

Abstract
Among Neuronal Ceroid Lipofuscinoses (NCLs), which are childhood fatal neurodegenerative disorders, the juvenile onset form (JNCL) is the most common. JNCL is caused by recessive mutations in the CLN3 gene. CLN3 encodes a lysosomal/endosomal transmembrane protein but its precise function is not completely known. We have previously reported that in baby hamster kidney (BHK) cells stably expressing myc-tagged human CLN3 (myc-CLN3), hyperosmotic conditions drastically increased myc-CLN3 mRNA and protein expression. In the present study, we analyzed the consequences of hyperosmolarity and increased CLN3 expression on cathepsin D activity and prosaposin processing using BHK cells transiently or stably expressing myc-CLN3. We found that hyperosmolarity increased lysotracker staining of lysosomes and elevated the levels of myc-CLN3 and lysosome-associated membrane protein-1 (LAMP1). Hyperosmolarity, independently of the expression level of myc-CLN3, decreased the levels of prosaposin and saposin D, which are protein cofactors in sphingolipid metabolism. The lysosomal enzyme cathepsin D (CTSD) mediates the proteolytic cleavage of prosaposin precursor into saposins A–D. Myc-CLN3 colocalized with CTSD and activity of CTSD decreased as myc-CLN3 expression increased and clearly decreased under hyperosmotic conditions. Nevertheless, levels of CTSD measured by Western blotting were not altered under any studied condition.
Our results suggest a direct involvement of CLN3 in the regulation of CTSD activity.
Keywords: Neuronal Ceroid Lipofuscinoses, Batten disease, CLN3 disease, cathepsin D, prosaposin, saposins, hyperosmolarity
INTRODUCTION
Neuronal Ceroid Lipofuscinoses (NCLs), in spite of a global prevalence of only 1 in 100,000 live births, are the most common group of autosomal recessive neurodegenerative disorders of childhood. Among the NCLs, the most frequent subtype is CLN3 disease, formerly known as juvenile neuronal ceroid lipofuscinosis [Kwon et al., 2011] or juvenile Batten disease [Kovacs et al., 2012], which is caused by mutations in the CLN3 gene [Consortium, 1995; Williams and Mole, 2012]. Despite the large number of studies concerning the consequences of mutant CLN3 in cells from juvenile CLN3 disease patients, cells derived from CLN3-deficient mouse models and genetic deletions in other model systems, the primary function and intracellular localization of the CLN3 protein has not been fully elucidated, yet. Recent studies using Förster Resonance Energy Transfer microscopy (FRET) analysis have confirmed that mammalian CLN3 is a 438 amino acid multi-pass transmembrane protein with N and C terminals in the cytosol [Ratajczak et al., 2014]. CLN3 has been primarily found in endosomes and lysosomes with a direct involvement in endocytic traffic [Cotman and Staropoli, 2012; Kollmann et al., 2013; Uusi-Rauva et al., 2012]. Other studies with mutated Btn1p, the orthologue protein of CLN3 in yeast, as well as subsequent analyses in patients’ fibroblasts and human embryonic kidney cells with mutated CLN3 have shown disturbances in the maintenance of vacuolar/lysosomal pH [Chattopadhyay et al., 2000; Golabek et al., 2000; Pearce et al., 1999; Vidal-Donet et al., 2013]. A recent study in the amoeba Dictyostelium discoideum has localized GFP-tagged CLN3 in the contractile vacuole system, an organelle that plays a central role in cell osmoregulation [Huber et al., 2014]. Further evidence of an osmoregulative role for CLN3 in renal control of water and K+ balance was found in mouse by Stein et al. [Stein et al., 2010]. Using a Cln3 reporter mouse harboring a nuclear-localized bacterial β-galactosidase (β-Gal) gene driven by the native Cln3 promoter this group detected β-Gal in medullary collecting duct principal kidney cells, with increased expression along the medullary osmotic gradient. Reporter mouse-derived renal epithelial cultures also demonstrated a tonicity-dependent increase in β-Gal expression [Stein et al., 2010]. Following this report, our group further researched the role of CLN3 in osmoregulation. Because of the lack of an antibody that specifically detects endogenous CLN3, an N-terminally myc-tagged human CLN3 was stably expressed in baby hamster kidney (BHK) cells. In this cell model, hyperosmolarity (800 mOsm), achieved by either NaCl/urea or sucrose, dramatically increased mRNA and protein levels of myc-CLN3 as determined by quantitative real-time PCR and Western blotting, and changed the subcellular localization of myc-CLN3 as showed by immunofluorescence [Getty et al., 2013].
NCLs are lysosomal storage diseases and most subtypes share common storage material consisting of accumulation of subunit c of ATP synthase and distinct lipoproteins. In the infantile and congenital NCL subtypes the main components of storage material are sphingolipid activator proteins (SAPs or saposins) A and D [Siintola et al., 2006; Tyynela et al., 1993] that are essential for lysosomal hydrolysis of sphingolipids. Saposins are derived from a single precursor, prosaposin (PSAP). PSAP exists as a soluble protein and as an integral lysosomal membrane protein. After reaching lysosomes, soluble PSAP is converted to saposins by lysosomal proteases [Hiraiwa et al., 1997; Salvioli et al., 2008]. Cathepsin D (CTSD) is the most widely studied of PSAP converting lysosomal enzymes. CTSD is a major component of lysosomes and functions as a highly active endopeptidase, playing an obvious role in the degradation and turnover of cellular components under nutrient-deficient conditions [Rawlings and Barrett, 1995]. CTSD deficiency also induces ATP synthase subunit c and ceroid lipofuscin accumulation in mouse neurons [Koike et al., 2000], which are typical features of NCL pathology.
The obvious commonalities between different types of NCL led us to research the relationship of CLN3 to proteins involved in the metabolism of lipid derivatives accumulated in other subtypes of NCL. Our results in BHK cells stably or transiently expressing myc-CLN3 suggest a direct involvement of CLN3 in the regulation of CTSD activity.
MATERIALS AND METHODS
PLASMIDS
The pUB6-B plasmid expressing an N-terminally myc-tagged human CLN3 under the control of the human ubiquitin C promoter was generated previously [Getty et al., 2013]. The pUB6-B empty plasmid (Life Technologies, Grand Island, NY) served as a negative control.
CELL CULTURE
The two baby hamster kidney (BHK) cell lines, one stably transfected with the pUB6-B plasmid expressing an N-terminally myc-tagged human CLN3 (C19), the other stably transfected with the empty pUB6-B plasmid (C2), were generated previously [Getty et al., 2013]. The pUB6-B expression vector provides resistance against the antibiotic, blasticidin. Therefore, C19 and C2 BHK cells were cultured under blasticidin selection, in high glucose Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), MEM non-essential amino acids (NEAA) (100 mM), penicillin-streptomycin (100 U/ml/100 mg/ml) (all from Hyclone, ThermoFisher Scientific, Waltham, MA), 1X GlutaMax (ThermoFisher Scientific, Waltham, MA), and blasticidin (50 μg/ml). The culture medium was changed every 2–3 days. BHK cells taken out of liquid nitrogen storage were grown under blasticidin (50 μg/ml) selection for a minimum of 2 weeks before use in any experiments. No cells were used beyond passage 15, or approximately 8 weeks in culture, to prevent genetic drift or contamination of the cultures.
TRANSIENT TRANSFECTION
The same plasmids were used for transient transfection as for the generation of the stably transfected C19 and C2 cell lines: pUB6-B plasmid expressing an N-terminally myc-tagged human CLN3 and the empty pUB6-B plasmid. Cells were plated in 6-well plates the day before to reach 80–90% confluency on the day of transfection. Empty vector and myc-CLN3 containing plasmid DNA contents were quantified using Nanodrop technology (Epoch Microplate Spectrophotometer, BioTek Instruments, Winooski, VT) and, for each well of cells to be transfected, 2.5 μg of DNA was diluted in 100 μl of Opti-MEM I Reduced Serum Medium without serum (ThermoFisher Scientific, Waltham, MA) in separate Eppendorf tubes. PLUS Reagent (ThermoFisher Scientific, Waltham, MA) was added to each sample and contents were gently mixed and incubated for 15 min at room temperature. Lipofectamine LTX Reagent (ThermoFisher Scientific, Waltham, MA) was added into the above diluted Opti-MEM:DNA solution, and tubes were gently mixed and incubated for 30 min at room temperature to allow the formation of DNA-Lipofectamine LTX Reagent complexes. Then, 100 μl of the DNA-Lipofectamine LTX Reagent complexes from empty vector and myc-CLN3 containing plasmids were added directly to each culture well, mixed gently for 5 min and then cells were incubated at 37°C in a CO2 incubator for 18–24 hours post-transfection before assaying for myc-CLN3 expression by Western blotting.
CELL CULTURE UNDER HYPEROSMOLARITY CONDITIONS
Cells in complete, blasticidin-containing medium were plated in 10-cm culture dishes (50,000 cells/dish) for immunoblot, and on poly-D-lysine (10 μg/ml) coated coverslips in 24-well plates (1,700 cells/well) for immunofluorescent staining. Cells were allowed to adhere overnight. Each subsequent day, the growth medium was replaced with complete medium (isotonic, 300 mOsm) supplemented with increasing osmolarity. Osmolarity was increased at 100 mOsm intervals to 500 mOsm, 600 mOsm, or 800 mOsm by the addition of NaCl plus urea (1.5:1 molar ratio).
ANTIBODIES
The rabbit anti-myc-tag antibody (2272) was obtained from Cell Signaling Technologies (Danvers, MA). Rabbit anti-PSAP (ab68466), rabbit anti-Cathepsin D (ab75852) and goat anti-Myc tag antibody (ab9132) antibodies were obtained from Abcam (Eugene, OR). Rabbit anti-SAP D antibody (sc-27024-R) and rat anti-LAMP1 antibody (sc-19992) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-β-actin antibody was purchased from Sigma-Aldrich (Saint Louis, MO). Mouse antibody against GM130 (610822) were purchased from BD Biosciences (San Jose, CA). Alexa Fluor 488- and 647-conjugated secondary antibodies and DAPI nuclear stain were acquired from ThermoFisher Scientific (Waltham, MA).
WESTERN BLOTTING
Protein samples were prepared from equal numbers of cells grown in 10-cm culture dishes (Corning Inc., Corning, NY). Cells were washed three times with ice-cold PBS and then scraped into 1 ml of PBS, followed by centrifugation at 200 g. Cell pellets were treated for 30 min with non-denaturing lysis buffers containing either 1% Triton X-100 (in 50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 5 mM EDTA, pH 7.4) or 1% n-Dodecyl β-D-maltoside (in 50 mM sodium phosphate buffer, pH 7.4) to extract proteins on ice. The 1% DDM-containing lysis buffer was used to efficiently extract myc-CLN3 and GM130 as we have previously shown [Getty et al., 2013]. Following a high speed spin (14,000 g), the post-nuclear supernatant was collected, and the protein concentration was measured using the Pierce 660 nm Protein Assay (ThermoFisher Scientific, Waltham, MA).
Samples were heated in Laemmli buffer at 37°C for 15 min when looking to detect myc-CLN3 and GM130 or at 100°C for 5 min for PSAP, SAP D, LAMP1, CTSD and β-actin. Proteins were resolved on 10, 12 or 15% polyacrylamide gels. After SDS-PAGE, proteins were transferred to nitrocellulose membranes (Millipore, Billerica, MA) using the standard wet transfer method at 100 V for 90 min. Membranes were then incubated in blocking buffer [5% milk in 100 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Tween-20 (TBST)] for 1 hour. Primary antibodies were applied in blocking buffer overnight at 4°C. According to the manufacturer’s instructions, for incubation with the rabbit anti-myc antibody, the buffer contained 5% BSA instead of milk. After the overnight incubation with the primary antibody, membranes were washed 3 times, for 5 min each with TBST, and incubated with a secondary antibody conjugated with horseradish peroxidase (GE Healthcare) in blocking buffer for 1 hour at room temperature. After washing the membranes with TBST 4 times (5 min each), signal was detected by chemiluminescence using ECL Plus Western Blotting Detection kit (GE Healthcare Life Sciences, Piscataway, NJ) and a BioSpectrum UVP Imaging system (Upland, CA).
IMMUNOFLUORESCENCE
Cells were grown on poly-D-lysine (10 μg/ml) coated coverslips. Cells were fixed in 4% paraformaldehyde (in PBS supplemented with Mg2+ and Ca2+) for 20 min at room temperature, permeabilized with 0.1% Triton X-100 (in PBS containing 0.05% sodium azide) for 5 min at room temperature, blocked in PBS containing 10% goat serum, 2% bovine serum albumin (BSA) and 0.05% sodium azide for 1 hour. After washing with PBS containing 2% BSA and 0.05% sodium azide (PBS-2%BSA-azide) cells were incubated with the primary antibodies in DPBS-2%BSA-azide overnight at 4°C. After washing with DPBS-2%BSA-azide, compatible Alexa Fluor 488-and 647-conjugated secondary antibodies diluted in DPBS-2%BSA-azide were applied for one 1 h at room temperature. Cells then were washed with PBS containing 0.05% sodium azide, mounted in ProLong Anti-Fade mounting medium (ThermoFisher Scientific, Waltham, MA) and sealed with clear nail polish.
Images were acquired using a Nikon Eclipse 90i confocal microscope (Nikon instruments, Inc., Melville, NY) with a CoolSNAP HQ camera (Roper Scientific GmbH, Germany) and NIS-Elements software package.
FLOW CYTOMETRY
After culturing BHK cells as described previously under hyperosmotic or isotonic conditions with the addition of bafilomycin (100 nM) or DMSO for the last 6 hours of the experiment, or after transient transfection with the myc-CLN3-expressing plasmid or the empty vector, the culture medium was replaced with prewarmed (37°C) medium containing 1 μM lysotracker and cells were incubated for 30 min. Afterwards, cells were trypsinized, transferred to Eppendorf tubes and centrifuged at 300 g for 5 min. Supernatant was discarded, the cells were resuspended in PBS and analyzed by flow cytometry (Exc. 647 nm, Em. 668 nm) at a BD Accuri C6 Flow Cytometer (BD Biosciences).
CTSD ENZYME ACTIVITY ASSAY
CTSD activity was measured using the fluorescence-based assay kit #KA0767 by Abnova (Walnut, CA) as described by the manufacturer. Briefly, cells were lysed using the provided lysis buffer, protein concentration was determined using the Pierce 660 nm Protein Assay and equal amounts of proteins were loaded in Corning Costar 96-Well Black Clear-Bottom Plates (Corning, NY). The release of the fluorescently labelled cathepsin-D substrate (GKPILFFRLK(Dnp)-D-R-NH2 labeled with MCA), as indicative of CTSD activity was measured by a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA) at Ex/Em = 328/460 nm.
STATISTICAL ANALYSIS
Statistical significance was determined by 1-way ANOVA with Bonferroni’s post-test for multiple comparisons using GraphPad Prism 6 (GraphPad Software, San Diego, CA).
RESULTS
We have previously reported that in baby hamster kidney (BHK) cells stably expressing myc-tagged human CLN3 (myc-CLN3) under the control of the human ubiquitin promoter, hyperosmotic conditions drastically increased myc-CLN3 protein expression and also enhanced the lysosomal localization of myc-CLN3 [Getty et al., 2013]. In the present study, we analyzed the consequences of hyperosmolarity and increased CLN3 expression on cathepsin D activity and prosaposin processing in BHK cells. Transient transfection of BHK cells with the myc-CLN3-expressing plasmid provided high expression level of myc-CLN3, similar to that induced by hyperosmolarity in BHK cells stably transfected with the myc-CLN3-expressing plasmid (C19 cells) (Fig. 1C, Fig. 2A–B). In this way, the effects of highly increased CLN3 expression under hyperosmolarity (C19 cells) and isotonic conditions (transient transfection) could be compared.
Fig. 1. Hyperosmolarity increases lysotracker staining of lysosomes and elevates CLN3 and LAMP1 protein levels.

(A) Mean fluorescence intensity values of Lysotracker deep red staining in BHK cells stably transfected with either a myc-CLN3-expressing plasmid (C19) or the empty vector (C2), under hyperosmotic (HO) or isotonic (IT) conditions. Bafilomycin, a specific inhibitor of lysosomal ATP-ase, was used to verify that the majority of labeled organelles were lysosomes (B) Mean fluorescence intensity values of Lysotracker deep red staining in BHK cells transiently transfected with a myc-CLN3-expressing plasmid (CLN3) or the empty vector (Vector). (C) Western blot showing the effective transient transfection of BHK cells with the myc-CLN3-expressing plasmid. An anti-myc antibody was used to detect myc-CLN3 and β-actin served as the loading control. (A–B) Symbols represent the values from three separate experiments (each symbol is the average of triplicated measurements) and the bars indicate the means.
Fig. 2. Hyperosmolarity increases myc-CLN3 and LAMP1 protein levels, and decreases prosaposin (PSAP) and saposin D (SAP D) protein levels.

(A) Representative Western blots for myc-CLN3, LAMP 1, GM130, PSAP, SAP D and β-actin in total cell extracts from BHK cells stably transfected with either a myc-CLN3-expressing plasmid (C19) or the empty vector (C2), under hyperosmotic (HO) or isotonic (IT) conditions, and from BHK cells transiently transfected with the myc-CLN3-expressing plasmid (CLN3) or the empty vector (Vector). Cells were incubated with or without bafilomycin, an inhibitor of lysosomal function. (B–C) Densitometric quantification of band intensities from the Western blots. (B) Myc-CLN3 and LAMP 1 band intensities were normalized to the band intensities of GM130 (loading control), a Golgi membrane protein. (C) PSAP and SAP D band intensities were normalized to the band intensities of β-actin (loading control). Symbols represent the individual values from three separate experiments and the bars indicate the means.
HYPEROSMOLARITY INCREASES LYSOTRACKER STAINING OF LYSOSOMES AND ELEVATES CLN3 AND LAMP1 PROTEIN LEVELS
As CLN3 has been widely associated to lysosomal homeostasis [Chattopadhyay et al., 2000; Golabek et al., 2000; Pearce et al., 1999; Vidal-Donet et al., 2013], we tested if hyperosmolarity, which significantly increases CLN3 expression, or increased expression of CLN3 alone (by transient transfection) induced lysosomal changes. BHK cells stably transfected with either the myc-CLN3-expressing plasmid (C19 cells) or the empty vector (C2 cells) were incubated under hyperosmotic (HO) or isotonic (IT) conditions. BHK cells transiently transfected with the myc-CLN3-expressing plasmid were incubated under isotonic conditions. Live cells were stained with Lysotracker deep red, a deep red-fluorescent dye for labeling and tracking acidic organelles, and were analyzed by flow cytometry (Exc. 647 nm, Em. 668 nm). Bafilomycin, a specific inhibitor of lysosomal ATP-ase, was used to verify that the majority of labeled organelles were lysosomes [Yoshimori et al., 1991]. Under isotonic conditions, lysotracker staining was markedly higher in C19 cells compared to C2 cells (Fig. 1A). Lysotracker staining of lysosomes increased dramatically under hyperosmotic conditions in C2 as well as in C19 cells (Fig. 1A); however, the increase was much higher in C19 cells. Increased expression of myc-CLN3 alone by transient transfection did not affect lysotracker staining (Vector vs. CLN3, Fig. 1B–C). The enhanced lysotracker staining can be due to an increase in the number or size of lysosomes, decreased lysosomal pH, or a combination of these three.
In C19 cells stably expressing myc-CLN3, hyperosmolarity considerably increased myc-CLN3 expression (Fig. 2A–B), confirming previous results [Getty et al., 2013]. Bafilomycin (for 6 h) did not significantly change myc-CLN3 protein levels in BHK cells transiently or stably transfected with the myc-CLN3-expressing plasmid (CLN3 and C19, respectively in Fig. 2A–B), indicating that myc-CLN3 is not degraded in lysosomes in these cells within 6 h. In the stably transfected BHK cells (C2 and C19), under hyperosmotic conditions, lysosomal-associated membrane protein 1 (LAMP-1), a type I transmembrane glycoprotein that resides primarily across lysosomal membranes [Carlsson and Fukuda, 1989], was also clearly increased (Fig. 2A–B). GM130, a cis-Golgi membrane protein, was used as a housekeeping protein in the Western blots for myc-CLN3 and LAMP-1 (Fig. 2A), and levels of myc-CLN3 and LAMP-1 were normalized to GM130 levels in the densitometric quantification (Fig. 2B). Our previous study [Getty et al., 2013] demonstrated that under hyperosmotic conditions the protein level of GM130 did not change and the GM130 band separated well from the myc-CLN3 band, making GM130 suitable as a loading control in Western blotting experiments. Furthermore, GM130, as a membrane protein, is a more appropriate loading control for membrane proteins such as CLN3 and LAMP1.
HYPEROSMOLARITY DECREASES PROSAPOSIN (PSAP) AND SAPOSIN D (SAP D) PROTEIN LEVELS INDEPENDENTLY OF THE EXPRESSION LEVEL OF MYC-CLN3
To examine the possible connection of CLN3 to proteins involved in the metabolism of lipid derivatives accumulated in other subtypes of NCL, and to test the involvement of sphingolipid metabolism in the adaptation to hyperosmolarity, PSAP levels along with SAP D levels were assessed by Western blotting in BHK cells stably transfected with the myc-CLN3-expressing plasmid (C19) or the empty vector (C2). PSAP and SAP D levels were decreased under hyperosmotic conditions in both C2 and C19 cells (Fig. 2A, C), independently of the expression level of myc-CLN3 (Fig. 2A, B). Under isotonic conditions, transient transfection of BHK cells with the myc-CLN3-expressing plasmid did not result in a statistically significant decrease in PSAP and SAP D levels (Vector vs. CLN3 in Fig. 2A, C). β-actin, a cytoskeletal protein, was used as a housekeeping protein in the Western blots for PSAP and SAP D (Fig. 2A), and levels of PSAP and SAP D were normalized to β-actin levels in the densitometric quantification (Fig. 2C).
CATHEPSIN D (CTSD) COLOCALIZES WITH MYC-CLN3 BUT HYPEROSMOLARITY DOES NOT CHANGE CTSD PROTEIN LEVEL
Since CTSD is a well-known PSAP converting lysosomal enzyme and CTSD impairment is known to result in lipofuscin accumulation, we studied the possible interaction of CLN3 with CTSD using immunofluorescent staining in BHK cells stably expressing myc-CLN3. Both proteins have been located in lysosomes but CLN3 is a transmembrane protein [Ratajczak et al., 2014] whereas CTSD is a soluble protein [Rawlings and Barrett, 1995]. The confocal microscopic images in Fig. 3A show that CTSD and myc-CLN3 colocalized under both isotonic and hyperosmotic conditions. While myc-CLN3 staining markedly increased under hyperosmotic conditions as compared to isotonic conditions, the increase in CTSD staining was not statistically significant (Fig. 3A–B).
Fig. 3. Cathepsin D (CTSD) colocalizes with myc-CLN3 but hyperosmolarity does not change CTSD protein level.

(A) Representative confocal images of immunofluorescently stained BHK cells stably expressing myc-CLN3 (C19), after incubation under hyperosmotic or isotonic conditions. Merged images were obtained by Image J software by the AND math command of the two source images, and aqua color indicates the colocalization of myc-CLN3 and CTSD. Scale bar indicates 50 μm. Experiments were performed in triplicate. (B) Densitometric analysis of CTSD and myc-CLN3 immunofluorescence intensities in C19 cells. Immunofluorescence intensities were measured in 3 microscopic images for each condition, normalized to the number of cells [250 cells under isotonic conditions (IT) and 340 cells under hyperosmotic conditions (HO)], and expressed as percentage of C19 cells under isotonic conditions. (C) Representative Western blots for myc-CLN3, CTSD and GM130 (loading control) in total cell extracts from BHK cells stably transfected with either a myc-CLN3-expressing plasmid (C19) or the empty vector (C2), under hyperosmotic (HO) or isotonic (IT) conditions. Cells were incubated without or with cycloheximide (100 μM, for 6 h), an inhibitor of protein synthesis. (D) Densitometric quantification of band intensities from the Western blots. Myc-CLN3 and CTSD band intensities were normalized to the band intensities of GM130 (loading control), a Golgi membrane protein. Symbols represent the individual values from three separate experiments and the bars indicate the means.
To further examine CTSD protein levels under hyperosmotic conditions we used Western blotting. Hyperosmolarity (HO), as compared to isotonic (IT) conditions, did not change immature CTSD expression in BHK cells stably transfected with either the myc-CLN3-expressing plasmid (C19) or the empty vector (C2) (Fig. 3C–D). In order to discern if CTSD or CLN3 protein synthesis or degradation processes were affected, cells were incubated with cycloheximide (100 μM), an inhibitor of protein biosynthesis, for 6 h. Immature CTSD levels were not affected by cycloheximide (Fig. 3C–D). Interestingly, cycloheximide decreased myc-CLN3 protein level under hyperosmotic conditions but did not change it under isotonic conditions, indicating that the observed increase in myc-CLN3 level under hyperosmotic conditions is due to increased synthesis rather than decreased protein degradation (Fig. 3C–D).
DECREASED CTSD ACTIVITY IN BHK CELLS EXPRESSING MYC-CLN3
To further investigate the possible interaction between myc-CLN3 and CTSD, we studied CTSD activity in BHK cells stably transfected with either the myc-CLN3-expressing plasmid (C19) or the empty vector (C2), incubated under hyperosmotic (HO) or isotonic (IT) conditions, as well as in BHK cells transiently transfected with the myc-CLN3-expressing plasmid and incubated under isotonic conditions. CTSD activity was measured in cell lysates by a fluorescence-based assay (Abnova). Under isotonic conditions, CTSD activity was significantly lower in C19 cells (stably expressing myc-CLN3) than in C2 control cells (not expressing myc-CLN3) (Fig. 4). Hyperosmolarity decreased CTSD activity in C2 cells and further decreased in C19 cells (Fig. 4). However, transient expression of myc-CLN3 under isotonic conditions also significantly decreased the enzyme activity of CTSD (Vector vs. CLN3 in Fig. 4), indicating a role of CLN3 in the regulation of CTSD activity under isotonic conditions.
Fig. 4. Decreased CTSD activity in BHK cells expressing myc-CLN3.
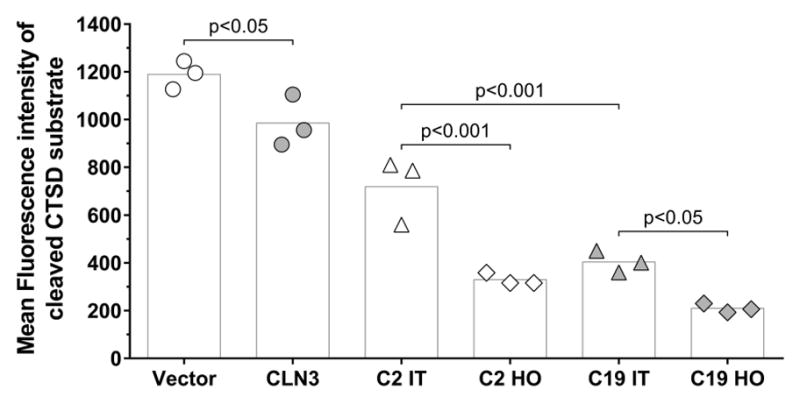
Mean fluorescence intensity values of cleaved CTSD substrate in BHK cells stably transfected with either a myc-CLN3-expressing plasmid (C19) or the empty vector (C2), under hyperosmotic (HO) or isotonic (IT) conditions, and from BHK cells transiently transfected with the myc-CLN3-expressing plasmid (CLN3) or the empty vector (Vector). CTSD activity was measured in cell lysates by a fluorescence-based assay (Abnova). Symbols represent the values from three separate experiments (each symbol is the average of triplicated measurements) and the bars indicate the means.
DISCUSSION
We have previously studied the osmoregulation of CLN3 expression and localization using baby hamster kidney (BHK) cells stably expressing myc-tagged human CLN3. Along an increase in human CLN3 mRNA and protein levels, immunofluorescent staining for the myc tag indicated that the subcellular localization of myc-CLN3 changed dramatically: under isosmolar conditions (300 mOsm), it was found in a punctate vesicular pattern surrounding the nucleus with prominent Golgi and lysosomal localizations but hyperosmolarity (800 mOsm) enhanced its lysosomal localization [Getty et al., 2013]. Nevertheless, this past study did not clarify if the change in CLN3 subcellular distribution was a consequence of increased osmolarity or the increased CLN3 expression, and did not examine the possible lysosomal functions associated to the increased CLN3 expression.
In the present study, we analyzed the consequences of hyperosmolarity and increased CLN3 expression on lysotracker staining of lysosomes, CTSD activity and prosaposin processing using BHK cells transiently or stably expressing myc-CLN3. Western blotting confirmed that osmotic stress significantly increased myc-CLN3 expression. We also found that under isotonic conditions, lysotracker staining was markedly higher in BHK cells stably expressing myc-CLN3 compared to control cells, and hyperosmolarity increased lysotracker staining of lysosomes in both control and myc-CLN3 expressing BHK cells (Fig. 1A); however, the increase was much higher in myc-CLN3 expressing cells. These results indicated a positive correlation between CLN3 expression level and lysotracker staining intensity. Increased expression of myc-CLN3 alone by transient transfection, however, did not affect lysotracker staining (Fig. 1B), suggesting that a short-term increase of CLN3 expression (until 18–24 h post-transfection) is not able to induce the same lysosomal changes as the continuously enhanced CLN3 expression in BHK cells stably expressing myc-CLN3.
The enhanced lysotracker staining in our study could result from an increase in the number or size of lysosomes, decreased lysosomal pH, or a combination of these three. The observed increase in lysotracker staining could also be due to an increase in autophagy, a mechanism of degradation of dysfunctional cellular organelles and excess proteins, where an autophagic vacuole is formed and fused with late endosomes and lysosomes [Shacka et al., 2008]. Supporting this notion, a recent study showed that hyperosmotic stress (up to 600 mOsm by adding NaCl) in rat notochordal cells activated autophagy via the Ca2+-dependent AMPK/mTOR pathway [Jiang et al., 2015]. Furthermore, Fumarola et al. showed in Jurkat cells that hyperosmotic conditions (up to 600 mOsm by adding sorbitol) induced deactivation of the mTOR pathway and consequently activated autophagy [Fumarola et al., 2005]. Abnormalities in autophagy have also been reported in CLN3-deficient cells [Lojewski et al., 2014; Vidal-Donet et al., 2013].
Our results indicate a role of CLN3 in the regulation of CTSD activity. Under physiological, isotonic conditions, BHK cells with either transient or stable myc-CLN3 expression had significantly decreased CTSD enzyme activity (Fig. 4).
Ceramides are proapoptotic molecules that are originated by de novo synthesis from serine and palmitate or by breakdown of sphingomyelin via different sphingomyelinases [Hannun and Obeid, 2008; Ogretmen and Hannun, 2001]. Acid sphingomyelinase-derived ceramide specifically binds to and activates CTSD [Heinrich at al., 1999]. Several reports have related impaired CLN3 expression to the accumulation of ceramide [Kang et al., 2013; Makoukji et al., 2015; Rusyn et al., 2008]. Rusyn et al. reported that wild type CLN3, but not mutant CLN3, binds galactosylceramide, and suggested that CLN3 may function as a galactosylceramide transporter [Rusyn et al., 2008]. More recently, a study determined the gene expression of CLN3 and 28 enzymes involved in sphingolipid metabolism in non-tumor vs. tumor samples from fresh and formalin-fixed/paraffin-embedded breast tissue [Makoukji et al., 2015]. CLN3 mRNA was overexpressed in the analyzed breast tumor samples, and the mRNA levels of ceramide synthases 2 and 6, sphingolipid delta(4)-desaturase (DEGS2), and acidic sphingomyelinase (SMPD1) were also higher in breast cancer than in control tissue [Makoukji et al., 2015], suggesting a link between CLN3 and sphingolipid metabolism.
Prosaposin (PSAP) is the lysosomal precursor of four Sphingolipid Activator Proteins or saposins (SAP A,B,C and D) that act as cofactors in the catabolism of glycosphingolipids to generate ceramides [Kishimoto et al., 1992]. Saposins A and D are the main components of storage material in the infantile and congenital forms of NCLs [Siintola et al., 2006; Tyynela et al., 1993]. Additionally, lysosomal storage disorders caused by deficiency in enzymes activated by saposins can be mimicked by mutations in the saposin(s) involved. For instance, saposin A deficiency can lead to progressive encephalopathy and abnormal myelination in the cerebral white matter, as seen in Krabbe disease [Matsuda et al., 2001], and saposin C deficiency can reproduce the neurological symptoms found in Gaucher disease [Horowitz and Zimran, 1994]. In the present study, we found that levels of the precursor PSAP and the mature SAP D protein were decreased by hyperosmolarity (Fig. 2). CTSD is responsible for the cleavage of the precursor PSAP and the generation of SAPs in the lysosomes [Hiraiwa et al., 1997]. Although, CTSD activity was decreased in the cells expressing myc-CLN3 (Fig. 4), we did not observe any changes in PSAP to SAP D conversion (Fig. 2). Besides CTSD, mesotrypsin and caspase-14 can also process PSAP, as a recent study revealed [Yamamoto-Tanaka et al., 2014], and these proteases may compensate for the decreased CTSD activity in our myc-CLN3-expressing cells.
In summary, our results suggest that CLN3 is involved in the regulation of CTSD activity.
Acknowledgments
Grant sponsor: NIH; Grant numbers: NS044310, NS067147
Grant sponsor: Luke and Rachel Batten Foundation; Grant number: N/A
This work was supported in part by the Luke and Rachel Batten Foundation and the National Institutes of Health (NS044310, NS067147).
Abbreviations
- CTSD
cathepsin D
- HO
hyperosmotic
- IT
isotonic
- JNCL
juvenile Neuronal Ceroid Lipofuscinosis
- LAMP1
lysosome-associated membrane protein-1
- NCLs
Neuronal Ceroid Lipofuscinoses
- PSAP
prosaposin
- SAP
saposin
- SMASE
sphingomyelinase
Footnotes
CONFLICT OF INTEREST
None of the authors of this paper has conflict of interest relating to the publication.
References
- Carlsson SR, Fukuda M. Structure of human lysosomal membrane glycoprotein 1. Assignment of disulfide bonds and visualization of its domain arrangement. J Biol Chem. 1989;264:20526–31. [PubMed] [Google Scholar]
- Chattopadhyay S, Muzaffar NE, Sherman F, Pearce DA. The yeast model for batten disease: mutations in BTN1, BTN2, and HSP30 alter pH homeostasis. J Bacteriol. 2000;182:6418–23. doi: 10.1128/jb.182.22.6418-6423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium TIBD. Isolation of a novel gene underlying Batten disease, CLN3. Cell. 1995;82:949–57. doi: 10.1016/0092-8674(95)90274-0. [DOI] [PubMed] [Google Scholar]
- Cotman SL, Staropoli JF. The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post-Golgi trafficking. Clin Lipidol. 2012;7:79–91. doi: 10.2217/clp.11.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumarola C, La Monica S, Guidotti GG. Amino acid signaling through the mammalian target of rapamycin (mTOR) pathway: Role of glutamine and of cell shrinkage. J Cell Physiol. 2005;204:155–65. doi: 10.1002/jcp.20272. [DOI] [PubMed] [Google Scholar]
- Getty A, Kovacs AD, Lengyel-Nelson T, Cardillo A, Hof C, Chan CH, Pearce DA. Osmotic stress changes the expression and subcellular localization of the Batten disease protein CLN3. PLoS One. 2013;8:e66203. doi: 10.1371/journal.pone.0066203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golabek AA, Kida E, Walus M, Kaczmarski W, Michalewski M, Wisniewski KE. CLN3 protein regulates lysosomal pH and alters intracellular processing of Alzheimer’s amyloid-beta protein precursor and cathepsin D in human cells. Mol Genet Metab. 2000;70:203–13. doi: 10.1006/mgme.2000.3006. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–50. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- Heinrich M, Wickel M, Schneider-Brachert W, Sandberg C, Gahr J, Schwandner R, Weber T, Saftig P, Peters C, Brunner J, Krönke M, Schütze S. Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J. 1999;18:5252–63. doi: 10.1093/emboj/18.19.5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraiwa M, Martin BM, Kishimoto Y, Conner GE, Tsuji S, O’Brien JS. Lysosomal proteolysis of prosaposin, the precursor of saposins (sphingolipid activator proteins): its mechanism and inhibition by ganglioside. Arch Biochem Biophys. 1997;341:17–24. doi: 10.1006/abbi.1997.9958. [DOI] [PubMed] [Google Scholar]
- Horowitz M, Zimran A. Mutations causing Gaucher disease. Hum Mutat. 1994;3:1–11. doi: 10.1002/humu.1380030102. [DOI] [PubMed] [Google Scholar]
- Huber RJ, Myre MA, Cotman SL. Loss of Cln3 function in the social amoeba Dictyostelium discoideum causes pleiotropic effects that are rescued by human CLN3. PLoS One. 2014;9:e110544. doi: 10.1371/journal.pone.0110544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang LB, Cao L, Yin XF, Yasen M, Yishake M, Dong J, Li XL. Activation of autophagy via Ca(2+)-dependent AMPK/mTOR pathway in rat notochordal cells is a cellular adaptation under hyperosmotic stress. Cell Cycle. 2015;14:867–79. doi: 10.1080/15384101.2015.1004946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Kim JB, Heo TH, Kim SJ. Cell cycle arrest in Batten disease lymphoblast cells. Gene. 2013;519:245–50. doi: 10.1016/j.gene.2013.02.022. [DOI] [PubMed] [Google Scholar]
- Kishimoto Y, Hiraiwa M, O’Brien JS. Saposins: structure, function, distribution, and molecular genetics. J Lipid Res. 1992;33:1255–67. [PubMed] [Google Scholar]
- Koike M, Nakanishi H, Saftig P, Ezaki J, Isahara K, Ohsawa Y, Schulz-Schaeffer W, Watanabe T, Waguri S, Kametaka S, Shibata M, Yamamoto K, Kominami E, Peters C, von Figura K, Uchiyama Y. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J Neurosci. 2000;20:6898–906. doi: 10.1523/JNEUROSCI.20-18-06898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmann K, Uusi-Rauva K, Scifo E, Tyynela J, Jalanko A, Braulke T. Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim Biophys Acta. 2013;1832:1866–81. doi: 10.1016/j.bbadis.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Kovacs AD, Saje A, Wong A, Ramji S, Cooper JD, Pearce DA. Age-dependent therapeutic effect of memantine in a mouse model of juvenile Batten disease. Neuropharmacology. 2012;63:769–75. doi: 10.1016/j.neuropharm.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JM, Adams H, Rothberg PG, Augustine EF, Marshall FJ, Deblieck EA, Vierhile A, Beck CA, Newhouse NJ, Cialone J, Levy E, Ramirez-Montealegre D, Dure LS, Rose KR, Mink JW. Quantifying physical decline in juvenile neuronal ceroid lipofuscinosis (Batten disease) Neurology. 2011;77:1801–7. doi: 10.1212/WNL.0b013e318237f649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lojewski X, Staropoli JF, Biswas-Legrand S, Simas AM, Haliw L, Selig MK, Coppel SH, Goss KA, Petcherski A, Chandrachud U, Sheridan SD, Lucente D, Sims KB, Gusella JF, Sondhi D, Crystal RG, Reinhardt P, Sterneckert J, Scholer H, Haggarty SJ, Storch A, Hermann A, Cotman SL. Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum Mol Genet. 2014;23:2005–22. doi: 10.1093/hmg/ddt596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makoukji J, Raad M, Genadry K, El-Sitt S, Makhoul NJ, Saad Aldin E, Nohra E, Jabbour M, Sangaralingam A, Chelala C, Habib RH, Boulos F, Tfayli A, Boustany RM. Association between CLN3 (Neuronal Ceroid Lipofuscinosis, CLN3 Type) Gene Expression and Clinical Characteristics of Breast Cancer Patients. Front Oncol. 2015;5:215. doi: 10.3389/fonc.2015.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda J, Vanier MT, Saito Y, Tohyama J, Suzuki K, Suzuki K. A mutation in the saposin A domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Hum Mol Genet. 2001;10:1191–9. doi: 10.1093/hmg/10.11.1191. [DOI] [PubMed] [Google Scholar]
- Ogretmen B, Hannun YA. Updates on functions of ceramide in chemotherapy-induced cell death and in multidrug resistance. Drug Resist Updat. 2001;4:368–77. doi: 10.1054/drup.2001.0225. [DOI] [PubMed] [Google Scholar]
- Pearce DA, Ferea T, Nosel SA, Das B, Sherman F. Action of BTN1, the yeast orthologue of the gene mutated in Batten disease. Nat Genet. 1999;22:55–8. doi: 10.1038/8861. [DOI] [PubMed] [Google Scholar]
- Ratajczak E, Petcherski A, Ramos-Moreno J, Ruonala MO. FRET-assisted determination of CLN3 membrane topology. PLoS One. 2014;9:e102593. doi: 10.1371/journal.pone.0102593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ. Families of aspartic peptidases, and those of unknown catalytic mechanism. Methods Enzymol. 1995;248:105–20. doi: 10.1016/0076-6879(95)48009-9. [DOI] [PubMed] [Google Scholar]
- Rusyn E, Mousallem T, Persaud-Sawin DA, Miller S, Boustany RM. CLN3p impacts galactosylceramide transport, raft morphology, and lipid content. Pediatr Res. 2008;63:625–31. doi: 10.1203/PDR.0b013e31816fdc17. [DOI] [PubMed] [Google Scholar]
- Salvioli R, Ricci-Vitiani L, Tatti M, Scarpa S, De Maria R, Vaccaro AM. The secretion and maturation of prosaposin and procathepsin D are blocked in embryonic neural progenitor cells. Biochim Biophys Acta. 2008;1783:1480–9. doi: 10.1016/j.bbamcr.2008.01.033. [DOI] [PubMed] [Google Scholar]
- Shacka JJ, Roth KA, Zhang J. The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front Biosci. 2008;13:718–36. doi: 10.2741/2714. [DOI] [PubMed] [Google Scholar]
- Siintola E, Partanen S, Stromme P, Haapanen A, Haltia M, Maehlen J, Lehesjoki AE, Tyynela J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain. 2006;129:1438–45. doi: 10.1093/brain/awl107. [DOI] [PubMed] [Google Scholar]
- Stein CS, Yancey PH, Martins I, Sigmund RD, Stokes JB, Davidson BL. Osmoregulation of ceroid neuronal lipofuscinosis type 3 in the renal medulla. Am J Physiol Cell Physiol. 2010;298:C1388–400. doi: 10.1152/ajpcell.00272.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyynela J, Palmer DN, Baumann M, Haltia M. Storage of saposins A and D in infantile neuronal ceroid-lipofuscinosis. FEBS Lett. 1993;330:8–12. doi: 10.1016/0014-5793(93)80908-d. [DOI] [PubMed] [Google Scholar]
- Uusi-Rauva K, Kyttala A, van der Kant R, Vesa J, Tanhuanpaa K, Neefjes J, Olkkonen VM, Jalanko A. Neuronal ceroid lipofuscinosis protein CLN3 interacts with motor proteins and modifies location of late endosomal compartments. Cell Mol Life Sci. 2012;69:2075–89. doi: 10.1007/s00018-011-0913-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Donet JM, Carcel-Trullols J, Casanova B, Aguado C, Knecht E. Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS One. 2013;8:e55526. doi: 10.1371/journal.pone.0055526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. 2012;79:183–91. doi: 10.1212/WNL.0b013e31825f0547. [DOI] [PubMed] [Google Scholar]
- Yamamoto-Tanaka M, Motoyama A, Miyai M, Matsunaga Y, Matsuda J, Tsuboi R, Hibino T. Mesotrypsin and caspase-14 participate in prosaposin processing: potential relevance to epidermal permeability barrier formation. J Biol Chem. 2014;289:20026–38. doi: 10.1074/jbc.M113.543421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991;266:17707–12. [PubMed] [Google Scholar]