

Abstract
Wolbachia pipientis, an obligate intracellular bacterium associated with arthropods and filarial worms, is a target for filarial disease treatment and provides a gene drive agent for insect vector population suppression/replacement. We compared proteomes of Aedes albopictus mosquito C/wStr1 cells persistently infected with Wolbachia strain wStr, relative to uninfected C7–10 control cells. Among approximately 2,500 proteins, iTRAQ data identified 815 differentially abundant proteins. As functional classes, energy and central intermediary metabolism proteins were elevated in infected cells, while suppressed proteins with roles in host DNA replication, transcription and translation suggested that Wolbachia suppresses pathways that support host cell growth and proliferation. Vacuolar ATPase subunits were strongly elevated, consistent with high densities of Wolbachia contained individually within vacuoles. Other differential level proteins had roles in ROS neutralization, protein modification/degradation and signaling, including hypothetical proteins whose functions in Wolbachia infection can potentially be manipulated by RNAi interference or transfection. Detection of flavivirus proteins supports further analysis of poorly understood, insect-specific flaviviruses and their potential interactions with Wolbachia, particularly in mosquitoes transinfected with Wolbachia. This study provides a framework for future attempts to manipulate pathways in insect cell lines that favor production of Wolbachia for eventual genetic manipulation, transformation and transinfection of vector species.
Keywords: Mosquito cell lines, Aedes albopictus, Wolbachia pipientis, intracellular bacterium, flavivirus, Transinfection
1. Introduction
The alphaproteobacterium, Wolbachia pipientis (Rickettsiales; Anaplasmataceae) is an obligate intracellular endosymbiont originally described in ovaries of Culex pipiens mosquitoes [1], and now known to be widely distributed among arthropods, including an estimated 40% of Culicine mosquitoes [2]. In mosquitoes, Wolbachia infection is associated with cytoplasmic incompatibility (CI), the reproductive distortion in which eggs of uninfected females fail to hatch if fertilized by sperm from an infected male [3, 4]. Because Wolbachia confers a reproductive advantage to infected females, the bacterium provides an attractive candidate for disease control through population suppression/eradication, first demonstrated nearly 50 years ago in Myanmar (Burma) with the filariasis vector, Culex pipiens fatigans [5, 6].
Interest in exploiting Wolbachia as a gene drive agent for control of vector-borne disease [4] has been stimulated by advances in genomics, including successful transformation of related intracellular bacteria [7]. In addition, Wolbachia reduces pathogen transmission following artificial introduction (transinfection) into mosquito species that do not harbor Wolbachia in nature, such as the Aedes aegypti vector of yellow fever, dengue, and Zika viruses and Anopheles vectors of human malaria [8, 9, 10, 11, 12]. Successful cage and field trials using Wolbachia-infected mosquitoes [13, 14, 15] strongly support intensified efforts to develop techniques for in vitro manipulation and genetic transformation of Wolbachia to facilitate its use as a biocontrol agent for vector-borne disease [2, 16, 17].
Cell lines that maintain high levels of Wolbachia provide a source of bacteria for genetic manipulation and subsequent recovery and amplification of transformants for introduction into target mosquitoes. Infected cell lines further provide an in vitro approach to investigating interactions with host cells, such as control of oxidative stress [18, 19], iron metabolism [20], chromatin remodeling [21, 22] and the molecular basis for CI [23]. The C/wStr1 cell line maintains a robust infection with the CI-inducing Wolbachia strain wStr [24, 25], which replicates in individual vacuoles surrounded by a host-derived membrane [26]. Among 790 Wolbachia proteins, we established a molecular ‘footprint’ of wStr infection dominated by chaperones, stress response and Wolbachia surface proteins [27].
Here we provide a quantitative proteomic analysis of the mosquito host cell response to wStr that suggests Wolbachia conforms to an emerging paradigm based on pathogenic intracellular bacteria in vertebrates that evade innate immunity responses and enhance access to host nutritional reserves [28, 29].
2. Materials and methods
2.1. Cultivation of cells, subcellular fractionation and preparation of protein extracts
Aedes albopictus C7–10 (control) and C/wStr1 (Wolbachia-infected) cells were maintained in Eagle’s minimal medium supplemented with glucose, non-essential amino acids, glutamine, vitamins, penicillin/streptomycin and 5% fetal bovine serum at 28°C in a 5% CO2 atmosphere as described previously [25, 27, 30]. For proteomic analysis, unsynchronized populations of exponentially growing cells from six 25-cm2 flasks were recovered by gentle pipetting, followed by centrifugation of pooled cells to remove medium. Biological replicates included cells from passages (p) 9, 16 and 35; data from all samples were combined for the final analysis. Cells were washed with serum-free medium, and subcellular fractions were prepared by sonication, filtration, centrifugation and sucrose gradient fractionation as detailed previously [27] and summarized at left in Fig. 1.
Fig. 1.
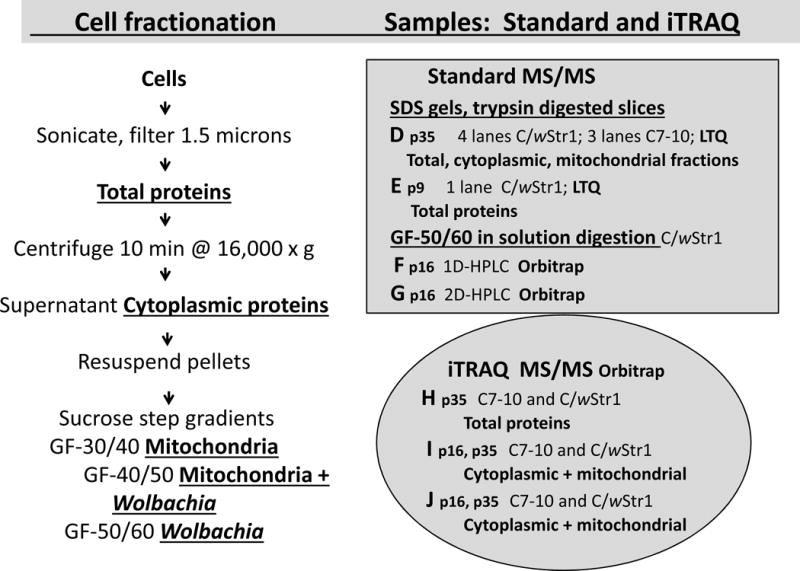
Sample preparation for MS/MS analysis. Samples were prepared according to the flow chart at left as detailed earlier by Baldridge et al. [27]. Underlined, bold text designates fractions used in the analyses at right. The shaded rectangle shows samples D (p16), E (p9), F (p16) and G (p16), from which host proteins were identified in a standard analysis, and the shaded oval represents samples H (p35), I, (p16 and p35) and J (p16 and p35) used for iTRAQ analysis; note that “p” designates passage number. Samples for MS data sets D and E were separated by SDS-PAGE and recovered from gel slices that were trypsin-digested in situ for protein identification by LTQ MS/MS. Data sets F and G were derived from the Wolbachia-enriched gradient fraction GF-50/60 digested in-solution with trypsin for HPLC separation of peptides and protein identification by Orbitrap MS/MS [27]. For the iTRAQ analysis, aliquots of total, cytoplasmic or mitochondrial fractions (pooled GF-30/40 and GF-40/50) from C7–10 and C/wStr1 cells were labeled with isobaric tags in three 4-plex reactions for protein identification by Orbitrap MS/MS.
2.2. Protein preparation
In an initial analysis of samples previously used to enumerate Wolbachia proteins (labeled D, E, F, G in Fig. 1; standard MS/MS shaded rectangle), Aedes host cell proteins were recovered from SDS PAGE gel lanes cut into 22 gel slices, subjected to in gel digestion with trypsin and analyzed by LC-MS/MS on an LTQ mass spectrometer (D and E). Samples F and G contained proteins recovered from the Wolbachia-enriched sucrose gradient fraction GF-50/60 that were digested in solution with trypsin for separation of peptides by reversed-phase high pressure liquid chromatography (HPLC) and identification on an Orbitrap mass spectrometer [27]. In a parallel quantitative analysis using the iTRAQ® peptide isobaric labeling technology and the Orbitrap Velos system, single samples of Wolbachia-infected C/wStr1 and uninfected C7–10 cell total cellular proteins (H in shaded ellipse, Fig. 1) and two samples each of cytoplasmic and mitochondrial proteins (I and J) were used for protein identification and quantitation.
The samples used for iTRAQ were prepared and labeled with isobaric tags at the Center for Mass Spectrometry and Proteomics at the University of Minnesota as described [31] with minor modifications. In brief, proteins were extracted with 8 M urea containing 0.2% SDS, 0.4 M triethylammonium bicarbonate pH 8.5 (TEAB) and 20% methanol, alkylated for 15 min at room temperature in 8 mM methyl methanesulfonate and digested with trypsin. The samples were cleaned, vacuum-dried and resuspended in 0.5 M TEAB, pH 8.5 to a final concentration of 2 μg/μl for labeling with isobaric tags (iTRAQ 4-plex reagent; AB Sciex, CA) and multiplexing. Data set H (see Fig. 1) was derived from 60 μg of C/wStr1 total cellular peptides labeled with isobaric tags 116 or 117 multiplexed with 60 μg C7–10 peptides labeled with tags 114 or 115. Data set I was derived from 19 μg of C/wStr1 peptides labeled with tags 115 (cyto) and 117 (mito) multiplexed with 19 μg C7–10 peptides labeled with tags 114 (cyto) and 116 (mito). Data set J was derived from a second preparation of C/wStr1 peptides labeled with tags 114 (cyto) and 116 (mito) multiplexed with C7–10 peptides labeled with tags 115 (cyto) and 117 (mito).
2.3. Mass spectrometry and protein identification
Tandem mass spectra were extracted by Sequest (Thermo Fisher Scientific, San Jose, CA, USA; version SRF v.3 or version 27, rev. 12) and searched against an rs_wolbachia_aedes_v200808_cRAP_flavivirusREV database that contained 74,570 protein entries from the A. aegypti, Wolbachia and flavivirus genomes as described previously [27]. We used the A. aegypti genome for validation of host proteins because the genomic data from the A. albopictus laboratory colony strain Foshan [32] was not yet available.
Scaffold (version 4.2.1, Proteome Software Inc., Portland, OR) was used to validate peptide detection and protein identifications from data sets D - G (Fig. 1). Peptide identifications were accepted at ≥ 95.0% probability by the Peptide Prophet algorithm [33]. Protein identifications assigned by the Protein Prophet algorithm [34] were accepted at ≥ 99% probability based on detection of at least two unique peptides. Sequest parameters, protein sequence database descriptions and program settings were detailed previously [27]. False discovery rates (FDRs) are reported in supplemental Table S1.
We searched the Orbitrap peptide tandem MS data sets (H, I and J; Fig. 1) from the iTRAQ® experiment with Protein Pilot 4.5 (Sciex, Foster City, CA) [35]. Search parameters included: cysteine MMTS; iTRAQ 4plex (Peptide Labeled); trypsin; instrument Orbi MS (1–3 ppm) Orbi MS/MS; biological modifications ID focus; thorough search effort; autobias correction; detected protein threshold > 0.05 (10%) and FDR analysis with reversed database. We applied a 1% global FDR to the protein summary report [36] and identified 2016 unique proteins, of which 815 had differential abundance in C/wStr1 relative to C7–10 protein extracts, and are reported in all tables as elevated (ratio > 1) or suppressed (ratio < 1).
Proteins were sorted into functional classes based on features such as NCBI website annotations, gene ontology, Keggs pathways and presence of conserved domains. Proteins with multiple activities/roles were subjectively assigned to a single functional class. Hypothetical proteins were classified as Function unknown unless protein sequence similarities or conserved domains revealed with Protein Cluster and BLASTp tools (http://BLAST.ncbi.nim.nih.gov) suggested alternate designations.
2.4. Statistical analysis
All tests of association were performed with SAS v9.4 (Cary, NC; http://www.sas.com/enus/home.html/). Details are described in legends to figures, tables and supplementary data.
2.5. Transmission electron microscopy
C/wStr1 cells were grown to near confluence in 25 cm2 tissue culture flasks. The medium was carefully removed and cells were fixed overnight at 4°C in 2.5% glutaraldehyde in 0.1 M sodium cacodylate, pH 7.4. Cells were resuspended in fresh fixative, collected by centrifugation and post-fixed with 1% osmium tetroxide in 0.1 M sodium cacodylate buffer. The sample was dehydrated and embedded in Embed 812 resin, trimmed and sectioned; sections (60–70 nm) were contrasted with 5% uranyl acetate and Santos’ lead citrate. Sections were observed with a JEOL 1200 EX II transmission electron microscope (JEOL LTD, Tokyo, Japan).
3. Results
3.1. Overview
A. albopictus C/wStr1 cells infected with Wolbachia CI-inducing strain wStr from the planthopper, Laodelphax striatellus were grown in Eagle’s medium supplemented with heat-inactivated fetal bovine serum. The robust infection (Fig. 2) was characterized by abundant cytoplasmic bacteria of variable morphology, enveloped individually within host cell membranes of unknown origin [26] as described previously for Wolbachia isolated from mosquito ovaries [1, 37]. For MS/MS analysis, proteins were recovered after cell fractionation and SDS-PAGE, and identities were assigned by searching a database that included host and Wolbachia proteins based on annotated genomes. In an initial analysis, we identified 790 wStr proteins and estimated their relative abundances using a statistical model of the relationship between protein mass and MS/MS-detected peptide count [27]. Here we report the host cell proteome of 2,471 proteins (hereafter referred to as Aedes proteins) from the same sample set supplemented by an additional quantitative iTRAQ analysis that identified a subset of 815 proteins with differential abundances in C/wStr1 cells relative to uninfected C7–10 cells. In the text below, we interpret the results in the context of potential molecular interactions between wStr and its host cells.
Fig. 2.
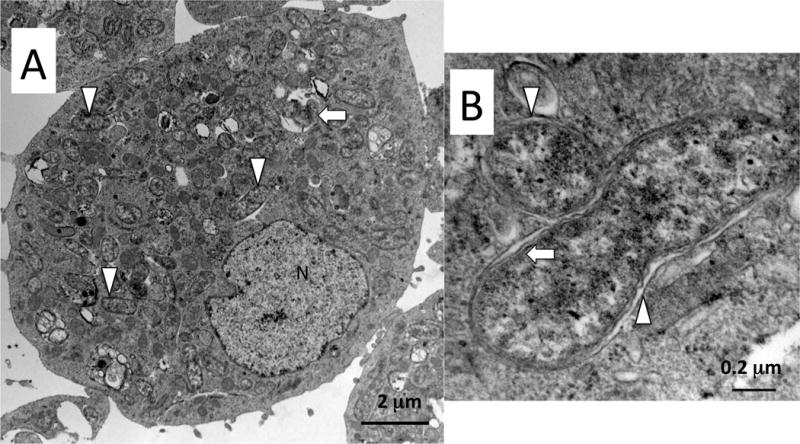
Representative transmission electron micrographs of C/wStr1 cells at near confluency. A) Host cell containing coccoid and elongate (arrowheads) wStr whose profiles range from ≈ 0.4–1.1 microns. Some inclusions appear to contain Wolbachia undergoing degradation (arrow). N, nucleus. B) Wolbachia in vertical and horizontal cross-section. Note the separation between the host-derived vacuolar membrane (arrowheads) and the bacterial cell wall, electron-lucent periplasmic space and inner periplasmic membrane (arrow).
3.2. Derivation of an Aedes host cell proteome from C/wStr1 and uninfected C7–10 cells
We derived an Aedes host cell proteome using two data sets (Table 1). From sample sets D, E, F, and G, designated as the standard analysis in Fig. 1, we defined a wStr proteome of 790 proteins (italicized entries at right in Table 1) previously described in detail [27]. Here we focus on 9,848 identifications of peptides matched to host proteins which, after subtracting overlapping accessions, corresponded to 1,859 unique Aedes proteins. An additional iTRAQ analysis (data sets H, I and J in Fig. 1) recovered 3,177 protein identifications that corresponded to 2016 unique host proteins (Table 1). For consensus totals, proteins with ≥ 97% BLASTp sequence identity were assigned a single accession number. The Standard and iTRAQ data sets showed considerable overlap, with an aggregate total of 2,471 unique Aedes proteins identified with a confidence level of ≥ 99% from 13,025 identifications. The consensus proteins are reported in an EXCEL spreadsheet (Table S1, Aedes proteome sheet 1) by name, accession, molecular mass, identified peptide numbers, percent protein sequence coverage, protein functional class and comments. The Aedes proteome includes 560 hypothetical proteins, of which we assigned 300 to a functional class based on conserved domains and BLASTp analyses that suggested a cellular function.
Table 1.
Cumulative Aedes albopictus and Wolbachia strain wStr protein identifications.
| Aedes albopictus proteins | Wolbachia wStr proteins | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Data seta | Total Cellb | Cyto.c Mitod | Wolb.e enrich | Total Aedesf | Tot. Cellb | Cytoc Mitod | Wolb.e enrich | Total wStrf |
| D (gel, C7–10) | 1373 | 2271 | – | 3644 | – | – | – | – |
| D (gel, C/wStr1) | 1199 | 1791 | 859 | 3849 | 391 | 746 | 521 | 1658 |
| E (gel, C/wStr1) | 820 | – | – | 820 | 404 | – | – | 404 |
| F, G (C/wStr1) | – | – | 1535 | 1535 | – | – | 851 | 851 |
| All data sets | 3392 | 4062 | 2394 | 9848 | 795 | 746 | 1372 | 2913 |
| Consensus | 1859 | 790 | ||||||
|
|
||||||||
| H (iTRAQ) | 1616 | 1616 | 110 | 110 | ||||
| I (iTRAQ) | – | 968 | – | 968 | 241 | 284 | – | 525 |
| J (iTRAQ) | – | 593 | – | 593 | – | – | – | – |
| All data sets | 1616 | 1561 | – | 3177 | 241 | 394 | – | 635 |
| Consensus | 2016 | 327 | ||||||
| Aggregate total | 2471 | 790 | ||||||
See Figure 1 for protein extracts included in each data set.
Total cellular proteins.
Cytoplasmic subcellular fraction proteins.
Mitochondrial subcellular fraction proteins.
Wolbachia-enriched subcellular fraction proteins.
All fractions combined. Total values for data sets D–G and H–J, respectively indicate the aggregate number of identifications from all cellular fractions. Consensus values indicate the total number of unique proteins identified from all data sets. Aggregate total indicates number of consensus proteins from combined Standard and iTRAQ data sets. Note that iTRAQ protein identifications were derived from equal mass mixtures of C/wStr1 and C7–10 protein extracts. Values in italic font were previously described in detail [27].
3.3. Validation of Wolbachia infection in iTRAQ samples
In Table 1, data sets H, I and J from C/wStr1 cells were expected to contain Wolbachia proteins entirely absent from C7–10 cells, and thus excluded from iTRAQ-based quantification. Analysis of the peptide spectra identified 327 wStr proteins (Table S2, sheet 1), compared to 790 proteins previously described from the standard analysis [27]. A univariable statistical model of the relationship between protein mass and MS-detected peptide count showed that log MW was a relatively weak (r-squared = 0.1394) but statistically significant (P < 0.0001) predictor of peptide count: log(peptides) = −0.23728 + 0.46866*log(mw). Based on that analysis, the majority of the 26 most abundant wStr proteins (Table S2, sheet 2) were from functional classes with the highest mean protein abundance levels (Suppl. Fig. S1).
3.4. Host proteins: Functional groups based on standard and iTRAQ data
Fig. 3 compares the functional class distribution of proteins from the standard (infected C/wStr1 cells, black bars; uninfected C710 cells, open bars) and iTRAQ (gray bars) samples normalized as fractional percentages of the largest class (protein modification/chaperones). In the standard analysis, differences in the number of proteins in C/wStr1 and C7–10 cells suggested that infected cells had a higher representation of proteins involved in energy, Transporters/Inorganic ion, lipid, secondary and amino acid metabolism (black diamonds in Fig. 3), compared to lower representation of proteins involved in motility/trafficking/secretion, Transcription/RNA modification, signal transduction, DNA replication/cell division, carbohydrate and nucleotide metabolism (white diamonds). With the exceptions of transcription and energy metabolism, distribution of proteins identified by iTRAQ approximated those of C/wStr1 values from the standard analysis (Fig. 3, compare black and gray columns).
Fig. 3.
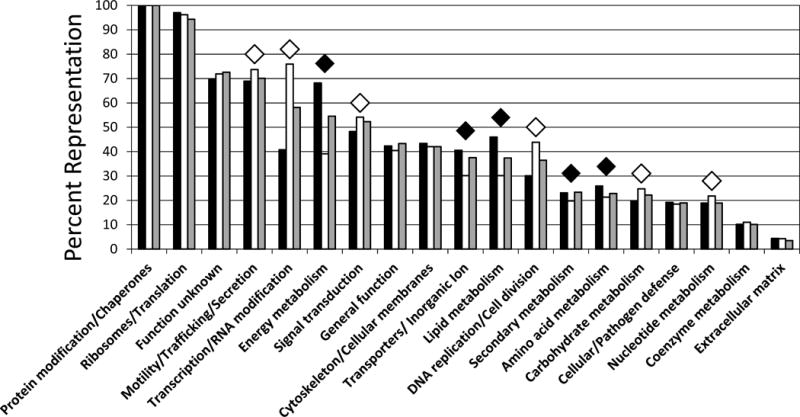
Distribution of identified Aedes host proteins by functional class. Individual protein identifications (8,313) in standard data sets D and E (see Table 1 and Fig. 1) and iTRAQ data sets H, I and J (3,177 identifications) were grouped into functional classes and normalized to numbers in the largest class, Protein modification/chaperones, defined as 100%. Functional classes were sorted left to right based on decreasing percentages in iTRAQ (gray bars). Diamond symbols indicate functional classes in which the ratio of C/wStr1 (black bars) relative to C7–10 (white bars) ranged from 0.5–0.9 (white diamonds) or 1.2–1.7 (black diamonds). Note that over 30% of proteins in the small Inorganic ion metabolism class (Table S1, sheet 1) have roles in ion transport and have been merged with the larger transporters class.
Relative protein abundance based on ratios from isobaric tags differed for 815 proteins representing 33% of the 2,471 unique Aedes proteins. In Table S3, sheet 1, we provide the iTRAQ raw data for those 815 proteins. Where data were available from multiple samples, ratios were consistent among 305 proteins with elevated abundance in infected C/wStr1 cells and 495 with suppressed abundance. Although individual protein levels varied widely within each functional class, only 15 proteins had conflicting ratios in one or more extracts (Table S3, sheet 2).
Each iTRAQ ratio was log-transformed to support statistical analysis of differences in protein abundance by functional class. A univariable linear regression analysis of the mean log(ratio) of protein abundance, using protein functional class as a variable and the function unknown class as the referent, showed that differences between functional classes were statistically significant (F=15.84, P < 0.0001) with beta coefficients ranging from 0.54716 to −0.10658 (Table 2) correlating positively with mean log protein abundance (range: 0.23 to −0.42). Based on visual inspection of mean iTRAQ ratios on a linear scale (Fig. 4), we arbitrarily identified a cluster of six functional classes with average ratios ranging from 1.2 to 1.4-fold higher in C/wStr1 than control cells (Fig. 4; diamonds on or above line c), which suggested that Wolbachia infection was associated with increased energy, lipid, carbohydrate, amino acid, secondary and nucleotide metabolism. Five functional classes had average ratios from 1.0 to 1.2 (Fig. 4; diamonds between lines b and c), and included co-enzyme metabolism, motility/trafficking/secretion, general function, protein modification/chaperones and transporters/inorganic ion. The signal transduction and cytoskeleton/cell membrane classes were relatively unchanged. Five classes had ratios < 1 (Fig. 4; diamonds between lines a and b): cellular/pathogen defense, function unknown, transcription/RNA modification, ribosomes/translation and DNA replication/cell division. Based on these ratios, the host response to C/wStr1 infection was characterized by higher abundance of proteins that support energy production and central intermediary metabolism, reduced abundance of proteins involved in DNA replication, transcription and translation, and relatively stable levels of proteins that participate in cellular maintenance.
Table 2.
Statistical summary of Aedes proteins with differential iTRAQ ratios in C/wStr1 relative to C7–10 cells.
| Protein functional class | # Proteins | % Total | Na | Meanb | Betac |
|---|---|---|---|---|---|
| Lipid metabolism | 39 | 4.79 | 61 | 0.23 (0.44) | 0.54716 |
| Energy metabolism | 61 | 7.48 | 116 | 0.22 (0.52) | 0.53115 |
| Amino acid metabolism | 26 | 3.19 | 45 | 0.14 (0.45) | 0.45108 |
| Carbohydrate metabolism | 25 | 3.07 | 49 | 0.11 (0.61) | 0.42181 |
| Nucleotide metabolism | 21 | 2.58 | 35 | 0.09 (0.50) | 0.40186 |
| Secondary metabolism | 25 | 3.07 | 41 | 0.01 (0.67) | 0.32779 |
| Coenzyme metabolism | 14 | 1.72 | 19 | 0.01 (0.50) | 0.31945 |
| Motility/Trafficking/Secretion | 59 | 7.24 | 96 | –0.02 (0.52) | 0.28941 |
| Protein modification/Chaperones | 87 | 10.67 | 139 | –0.03 (0.45) | 0.28605 |
| General function | 45 | 5.52 | 61 | –0.04 (0.52) | 0.27744 |
| Transporters/Inorganic ion metabolisme | 34 | 4.17 | 48 | –0.08 (0.47) | 0.23015 |
| Signal transduction | 39 | 4.79 | 52 | –0.12 (0.45) | 0.19667 |
| Cytoskeleton/Cellular membranesf | 51 | 6.26 | 91 | −0.14 (0.47) | 0.17712 |
| Cellular/Pathogen defense | 22 | 2.70 | 34 | −0.21 (0.59) | 0.10285 |
| Transcription/RNA modification | 52 | 6.38 | 76 | −0.34 (0.27) | −0.02330 |
| Ribosomes/Translation | 110 | 13.50 | 192 | −0.37 (0.23) | −0.05360 |
| DNA replication/Cell division | 44 | 5.40 | 69 | −0.42 (0.27) | −0.10658 |
| Function unknownd | 61 | 7.48 | 83 | −0.31 (0.56) | 0 (referent)d |
Number of differential iTRAQ ratios from MS data sets H, I and J; Total of 1307 ratios representing 815 proteins (Table S3, sheet 1).
Mean (standard deviation) of log transformed iTRAQ ratios in C/wStr1 relative to C7–10 extracts.
Beta coefficient of functional class in univariable linear regression model; mean of all classes excluding function unknown is 0.24314.
Function unknown was modeled as the referent class.
Transporter and Inorganic ion metabolism functional classes merged.
Cytoskeleton/Cellular membrane and Extracellular matrix functional classes merged.
Fig. 4.
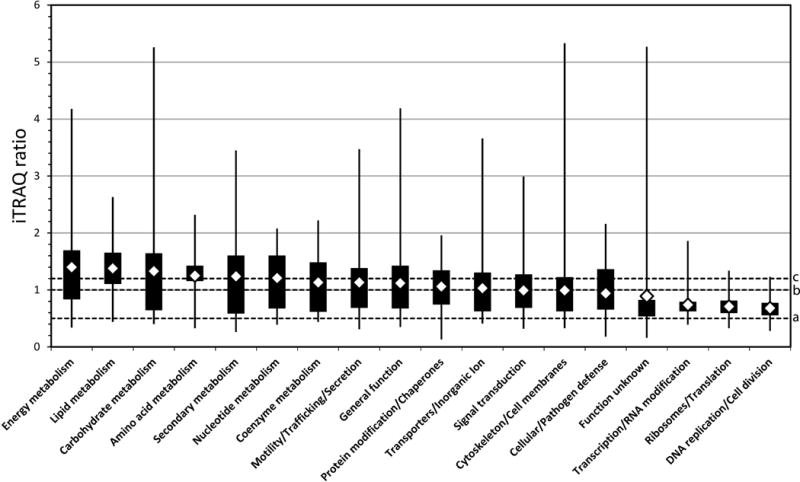
Distribution of Aedes host proteins with differential iTRAQ ratios by functional class. Average ratios by functional class were calculated from 1,307 observations (representing 815 consensus proteins listed in Table S4). Ratios are depicted in decreasing order on a linear scale, which was converted to log scale for statistical analyses described in the text. Diamonds indicate mean ratios (listed in Table 4), filled boxes represent first through third quartiles, and bars indicate minimum and maximum values. A small number of proteins with differential ratios in the inorganic ion and extracellular matrix class were included in the transporter and cytoskeleton/cell membrane classes, respectively.
3.5. Individual protein abundance levels and partitioning of metabolic resources
Table S4 summarizes Table S3, showing protein functional classes on individual sheets, and average iTRAQ ratios (calculated from Table S3, columns E, H, K, N and Q). Ratios > 0 indicate elevated abundance in infected C/wStr1 cells, while suppressed proteins have ratios < 0. In general, arithmetic averages giving equal weight to each of 815 proteins (Table 3) were in good agreement with the statistical analysis based on log-transformed values from 1307 observations (shown in parentheses in Table 3). Of the 815 proteins, 271 (33%) had ratios > 1.2, including individual proteins in all of the functional categories except DNA/cell division. Thirty-seven proteins (4.5%) had ratios ≥ 2, and these top-37 proteins were also distributed within several functional categories (Table 4). Below, we highlight some proteins, emphasizing those with increased abundance in infected cells, and focus the Discussion section on the top-37 proteins in the context of potential interactions and avenues for pharmacologic manipulation.
Table 3.
Summary of iTRAQ protein ratios from Table S4.
| Protein functional classa | Sheetb | Total (%)c | Ave.d | Rangee | > 1.2 (%)f | ≥ 2 (%)g |
|---|---|---|---|---|---|---|
| Energy metabolism | 1 | 61 (7.5%) | 1.24 (1.40) | 0.35–2.65 | 35 (57%) | 5 (8%) |
| Lipid metab. | 2 | 39 (4.8%) | 1.42 (1.38) | 0.48–2.63 | 29 (74%) | 4 (10%) |
| Carbohydrate metab. | 3 | 25 (3.1%) | 1.24 (1.33) | 0.42–3.06 | 16 (64%) | 2 (8%) |
| Amino acid metab. | 4 | 26 (3.2%) | 1.20 (1.25) | 0.33–1.79 | 17 (65%) | 0 |
| Secondary metab. | 5 | 25 (3.1%) | 1.24 (1.24) | 0.36–2.91 | 13 (52%) | 4 (16%) |
| Nucleotide metab. | 6 | 21 (2.6%) | 1.22 (1.21) | 0.46–2.07 | 13 (62%) | 2 (10%) |
| Coenzyme metab. | 7 | 14 (1.7%) | 1.11 (1.13) | 0.44–2.09 | 7 (50%) | 1 (7%) |
| Motility/Trafficking/Secretion | 8 | 59 (7.2%) | 1.10 (1.13) | 0.47–3.23 | 21 (36%) | 6 (10%) |
| General function | 9 | 45 (5.5%) | 1.10 (1.12) | 0.52–4.19 | 16 (36) | 3 (7%) |
| Protein modifn/Chaperones | 10 | 87 (10.7%) | 1.06 (1.06) | 0.27–1.85 | 38 (44%) | 0 |
| Transporters/Ions | 11 | 34 (4.2%) | 1.11 (1.03) | 0.42–3.66 | 14 (41%) | 1 (3%) |
| Signal transduction | 12 | 39 (4.8%) | 1.02 (0.99) | 0.35–2.99 | 14 (36%) | 2 (5%) |
| Cytoskeleton/Cell. membranes | 13 | 51 (6.2%) | 1.00 (0.99) | 0.40–5.33 | 13 (25%) | 2 (4%) |
| Cellular/Pathogen defense | 14 | 22 (2.7%) | 1.01 (0.94) | 0.18–2.10 | 9 (41%) | 1 (4%) |
| Function unknown | 15 | 61 (7.5%) | 0.88 (0.89) | 0.29–5.01 | 12 (20%) | 4 (7%) |
| Transcription/RNA modifn. | 16 | 52 (6.4%) | 0.73 (0.74) | 0.49–1.79 | 1 (2%) | 0 |
| Ribosomes/Translation | 17 | 110 (13.5%) | 0.72 (0.71) | 0.33–1.34 | 3 (3%) | 0 |
| DNA replication/Cell division | 18 | 44 (5.4%) | 0.69 (0.68) | 0.45–1.18 | 0 | 0 |
| Aggregate total | 815 (100%) | 271 (33%) | 37 (4.5%) |
Protein functional class from Table S4.
Sheet numbers in Table S4.
Total number of proteins, with percentages.
Arithmetic average of protein ratios (C/wStr1 over C7–10) with equal weight given to each of 815 proteins. (see Table S4). Weighted averages based on 1307 observations (see Fig. 4) are given in parentheses.
Range of protein ratios.
Number of proteins with ratios > 1.2, with percentages.
Number of proteins with ratios > 2.0, with percentages.
Table 4.
Top-37 Aedes iTRAQ proteins.
| Identified proteins | Accessiona | Ratiob | Functional classc |
|---|---|---|---|
| neurotactin | gi|157121167 | 5.33 | Cytoskeleton/Cellular membranes |
| hypothetical protein AAEL001471 | gi|157119483 | 5.01 | Function unknown |
| apoptosis protein | gi|157124948 | 4.19 | General function |
| hypothetical protein AAEL010818 | gi|157127471 | 3.66 | Transporters/Inorganic Ion metabolism |
| V-ATPase V0 116 kDa subunit i | gi|157138700 | 3.23 | Motility/Trafficking/Secretion |
| glycogen phosphorylase | gi|157108521 | 3.06 | Carbohydrate metabolism |
| hypothetical protein AAEL010678 | gi|157126928 | 2.99 | Signal transduction |
| cytochrome P450 | gi|157105117 | 2.91 | Secondary metabolism |
| V-ATPase V1 subunit C | gi|157109023 | 2.81 | Motility/Trafficking/Secretion |
| V-ATPase V1 subunit H | gi|157113604 | 2.75 | Motility/Trafficking/Secretion |
| V-ATPase V1 subunit D | gi|157124332 | 2.73 | Motility/Trafficking/Secretion |
| malate dehydrogenase | gi|157116681 | 2.65 | Energy metabolism |
| 3-hydroxyl-coa dehydrogenase | gi|157132312 | 2.63 | Lipid metabolism |
| hypothetical protein AAEL013851 | gi|157137775 | 2.61 | General function |
| 3-hydroxyl-coa dehydrogenase | gi|157117489 | 2.52 | Lipid metabolism |
| CRAL/TRIO domain-containing protein | gi|157135818 | 2.50 | General unknown |
| cytochrome p450 | gi|157167202 | 2.50 | Secondary metabolism |
| phophoenolpyruvate carboxykinase | gi|157103343 | 2.48 | Energy metabolism |
| cytochrome c oxidase subunit iv | gi|157108935 | 2.43 | Energy metabolism |
| lethal(2)essential for life protein, l2efl | gi|157135547 | 2.39 | Function unknown |
| citrate synthase | gi|157133341 | 2.24 | Energy metabolism |
| hypothetical protein AAEL002350 | gi|157128286 | 2.22 | Function unknown |
| V-ATPase subunit E | gi|157131212 | 2.20 | Motility/Trafficking/Secretion |
| hypothetical protein AAEL002861 | gi|157132592 | 2.17 | Function unknown |
| choline-phosphate cytidylyltransferase a, b | gi|157129766 | 2.12 | Lipid metabolism |
| phosphorylase kinase, gamma, partial | gi|157137241 | 2.12 | Signal transduction |
| glutathione peroxidase | gi|157118772 | 2.10 | Cellular/Pathogen defense |
| beta-alanine synthase, putative | gi|157112908 | 2.09 | Coenzyme metabolism |
| zeta coat protein | gi|157134570 | 2.08 | Motility/Trafficking/Secretion |
| adenosine diphosphatase | gi|157110697 | 2.07 | Nucleotide metabolism |
| annexin B9 | gi|157129014 | 2.05 | Cytoskeleton/Cellular membranes |
| aldehyde dehydrogenase, putative | gi|157131682 | 2.05 | Secondary metabolism |
| phosphofructokinase | gi|157114499 | 2.04 | Energy metabolism |
| short chain type dehydrogenase | gi|157107865 | 2.04 | Lipid metabolism |
| adenylate kinase 3, putative | gi|157106105 | 2.02 | Nucleotide metabolism |
| estradiol 17 beta-dehydrogenase | gi|157114880 | 2.02 | Secondary metabolism |
| phophoglucomutase | gi|157124898 | 2.00 | Carbohydrate metabolism |
Host proteins with iTRAQ ratios ≥ 2.00 from Table S4. V-ATPase subunits are shaded dark gray, and hypothetical proteins are shaded light gray.
NCBI accession number.
iTRAQ ratio (C/wStr1 over C7–10).
Protein functional class.
3.5.1. Energy metabolism
Over 7.5% of the differentially abundant proteins have roles in energy production (Table S4, sheet 1), of which 57%, including the glycolytic and most of the TCA cycle enzymes, were expressed at ratios exceeding the 1.2 threshold (Fig. 4). Ratios for key regulatory enzymes, phosphofructokinase (2.04) and pyruvate kinase (1.45) were elevated, as was lactate dehydrogenase (1.99), which interconverts lactate and pyruvate, with concomitant interconversion of NAD+ and NADH. Pyruvate dehydrogenase (1.67) converts pyruvate to acetyl-CoA, which enters the TCA cycle for further metabolism by citrate synthase (2.24) and malate dehydrogenase (1.31). Cytoplasmic malate dehydrogenase (2.65) is critical to the malate-aspartate shuttle, which regenerates NADH in the mitochondrial matrix, maximizing production of ATP from glucose.
3.5.2. Lipid metabolism
Of 39 proteins involved in lipid metabolism (Table S4, sheet 2), 74% had ratios > 1.2, and 10% were > 2.0, including 17 proteins (1.17 – 2.63) that participate in fatty acid metabolism in the cytoplasm, mitochondria or peroxisomes. These included two cytoplasmic acyl-CoA oxidases (1.43 and 1.56) as well as two mitochondrial acyl-CoA dehydrogenases (1.54 and 1.36) that initiate β-oxidation of fatty acids to provide reducing equivalents to the mitochondrial electron transport chain and acetyl-CoA to the TCA cycle. An elevated cytoplasmic glycerol-3-phosphate dehydrogenase (1.65) is a key enzymatic link between glycolysis and lipid metabolism which, in conjunction with the mitochondrial glycerol-3-phosphate dehydrogenase (1.55), functions in the glycerol phosphate shuttle, involved in regeneration of the cytosolic NAD+ pool depleted by glycolysis. Sulfide quinone reductase (1.60) links sulfur amino acid and lipid metabolism/storage, while a sterol carrier protein (1.56) has a thiolase domain that influences intracellular lipid circulation. Six additional elevated proteins (1.11–1.85) have roles in transport of lipids and steroids that likely intersect with secondary metabolism of steroids.
3.5.3. Carbohydrate metabolism
Of 25 carbohydrate metabolism proteins (Table S4, sheet 3), several enzymes that participate in glycogen synthesis and degradation had increased ratios, suggesting that turnover of glycogen is an important feature of the Wolbachia infection. Key enzymes included the biosynthetic enzymes glycogen synthetase (1.74) and starch branching enzyme (1.69), and degradative enzymes glycogen phosphorylase (3.06) and phosphorylase b kinase (1.37). The ratio for glycogen phosphorylase is among the highest observed in this study, suggesting that generation of glucose, potentially as a precursor for increased synthesis of amino acids, supports Wolbachia maintenance and replication. Seven enzymes that function in the anabolic gluconeogenesis and pentose phosphate pathways that parallel glycolysis were elevated, including phosphoglucomutase (2.0) and 6-phosphogluconate dehydrogenase (1.86).
3.5.4. Amino acid metabolism
The Aedes proteome contained 51 proteins with primary roles in amino acid metabolism (Table S1, entries 2–52) of which 26 showed differential ratios in C/wStr1 cells (Table S4, sheet 4). Activities of glutamine (1.72) and asparagine (1.32) synthetases, glutamate dehydrogenase (1.66) and aspartate aminotransferases (1.55 and 1.72) link amino acid metabolism to the TCA cycle through α- ketoglutarate and oxaloacetate intermediates. Although glutamate cysteine ligase (1.23) was above 1.2, glutamate synthase (0.68) and cysteine desulfurylase (0.79), which have important roles in glutathione and sulfur metabolism, were among seven suppressed proteins (0.33–0.79). Upregulation of enzymes that generate glutamine and asparagine may compensate for depletion of host amino acid pools by Wolbachia, which lacks some amino acid biosynthetic pathways [38]. Glutamine is the most abundant amino acid in mammalian cells and is implicated in a number of signaling pathways, while the promoter for asparagine synthetase contains one of the better-known amino acid response elements [39, 40]. Although the mechanisms by which amino acid abundance regulates intracellular metabolism are poorly understood in mammalian cells, and have not been explored in insect cell lines, the likelihood that Wolbachia infection is associated with an overall deficiency in host amino acid pools is consistent with the general suppression of proteins that participate in transcription, translation and DNA replication.
3.5.5. Secondary metabolism
A strongly elevated farnesyl-pyrophosphate synthetase (1.95, Table S4, sheet 5) catalyzes an initial step in biosynthesis of squalene, dolichol, terpenes, steroids and juvenile hormone (JH), a terpene-based hormone unique to insects. An epoxide hydrolase (1.78) participates in regulating titers of JH by degrading it to an inactive diol. The Aedes homolog of estradiol 17-beta-dehydrogenase (2.02; 20-hydroxyecdysone dehydrogenase) converts ecdysone to a more active form, which in concert with JH regulates metabolism, growth and development in insects. Elevated cytochrome P450 enzymes (1.17 and 2.5) have insect homologs that are involved in steroid, terpenoid and xenobiotic metabolism. An elevated P450 homolog of Cyp304a1 (2.91) is involved in the stress response to DNA damage in Drosophila. Adrenodoxin, which functions as the mitochondrial P450 reductase, was suppressed (0.38). Enzymes with likely secondary functions in vitamin metabolism and lipid peroxidation included an aldehyde dehydrogenase (2.05), while an elevated leukotriene A-4 hydrolase (1.41) participates in metabolism of phopholipids, arachidonic acid and eicosanoids in signaling pathways that affect kinases and other regulatory enzymes.
3.5.6. Nucleotide metabolism
Among 21 proteins, 13 had ratios above 1.2 (Table S4, sheet 6), including adenosine diphosphatase (2.07) and several nucleotide kinases that maintain nucleotide pools and influence activity of numerous enzymes as substrates or through regulatory interactions such as allosteric regulation of phosphofructokinase by ATP and AMP. Both subunits of ribonucleoside-diphosphate reductase required for synthesis of dNTP precursors were elevated (1.13 and 1.58). Adenylsulfate kinase (1.5) functions in the assimilation of sulfate and its incorporation into cysteine, methionine, glutathione, sulfolipids and iron-sulfur clusters, which are critical components of many proteins in energy production, electron transport, oxidative phosphorylation and other functions. Although most proteins with roles in purine and pyrimidine biosynthesis were suppressed, inosine-5-monophosphate dehydrogenase (1.26), which catalyzes the first step in guanine nucleotide synthesis and has regulatory effects in energy transfer, signal transduction and DNA/RNA synthesis, was elevated. The six suppressed enzymes (0.46 – 0.72) included adenylosuccinate synthetase, which catalyzes the first step in adenine nucleotide synthesis.
3.5.7. Co-enzyme metabolism
Nicotinate phophoribosyltransferase (1.34), which catalyzes the rate-limiting step in NAD salvage synthesis was elevated (Table S4, sheet 7), while hypothetical host protein AAEL002178 (0.83) and methylenetetrahydrofolate dehydrogenase (0.73), with roles in folate and glyoxylate metabolism, were among seven suppressed proteins (0.44–0.85).
3.5.7. Motility secretion and intracellular trafficking
Nine of 13 V-type H+ ATPase subunits were elevated in the Aedes proteome (1.47–3.23; Table S4, sheet 8), including ATPase A (1.84) and the regulatory C (2.81) subunits of its V1 catalytic component. The V1 component hydrolyzes ATP, driving proton-pumping activity of the membrane-bound V0 assembly. Two isoforms of the V0 regulatory subunit were elevated (3.23 and 1.47) and potentially contribute to transport of nutrients into vacuoles occupied by Wolbachia. An elevated membrane traffic protein (1.86) contains an F-Bar dimerization module that influences membrane conformation. Among 23 proteins involved in ER/Golgi vesicle trafficking, only four were elevated (1.26–1.39), and 19 were suppressed proteins (0.31–0.78), including ERp44, a stress-induced ER protein that prevents secretion of proteins with unpaired cysteines and also modulates inositol triphosphate-dependent release of Ca2+. Among 17 proteins involved in endosome, lysosome and autophagy processes, four were elevated, (1.2–2.08), including saposin (1.52), which facilitates lysosomal degradation of glycosphingolipds. The suppressed proteins included Atg3 (0.71) and Atg5 (0.72), which have roles in the maturation of autophagosomes in canonical autophagy. In contrast, non-canonical Atg-5 independent autophagy is critical in the life cycle of some intracellular microbes, such as Francisella tularensis replication, infection by Brucella abortus, and entry into vacuoles by Mycobacterium marinum [28]. Other autophagy-related proteins, including clathrin heavy chain (0.89) and five coated pit and vesicle formation proteins, two dynein (0.93, 0.87) and three myosin motor proteins (0.76, 0.69, 0.60) were suppressed, in contrast to the elevated kinesin light (1.36) and heavy chain (1.24) subunits that carry cargo in anterograde transport to microtubule positive ends.
3.5.8. General function
Elevated general function proteins (Table S4, sheet 9) included a protein with a potential role in apoptosis (4.19), six hypothetical host proteins (1.28–2.61) with likely functions in metabolite processing and proteolysis, and bleomycin hydrolase (1.58), a cytoplasmic cysteine peptidase that forms a hexameric barrel ring reminiscent of the 26S proteasome cylindrical complex. A CRAL/TRIO domain protein with elevated levels (2.5) has general functions in lipophilic substrate transfer and in Ras/Rho GTPase family signaling pathways that regulate cell growth, cytoskeletal/membrane organization, motility and intracellular trafficking.
3.5.9. Protein modification, degradation and chaperones
Cytosolic chaperones, proteases and components of the 26S proteasome were generally elevated in C/wStr1 cells, consistent with a stress response accompanied by increased protein degradation (Table S4, sheet 10). Fourteen chaperones were elevated (1.14–1.68), including Hsp 10, 20, 60 and 90. Six proteases (1.15–1.85), including the major lysosomal protease, cathepsin D (1.58) were elevated, and twenty proteins (1.08–1.58) related to ubiquitin conjugation, activation, regulation and structure of the 26S proteasome complex were increased. Upregulation of proteasome subunits in newly-infected cells [41] suggests that Wolbachia may obtain amino acids from host proteasomal activity, as has been documented for Legionella pneumophila using a mutant that lacks the AnkB F-box effector protein [42]. Ratios for seven proteins that function in post-translational protein modification (1.21–1.72), including a UDP-glucosyltransferase (1.43) were increased, while suppressed proteins (0.46–0.88) primarily function in glycosylation, glycan formation and side chain modifications in the ER and Golgi. Calnexin (1.50) and calreticulin (1.31), which prevent maturation of mis-folded proteins in the ER, were elevated, but eight additional proteins with roles in protein folding and repair were suppressed (0.36–0.73).
3.5.10. Transporters and Inorganic ions
Differentially expressed proteins (Table S4, sheet 11) featured five hypothetical host proteins, of which two, AAEL010118 (3.66) and AAEL010816 (1.69), had strong BLASTp similarities to mitochondrial calcium uptake and phosphate carrier proteins. Transport proteins with increased ratios included a calcium-transporting ATPase (1.19), an inorganic pyrophosphatase (1.23), a mitochondrial phosphate carrier protein (1.37), and a mitochondrial solute carrier with ADP/ATP translocase activity (1.29). Suppressed proteins (0.42–0.75) included plasma membrane calcium (0.66) and sodium/potassium ATPases (0.67), a potassium/chloride symporter (0.60) from a family that transports amino acids and sugars, and an equilibrative nucleoside transporter (0.50), which transports nucleoside substrates into cells.
3.5.11. Signal transduction
Seven GTPase/GTP binding proteins were elevated (1.16–1.40, Table S4, sheet 12) and nine were suppressed (0.58–0.87), including hypothetical protein AAEL005197 (0.61) with domain and BLASTp identities to a RagA GTPase that activates mTORC1 associated with the vacuolar ATPase complex on the lysosomal surface in response to amino acid signaling. The mTORC1 complex also responds to membrane receptor and phoshatidylinositol kinase signaling mediated by proteins such as an elevated inositol 1,4,5-triphosphate receptor (1.24), and hypothetical protein AAEL010678 (2.99), with a putative phopholipase C domain specific for phosphoinositides. Calcyphosine/tpp (1.34) participates in both phosphatidylinositol and cAMP-mediated signaling. One of two elevated cAMP-dependent kinase regulatory subunits (1.83) is a critical component of pathways that regulate glycogen, sugar and lipid metabolism that intersect with mTOR signaling. Protein phosphatase-2b, which functions as the catalytic subunit of calcineurin, a Ca2+-calmodulin-dependent serine/threonine phosphatase that participates in signaling pathways, was elevated (1.38) and, in contrast to suppressed levels of phosphatase-1, (0.71) −2a (0.71) and −2c (0.82) with roles in regulation of glycogen metabolism, transcription and translation.
3.5.12. Cytoskeleton and cellular membranes
A strongly elevated annexin B9 (2.05; Table S4, sheet 13) is required for multivesicular body function and endosomal trafficking in Drosophila, and two annexin B10 proteins with actin-binding sites were also elevated (1.42 and 1.77). Annexins mediate Ca2+-dependent membrane and protein interactions in endocytosis, signaling, motility and vesicle transport and lipid raft formation. Two flotillins (1.40, 1.54) and a hypothetical stomatin-like protein (AAEL004490; 1.42) with roles in lipid raft and caveolae formation were elevated. The Aedes proteome contains six actin isoforms (Table S1, entries 201–206), but none had ratios > 1.0. Coracle protein, which anchors other proteins to actin, was suppressed (0.62), as were 12 other proteins involved in actin filament formation, stabilization and membrane interactions (0.49 to 0.79; see the red entries in Table S4, sheet 13), while eight were elevated (range 1.0–1.45). Tubulin and putative microtubule-associated proteins were all suppressed, as were a cation-transporting ATPase (0.76) and a phospholipid scramblase (0.74), which influence membrane dynamics by mediating phospholipid transfer between leaflets.
3.5.13. Cellular and pathogen defense
Superoxide dismutases (1.84 and 1.88), glutathione peroxidases (2.10 and 1.51) and thioredoxin reductases (1.02 and 1.24), involved in neutralization of reactive oxygen species, were elevated, while catalase was suppressed (0.74.) Levels of peroxiredoxins (1.21, 0.74, 0.68, 0.46) involved in thiol-dependent ROS defense were mixed (Table S4, sheet 14).
3.5.14. Function unknown
Although only 12 of 61 proteins with unknown or putative functions had elevated levels in C/wStr1 protein extracts (Tables S4, sheet 15), they included the protein with the highest expression level, AAEL001471 (5.01), with a putative function in protein modification/degradation. Hypothetical protein AAEL004129 (0.56) has an ArfGap domain and sequence similarity to putative GTPase activating proteins for Arf, a regulator of mTORC1.
3.5.15. Transcription, translation and cell division
Proteins with the lowest iTRAQ ratios, representing processes that are suppressed in infected cells, included those involved in transcription (52 proteins; Table S4, sheet 16), translation (110 proteins; sheet 17) and DNA replication/cell division (44 proteins; sheet 18). Among proteins with roles in transcription and RNA modification, three were elevated, including a single DEAD box ATP-dependent RNA helicase (1.79), which is a homolog of yeast dbp2, an enzyme that associates with chromatin, regulates fidelity of transcription initiation and influences ribonucleoprotein assembly by altering RNA structure. Seven additional DEAD box RNA helicases were among 49 suppressed proteins. The ribosomes/translation class included 110 proteins, of which six had elevated levels including serine (1.22), alanine (1.22) and glycine (1.18) tRNA synthetases. All cytosolic and mitochondrial ribosomal proteins were suppressed (0.45–0.86), as were ribosomal maturation and translation factors, and most tRNA synthetases and tRNA modification enzymes. Among 44 proteins with diverse roles in DNA replication, repair, packaging and cell division, all but a DNA topoisomerase (1.18) were suppressed. In aggregate, of 206 differentially expressed proteins with roles in DNA replication, cell division, transcription, RNA processing or translation, only 10 had increased levels in infected C/wStr1 cells.
3.6. Aedes hypothetical proteins
Of 560 hypothetical proteins in the Aedes proteome, 300 were assigned to a putative functional class. Of these, 75 are distributed throughout 17 of the 18 sheets showing iTRAQ ratios (Table S4, shaded as yellow entries). In addition, sheet 15 (function unknown) includes 52 hypothetical proteins among 61 entries. Thus, 136 (17%) of the 815 iTRAQ proteins are poorly known, of which 93% are hypothetical proteins. Table 5 highlights Aedes hypothetical proteins with C/wStr1 relative expression levels > 1.5 (shaded in gray; 9.4%) or < 0.5 (unshaded, 10.2%). Hypothetical protein AAEL001471 was strongly elevated (5.01) and contained zinc finger and UBA_SQSTM UBL domains found in ubiquitin-binding protein p62, a scaffolding protein that regulates endosomal signaling pathways and plays an important role in ubiquitin-mediated phagosomal clearance of intracellular Listeria and Salmonella. A potential role for this protein is consistent with known deficiencies in Wolbachia’s amino acid biosynthetic capacities and dependence on host amino acids. Likewise, a zinc-dependent peptidase (1.56) has potential function in provisioning Wolbachia with amino acids. Two phospholipases (2.99 and 1.86) also had high ratios, suggesting that degradation of host lipids also contributes to Wolbachia’s maintenance. Among the suppressed hypothetical proteins, three with potential roles in chromosome maintenance (0.48) and cell cycle regulation (0.40 and 0.35) were noteworthy in the context of an overall suppression of proteins that participate in DNA replication and cell division.
Table 5.
Aedes hypothetical proteins with high and low iTRAQ ratios.
| Hypothetical proteinsa | Accessionb | kDac | Ratio (avg)d | Functional classe | Comments/pathways |
|---|---|---|---|---|---|
| AAEL001471 | gi|157119483 | 82 | 5.01 | Function unknown | Zinc finger, ubiquitin-binding, signaling |
| AAEL010818 | gi|157127471 | 22 | 3.66 | Transporters/Inorganic Ion | homology to mitochondrial phosphate carrier protein |
| AAEL010678 | gi|157126928 | 44 | 2.99 | Signal transduction | phospholipase C-like phosphodiesterases domain |
| AAEL013851 | gi|157137775 | 27 | 2.61 | General function | modifies proteins associated with ribosomes, translation |
| AAEL002350 | gi|157128286 | 18 | 2.22 | Function unknown | DU1075 domain of unknown function |
| AAEL002861 | gi|157132592 | 48 | 2.17 | Function unknown | saccharopine dehydrogenase; lysine metabolism in fungi |
| AAEL003651 | gi|157137711 | 57 | 1.86 | Lipid metabolism | phopholipases associated with Golgi membranes |
| AAEL010116 | gi|157125198 | 60 | 1.69 | Transporters/Inorganic Ion | mitochondrial Calcium uptake protein; calcium sensor |
| AAEL008454 | gi|157118896 | 23 | 1.61 | Secondary metabolism | catalyses isochorismate to 2,3-dihydroxybenzoate and pyruvate |
| AAEL012863 | gi|157133791 | 27 | 1.61 | General function | probable member RIM1 Acetyltransferases COG0456 |
| AAEL008862 | gi|157119998 | 113 | 1.56 | General function | probable zinc-dependent peptidase |
| AAEL006343 | gi|157113179 | 69 | 1.55 | Function unknown | VCBS repeat domain; immunomodulatory protein |
| AAEL007236 | gi|157115555 | 32 | 0.5 | Secondary metabolism | Farnesoic acid O-methyl transferase. |
| AAEL014011 | gi|157138509 | 93 | 0.49 | Function unknown | ankyrin domain |
| AAEL002691 | gi|157131093 | 143 | 0.48 | Function unknown | SMC domain for chromosome maintenance |
| AAEL006364 | gi|157113255 | 43 | 0.48 | Carbohydrate metabolism | Amino sugar and nucleotide sugar metabolism |
| AAEL014194 | gi|157103922 | 27 | 0.46 | Function unknown | similarity to Microtubule-associated |
| AAEL004180 | gi|157104866 | 51 | 0.44 | Coenzyme metabolism | probable ubiquinone monooxygenase |
| AAEL004958 | gi|157108115 | 62 | 0.43 | Secondary metabolism | Farnesoic acid O-methyl transferase domain |
| AAEL002115 | gi|157126696 | 56 | 0.42 | Function unknown | Zinc finger domain |
| AAEL005141 | gi|157108846 | 26 | 0.4 | Function unknown | signal peptide membrane domain |
| AAEL011155 | gi|157128487 | 44 | 0.4 | Function unknown | has DUF155 domain found in some yeast cyclins |
| AAEL014816 | gi|157107145 | 54 | 0.39 | Function unknown | 70% similar to acidic membrane protein |
| AAEL008569 | gi|157119197 | 25 | 0.35 | Signal transduction | signal transduction protein; cyclin-dependent kinase cdk5 |
| AAEL008658 | gi|157119386 | 82 | 0.29 | Function unknown | similar to UPF0183 proteins of unknown function |
NCBI protein definition; proteins from Table S4 with ratios > 1.5 shaded gray.
NCBI protein accession number.
Calculated molecular mass in kilodaltons.
iTRAQ protein ratio (C/wStr1 over C7–10).
Protein functional class.
3.7. Flavivirus proteins
We detected unique peptides corresponding to flavivirus capsid, membrane and envelope structural proteins, as well as seven non-structural proteins from flaviviruses associated with mosquitoes and ticks. The majority of those peptides were present in total cellular and cytoplasmic extracts from both C/wStr1-infected and control cells subjected to gel fractionation to maximize protein detection (data sets D and E in Fig. 1), while the Wolbachia-enriched data sets F and G contained the fewest viral peptides. Table 6 summarizes the 95% confidence peptides matched to proteins from 10 better-known flaviviruses associated with hemorrhagic or encephalitic disease in humans, as well as flaviviruses unique to mosquitoes. Unique peptides ranged from three (1.6% coverage) for the insect-specific CFA described from an Aedes aegypti mosquito cell line [43], to 34 peptides that matched all WNV proteins except NS4A and NS4B, or 15.4% sequence coverage of the approximately 3400 amino acids within the viral polyprotein (Fig. S2).
Table 6.
MS/MS peptides in C7–10 and C/wStr1 extracts matched to selected Flavivirus polyprotein sequences.
| Flavivirus | Accessiona | Peptidesb | Coveragec | Vectorsd | Hostse |
|---|---|---|---|---|---|
| DENV4; Dengue | GI: 119390884 | 16 | 7.2 |
Ae. aegypti Ae. albopictus |
primates |
| YFV; Yellow fever | GI: 383464646 | 15 | 6.7 |
Ae. aegypti Hemagogus spp. |
primates |
| ZIK; Zika | GI: 146411781 | 11 | 5.3 |
Ae. aegypti Ae. albopictus |
primates |
| JEV; Japanese encephalitis | GI: 12964701 | 27 | 13.5 |
Aedes spp. Culex spp. Anopheles spp. |
mammals birds |
| WNV; West Nile virus | GI: 90025136 | 34 | 15.4 |
Aedes spp. Culex spp. |
mammals birds |
| POW; Powassan virus | GI: 730353 | 7 | 3.6 |
Aedes spp. Culex spp. |
mammals birds |
| TBEV; Tick-borne encephalitis | GI: 1066075 | 16 | 6.9 |
Ixodes ticks Dermacentor ticks |
mammals |
| AeFV; Aedes Flavivirus | GI: 251823440 | 11 | 4.9 |
Ae. aegypti Ae. flavopictus Culex pipiens |
mosquitoes |
| CxFV; Culex flavivirus | GI: 247893527 | 6 | 3.0 |
Culex pipiens Culex tarsalis Culex nigripalpus Culex spp. |
mosquitoes |
| CFA; Cell fusion agent | GI: 464429 | 3 | 1.6 |
Ae. aegypti Ae. albopictus Culex spp. |
mosquitoes |
Genbank accession numbers for polyprotein sequences, which undergo post-translational cleavage to produce three structural and seven nonstructural proteins (see Fig. S2).
Total number of 95% confidence unique peptides.
Percent polyprotein sequence coverage.
Includes major arthropod vectors.
Principal vertebrate hosts.
4. Discussion
An important goal of our efforts to explore interactions between Wolbachia and insect cell lines that support its replication is development of protocols for mass production of this obligate intracellular bacterium, potentially manipulated by genetic technologies, for introduction into target insects. Although Wolbachia typically replicates in the reproductive tissues of its insect hosts [1, 6], successful transfer of Wolbachia strains between insect species [11] and into cultured cells [24, 25, 44, 45] indicates that Wolbachia do not require cell lines derived specifically from their host species or from reproductive tissues. For most insect cell lines, including C/wStr1, the tissue of origin is not known because tissue-specific markers are not available (reviewed in [46]). Because Wolbachia infects a wide range of insects and nematode worms, genetic manipulation will be facilitated by development of a “generic” host cell line that supports recovery and expansion of a wide range of Wolbachia strains. C/wStr1 mosquito cells maintain a persistent, high-density infection with Wolbachia strain wStr from the planthopper Laodelphax striatellus [24]. In these cells, individual bacteria are enveloped by a single host-derived membrane, as was described in pioneering observations with mosquito ovaries [1, 37, 47].
As an obligate intracellular bacterium, Wolbachia has a streamlined genome and reduced metabolic capabilities [38], making its growth and replication dependent on host cell precursors or products derived from breakdown of complex host cell macromolecules such as glycogen, lipids, and proteins. Evidence for competition between Wolbachia and host cells in culture includes a twofold increase in cell density when infected cells are plated in medium containing tetracycline/rifampicin, which suppress Wolbachia [25]. Infection of naive cells with Wolbachia is accompanied by increased ubiquitylation, suggesting use of proteasome-generated precursors [41]. A decrease in Wolbachia levels when culture medium is depleted of riboflavin suggests dependence on metabolically healthy host cells, as Wolbachia itself retains a metabolic capacity for riboflavin synthesis [48]. Differential responses to paraquat suggest that, relative to its host cell, Wolbachia is more sensitive to oxidative damage [49].
The present proteomic comparison of wStr infected and uninfected A. albopictus mosquito cells provides one of the first systematic investigations of a host cell response to Wolbachia infection. Among 2,471 unique proteins grouped according to functional class, a standard MS/MS analysis based on differences in number of protein identifications was in good agreement with quantitative iTRAQ data based on ratios of isobaric tags. To date, these data provide the most comprehensive proteomic analysis with mosquitoes (reviewed in [50]). For the majority of the 815 proteins detected by iTRAQ, increases or decreases in relative levels were within 2-fold, suggesting that C/wStr1 cells maintain homeostasis within a relatively narrow range of protein expression, providing sufficient amino acid and lipid resources for maintenance of a persistent Wolbachia infection despite suppression of host cell transcription, translation and cell division.
Aside from exploring overall metabolic change that accompanies a persistent wStr infection, we sought to identify potential targets that could be manipulated to provide a selective advantage to the infected cell. We envision use of genetic technologies to develop a cell line that exhibits improved growth after Wolbachia infection. Conditions under which Wolbachia provides positive selection would enhance efforts to develop efficient transformation procedures and allow recovery of genetically manipulated strains. Towards that end, we focus the following discussion on the 4.7% of proteins (Table 4; top-37 proteins) for which increased expression in infected cells exceeded a threshold ratio of 2.0, emphasizing pathways susceptible to pharmacological and/or genetic manipulation in host cells.
Neurotactin, a cell surface glycoprotein expressed during Drosophila development, had the highest ratio (5.33) for differential expression in Wolbachia-infected cells. In embryos of Drosophila, neurotactin occurs at sites of cell-cell contact, with eventual localization to nervous or endocrine systems. The protein contains an extracellular domain with homology to serine esterases, but lacks the active site and is highly susceptible to proteolytic degradation [51]. Although it is tempting to speculate that antisense expression of neurotactin might reduce the strongly adhesive properties of wAlbB-infected Aa23 cells [52], we note that, in a converse experiment with non-adhesive Drosophila S2 cells, transfection with the neurotactin gene did not increase cell-cell interaction [53].
Six top-37 proteins (Table 4) were hypothetical proteins, including AAEL001471, the second most highly elevated protein (5.01), which has domains suggestive of a role in ubiquitin-mediated proteolysis that would provide amino acids to Wolbachia. Many ubiquitin-related proteins were moderately elevated in infected cells. AAEL008862 (1.56) may encode a zinc-dependent peptidase. Two suppressed proteins AAEL011155 (0.40) and AAEL008569 (0.39) have motifs associated with cyclins, consistent with reduced expression of proteins involved in DNA replication and cell division (Table 5).
We speculate that hypothetical proteins associated with Wolbachia infection may be poorly known, in part because mosquitoes, unlike fleas and ticks, do not transmit bacterial pathogens. Mosquito cells respond to heat-killed extracellular bacteria by induction of various defensive proteins, including cecropins, defensins, transferrin and lysozyme [54], none of which are highly expressed in Wolbachia-infected cells. Deficiencies in the Wolbachia cell wall [38] may allow Wolbachia to avoid a typical immune response; alternatively, Wolbachia may secrete as yet unidentified effector proteins with protective functions. Elucidating functions of hypothetical proteins remains a key problem in molecular biology, exemplified by the recent finding that, of 473 genes comprising the genome of synthetic microbe Syn 3.0, 149 have unknown functions [55].
Subunits of the vacuolar, or V-type, H+ATPase complex account for 5 of the top-37 entries and constitute an obvious group of upregulated host proteins with a common function. V-ATPase shares a common ancestor with the F-ATPases of eubacteria, mitochondria and chloroplasts. In a previous study [27], we detected 7 subunits of the Wolbachia F0F1 ATPase, of which two were here among the most abundant wStr proteins (Table S2, sheet 2). We note that the F-ATPases produce ATP, while the V-ATPases utilize ATP.
V-ATPase is comprised of 14 subunits that form a cytosolic V1 complex associated with ATP hydrolysis, and a membrane-embedded V0 complex, which transports H+ into vesicles or into the extracellular fluid. Acidification of intracellular compartments contributes to a wide range of intracellular functions, including dissociation of ligands from receptors, proteolytic processing of peptides such as insulin, and degradation of macromolecules by acidic hydrolases, including proteases, nucleases, glycosidases and lipases. Dissociation of the V1 and V0 complexes occurs in response to glucose deprivation; reassembly is mediated by the glycolytic enzyme aldolase, functioning as a glucose sensor [56].
By convention, the soluble V1 subunits and membrane-associated V0 subunits of V-ATPase are distinguished by capital and lower-case letters, respectively. The 116 kDa V0 subunit i was most highly expressed (3.23), and V1 subunits C (2.81), D (2.73), E (2.20) and H (2.75) were also highly elevated in infected cells. It will be of particular interest to learn whether a Wolbachia-encoded effector molecule modulates expression of host-encoded V-ATPase components, which potentially facilitate transfer of nutrients from host cytoplasm to the bacterium. We note that RNAi-mediated reduction of vATPase subunit transcripts severely inhibited mTORC1 signaling in cultured Drosophila S2 cells [57].
The malaria parasite, Plasmodium falciparum exports a functional V-ATPase into the erythrocyte host cell, where it targets the plasma membrane and plays a role in maintenance of intracellular pH [58]. Suppression of V-ATPase with bafilomycin reduces dengue infection in adult mosquitoes [59] and in mosquito cell lines [60], presumably because acidification of the endosome is required for release of virions into the cytoplasm. Inhibitors of V-ATPases, such as bafilomycin and concanamycin, and of F-ATPases, such as oligomycin [61], provide potential tools for investigating V- and F-ATPases in establishment and maintenance of Wolbachia infection.
Finally, V-ATPase has been implicated in an amino acid signaling pathway that regulates autophagy [62]. Autophagy is constitutively involved in cellular homeostasis, and the best-studied “canonical” autophagy responds to AMP-activated protein kinase and mTOR inhibition. Formation of non-canonical pathogen-specific autophagosomes, or xenophagy, is initiated through different pathways, and recruits proteins that do not participate in the canonical pathway. Intracellular microbes including Anaplasma, Coxiella, Franciscella, Legionella, Mycobacterium, and Salmonella evade xenophagy while stimulating canonical autophagic processes that increase intracellular nutrient pools from which they benefit [28]. Anaplasma phagocytophilum, which like Wolbachia is classified in the Anaplasmataceae, modulates mitochondrial function and apoptosis through the secreted T4SS effector, Ats1. Ats1 shuttles between mitochondria and autophagosomes, delays apoptosis by inhibiting release of cytochrome c from mitochondria and may also have a role in autophagosome inititation [29].
One additional protein in the motility/trafficking/secretion class is the Zeta-coat protein, which resembles clathrin in formation of vesicles that transport molecules within cells. Recent evidence suggests that, in a green alga, formation of these vesicles may be reduced by treatment with the phosphatidylinositol kinase inhibitor, wortmannin [63]. We note, however, that application of wortmannin to C/wStr1 cells can be complicated by its short half-life in culture media (8–13 minutes), and off-target effects such as induction of nitric oxide production and protein phosphorylation (http://www.enzolifesciences.com/BML-ST415/wortmannin/).
A suite of six potentially related proteins is involved in carbohydrate metabolism. Activated by phosphorylase kinase (2.12), glycogen phosphorylase (3.06) is the rate-limiting enzyme in the conversion of glycogen to glucose 1-phosphate, which is converted to glucose 6-phosphate by phosphoglucomutase (2.00). Depending on the energy requirements of the cell, glucose 6-phosphate enters the glycolytic pathway to produce ATP, or the pentose phosphate pathway to produce ribose/NADPH. Phosphofructokinase (2.04) is the key regulatory enzyme in the glycolytic pathway, suggesting a glycolytic fate for glucose 6-phosphate. High expression of citrate synthase (2.24) provides support for upregulation of the TCA cycle, and phosphoenolpyruvate carboxykinase (2.48), which catalyzes the commitment step in gluconeogenesis, acts at the junction between the TCA cycle and glycolysis. Malate dehydrogenase (2.65) participates in the TCA cycle and in gluconeogenesis. Increased expression of pathways involved in carbohydrate metabolism may supplement pools of amino acids that cannot be synthesized by Wolbachia, as well as precursors for nucleic acid and lipid biosynthesis. The potential role of glycogen metabolism in cancer progression has stimulated interest in drugs such as 2-deoxyglucose and 1,4-dideoxy-1,4-amino-D-arabinitol, which target enzymes in glycogen metabolism [64].
Among the top 37 proteins, participants in oxidation-reduction reactions included six dehydrogenases (2.02–2.65), a cytochrome oxidase subunit (2.43), and glutathione peroxidase (2.1). The elevated levels of superoxide dismutases, peroxidases and thioredoxins indicated an oxidative stress response in infected cells, potentially associated with a cellular mechanism for Wolbachia suppression [18]. Studies with paraquat indicate that Wolbachia are more sensitive to oxidative damage than host cells [46], and an Ahp/Tsa antioxidant is among the most abundant wStr proteins (Table S2, sheet 2 and [27]). However, antioxidant gene expression and ROS levels did not vary significantly in tetracycline-cured Aa23 cells infected with wAlbB, wMel or wMelPop strains [65]. Other top-37 proteins included two 3-hydroxyacyl-CoA dehydrogenases (2.63, 2.52) and short chain dehydrogenase (2.04) involved in fatty acid metabolism, an aldehyde dehydrogenase (2.05) that may participate in detoxification, and a mosquito homolog of the estradiol 17 beta-dehydrogenase, which likely plays a role in metabolism of the insect steroid hormone 20-hydroxyecdysone, an inhibitor of the cell cycle in mosquito cells [66].
Although not among the top-37 proteins, we identified peptides from flaviviruses in both infected and uninfected A. albopictus cells. Over 70 flaviviruses representing nine serogroups have been described, including dual-host flaviviruses such as Yellow fever, Zika, Chikungunya and West Nile that are important pathogens in humans [67]. Insect-specific flaviviruses that do not infect vertebrates have been isolated from mosquitoes worldwide, and flavivirus-like sequences, including some that encode open reading frames, are integrated into mosquito genomes (reviewed in [43]). Although potential interaction between Wolbachia and endogenous viruses remains unexplored, we note that West Nile virus can be transmitted transovarially from female to offspring [68, 69], suggesting that in Wolbachia infected-mosquitoes, virus and bacterium co-occupy the ovarian niche. Cell fusion agent, discovered over 40 years ago in an A. aegypti cell line [70], occurs widely in field-caught and laboratory strains of A. aegypti [43], a species into which Wolbachia has been introduced by transinfection [11]. Mechanisms by which Wolbachia inhibits pathogen infections in transinfected mosquitoes can be variable and are not well understood [12, 71]. Likewise, superinfection exclusion (homologous interference) between mosquito-specific and dual host flaviviruses in cell lines is variable [43], suggesting a need for systematic investigation of potential interaction between endogenous flaviviruses and the better-known human pathogens in the mosquito host.
In aggregate, proteomic evidence for altered autophagy, metabolic signaling and protein degradation pathways, coupled with elevated vATPase, amino acid metabolism and TCA cycle proteins suggested that wStr’s replication within host derived vacuoles evades xenophagy and sequesters host-derived amino acids. Elevated levels of proteases support earlier evidence for enhanced proteasome activity in A. albopictus cells infected with wAlbB [41] and potentially increase access to host amino acid pools. An integrated metabolomic, transcriptomic and proteomic analysis of the tick ISE6 cell line infected with Wolbachiäs relative, A. phagocytophilum, likewise suggests that infection is accompanied by changes in glucose metabolism, protein processing, and manipulation of autophagy [72]. A wide array of altered-level chaperones and protein folding, modification and trafficking proteins suggests a similar dynamic in C/wStr1 cells. Wolbachia-infected cells provide direct evidence for differential expression of several hypothetical Aedes proteins and a potential system for investigating their functions. Finally, we have identified targets that could be explored by RNAi knockdown of gene expression or in genetically manipulated cell lines. In future experiments, it will be of particular interest to explore effects of available pharmaceutical agents on Wolbachia growth kinetics, abundance and ability to infect cell lines, and to compare expression of proteins in cell lines to that of infected tissues in the insect host.
Supplementary Material
Fig. S1. Distribution of wStr proteins identified in iTRAQ MS data sets (H, I and J) by protein functional class and relative abundance as determined by a univariable statistical model of the relationship between protein mass and MS-detected peptide numbers expressed as Studentized residuals. SR values are a measure of the difference between expected and observed log peptide count adjusted by estimated standard error, in which 0 corresponds to average abundance. Diamond symbols indicate mean SR values; filled boxes represent first through third quartiles of SR values; bars indicate minimum and maximum values with exception of circles representing outliers, whose values are >1.5 * IQR from the median.
Fig. S2. MS/MS detection of peptides that occur in selected flavivirus polyproteins. Panel A) Flaviviruses are single-stranded, positive sense RNA viruses. The viral genome encodes an approximately 3,300–3,400 amino acid polyprotein that is processed into three structural and seven non-structural (NS) proteins. For some polyproteins, annotation of processed, mature proteins is incomplete. Panel B) Accessions are listed in Black, followed by specific peptides from color-coded data sets. Blue peptides, data set D; green, data set E; violet, data sets F and G; red, data set H, as shown in Fig. 1. Amino acid coordinates in the polyprotein appear at left of the detected peptides. Peptides without terminal R/K trypsin sites are preceded or followed by asterisks (*). Where mismatches (X/Y) occur, amino acids X in black font occur in the most commonly detected polyprotein accession listed at top, and colored font designates amino acids Y in the related virus accession indicated at right.
Table S1. The Aedes total proteome derived from standard and iTRAQ MS/MS data. In the “Aedes proteome” first sheet, all proteins identified in the standard (D – G) and iTRAQ (H – J) data sets (Fig. 1) are reported in corresponding columns D – J with the number of detected peptides separated by a “ \ ” symbol from the corresponding percent protein sequence coverage. Columns A, B and C: protein identity, NCBI accession number and calculated molecular mass in kiloDaltons. Shading designates proteins present only in the standard (light gray) or iTRAQ (dark gray) data; proteins common to both data sets are not shaded. The “Gel CwStrC7-10” sheet reports the gel lane breakdown of the subset of 1507 proteins in Column D of the “Aedes proteome” sheet. The “Data sets F and G only ” sheet reports 81 host proteins identified only in data sets F and G. The “FDRs” sheet shows false discovery rates for datasets D – G.
Table S2.Wolbachia strain wStr proteins identified from iTRAQ MS data sets H, I and J. In sheet 1, columns A, B, C and D show protein identity, accession number, functional category and calculated molecular mass in kiloDaltons. Columns E and F show data from data sets I and J, respectively, in Fig 1 (cytoplasmic plus mitochondrial fractions from cells at passages 16 (E) and 35 (F); column G shows samples from data set H (total protein, passage 35) in Fig. 1. Values indicate numbers of detected peptides separated by a “ \ ” symbol from the corresponding percent protein sequence coverage. Columns I, J and K represent calculated Studentized residuals (SR), which are averaged and rounded to two significant figures in column H ranked from high (3.89) to low (−1.58), where 0 indicates average abundance. In sheet 2, the 26 abundant and highly abundant wStr proteins from the iTRAQ samples are compared to the previously defined wStr SDS PAGE footprint and abundant proteins [27].
Table S3.Aedes host proteins with iTRAQ ratios indicating differential abundance in C/wStr1 relative to C7-10 cells. Sheet 1) Columns A, B, and C: protein identity, accession number, and calculated molecular mass in kiloDaltons. For cytoplasmic and mitochondrial fractions, two sets of extracts were prepared at cell passage 16 and passage 35. Sets of three columns (D/E/F; G/H/I; J/K/L; M/N/O; and P/Q/R) represent data sets H and I at passage 35 and J at passage 16. Passage numbers are included to facilitate book-keeping and differences are not considered significant. Within each set of three columns, D, G, J, M and P show the number of unique peptides separated by a “ \ ” symbol from the corresponding percent protein sequence coverage. Columns E, H, K, N, and Q show C/wStr1 over C7-10 iTRAQ ratios reported by Protein Pilot; F, I, L, O and R show logex values used for statistical analysis (see Table 3). Column S shows logex values averaged over all samples for a given accession. Proteins are ordered alphabetically according to functional categories (Column T), with comments related to cellular functions (Column U). Sheet 2 lists 15 proteins for which agreement was poor among the five sample sets. Sheet 3 shows Protein Pilot file names and isobaric tag labels of protein extracts.
Table S4. Summary of Aedes host protein iTRAQ ratios. Sheets 1 to 18 show proteins within a single functional class from Table S3, with identity (column A) and accession number (B). The sheets are provided in the order of decreasing average values defined in Fig. 4. Each subclassification (column C) within a functional class is color-coded, and hypothetical proteins are shaded in yellow. Column D shows average Protein Pilot iTRAQ ratios, used for sorting. Entries shaded gray have a ratio greater than 1.00. Column headings E through I indicate cytoplasmic (Cyto) mitochondrial (Mito) or total (Tot) protein samples, with passage number (p16, p35) for bookkeeping purposes.
Acknowledgments
This work was supported by grant 5 R01 AI 081322 from the National Institutes of Health and by the University of Minnesota Agricultural Experiment Station, St. Paul, MN.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors have no conflicts of interest.
References
- 1.Hertig M. The rickettsia, Wolbachia pipientis (gen. et sp.n) and associated inclusions of the mosquito. Culexpipiens Parasitology. 1936;38:453–486. [Google Scholar]
- 2.Bourtzis K, Dobson SL, Xi Z, Rasgon JL, Calvitti M, Moreira LA, et al. Harnessing mosquito-Wolbachia symbiosis for vector and disease control. Acta Trop. 2014;132:S150–63. doi: 10.1016/j.actatropica.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Bourtzis K, Braig HR, Karr TL. Cytoplasmic incompatibility. In: Bourtzis K, Miller T, editors. Insect Symbiosis. Vol. 1. CRC Press; New York: 2003. pp. 217–246. [Google Scholar]
- 4.Sinkins SP, Gould F. Gene drive systems for insect disease vectors. Nat Rev Genet. 2006;7:427–435. doi: 10.1038/nrg1870. [DOI] [PubMed] [Google Scholar]
- 5.Laven H. Eradication of Culex pipiens fatigans through cytoplasmic incompatibility. Nature. 1967;216:383–384. doi: 10.1038/216383a0. [DOI] [PubMed] [Google Scholar]
- 6.Yen JH, Barr AR. New hypothesis of the cause of cytoplasmic incompatibility in Culex pipiens L. Nature. 1971;232:657–658. doi: 10.1038/232657a0. [DOI] [PubMed] [Google Scholar]
- 7.Beare PA, Sandoz KM, Omsland A, Rockey DD, Henzein RA. Advances in genetic manipulation of obligate intracellular bacterial pathogens. Front Microbiol. 2011;2:97. doi: 10.3389/fmicb.2011.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kambris Z, Cook PE, Phuc HK, Sinkins SP. Immune activation by life-shortening Wolbachia and reduced filarial competence in mosquitoes. Science. 2009;326:134–6. doi: 10.1126/science.1177531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeap HL, Mee P, Walker T, Weeks AR, O’Neill SL, Johnson P, et al. Dynamics of the “popcorn” Wolbachia infection in outbred Aedes aegypti informs prospects for mosquito vector control. Genetics. 2011;187:83–95. doi: 10.1534/genetics.110.122390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bian G, Joshi D, Dong Y, Lu P, Zhou G, Pan X, et al. Wolbachia invades Anopheles stephensi populations and induces refractoriness to Plasmodium infection. Science. 2013;340:748–751. doi: 10.1126/science.1236192. [DOI] [PubMed] [Google Scholar]
- 11.Hughes GL, Rasgon JL. Transinfection: a method to investigate Wolbachia-host interactions and control arthropod-borne disease. Insect Mol Biol. 2014;23:141–151. doi: 10.1111/imb.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson KN. The impact of Wolbachia on virus infection in mosquitoes. Viruses. 2015;7:5705–5715. doi: 10.3390/v7112903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker T, Johnson PH, Moreira LA, Iturbe-Ormaetxe I, Frentiu FD, McMeniman CJ, et al. The wMel Wolbachia strain blocks dengue and invades caged Aedes aegypti populations. Nature. 2011;476:450–453. doi: 10.1038/nature10355. [DOI] [PubMed] [Google Scholar]
- 14.Hoffmann AA, Montgomery BL, Popovici J, Iturbe-Ormaetxe I, Johnson PH, Muzzi F, et al. Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature. 2011;476:454–57. doi: 10.1038/nature10356. [DOI] [PubMed] [Google Scholar]
- 15.O’Connor L, Plichart C, Sang AC, Breisforad CL, Bossin HC, Dobson SL. Open release of male mosquitoes infected with a Wolbachia biopesticide: field performance and infection containment. PLoS Negl Trop Dis. 2012;6:e1797. doi: 10.1371/journal.pntd.0001797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricci I, Valzano M, Ulisse U, Epis S, Cappelli A, Favia G. Symbiotic control of mosquito borne disease. Pathog Glob Health. 2012;106:380–85. doi: 10.1179/2047773212Y.0000000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LePage D, Bordenstein SR. Wolbachia: Can we save lives with a great pandemic? Trends Parasitol. 2013;29:385–393. doi: 10.1016/j.pt.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brennan LJ, Keddie BA, Braig HR, Harris HL. The endosymbiont Wolbachia pipientis induces the expression of host antioxidant proteins in an Aedes albopictus cell line. PLoS ONE. 2008;3:e2083. doi: 10.1371/journal.pone.0002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zug R, Hammerstein P. Wolbachia and the insect immune system: what reactive oxygen species can tell us about the mechanisms of Wolbachia-host interactions. Front Microbiol. 2015;6:1201. doi: 10.3389/fmicb.2015.01201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gill AC, Darby AC, Makepeace BL. Iron necessity: the secret of Wolbachiäs success? PloS Negl Trop Dis. 2014;8:e3224. doi: 10.1371/journal.pntd.0003224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riparbelli MG, Giordano R, Ueyama M, Callaini G. Wolbachia-mediated male killing is associated with defective chromatin remodeling. PloS One. 2012;7:e30045. doi: 10.1371/journal.pone.0030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan LL, Chen X, Zong Q, Zhao T, Wang JL, Zheng Y, et al. Quantitative proteomic analyses of molecular mechanisms associated with cytoplasmic incompatibility in Drosophila melanogaster induced by Wolbachia. J Proteome Res. 2015;14:3835–47. doi: 10.1021/acs.jproteome.5b00191. [DOI] [PubMed] [Google Scholar]
- 23.Beckmann JF, Fallon AM. Detection of the Wolbachia protein WPIP0282 in mosquito spermathecae: implications for cytoplasmic incompatibility. Insect Biochem Mol Biol. 2013;43:867–78. doi: 10.1016/j.ibmb.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noda H, Miyoshi T, Koizumi Y. In vitro cultivation of Wolbachia in insect and mammalian cell lines. In Vitro Cell Dev Biol Anim. 2002;38:423–427. doi: 10.1290/1071-2690(2002)038<0423:IVCOWI>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 25.Fallon AM, Baldridge GD, Higgins LA, Witthuhn BA. Wolbachia from the planthopper Laodelphax striatellus establishes a robust, persistent, streptomycin-resistant infection in clonal mosquito cells. In Vitro Cell Dev Biol Anim. 2013;49:66–73. doi: 10.1007/s11626-012-9571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serbus LR, Casper-Lindley C, Landmann F, Sullivan W. The genetics and cell biology of Wolbachia-host interactions. Annu Rev Genet. 2008;42:683–707. doi: 10.1146/annurev.genet.110306.130354. [DOI] [PubMed] [Google Scholar]
- 27.Baldridge GD, Baldridge AS, Witthuhn BA, Higgins LA, Markowski TW, Fallon AM. Proteomic profiling of a robust Wolbachia infection in an Aedes albopictus mosquito cell line. Mol Microbiol. 2014;94:537–56. doi: 10.1111/mmi.12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steele S, Brunton J, Kawula T. The role of autophagy in intracellular pathogen nutrient acquisition. Front Cell Infect Microbiol. 2015;5:51. doi: 10.3389/fcimb.2015.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Escoll P, Roland M, Buchreiser C. Modulation of host autophagy during bacterial infection: Sabotaging host munitions for pathogen nutrition. Front Immunol. 2016;7:81. doi: 10.3389/fimmu.2016.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shih KM, Gerenday A, Fallon AM. Culture of mosquito cells in Eagle’s medium. In Vitro Cell Develop Biol Anim. 1998;34:629–630. doi: 10.1007/s11626-996-0010-1. [DOI] [PubMed] [Google Scholar]
- 31.Oliva Chavez AS, Fairman JW, Felsheim RF, Nelson CM, Herron MJ, Higgins LA, et al. An 0-Methyltransferase Is required for Infection of tick cells by Anaplasma phagocytophilum. PloS Pathog. 2015;11(11):e1005248. doi: 10.13/journal.ppat.1005248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen XG, Jiang X, Gu J, Xu M, Wu Y, Deng Y, et al. Genome sequence of the Asian tiger mosquito, Aedes albopictus, reveals insights into its biology, genetics, and evolution. Proc Nat Acad Sci USA. 2015;112:5907–5915. doi: 10.1073/pnas.1516410112.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keller A, Nesvizhskii A, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database seach. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 34.Nesvizhskii A, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 35.Shilov IM, Seymour SL, Patel AA, Loboda A, Tang WH, Kaeting SP, et al. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol Cell Proteomics. 2007;6:1638–1655. doi: 10.1074/mcp.T600050-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Tang WH, Shilove IV, Seymour SL. Nonlinear fitting method for determining local false discovery rates from decoy database searches. J Proteome Res. 2008;7:3661–3667. doi: 10.1021/pr070492f. [DOI] [PubMed] [Google Scholar]
- 37.Wright JD, Sjostrand FS, Portaro JK, Barr AR. The ultrastructure of the rickettsia-like microorganism Wolbachia pipientis and asssociated virus-like bodies in the mosquito Culex pipiens. J Ultrastructure Res. 1978;63:79–85. doi: 10.1016/s0022-5320(78)80046-x. [DOI] [PubMed] [Google Scholar]
- 38.Wu M, Sun LV, Vamathevan J, Riegler M, Deboy R, Brownlie JC, et al. Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile gentic elements. PLoS Biol. 2004;2 doi: 10.1371/journal.pbio.002006939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fafournoux P, Bruhat A, Jousse C. Amino acid regulation of gene expression. Biochem J. 2000;351:1–12. doi: 10.1042/0264-6021:3510001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brasse-Lagnel C, Lavoinne A, Husson A. Control of mammalian gene expression by amino acids, especially glutamine. FEBS J. 2009;276:1826–44. doi: 10.1111/j.1742-4658.2009.06920.x. [DOI] [PubMed] [Google Scholar]
- 41.Fallon AM, Witthuhn BA. Proteasome activity in a naïve mosquito cell line infected with Wolbachia pipientis wAlbB. In Vitro Cell Dev Biol Anim. 2009;45:460–466. doi: 10.1007/s11626-009-9193-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Price CT, Al-Khodor S, Al-Quadan T, Santic M, Habyarimana F, Kalia A, et al. Molecular mimicry by an F-box effector of Legionella pneumophila hijacks a conserved polyubiquitin machinery within macrophages and protozoa. PLoS Pathog. 2009;5:e1000704. doi: 10.1371/journal.ppat.1000704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blitvich BJ, Firth A. Insect-specific flaviviruses: A systematic review of their discovery, host range, mode of transmission, superinfection exclusion potential and genomic organization. Viruses. 2015;7:1927–1959. doi: 10.3390/v7041927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Neill SL, Pettigrew M, Sinkins SP, Braig HR, Andreadis TG, Tesh RB. In vitro cultivation of Wolbachia pipientis in an Aedes albopictus cell line. Insect Mol Biol. 1997;6:33–39. doi: 10.1046/j.1365-2583.1997.00157.x. [DOI] [PubMed] [Google Scholar]
- 45.Dobson SL, Marsland EJ, Veneti Z, Bourtzis K, O’Neill SL. Characterization of Wolbachia host range via the in vitro establishment of infections. Applied Environ Microbiol. 2002;68:656–660. doi: 10.1128/AEM.68.2.656-660.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fallon AM. Transfection of cultured mosquito cells. In: Crampton JM, Beard CB, Louis C, editors. The Molecular Biology of Insect Disease Vectors. Chapman and Hall; New York: 1997. pp. 430–443. [Google Scholar]
- 47.Wright JD, Barr R. The ultrastructure and symbiotic relationships of Wolbachia of mosquitoes of the Aedes scutellaris group. J Ultrastructure Res. 1980;72:52–64. doi: 10.1016/s0022-5320(80)90135-5. [DOI] [PubMed] [Google Scholar]
- 48.Fallon AM, Baldridge GB, Carroll EM, Kurtz CM. Depletion of host cell riboflavin reduces Wolbachia levels in cultured mosquito cells. In Vitro Cell Dev Biol Anim. 2014;50:707–713. doi: 10.1007/s11626-014-9758-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fallon AM, Kurtz CM, Carroll EM. The oxidizing agent, paraquat, is more toxic to Wolbachia than to mosquito host cells. In Vitro Cell Dev Biol Anim. 2013;49:501–507. doi: 10.1007/s11626-013-9634-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sehrawat N, Gakhar SK. Mosquito proteomics: present and future perspectives. Res Biotechnol. 2014;5:25–33. 2014. [Google Scholar]
- 51.Hortsch M, Goodman CS. Cell and substrate adhesion molecules in Drosophila. Annu Rev Cell Biol. 1991;7:505–557. doi: 10.1146/annurev.cb.07.110191.002445. [DOI] [PubMed] [Google Scholar]
- 52.Fallon AM. Cytological properties of an Aedes albopictus mosquito cell line infected with Wolbachia strain wAlbB. In Vitro Cell Dev Biol-Anim. 2008;44:154–161. doi: 10.1007/s11626-008-9090-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barthalay Y, Hipeau-Jacquotte R, de la Escalera S, Jiminez F, Piovant M. Drosophila neurotactin mediates heterophilic cell adhesion. EMBO J. 1990;9:3603–09. doi: 10.1002/j.1460-2075.1990.tb07571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fallon AM, Sun D. Exploration of mosquito immunity using cells in culture. Insect Biochem Mol Biol. 2001;31:263–278. doi: 10.1016/s0965-1748(00)00146-6. [DOI] [PubMed] [Google Scholar]
- 55.Hutchison CA, Chuang R-Y, Noskov VN, Assad-Garcia N, Deerinck TJ, Ellisman MH, et al. Design and synthesis of a minimal bacterial genome. Science. 2016(351):1414. doi: 10.1126/science.aad6253. [DOI] [PubMed] [Google Scholar]
- 56.Beyenbach KW, Wieczorek H. The V-type H+ ATPase: molecular structure and function, physiological roles and regulation. J Exp Biol. 2006;209:577–589. doi: 10.1242/jeb.02014. [DOI] [PubMed] [Google Scholar]
- 57.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marchesini N, Vieira M, Luo S, Moreno SNJ, Docampo R. A malaria parasite-encoded Vacuolar H+-ATPase is targeted to the host erythrocyte. J Biol Chem. 2005;280:36841–47. doi: 10.1074/jbc.M507727200. [DOI] [PubMed] [Google Scholar]
- 59.Kang S, Shields AR, Jupatanakul N, Dimopoulos G. Suppressing Dengue-2 infection by chemical inhibition of Aedes aegypti host factors. PLoS Negl Trop Dis. 2014;8:e3084. doi: 10.1371/journal.pntd.0003084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sessions OM, Barrows NJ, Souza-Neto JA, Robinson TJ, Hershey CL, Rodgers MA, et al. Discovery of insect and human dengue virus host factors. Nature. 2009;458:1047–1050. doi: 10.1038/nature07967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hao W, Chang CP, Tsao CC. Oligomycin-induced bioenergetic adaptation in cancer cells with heterogeneous bioenergetic organization. J Biol Chem. 2010;285:12647–54. doi: 10.1074/jbc.M109.084194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meijer AJ, Lorin S, Blommaart EF, Codogno P. Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino acids. 2015;47:2037–63. doi: 10.1007/s00726-014-1765-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Foissner I, Sommer A, Hoeftberger M, Hoepflinger MC, Absolonova M. Is wortmannin-induced reorganization of the trans-Golgi network the key to explain charasome formation? Front Plant Sci. 2016;7:756. doi: 10.3389/fpls.2016.00756. doi:10.3389.fpls.2016.00756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zois CE, Harris AL. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J Mol Med. 2016;94:137–154. doi: 10.1007/s00109-015-1377-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Molloy JC, Sinkins SP. Wolbachia do not induce reactive oxygen species-dependent immune pathway activation in Aedes albopictus. Viruses. 2015;7:4624–39. doi: 10.3390/v7082836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerenday A, Fallon AM. Increased levels of the cell cycle inhibitor protein, dacapo, accompany 20-hydroxyecdysone-induced G1 arrest in a mosquito cell line. Arch Insect Biochem Physiol. 2011;78:61–73. doi: 10.1002/arch.20440.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang Y-J, Higgs S, Horne KM, Vanlandingham DL. Flavivirus-mosquito interactions. Viruses. 2014;6:4703–30. doi: 10.3390/v6114703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goddard LB, Roth AE, Reisen WK, Scott TW. Vertical transmission of West Nile virus by three California Culex (Diptera: Culicidae) species. J Med Entomol. 2003;40:743–746. doi: 10.1603/0022-2585-40.6.743. [DOI] [PubMed] [Google Scholar]
- 69.Mishra AC, Mourya DT. Transovarial transmission of West Nile virus in Culex vishnui mosquito. Indian J Med Res. 2001;114:212–214. [PubMed] [Google Scholar]
- 70.Stollar V, Thomas VL. An agent in the Aedes aegypti cell line (Peleg) which causes fusion of Aedes albopictus cells. Virology. 1975;64:367–377. doi: 10.1016/0042-6822(75)90113-0. [DOI] [PubMed] [Google Scholar]
- 71.Rainey SM, Shah P, Kohl A, Dietrich I. Understanding the Wolbachia-mediated inhibition of arboviruses in mosquitoes: progress and challenges. J Gen Virol. 2014;95:517–530. doi: 10.1099/vir.0.057422-0. [DOI] [PubMed] [Google Scholar]
- 72.Villar M, Ayllon N, Alberdi P, Moreno M, Tobes R, Mataeos-Hernandez L, et al. Integrated metabolomics, transcriptomics and proteomics identifies metabolic pathways affected by Anaplasma phagocytophilum infection in tick cells. Mol Cell Proteomics. 2015;14:3154–72. doi: 10.107/mcp.M115.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Distribution of wStr proteins identified in iTRAQ MS data sets (H, I and J) by protein functional class and relative abundance as determined by a univariable statistical model of the relationship between protein mass and MS-detected peptide numbers expressed as Studentized residuals. SR values are a measure of the difference between expected and observed log peptide count adjusted by estimated standard error, in which 0 corresponds to average abundance. Diamond symbols indicate mean SR values; filled boxes represent first through third quartiles of SR values; bars indicate minimum and maximum values with exception of circles representing outliers, whose values are >1.5 * IQR from the median.
Fig. S2. MS/MS detection of peptides that occur in selected flavivirus polyproteins. Panel A) Flaviviruses are single-stranded, positive sense RNA viruses. The viral genome encodes an approximately 3,300–3,400 amino acid polyprotein that is processed into three structural and seven non-structural (NS) proteins. For some polyproteins, annotation of processed, mature proteins is incomplete. Panel B) Accessions are listed in Black, followed by specific peptides from color-coded data sets. Blue peptides, data set D; green, data set E; violet, data sets F and G; red, data set H, as shown in Fig. 1. Amino acid coordinates in the polyprotein appear at left of the detected peptides. Peptides without terminal R/K trypsin sites are preceded or followed by asterisks (*). Where mismatches (X/Y) occur, amino acids X in black font occur in the most commonly detected polyprotein accession listed at top, and colored font designates amino acids Y in the related virus accession indicated at right.
Table S1. The Aedes total proteome derived from standard and iTRAQ MS/MS data. In the “Aedes proteome” first sheet, all proteins identified in the standard (D – G) and iTRAQ (H – J) data sets (Fig. 1) are reported in corresponding columns D – J with the number of detected peptides separated by a “ \ ” symbol from the corresponding percent protein sequence coverage. Columns A, B and C: protein identity, NCBI accession number and calculated molecular mass in kiloDaltons. Shading designates proteins present only in the standard (light gray) or iTRAQ (dark gray) data; proteins common to both data sets are not shaded. The “Gel CwStrC7-10” sheet reports the gel lane breakdown of the subset of 1507 proteins in Column D of the “Aedes proteome” sheet. The “Data sets F and G only ” sheet reports 81 host proteins identified only in data sets F and G. The “FDRs” sheet shows false discovery rates for datasets D – G.
Table S2.Wolbachia strain wStr proteins identified from iTRAQ MS data sets H, I and J. In sheet 1, columns A, B, C and D show protein identity, accession number, functional category and calculated molecular mass in kiloDaltons. Columns E and F show data from data sets I and J, respectively, in Fig 1 (cytoplasmic plus mitochondrial fractions from cells at passages 16 (E) and 35 (F); column G shows samples from data set H (total protein, passage 35) in Fig. 1. Values indicate numbers of detected peptides separated by a “ \ ” symbol from the corresponding percent protein sequence coverage. Columns I, J and K represent calculated Studentized residuals (SR), which are averaged and rounded to two significant figures in column H ranked from high (3.89) to low (−1.58), where 0 indicates average abundance. In sheet 2, the 26 abundant and highly abundant wStr proteins from the iTRAQ samples are compared to the previously defined wStr SDS PAGE footprint and abundant proteins [27].
Table S3.Aedes host proteins with iTRAQ ratios indicating differential abundance in C/wStr1 relative to C7-10 cells. Sheet 1) Columns A, B, and C: protein identity, accession number, and calculated molecular mass in kiloDaltons. For cytoplasmic and mitochondrial fractions, two sets of extracts were prepared at cell passage 16 and passage 35. Sets of three columns (D/E/F; G/H/I; J/K/L; M/N/O; and P/Q/R) represent data sets H and I at passage 35 and J at passage 16. Passage numbers are included to facilitate book-keeping and differences are not considered significant. Within each set of three columns, D, G, J, M and P show the number of unique peptides separated by a “ \ ” symbol from the corresponding percent protein sequence coverage. Columns E, H, K, N, and Q show C/wStr1 over C7-10 iTRAQ ratios reported by Protein Pilot; F, I, L, O and R show logex values used for statistical analysis (see Table 3). Column S shows logex values averaged over all samples for a given accession. Proteins are ordered alphabetically according to functional categories (Column T), with comments related to cellular functions (Column U). Sheet 2 lists 15 proteins for which agreement was poor among the five sample sets. Sheet 3 shows Protein Pilot file names and isobaric tag labels of protein extracts.
Table S4. Summary of Aedes host protein iTRAQ ratios. Sheets 1 to 18 show proteins within a single functional class from Table S3, with identity (column A) and accession number (B). The sheets are provided in the order of decreasing average values defined in Fig. 4. Each subclassification (column C) within a functional class is color-coded, and hypothetical proteins are shaded in yellow. Column D shows average Protein Pilot iTRAQ ratios, used for sorting. Entries shaded gray have a ratio greater than 1.00. Column headings E through I indicate cytoplasmic (Cyto) mitochondrial (Mito) or total (Tot) protein samples, with passage number (p16, p35) for bookkeeping purposes.