173069.
Footnotes
Conflict of Interest: There are no conflicts of interest or additional funding/contributions to be disclosed.
References
- Bliss CI. The toxicity of poisons applied jointly. Ann Appl Biol. 1939;26:585–615. [Google Scholar]
- Carlsson S, Vickers AJ, Roobol M, Eastham J, Scardino P, Lilja H, Hugosson J. Prostate cancer screening: facts, statistics, and interpretation in response to the US Preventive Services Task Force Review. J Clin Oncol. 2012;30:2581–4. doi: 10.1200/JCO.2011.40.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho MA, Zecchin KG, Seguin F, Bastos DC, Agostini M, Rangel AL, Veiga SS, Raposo HF, Oliveira HC, Loda M, Coletta RD, Graner E. Fatty acid synthase inhibition with Orlistat promotes apoptosis and reduces cell growth and lymph node metastasis in a mouse melanoma model. Int J Cancer. 2008;123:2557–65. doi: 10.1002/ijc.23835. [DOI] [PubMed] [Google Scholar]
- Davidson MH, Hauptman J, DiGirolamo M, Foreyt JP, Halsted CH, Heber D, Heimburger DC, Lucas CP, Robbins DC, Chung J, Heymsfield SB. Weight control and risk factor reduction in obese subjects treated for 2 years with orlistat: a randomized controlled trial. JAMA. 1999;281:235–42. doi: 10.1001/jama.281.3.235. [DOI] [PubMed] [Google Scholar]
- Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, Cascante M. Targeting cell cycle regulation in cancer therapy. Pharmacol Ther. 2013;138:255–71. doi: 10.1016/j.pharmthera.2013.01.011. [DOI] [PubMed] [Google Scholar]
- Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, Nelson CC. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. 2004;64:2212–21. doi: 10.1158/0008-5472.can-2148-2. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Houck K, Jakeman L, Leung DW. Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocr Rev. 1992;13:18–32. doi: 10.1210/edrv-13-1-18. [DOI] [PubMed] [Google Scholar]
- Field FJ, Born E, Murthy S, Mathur SN. Polyunsaturated fatty acids decrease the expression of sterol regulatory element-binding protein-1 in CaCo-2 cells: effect on fatty acid synthesis and triacylglycerol transport. Biochem J. 2002;368:855–64. doi: 10.1042/BJ20020731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J. Diagnostic and therapeutic applications of angiogenesis research. C R Acad Sci III. 1993;316:909–18. [PubMed] [Google Scholar]
- Ford OH, 3rd, Gregory CW, Kim D, Smitherman AB, Mohler JL. Androgen receptor gene amplification and protein expression in recurrent prostate cancer. J Urol. 2003;170:1817–21. doi: 10.1097/01.ju.0000091873.09677.f4. [DOI] [PubMed] [Google Scholar]
- Fujiwara J, Sowa Y, Horinaka M, Koyama M, Wakada M, Miki T, Sakai T. The anti-obesity drug orlistat promotes sensitivity to TRAIL by two different pathways in hormone-refractory prostate cancer cells. Int J Oncol. 2012;40:1483–91. doi: 10.3892/ijo.2012.1353. [DOI] [PubMed] [Google Scholar]
- Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, Mo YY, Hirota S, Hosobe S, Tsukada T, Miura K, Kamada S, Saito K, Iiizumi M, Liu W, Ericsson J, Watabe K. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008;68:1003–11. doi: 10.1158/0008-5472.CAN-07-2489. [DOI] [PubMed] [Google Scholar]
- Furuya Y, Akimoto S, Yasuda K, Ito H. Apoptosis of androgen-independent prostate cell line induced by inhibition of fatty acid synthesis. Anticancer Res. 1997;17:4589–93. [PubMed] [Google Scholar]
- Gabrielson EW, Pinn ML, Testa JR, Kuhajda FP. Increased fatty acid synthase is a therapeutic target in mesothelioma. Clin Cancer Res. 2001;7:153–7. [PubMed] [Google Scholar]
- Galinanes M, Mullane KM, Bullough D, Hearse DJ. Acadesine and myocardial protection. Studies of time of administration and dose-response relations in the rat. Circulation. 1992;86:598–608. doi: 10.1161/01.cir.86.2.598. [DOI] [PubMed] [Google Scholar]
- Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005;65:3980–5. doi: 10.1158/0008-5472.CAN-04-3995. [DOI] [PubMed] [Google Scholar]
- Giro-Perafita A, Palomeras S, Lum DH, Blancafort A, Vinas G, Oliveras G, Perez-Bueno F, Sarrats A, Welm AL, Puig T. Preclinical Evaluation of Fatty Acid Synthase and EGFR Inhibition in Triple-Negative Breast Cancer. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-3133. [DOI] [PubMed] [Google Scholar]
- Goldfarb M. The fibroblast growth factor family. Cell Growth Differ. 1990;1:439–45. [PubMed] [Google Scholar]
- Grivas N, Goussia A, Stefanou D, Giannakis D. Microvascular density and immunohistochemical expression of VEGF, VEGFR-1 and VEGFR-2 in benign prostatic hyperplasia, high-grade prostate intraepithelial neoplasia and prostate cancer. Cent European J Urol. 2016;69:63–71. doi: 10.5173/ceju.2016.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. The AMP-activated protein kinase pathway–new players upstream and downstream. J Cell Sci. 2004;117:5479–87. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- He L, Zhou X, Huang N, Li H, Tian J, Li T, Yao K, Wu G, Nyachoti CM, Kim SW, Yin Y. The Mechanism of AMPK Regulation on Glucose, Lipid and Protein Metabolic Homeostasis and Its Related Physiologically Nutritional Significance. Curr Protein Pept Sci. 2016 doi: 10.2174/1389203717666160627071125. [DOI] [PubMed] [Google Scholar]
- Heiligtag SJ, Bredehorst R, David KA. Key role of mitochondria in cerulenin-mediated apoptosis. Cell Death Differ. 2002;9:1017–25. doi: 10.1038/sj.cdd.4401055. [DOI] [PubMed] [Google Scholar]
- Jayakumar A, Tai MH, Huang WY, al-Feel W, Hsu M, Abu-Elheiga L, Chirala SS, Wakil SJ. Human fatty acid synthase: properties and molecular cloning. Proc Natl Acad Sci U S A. 1995;92:8695–9. doi: 10.1073/pnas.92.19.8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Kemmerer M, Finkernagel F, Cavalcante MF, Abdalla DS, Muller R, Brune B, Namgaladze D. AMP-Activated Protein Kinase Interacts with the Peroxisome Proliferator-Activated Receptor Delta to Induce Genes Affecting Fatty Acid Oxidation in Human Macrophages. PLoS One. 2015;10:e0130893. doi: 10.1371/journal.pone.0130893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WH, Lee JW, Suh YH, Lee HJ, Lee SH, Oh YK, Gao B, Jung MH. AICAR potentiates ROS production induced by chronic high glucose: roles of AMPK in pancreatic beta-cell apoptosis. Cell Signal. 2007;19:791–805. doi: 10.1016/j.cellsig.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Knudsen KE, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res. 2009;15:4792–8. doi: 10.1158/1078-0432.CCR-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi N, Inoue M, Iwasaki M, Sasazuki S, Tsugane AS. Dairy product, saturated fatty acid, and calcium intake and prostate cancer in a prospective cohort of Japanese men. Cancer Epidemiol Biomarkers Prev. 2008;17:930–7. doi: 10.1158/1055-9965.EPI-07-2681. [DOI] [PubMed] [Google Scholar]
- Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL, Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001;61:3550–5. [PubMed] [Google Scholar]
- Mavrou A, Oltean S. SRPK1 inhibition in prostate cancer: A novel anti-angiogenic treatment through modulation of VEGF alternative splicing. Pharmacol Res. 2016;107:276–81. doi: 10.1016/j.phrs.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–77. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- Menendez JA, Vellon L, Lupu R. Antitumoral actions of the anti-obesity drug orlistat (XenicalTM) in breast cancer cells: blockade of cell cycle progression, promotion of apoptotic cell death and PEA3-mediated transcriptional repression of Her2/neu (erbB-2) oncogene. Ann Oncol. 2005;16:1253–67. doi: 10.1093/annonc/mdi239. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–9. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- Osada S, Naganawa A, Misonou M, Tsuchiya S, Tamba S, Okuno Y, Nishikawa J, Satoh K, Imagawa M, Tsujimoto G, Sugimoto Y, Nishihara T. Altered gene expression of transcriptional regulatory factors in tumor marker-positive cells during chemically induced hepatocarcinogenesis. Toxicol Lett. 2006;167:106–13. doi: 10.1016/j.toxlet.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Pizer ES, Chrest FJ, DiGiuseppe JA, Han WF. Pharmacological inhibitors of mammalian fatty acid synthase suppress DNA replication and induce apoptosis in tumor cell lines. Cancer Res. 1998;58:4611–5. [PubMed] [Google Scholar]
- Pizer ES, Thupari J, Han WF, Pinn ML, Chrest FJ, Frehywot GL, Townsend CA, Kuhajda FP. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res. 2000;60:213–8. [PubMed] [Google Scholar]
- Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M. Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS Genet. 2008;4:e1000133. doi: 10.1371/journal.pgen.1000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R, Meuth M. Chk1 and p21 cooperate to prevent apoptosis during DNA replication fork stress. Mol Biol Cell. 2006;17:402–12. doi: 10.1091/mbc.E05-07-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski MC, Pouwer RH, Gunter JH, Lubik AA, Quinn RJ, Nelson CC. The fatty acid synthase inhibitor triclosan: repurposing an anti-microbial agent for targeting prostate cancer. Oncotarget. 2014;5:9362–81. doi: 10.18632/oncotarget.2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer H, Engel S, Milosevic N, Sharifpanah F, Wartenberg M. Activation of AMP-kinase by AICAR induces apoptosis of DU-145 prostate cancer cells through generation of reactive oxygen species and activation of c-Jun N-terminal kinase. Int J Oncol. 2012;40:501–8. doi: 10.3892/ijo.2011.1230. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281–92. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S, Witkowski A, Joshi AK. Structural and functional organization of the animal fatty acid synthase. Prog Lipid Res. 2003;42:289–317. doi: 10.1016/s0163-7827(02)00067-x. [DOI] [PubMed] [Google Scholar]
- Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994;353:33–6. doi: 10.1016/0014-5793(94)01006-4. [DOI] [PubMed] [Google Scholar]
- Swinnen JV, Van Veldhoven PP, Timmermans L, De Schrijver E, Brusselmans K, Vanderhoydonc F, Van de Sande T, Heemers H, Heyns W, Verhoeven G. Fatty acid synthase drives the synthesis of phospholipids partitioning into detergent-resistant membrane microdomains. Biochem Biophys Res Commun. 2003;302:898–903. doi: 10.1016/s0006-291x(03)00265-1. [DOI] [PubMed] [Google Scholar]
- Thupari JN, Kim EK, Moran TH, Ronnett GV, Kuhajda FP. Chronic C75 treatment of diet-induced obese mice increases fat oxidation and reduces food intake to reduce adipose mass. Am J Physiol Endocrinol Metab. 2004;287:E97–E104. doi: 10.1152/ajpendo.00261.2003. [DOI] [PubMed] [Google Scholar]
- Tong L. Acetyl-coenzyme A carboxylase: crucial metabolic enzyme and attractive target for drug discovery. Cell Mol Life Sci. 2005;62:1784–803. doi: 10.1007/s00018-005-5121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatadri R, Muni T, Iyer AK, Yakisich JS, Azad N. Role of apoptosis-related miRNAs in resveratrol-induced breast cancer cell death. Cell Death Dis. 2016;7:e2104. doi: 10.1038/cddis.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, Giri S, Andreelli F. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol. 2010;45:276–95. doi: 10.3109/10409238.2010.488215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakil SJ. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry. 1989;28:4523–30. doi: 10.1021/bi00437a001. [DOI] [PubMed] [Google Scholar]
- Yoshii Y, Furukawa T, Oyama N, Hasegawa Y, Kiyono Y, Nishii R, Waki A, Tsuji AB, Sogawa C, Wakizaka H, Fukumura T, Yoshii H, Fujibayashi Y, Lewis JS, Saga T. Fatty acid synthase is a key target in multiple essential tumor functions of prostate cancer: uptake of radiolabeled acetate as a predictor of the targeted therapy outcome. PLoS One. 2013;8:e64570. doi: 10.1371/journal.pone.0064570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecchin KG, Rossato FA, Raposo HF, Melo DR, Alberici LC, Oliveira HC, Castilho RF, Coletta RD, Vercesi AE, Graner E. Inhibition of fatty acid synthase in melanoma cells activates the intrinsic pathway of apoptosis. Lab Invest. 2011;91:232–40. doi: 10.1038/labinvest.2010.157. [DOI] [PubMed] [Google Scholar]
- Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009;9:407–16. doi: 10.1016/j.cmet.2009.03.012. [DOI] [PubMed] [Google Scholar]
- Zhao W, Sachsenmeier K, Zhang L, Sult E, Hollingsworth RE, Yang H. A New Bliss Independence Model to Analyze Drug Combination Data. J Biomol Screen. 2014;19:817–21. doi: 10.1177/1087057114521867. [DOI] [PubMed] [Google Scholar]
- Zhi J, Melia AT, Eggers H, Joly R, Patel IH. Review of limited systemic absorption of orlistat, a lipase inhibitor, in healthy human volunteers. J Clin Pharmacol. 1995;35:1103–8. doi: 10.1002/j.1552-4604.1995.tb04034.x. [DOI] [PubMed] [Google Scholar]