

Abstract
Flash nanoprecipitation (FNP) has proven to be a powerful tool for the rapid and scalable assembly of solid-core nanoparticles from block copolymers. The process can be performed using a simple confined impingement jets mixer and provides an efficient and reproducible method of loading micelles with hydrophobic drugs. To date, FNP has not been applied for the fabrication of complex or vesicular nanoarchitectures capable of encapsulating hydrophilic molecules or bioactive protein therapeutics. Here, we present FNP as a single customizable method for the assembly of bicontinuous nanospheres, filomicelles and vesicular, multilamellar and tubular polymersomes from poly(ethylene glycol)-bl-poly(propylene sulfide) block copolymers. Multiple impingements of polymersomes assembled via FNP were shown to decrease vesicle diameter and polydispersity, allowing gram-scale fabrication of monodisperse polymersomes within minutes. Furthermore, we demonstrate that FNP supports the simultaneous loading of both hydrophobic and hydrophilic molecules respectively into the polymersome membrane and aqueous lumen, and encapsulated enzymes were found to be released and remain active following vesicle lysis. As an example application, theranostic polymersomes were generated via FNP that were dual loaded with the immunosuppressant rapamycin and a fluorescent dye to link targeted immune cells with the elicited immunomodulation of T cells. By expanding the capabilities of FNP, we present a rapid, scalable and reproducible method of nanofabrication for a wide range of nanoarchitectures that are typically challenging to assemble and load with therapeutics for controlled delivery and theranostic strategies.
Keywords: Self-assembly, polymersome, flash nanoprecipitation, drug delivery, block copolymer
Introduction
Nanocarriers present a versatile method of controlled delivery for bioactive molecules that may otherwise be too hydrophobic or susceptible to degradation for therapeutic applications. A key parameter of nanocarrier design is the nanoarchitecture, which strongly influences in vivo transport, biodistribution, and cellular uptake [1–3]. The ability to tailor nanocarrier architecture has resulted in numerous advancements in targeted delivery, providing enhanced circulation time, membrane permeation and the simultaneous loading of multiple molecules that differ in water solubility [2, 4, 5]. The self-assembly of block-copolymers allows the formation of diverse soft nanoarchitectures, but presents several engineering challenges, namely: loading efficiency, scalability, repeatability and ease of fabrication. Flash nanoprecipitation (FNP) is a fabrication technique capable of addressing the majority of these issues, but has so far only been applied for the formation of solid-core nanoparticles and their loading with hydrophobic drugs [6, 7].
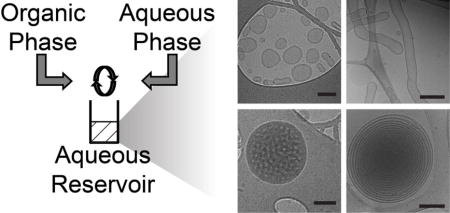
Protocols for FNP employ multi-stream mixers in which an organic solution of solubilized hydrophobic drug and an amphiphilic block copolymer dissolved in a water-miscible common solvent are impinged upon an aqueous solution briefly under turbulent conditions and subsequently introduced into an aqueous reservoir (Fig. 1A) [8, 9]. The supersaturated conditions generated by the turbulent mixing induces precipitation and nucleation of the hydrophobic solute and coprecipitation of the block copolymer for stabilization of monodisperse nanoparticles with hydrophobic drug cores [6, 7, 10]. Mixing occurs over millisecond timescales and is followed by transfer to a reservoir of aqueous nonsolvent to strip away solvent still associating with the aggregated drug and block copolymer. Modifications of this protocol has resulted in the rapid, scalable formation of a variety of nanocolloids with hydrophobic cores containing therapeutics and imaging agents with low water solubility [11–13], and only rarely for hydrophilic and ionic molecules [14]. While recent focus has been placed on this process of competitive aggregation for the scalable loading of nanoparticles with hydrophobic drugs, FNP was originally applied to achieve rapid changes in solvent quality for homogenous precipitation and self-assembly of block copolymers to investigate the mechanism and kinetics of micellization [15].
Fig. 1.
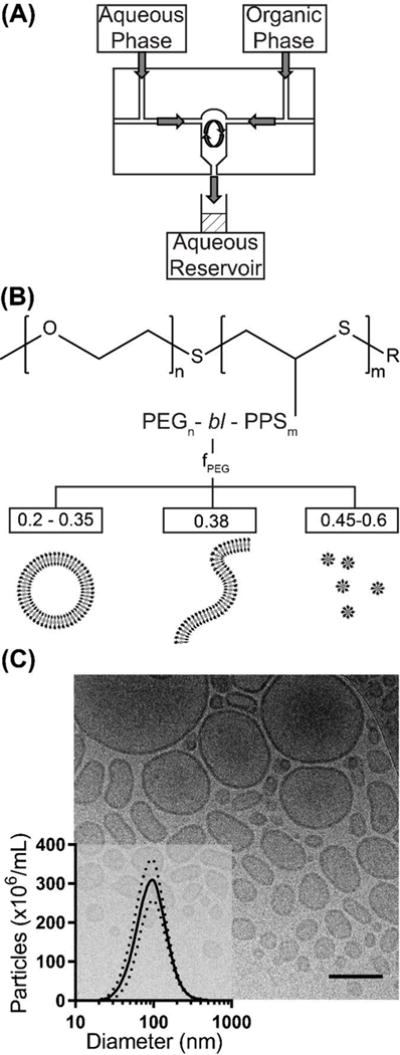
Overview of polymersome formation by flash nanoprecipitation (FNP). (A) A schematic of the CIJ mixer. (B) The structure of the diblock copolymer poly(ethylene glycol)-block-poly(propylene sulfide), and the weight fraction (fPEG) dependent nanostructures known to form using the thin film hydration method. (C) A representative cryoTEM image of polymersomes formed by FNP, scale bar = 300 nm. Inset is a size distribution of polymersomes measured by nanoparticle tracking analysis (NTA), n = 6. Standard deviation is represented by the dotted lines.
While micelles can form within nanoseconds, the combined entropically and enthalpically driven transition in aggregate morphology to filomicelles, bilayer sheets, and vesicles occurs over much longer timescales [16, 17]. The glass transition (Tg) of the amphiphile’s hydrophobic block influences chain flexibility and as a result the timescale of aggregate shape transformations, which can range from hours for glassy high Tg polymers to milliseconds for low Tg polymers [18–20]. Thus, the rapid mixing followed by an immediate increase in water content within the reservoir during FNP effectively minimizes the chain mobility of the hydrophobic copolymer blocks to stably lock the molecular orientation of the assembly, and this is particularly effective for high Tg polymers like polystyrene [6, 8, 15, 21, 22]. It was determined that the organic solvent must be removed from the assembly quickly to prevent nanoparticle instability and ripening, which can be achieved via flash solvent evaporation or using aqueous reservoirs with large volumes to decrease the solvent concentration [23, 24]. The use of a low Tg hydrophobic block in the rapid mixing context of FNP may allow access to more complex self-assembled soft nanoarchitectures such as polymer vesicles (i.e. polymersomes).
In the context of drug delivery, micelles and solid-core nanoparticles are limited to hydrophobic molecules, or must be covalently or electrostatically associated with hydrophilic molecules, generally on their surface. In contrast, some nanoarchitectures, like polymersomes, are amenable to the loading of hydrophilic molecules without chemical modification, which can be advantageous for maintaining the bioactivity of therapeutics. Structurally analogous to liposomes, polymersomes possess enhanced physical and chemical stability and have emerged as versatile drug delivery vehicles [25, 26]. Polymersomes are comprised of three separate topological regions: an inner aqueous cavity, a hydrophobic membrane and an external surface that together allow simultaneous transport of both water soluble and lipophilic payloads as well as incorporation of adhesive and targeting moieties. A key advantage of the inner lumen is the ability to encapsulate and protect sensitive biologics, such as enzymes and nucleic acids. The size and shape of these nanocarriers impacts their biodistribution, systemic clearance, cellular internalization, optical properties and overall therapeutic potential [1–3, 27, 28]. Aggregate morphology can be specified by synthesizing block copolymers with a particular hydrophilic weight fraction (typically >45% for polymersomes), kinetically trapping metastable intermediate structures during self-assembly or modulating the vesicles after formation often using shear forces or osmotic pressure gradients [18, 19, 22, 29–31]. These methods have resulted in a wide range of nanostructures with unique properties and applications, including tubule polymersomes, multilamellar nested vesicles and bicontinuous nanostructures [32–35].
High throughput assembly of soft nanoarchitectures remains a challenge, as most of these morphologies can require days for formation and represent only a small fraction of the assembled aggregate population. The most commonly used methods of polymersome formation from di- or tri-block polymers are diverse variations of thin film hydration, solvent dispersion and microfluidics [22, 36–38]. Of these methods, thin film hydration and microfluidics have proven to be most amenable to the loading of protein biologics, as they avoid extensive exposure of payloads to organic solvents that can disrupt protein conformation and activity [3, 39–41]. Unfortunately, thin film hydration cannot control for vesicle size and requires additional processing steps, primarily by extrusion through nanoporous membranes. Microfluidics is low throughput (μL/min) and fabricates primarily microscale vesicles due to restrictions on channel dimensions. Thus although numerous methods have been developed for polymersome formation, a need still exists for facile, rapid, high throughput methods capable of both specifying nanostructure morphology and loading diverse bioactive payloads. Poly(ethylene glycol)-bl-poly(propylene sulfide) (PEG-bl-PPS) is a highly versatile self-assembling block copolymer that can be engineered to stably form polymersomes and a variety of other nanoarchitectures. This ability is in part due to the low Tg of PPS (227 K), which permits high chain flexibility at room temperature and rapid transitions between metastable aggregate morphologies [42, 43]. Depending on the molecular weight ratio of the hydrophilic PEG and hydrophobic PPS blocks as well as the method of assembly, PEG-bl-PPS can assemble into micelles, vesicles and filomicelles [42, 44]. Even at relatively low MW, PEG-bl-PPS aggregates are highly stable in dilute aqueous solutions as evidenced by an octanol/water partition coefficient (logP) of 13.07 and a critical micelle concentration of 3 × 10−6 M [45]. This stability has allowed PEG-bl-PPS nanocarriers to support the delivery of a variety of therapeutic molecules and fluorophores both in vitro and in vivo [3, 39, 46, 47]. PEG-bl-PPS nanocarriers are non-inflammatory, and elicited cellular responses are induced by the transported payload and not the polymer itself [39]. Furthermore, the oxidation-sensitivity of PPS provides a means for controlled degradation and payload release via photo-oxidation or cell-derived ROS as well as systemic clearance of the block copolymer through the kidneys [3, 39, 42, 46]. Small scale assembly and loading of PEG-bl-PPS nanocarriers have been achieved via thin film hydration, direct hydration, solvent extraction, and freeze thaw-cycling, which, in the case of polymersomes, subsequently requires extensive extrusion to form monodisperse nanocarriers [39, 42, 44, 48].
We hypothesized that under the standard conditions of use, FNP may serve as a scalable and rapid method to assemble PEG-bl-PPS copolymers into diverse soft nanoarchitectures in addition to micelles. The rapid millisecond timescale of PEG-bl-PPS transitions between metastable intermediate morphologies more closely matches the mixing time of FNP, which may allow kinetic entrapment of nanostructures that are otherwise rarely observed outside of molecular dynamics models [16]. Here, we demonstrate that by adjusting the aqueous solution conditions, number of impingements and reservoir volume, FNP can rapidly assemble PEG-bl-PPS block copolymers into bicontinuous nanospheres, filomicelles and vesicular, multilamellar and tubular polymersomes. Many of these nanocarrier morphologies are difficult to achieve reproducibly and have not been previously reported for PEG-bl-PPS copolymer systems. We additionally demonstrate that multiple impingements of polymersomes allowed further modulation of the obtained nanostructure and a rapid method of decreasing polydispersity, which presents an alternative to more time-intensive extrusion. Furthermore, we found that diverse hydrophilic molecules, including proteins, could be stably encapsulated within polymersomes via FNP and retain their bioactivity following release. Co-loading of therapeutics with fluorescent dyes allowed rapid formation of theranostic polymersomes. As an immunotheranostic (i.e combining immunotherapy and diagnostics) application of this methodology, we demonstrate enhanced immunosuppression of T cells by low dosage subcutaneous (SC) injection of rapamycin loaded within fluorescent polymersomes. Flow cytometric analysis allowed association of the therapeutic effect with intracellular delivery to lymph node resident and splenic macrophages. Our results enhance the versatility of FNP and may provide a facile high throughput method to fabricate drug-loaded polymersomes for biomedical applications.
Results and Discussion
Formation of Polymersomes via Flash Nanoprecipitation
The rate of self-assembly strongly influences the nanoarchitecture of block copolymer aggregates, and for FNP, at a standard temperature, this is primarily determined by the polymer concentration, steric stabilization of the hydrophilic block, similarity in solubility parameters of the common solvent and non-solvent for both the hydrophilic and hydrophobic blocks, and the rate of removal of the solvent upon addition to the non-solvent reservoir [8, 15, 17, 23, 36]. Although many of these conditions have been optimized for specific block copolymer chemistries and drug loading, several standard practices have been universally adopted for FNP protocols to achieve stable kinetically trapped nanoparticles. These include the use of tetrahydrofuran (THF) and pure water respectively as the solvent and nonsolvent as well as large reservoir-to-solvent volume ratios [8, 9, 49, 50]. Matching Hildebrand solubility parameters is a good indicator of miscibility, and thus polymers and drugs with Hildebrand solubility parameters similar to that of THF will likely be suitable for standard FNP protocols. Decreasing the polymer concentration as well as solvent selection have been employed to both specify nanoparticle size and to enhance payload complexation with the block copolymer [6, 51]. We therefore hypothesized that the use of a diblock copolymer with an appropriate hydrophilic to hydrophobic block molecular weight ratio and low Tg may be induced to form polymersomes under the millisecond mixing conditions of FNP. The Tg and Hildebrand solubility parameter (δ) for PPS (δ = 17.9 MPa1/2) both suggested PEG-bl-PPS to be well suited for the kinetic entrapment of metastable aggregate morphologies using standard protocols for FNP in a confined impingement jets (CIJ) mixer, which involves the impingement of THF (δ = 18.6 MPa1/2) with an aqueous solution at a ffow rate of 1.2 × 10−6 m3/s (Fig. 1A) [9].
In order to investigate the potential for FNP to form polymersomes, we began by utilizing PEG17-bl-PPS30-Thiol, a polymer already demonstrated to form polymersomes via both thin film rehydration and solvent dispersion [3, 39, 44, 46]. This copolymer was dissolved in THF, impinged at 1.2 × 10−6 m3/s against 10 mM phosphate buffered saline (1xPBS) within a CIJ mixer and introduced into a 1xPBS aqueous reservoir (Fig. 1A, B). The resulting assemblies were analyzed via cryoTEM and nanoparticle tracking analysis (NTA) to respectively assess their morphology and size, and were found to consist of polymersomes (Fig. 1C). The use of a PBS salt solution served two purposes. First, vesicular nanoarchitectures maintain osmotic balance with downstream biological fluids in which they will be applied, or risk rupture. Second, we suspected that decreasing the PEG solubility and resulting rate of steric stabilization with a kosmotropic salt solution would allow increased chain mobility for vesicle assembly [6, 52, 53]. To further enhance chain mobility, we performed FNP under solvent conditions which normally result in unstable Ostwald ripening and coalescence. Specifically, the volume ratio of THF/aqueous dilution within the reservoir was only 1:6, while ratios of 1:20 are often suggested in other implementations of FNP for rapid solvent extraction to kinetically trap micellar nanostructures [9]. Use of these conditions (decreased PEG solubility and increased organic solvent concentration in the reservoir) likely provided a system of dynamic self-assembly that allowed continued assembly of the PEG17-bl-PPS30-Thiol into polymersomes within the aqueous reservoir. This was later verified by the observed loading of fluorescently tagged 10 kDa dextran from the reservoir into the polymersome lumen (Fig. S1A). Switching the solvent to dimethylformamide (δ = 24.8 MPa1/2) resulted in the formation of micelles for copolymers that were initially found to assemble vesicles when THF was used as the common solvent. This result would be expected due the lower similarity in solubility parameters between DMF and PPS relative to THF and PPS, which would lower chain flexibility and produce slower kinetics for nanostructure transitions. Slowing the transition kinetics through the use of less similar common solvents is known to promote micelle instead of vesicle formation for other block copolymer systems [17].
Multiple Impingements Reduce Polymersome Diameter and Polydispersity
Nanocarriers used in biomedical applications are typically expected to have a low polydispersity for therapeutic reproducibility and consistency in biodistribution and cellular targeting. The polymersomes formed by FNP (Table 1) demonstrated a polydispersity index (PDI) that ranged from 0.220 to 0.634, which is similar to polymersomes formed by solvent dispersion and thin film rehydration that require subsequent extrusion through membranes with nanoscale porosity to achieve monodisperse vesicles (Fig. 2A and S2). The extrusion process can be time-consuming, result in the loss of product, potentially reduce loading efficiency, and presents an opportunity for the introduction of contaminants and endotoxin. Shear flow has been demonstrated to be a viable and underexplored mechanism to influence the shape and polydispersity of metastable aggregate states [30]. Since vesicle uniformity has been found to improve with increasing shear rate, we hypothesized that repeated impingement of polymersomes within the CIJ mixer in the continued presence of organic solvent, promoting continued fluidity of the PPS membrane, may decrease vesicle polydispersity without the need for subsequent extrusion.
Table 1.
Relationship between PEG weight fraction (fPEG) and morphology. Com/Aq Solv = Common/Aqueous Solvents used during the impingement process. M = Micelles, FM = Filomicelles, BN = Bicontinuous Nanospheres, P = Polymersomes, MLP = Multilamellar Polymersomes, TP = Tubular Polymersomes. Predominant population(s) shown in bold.
| # | PEG DoP | PPS DoP | fPEG | End Capping | Com/Aqu Solv | D (nm) | PDI | Morphology |
|---|---|---|---|---|---|---|---|---|
| 1 | 17 | 75 | 0.119 | Thiol | THF/PBS | 143.13 | 0.62 | BN, M, P |
| 2 | 17 | 44 | 0.187 | Benzyl | THF/PBS | 116.53 | 0.24 | P |
| 3 3‡ |
17 | 38 | 0.21 | Thiol | THF/PBS THF/Water |
115.29 N/A |
0.63 N/A |
P, MLP*, TP* FM |
| 4 4† |
17 | 36 | 0.219 | Thiol | THF/PBS DMF/PBS |
80.51 41.53 |
0.37 0.31 |
P, MLP*, TP* M, P** |
| 5 | 45 | 96 | 0.219 | Benzyl | THF/PBS | 20.54 | 0.25 | M |
| 6 | 17 | 35 | 0.224 | Pyridyl Sulfide | THF/PBS | 68.58 | 0.22 | P, MLP*, TP* |
| 7 | 17 | 33 | 0.235 | Thiol | THF/PBS | 95.06 | 0.55 | P, MLP* |
| 8 | 17 | 30 | 0.252 | Thiol | THF/PBS | 97.96 | 0.52 | P, MLP* |
| 9 | 17 | 23 | 0.305 | Thiol | THF/PBS | 29.78 | 0.43 | M, P, FM |
| 10 | 45 | 44 | 0.38 | Benzyl | THF/PBS | 14.28 | 0.33 | M, FM |
| 11 | 45 | 38 | 0.415 | Phthalimide | THF/PBS | 19.03 | 0.29 | M |
| 12 | 45 | 24 | 0.529 | Benzyl | THF/PBS | 10.13 | 0.38 | M |
| 13 | 45 | 20 | 0.574 | Benzyl | THF/PBS | 12.75 | 0.3 | M |
| 14 | 45 | 12 | 0.692 | Benzyl | THF/PBS | 15.43 | 0.43 | M |
Population only found after multiple impingements.
Very rare population. DLS diameter and polydispersity data not available for samples predominantly composed of filomicelles, i.e. sample 3‡.
Fig. 2.
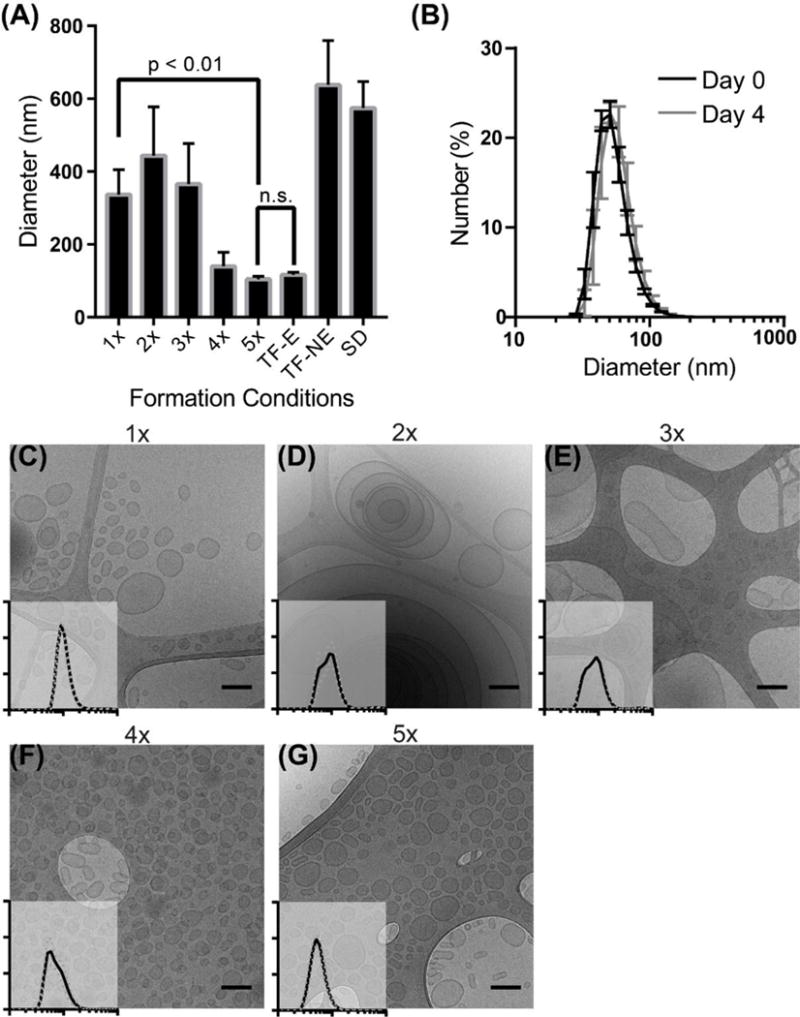
Fabrication of monodisperse polymersomes via flash nanoprecipitation. (A) DLS mean diameter of polymersomes formed after multiple impingements (1x–5x), or formed by thin film (TF) or solvent dispersion (SD) with (E) or without (NE) extrusion. Error bars are standard error, n = 5. (B) DLS size distribution of 5x impinged polymersomes the day of formation or after four days of storage at room temperature. Error bars are standard error, n = 3. (C–G) CryoTEM images of polymersomes formed after multiple impingements (1x–5x, respectively) with insets of DLS size distributions. X- and y- axes correspond to that of (B).
We found that multiple impingements through the CIJ mixer both decreased the mean polymersome diameter and lowered the PDI to levels achievable by extrusion (Fig. 2A and S2). To maintain high levels of the common solvent within hydrophobic PPS membranes for continued chain flexibility, we impinged a PEG-bl-PPS solution in THF against 1xPBS, collected the resulting mixture without diluting in a reservoir, evenly divided the THF/polymersome/PBS solution between the CIT mixer syringes and repeated the impingement. As no reservoir was used, the volume remained constant, and the impingement could be repeated multiple times over the course of a several minutes without sample loss. By the fifth impingement, there was no statistical difference in the diameter or PDI between FNP polymersomes and polymersomes formed by thin film hydration that were subsequently extruded through a 0.1 μm filter. Furthermore, 5x impinged polymersomes remained stable for days and demonstrated no detectable change in PDI (Fig. 2B). This change in the distribution of sizes can be observed by cryoTEM from the first impingement to the fifth and final impingement (Fig. 2C–G). While the first impingement generated primarily unilamellar and some rare multilamellar polymersomes ranging between 50 nm to nearly a micron in diameter, the 5x impinged polymersomes were monodisperse (PDI < 0.15) and all possessed a single bilayer (Fig. 2G). Interestingly, the 2x and 3x polymersome populations were composed of large, multilamellar, and/or tubular polymersomes (Fig. 2D, 2E, S5C and S5E). Tubular polymersomes have been predicted in simulations of polymersome formation under conditions of shear flow [30]. Multilamellar vesicles may be a result of multiple fusion events induced by increased polymersome collisions under conditions of turbulent flow while PPS chains remain fluid and swollen with THF. Since no changes in vesicle polydispersity or structure were observed after 5 impingements, it is possible that continued diffusion of THF out of the PPS domains reduced chain flexibility sufficiently to prevent polymersome fusion. In silico simulations of these events will be required to verify our hypotheses. We found this protocol of repeated impingements to support the gram-scale production of monodisperse polymersomes within a matter of minutes, which is a task that could require days to weeks to achieve by alternative fabrication methods such as thin film hydration and extrusion.
PEG-bl-PPS Copolymers Can Form Numerous Nanoarchitectures via Flash Nanoprecipitation
PEG-bl-PPS can self-assemble into several nanoarchitectures simply by tuning the PEG weight fraction (fPEG).[44] Using thin film hydration, fPEG between 0.20 and 0.30 form polymersomes, fPEG ~ 0.38 forms filomicelles and fPEG > 0.40 generally results in spherical micelles (Fig. 1B). In order to determine whether there is a similar connection between fPEG and morphology when polymersomes are formed by FNP, we synthesized the polymers listed in Table 1 and assessed their assembly into nanostructures using cryogenic electron microscopy (Fig. 3). These polymers were impinged against a 40 mM calcein solution in 1xPBS, as calcein is a water-soluble dye and its encapsulation would confirm the formation of vesicle structures, as opposed to the lipophilic cores of micelles and filomicelles. The diameters and calcein-loading ability of the assembled nanocarriers are shown in Fig. 3A. Flash nanoprecipitated polymersomes were found to occupy a space characterized by diameters ranging from 50–200 nm and PEG weight fractions from 0.187–0.305, similar to that for thin film rehydration (Fig. 3A, dotted oval). Copolymers 10–14 (fPEG > 0.40) were found to form primarily micelles, with a small population of filomicelles detected in the copolymer 10 sample. While copolymer 3 assembled polymersomes using PBS as the aqueous nonsolvent, switching to pure water resulted in the generation of filomicelles, likely due to the increased steric stabilization of the PEG corona (Fig. 3G and Table 1).
Fig. 3.
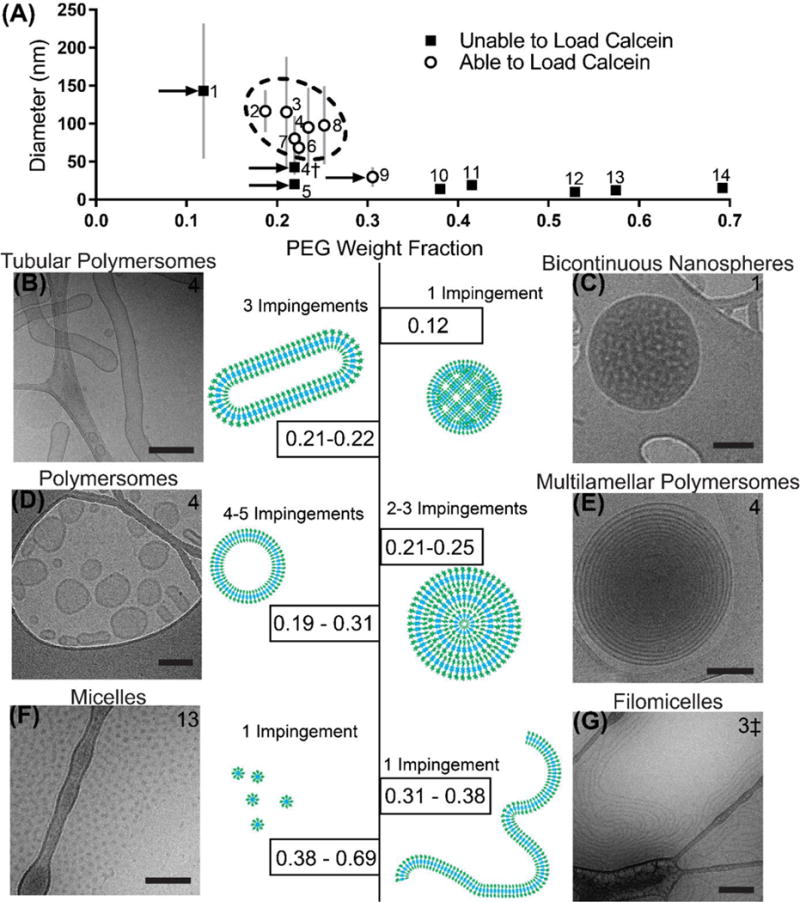
Relationship between PEG weight fraction and morphology. (A) Diameter of nanostructures formed via FNP from PEG-bl-PPS copolymers of varying block lengths. Error bars represent the standard deviation of the nanostructure populations (PDI × Mean Diameter). Dotted area represents polymersome-forming samples. Arrows point out samples of note. †Sample formed using DMF as the organic solvent, rather than THF. ‡Sample formed using water instead of 1xPBS. (B–G) Weight fractions of PEG responsible for forming specific nanostructures via flash nanoprecipitation, paired with cartoon and representative cryoTEM images. All scale bars = 100 nm, with the exception of scale bars within (B) and (E), which are 300 nm. Sample number is listed in the upper corner of each cryoTEM image, and the number of impingements used is listed for each morphology. See Table 1 for details of copolymers and Fig. S5 for low magnification images.
The three copolymers 1, 4, 5 and 9 (Fig. 3A, arrows), did not follow the previously described trends. Copolymer 1 failed to load calcein and was outside of the polymersome-forming range, yet had a diameter similar to that of polymersomes. CryoTEM imaging revealed these nanostructures to be monodisperse bicontinuous nanospheres (Fig. 3C and Fig. S5D), which is a nanostructure rarely formed from diblock copolymers in a controlled fashion [54]. FNP may therefore present a rapid and scalable alternative method of formation of polymeric biocontinuous nanostructures in simple aqueous solutions without the need for additives or complex block copolymer architectures (e.g. dendritic-linear [32], multiblock [55] and miktoarm stars [56]). Switching the solvent to dimethylformamide (δ = 24.8 MPa1/2) resulted in the formation of micelles from copolymer 4†, which was initially found to assemble vesicles when THF was instead used as the common solvent (Fig. 3A, Table 1). This result would be expected due to the lower similarity in solubility parameters between DMF and PPS relative to THF and PPS, which would lower chain flexibility and produce slower kinetics for nanostructure transitions. Slowing the transition kinetics through the use of less similar common solvents is known to promote micelle instead of vesicle formation for other block copolymer systems [17]. DLS and cryoTEM revealed copolymer 5 to form micelles even though its fPEG was within the polymersome-forming range (Fig. S3A). This discrepancy can be explained by the higher molecular weight (MW) of the PEG block (MW 2000) relative to the other polymersome forming copolymers (MW 750) (Table 1), as PEG steric stabilization increases with MW and strongly impacts nanostructure morphology [57]. Additionally, the longer PPS chain could result in higher degrees of entanglement and therefore decreased mobility. Copolymer 9 successfully loaded calcein, but had a diameter closer to that of micelles. CryoTEM found this sample to be a mix of morphologies, that included a low percentage of polymersomes and a dominant micelle population (Fig. S3B).
The alternative vesicular morphologies of multilamellar and tubular polymersomes (Fig. 3 and S5) were detected that were capable of loading calcein in addition to single-bilayered polymersomes. The multilamellar polymersomes represented >30% of assembled nanostructures formed from copolymers 3, 4, 6, 7, and 8 after 2x impingements (Fig. 3E and S5E and Table 1). These vesicles were easily distinguishable from the relatively common polymersome-within-polymersome structure, as they were characterized by numerous layers of nested vesicles, often reaching a dozen or more layers deep. Although initially absent, tubular polymers with lengths commonly exceeding a micron became a dominant population (>50% of assembled aggregates) after 3x impingements of copolymers 3, 4 and 6 (Fig. 3B, and Table 1). Tubular polymersome are difficult to assemble either quickly or as the dominant aggregate population, often requiring weeks to form and subsequent separation from more prevalent spherical polymersomes [29, 58, 59].
Polymersomes Formed via Flash Nanoprecipitation Effectively Load both Hydrophobic and Hydrophilic Molecules
A significant benefit of polymersomes over micelles and other lipophilic-core nanoparticles is that their aqueous lumen can encapsulate hydrophilic molecules while hydrophobic molecules can be simultaneously loaded within the vesicle bilayer [46, 60]. FNP has primarily been limited to the encapsulation of hydrophobic molecules with a preferred logP greater than 6 for stable nanoparticle formation [51, 61]. Ion-pairing of weakly hydrophobic and hydrophilic payloads with counter-ions to form hydrophobic salts for complexation with the block copolymer can allow nanoparticle assembly, but this method is not amenable to the loading of most hydrophilic biologics [6, 62]. To determine the influence of payload size and water solubility on loading efficiency into polymersomes, we tested molecules ranging from <1 kDa to >100 kDa and with varying hydrophobicity and Stokes radius: calcein, ethyl eosin, indocyanine green, rapamycin, lipophilic dye DiD, dextran (10 and 70 kDa), green fluorescent protein (GFP), ovalbumin, and alkaline phosphatase (Fig. 4A). With a logP of 1.6, calcein is relatively hydrophilic and small [63], resulting in the lowest loading efficiency (5.25%), possibly due to its diffusion from the interior of assembling polymersomes into the exterior aqueous reservoir before vesicle stabilization. In contrast, ethyl eosin, rapamycin, DiD, and indocyanine green presented very high loading efficiencies, as they can partition into the hydrophobic bilayers during polymersome assembly. The loading efficiency of water soluble macromolecules was significantly higher than for calcein and ranged between 16–43%, depending on the specific macromolecule (Fig. 4A). This loading efficiency compares favorably to other polymersome formation/loading techniques, including co-solvent evaporation[64], thin film rehydration[39, 65–67], direct hydration[48], and electroporation[68], especially considering the speed and ease of loading using FNP. A number of other variables besides the Stokes radius contribute to the ability for a molecule to permeate through an amphiphilic membrane, including molecular shape, hydrophobic affinity, and membrane thickness. Further improvement in the loading efficiency may be achieved by several means, including increasing the polymer concentration, decreasing the reservoir volume or modulating electrostatic interactions via changing the pH or ionic strength of solutions. Co-loading of hydrophilic and hydrophobic molecules into polymersomes was achieved simply by dissolving water soluble molecules in the aqueous stream and lipophilic molecules in the organic stream prior to impingement within the mixer. The co-loading efficiencies for hydrophilic-hydrophobic pairs of molecules are displayed in Table 2. Encapsulation of calcein (490 nm excitation) and DiI (540 nm excitation) respectively within the polymersome lumen and membrane produced a FRET emission from DiI when exposed to a 490 nm light source (Fig. S5). The ease of this co-loading process demonstrates that FNP is a powerful tool for the formation of nanocarriers loaded with diverse molecular payloads.
Fig. 4.
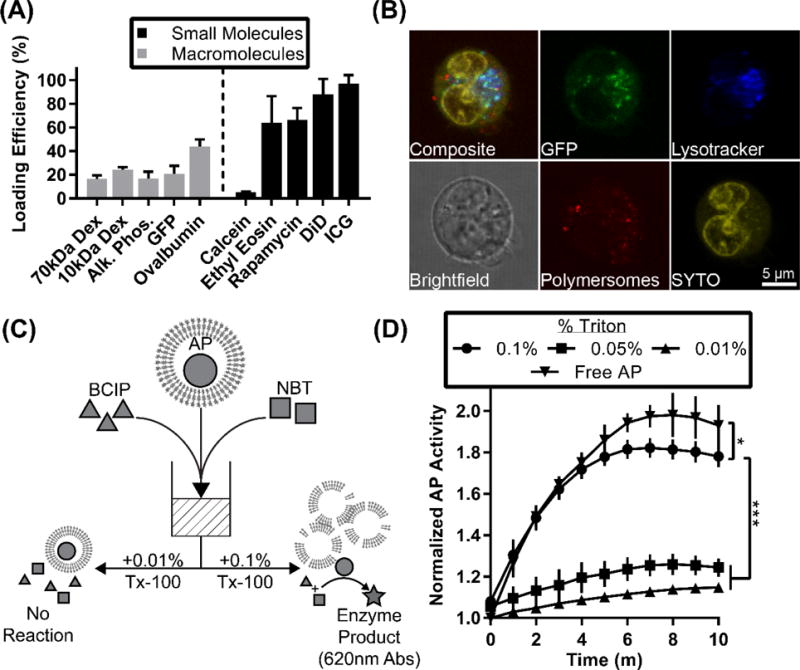
Loading of polymersomes with small molecules and macromolecules. (A) Loading efficiency of small molecules and macromolecules. (B) Live-cell confocal microscopy image of polymersome uptake and delivery of GFP in a bone marrow-derived dendritic cell. Polymersomes were loaded with the hydrophobic ethyl eosin (red) and hydrophilic GFP (green). Cells were additionally stained with SYTO 61 (yellow) and lysotracker (blue). Scale bar = 5 microns. (C) Graphical representation of experimental setup. Alkaline phosphatase (AP) is represented by circles, BCIP by triangles, and NBT by squares. The product of the enzymatic reaction, formazan, absorbs strongly at 620 nm and is represented by a star. (D) Time-course of enzyme activity assay. Y-axis represents fold increase over original absorbance reading. Error bars represent standard deviation, n = 4. Statistical significance determined by 2-way ANOVA, * p<0.05 and *** p<0.001.
Table 2.
Loading efficiency for dual-loading by flash nanoprecipitation. All samples fabricated in triplicate. LogP values given when available.
| Dual Loaded Cargoes | Hydrophilic Loading (%) | Hydrophobic Loading (%) | logP Values |
|---|---|---|---|
| TMR-Dextran 70kDa, ICG | 16.60 ± 2.98 | 97.12 ± 7.04 | N/A, 9.056 |
| Alkaline Phosphatase, Ethyl Eosin | 19.00 ± 5.62 | 64.91 ± 5.42 | N/A, 7.497 |
| Calcein, Ethyl Eosin | 5.06 ± 1.66 | 52.02 ± 2.65 | 1.608, 7.497 |
| Calcein, DiI | 2.54 ± 2.17 | 103.47 ± 12.11 | 1.608, 18.824 |
| GFP, Ethyl Eosin | 20.85 ± 6.74 | 63.71 ± 8.42 | N/A, 7.497 |
| Rapamycin, DiD | N/A | 65.59 ± 7.21 87.88 ± 13.11 |
6.181, 19.38 |
Flash Nanoprecipitation-Loaded Proteins Remain Biologically Active
One concern for the use of FNP with proteins is the mixing of organic solvent with the aqueous solution, which has the potential to denature the structure of proteins and decrease their bioactivity. A possible solution to this concern is to load the hydrophilic cargo within the reservoir, after the impingement has occurred but before the vesicles have fully assembled. To determine the efficiency of this method, we attempted to simultaneously load polymersomes with two separate fluorescent dextrans of the same molecular weight by dissolving one in the impinged aqueous stream and one in the reservoir at the same concentration. The highly water soluble macromolecule dextran (10 kDa) tagged with either cascade blue (CB) or fluorescein (F) was used. To avoid any potential influences of the attached fluorophore on the loading efficiency, we conducted the experiments in pairs, with the dextran-CB in the syringe and dextran-F in the reservoir, and vice versa. We found there to be a statistically significant increase in loading via syringe compared to reservoir, though loading by reservoir still occurred at detectable levels (Fig. S1A). We further investigated whether loading via reservoir or syringe resulted in a greater amount of functional protein by loading GFP, which has a conformation-dependent fluorescence. We found that GFP fluoresced at equal levels whether loaded by syringe or reservoir within polymersomes (Fig. S1B,C). The reservoir solution resulting from FNP of GFP in the aqueous phase syringe or reservoir without copolymer, GFP in the aqueous phase syringe or syringe with PEG-bl-PPS in the organic phase syringe, and PEG-bl-PPS in the organic phase syringe without GFP were compared for GFP fluorescence following separation by size exclusion chromatography (Fig. S1C,D). Analysis of the fluorescence area under the curve revealed the process of FNP to decrease GFP fluorescence by 22.8 ± 5.4%. However, equal levels of fluorescence were again found for GFP-loaded polymersomes when the GFP was loaded from the syringe relative to the reservoir, suggesting that higher levels of loading occurred via the syringe. These results therefore demonstrate that loading protein via the reservoir is preferable in cases where maintaining conformation is critical to macromolecule function, while loading via the syringe can achieve higher loading efficiencies at the expense of small decreases in protein bioactivity.
The activity of GFP in biologically-relevant systems was further explored by delivering GFP in vitro within polymersomes to bone marrow-derived dendritic cells (BMDCs). BMDCs were generated from bone marrow freshly collected from C57BL/6 mice, and after maturation were plated in the presence of polymersomes. The polymersomes were co-loaded with the hydrophobic dye ethyl eosin and hydrophilic GFP, and the cells were further stained with nuclear stain SYTO 61 and lysosome stain Lysotracker Blue prior to imaging on a confocal microscope. Punctae of GFP and ethyl eosin were found within cells, demonstrating that conformationally-active GFP could be delivered to live cells via polymersomes (Fig. 4B). These results supported previously published confocal microscopy images and verifies that FNP had no impact on the ability of PEG-bl-PPS polymersomes to deliver payloads to the cytoplasm of BMDCs [39].
To further confirm the continued biological activity of loaded proteins, we investigated the function of an enzyme, alkaline phosphatase (AP), encapsulated into polymersomes by FNP. AP removes phosphate groups from a number of substrates, including 5-bromo-4-chloro-3-indolyl-phosphate (BCIP), whose dephosphorylation is detectable by nitro blue tetrazolium (NBT) at an absorbance of 620 nm [69]. We therefore encapsulated AP into the lumen of polymersomes using FNP, and added them to a solution of BCIP and NBT. AP-polymersomes were lysed using Triton X-100 to allow the AP, BCIP and NBT to freely react in solution (Fig. 4C). At low concentrations of Triton X-100 a low level of background reactivity was observed in the system, which was likely due to a burst effect resulting from the release of hydrophilic reagents trapped within the vesicle membranes. The addition of 0.1% Triton X-100 resulted in the continued formation of significantly more product over a ten-minute period, verifying the retention of AP tertiary conformation and bioactivity following encapsulation within polymersomes by FNP. Free AP at a concentration equivalent to the final loading concentration within the polymersomes was also tested, and was found to have a marginally greater activity than the polymersome loaded-AP after addition of 0.1% Triton X-100, differing by only 6.28% after 10 min (Fig. 4D).
Low Dosage Subcutaneous Administration of Rapamycin Loaded into Polymersomes by FNP Reduces Splenic CD4+ and CD8+ T Cell Populations
To explore FNP as a method to fabricate polymersome formulations for the in vivo delivery of therapeutics, we loaded the model immunomodulatory drug rapamycin (Sirolimus), an FDA-approved immunosuppressant, into fluorescent polymersomes. Rapamycin has a low water solubility (logP = 6.181) and loaded readily into polymersomes with an efficiency of 65% (Fig. 4A). Rapamycin inhibits the mechanistic target of rapamycin (mTOR) kinase, which is a key regulator of cell growth, metabolism and proliferation and elicits cellular responses that are highly dependent on the cell type. Thus, identifying which cells are being influence by rapamycin is critical to understanding immune responses generated during its therapeutic use, and this may be achieved via an immunotheranostic delivery system. In the case of T cells, mTOR inhibition is known to decrease proliferation, migration and overall population levels for T cells, particularly CD4+CD25− T cell and effector CD8+ T cell subsets [70–73]. For dendritic cells, rapamycin has a suppressive effect on maturation and differentiation by inhibiting expression of co-stimulatory molecules and inflammatory cytokines [74–76]. Macrophages respond to free form rapamycin by polarizing towards a pro-inflammatory M1 phenotype, which can be strongly influenced by the cytokine environment at the time of stimulation [77]. Although T cells are not phagocytic and do not associate with polymersomes, their levels and activity can be modulated via the targeting of antigen presenting cells, such as phagocytic macrophages and dendritic cells [3]. PEG-bl-PPS nanocarriers are endocytosed by phagocytic immune cells in lymph nodes and spleen following SC administration [3, 78, 79], and we hypothesized that controlled delivery of rapamycin within polymersomes to these cells may enhance immunosuppression at lower dosages than what is typically employed therapeutically in mice while avoiding uptake of rapamycin by T cells. Rapamycin is administered to mice orally, intraperitoneally, and subcutaneously, at doses ranging from 75 μg/kg/day to >10 mg/kg/day [80–82]. Generally, an effective dose for sustained allograft survival in mice is considered to be 1.5–3 mg/kg/day [83–85].
To demonstrate the difference between rapamycin efficacy in its polymersome-loaded versus free form, we administered rapamycin to mice once every three days at an effective average dosage of only 0.33 mg/kg/day. After only three administrations of rapamycin loaded polymersomes, we found that splenic CD4+ and CD8+ T cells were significantly reduced in proportion to the total number of T cells compared to free rapamycin, which instead elicited no significant effects on T cells (Fig 5A, S6, flow cytometry gating strategy shown in S7). Similar decreases in T cell numbers following injection of rapamycin-loaded nanoparticles have been reported by others and may contribute to tolerogenic therapies [86]. CD8+ dendritic cells, a dendritic cell population known for superior cross presentation of antigen and resulting CD8+ T cell activation, were also significantly decreased in proportion to the total number of dendritic cells (5A, S6). The significant drop in CD4+ T cells and CD8+ T cells was accompanied by an increase in the proportion of ‘double negative’ (CD4− CD8−, DN) T cells (Fig 5B) and a decrease in the total number of T cells (Fig 5C). The increase in the proportion of DN T cells was matched by the overall decrease in T cells, resulting in no significant change in the proportion of DN T cells within the total immune cell population (Fig S8). Free rapamycin was found to have no significant impact on immune cell populations at this comparatively low dosage and brief course of treatment (Fig 5, S6). The decrease in CD4+ and CD8+ T cell populations is not likely to have occurred through direct intracellular delivery of rapamycin to T cells, as these cells showed no association with polymersomes as measured by DiD fluorescence (Fig. 5D). In contrast, phagocytic cells in both the lymph nodes and spleen, such as macrophages, Ly-6Chi monocytes, and dendritic cells all demonstrated significant uptake of polymersomes (Fig. 5D). This theranostic strategy thus implicates antigen presenting cells as the mediators of the observed T cell immunomodulation and demonstrates that flash nanoprecipitation can be employed for rapid fabrication and loading of soft nanoarchitectures capable of controlled delivery of imaging agents and therapeutic drugs in vivo.
Fig. 5.
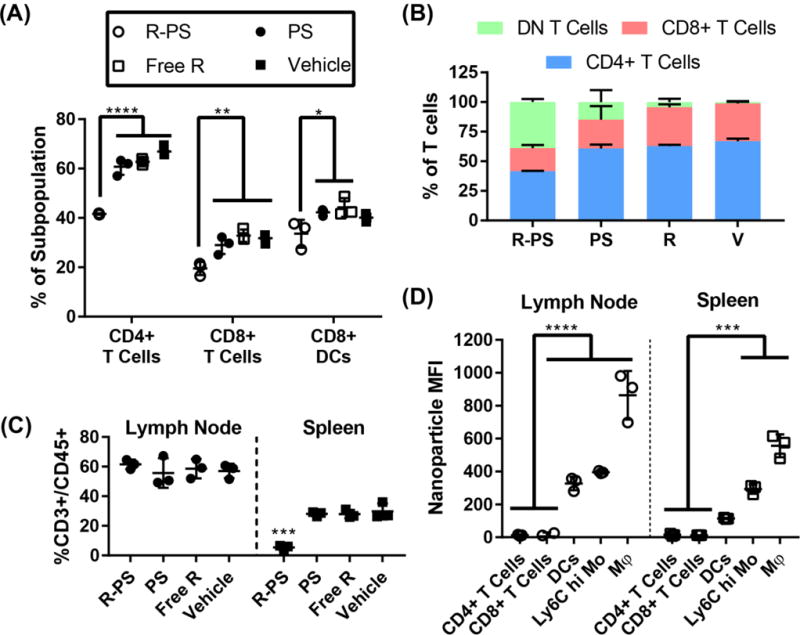
In vivo delivery of theranostic rapamycin/DiD-loaded polymersomes formed by flash nanoprecipitation. (A) Percentage of CD8+ T cells (CD45+ CD3+ CD4− CD8+) and CD4+ T cells (CD45+ CD3+ CD4+ CD8−) within the total T cell population (CD45+ CD3+) and percentage of CD8+ DCs (CD11c+ I-A/I-E+ CD8+) within the total DC (CD11c+) population. Treatment groups were rapamycin polymersomes (R-PS), free rapamycin, blank polymersomes, and vehicle (PBS). (B) T cell subpopulations as a percent of total T cell population for all four treatment groups. (C) T cells in the spleen and lymph nodes, as a percentage of CD45+ cells. (D) Median fluorescence intensity of the polymersome channel for selected cell populations in the spleen and lymph nodes of mice administered rapamycin/DiD-loaded polymersomes. N=3, statistical significance determined by Tukey’s multiple comparisons test, * p < 0.05, ** p < 0.01, ***p < 0.001, ****p < 0.0001.
Conclusion
Delivery of drugs in vivo, ranging from hydrophilic to hydrophobic and small molecule to protein biologics, remains a significant challenge for the field of biomedical research. Concerns over efficiency, stability, and scalability have plagued many fabrication processes for self-assembled soft nanoarchitectures, many of which hold vast and unrealized potential for advanced strategies of controlled delivery. Here, we have demonstrated that FNP, a rapid and scalable method of assembling solid-core block copolymer nanoparticles, can be used to assemble PEG-bl-PPS into monodisperse polymersomes and a variety of other soft nanoarchitectures, including tubular and multillamelar polymersomes, filomicelles and rare polymeric bicontinuous nanospheres. FNP is usually performed using block copolymers with high Tg hydrophobic blocks, like polystyrene, that undergo slow structural transitions and allow the formation of stable micellar aggregates. Our results suggest that matching the copolymer aggregation rate with the turbulent mixing time during FNP, which can be controlled by the flow rate through the mixing chamber, may allow FNP to be applied for the formation of diverse metastable nanostructures for other polymers in addition to PEG-bl-PPS. We have also found that FNP can effectively load (and co-load) vesicular nanoarchitectures with small molecules and proteins with a wide range of water solubility and molecular weights while maintaining their biological activity. We have demonstrated three examples of biological relevance: in vitro delivery of GFP to BMDCs, in vitro enzyme activity of alkaline phosphatase, and in vivo delivery of rapamycin to immune cells, the latter of which demonstrated immunosuppression of CD4+ and CD8+ T cells at a 5-fold lower dosage than is typically utilized. These findings significantly expand the capabilities of FNP and provide new routes for the high throughput nanofabrication of diverse therapeutic nanocarriers.
Materials and Methods
Materials
The following reagents were obtained from Sigma-Aldrich: poly(ethylene glycol) methyl ether MW 2000 (product 202509), poly(ethylene glycol) methyl ether MW 750 (product 202495), poly(ethylene glycol) MW 300 (product 202371), methanesulfonyl chloride (product 471259), triethylamine (product T0886), potassium carbonate (product 791776), thioacetic acid (product T30805), 0.5M sodium methoxide solution (product 403067), propylene sulfide (product P53209), acetic acid (product 695092), benzyl bromide (product B1 7905), N-(2-bromoethyl)phthalimide (product B66302), 2,2′-dipyridyl disulfide (product 143049), calcein (product C0875), ethyl eosin (product 199540), indocyanine green (product I2633), dichloromethane (product 320269), celite filter cel (product 22139), activated charcoal powder (product 161551), anhydrous tetrahydrofuran (product 401757), tetrahydrofuran (product 437638), ethanol (product 459844), methanol (product 179337), diethyl ether (product 346136), deuterated chloroform (product 151823), Tween 80 (product P8074), and sepharose 4B (product 4B200). The following reagents were purchased from ThermoFisher Scientific: tetramethylrhodamine-dextran 70 kDa (product D1818), fluorescein-dextran 10 kDa (product D1820), cascade blue-dextran 10 kDa (product D1976), Dil (product V22885), DiD (product D7757), ovalbumin texas red conjugate (product O23021), toluene (product T324), hexanes (product H292), HPLC-grade dimethylformamide (product AA22915K2), Lysotracker Blue DND-22 (product L7525), SYTO 61 (product S11343), ACK lysing buffer (product A1049201), HBSS (product 14175079) and 1xPBS tablets (product BP2944). Recombinant A. victoria GFP protein was a generous gift from the Jewett Lab at Northwestern University. Electron microscopy holey carbon 200 mesh copper grids were purchased from Electron Microscopy Sciences (product HC200CU). Rapamycin was purchased from Selleck Chemicals. Recombinant IL-4, recombinant GM-CSF, and all antibodies for flow cytometry were purchased from BioLegend.
Polymer synthesis
The synthesis of poly(ethylene glycol)-block-poly(propylene sulfide) (PEG-bl-PPS) was performed as described previously [39, 46]. Briefly, methyl ether PEG (MW 750 and 2000) were functionalized first with the mesylate leaving group, which was then reacted with thioacetic acid to form a protected PEG-thioacetate. Base activation of the thioacetate resulted in the formation of a thiolate anion, which was used as the initiator for ring opening polymerization of propylene sulfide. The reaction was completed with the addition of end capping groups (benzyl bromide or bromoethyl phthalimide), a disulfide-bonding group (dipyridyl disulfide), or the protonation of the thiolate anion with acetic acid, leaving a free thiol at the end of the polymer (PEG- bl-PPS-thiol). Degree of polymerization was assessed via H1 NMR (3H methyl ether, 3.36 singlet; 4H PEG -CH2-CH2-, wide peak 3.60–3.64; 1H CH2-CH-CH3 wide peak 2.56–2.65; 2H -CH-CH2-CH-CH3, wide peak 2.82–2.95, 3H -CH2-CH3 wide peak 1.30–1.38).
Nanocarrier Fabrication via Flash Nanoprecipitation
Nanocarriers were formed using the confined impingement jets (CIJ) mixer described by Han et al. [9] PEG-bl-PPS copolymers and hydrophobic molecules to be loaded within polymersomes were dissolved in 500 μL of tetrahydrofuran (THF) and placed into a 1 mL plastic disposable syringe. 500 μL of phosphate buffered saline (1xPBS) and any hydrophilic molecules to be loaded within the vesicle lumen were prepared in a second 1mL syringe. The two solutions were impinged against one another within the CIJ mixer by hand, at a rate of approximately 1 mL/s. The supersaturated solution exited the mixer into a 20 mL glass scintillation vial containing a 2.5 mL reservoir of 1xPBS. This product was then separated from unloaded molecules and THF on a sepharose 4B size exclusion column. For fluorescence measurements, samples were taken both before and after column purification to assess loading efficiency.
Alternative Nanocarrier Fabrication Techniques
As controls, polymersomes were formed by the standard solvent dispersion and thin film techniques. In the case of solvent dispersion, 20 mg of polymer was dissolved in 500 μL of THF, which was dripped into a stirring reservoir of 3 mL of 1xPBS. This resulted in an identical THF:1xPBS ratio as used for the FNP fabrication method. Hydrophobic molecules were dissolved in the requisite organic solvent and added to the polymer/THF solution prior to addition to 1xPBS. Hydrophilic molecules were first dissolved in the 1xPBS reservoir prior to the addition of the polymer/THF solution. In the case of thin film, 20mg of polymer was weighed into a 1.8 mL glass HPLC vial and dissolved in 750 μL of DCM, which was subsequently removed by vacuum desiccation for 6 hours. 1 mL of 1xPBS was then added to the HPLC vial, which was shaken at 1500 rpm overnight on a Multi-Therm shaker (Heidolph) at room temperature. Hydrophobic molecules, dissolved in their respective organic solvent, were added to the polymer/DCM solution, and were dried with the polymer in the vacuum desiccator. Hydrophilic molecules were dissolved first in the 1xPBS prior to its addition to the dried polymer deposit for hydration under shaking at room temperature.
Co-Loading Experiments
For calcein and ethyl eosin co-loading experiments, the aqueous solvent consisted of 0.4 mM calcein in 1xPBS. 10 μL of a 5 mg/mL ethyl eosin solution in ethanol was added to 490 μL of THF and 20 mg of polymer. For the TMR-dextran and ICG co-loading experiments, 2 mg of TMR-dextran (70 kDa) was dissolved in 500 μL 1xPBS. 50 μL of a 1 mg/mL ICG solution in ethanol was added to 450 μL of THF and 20 mg of polymer. For alkaline phosphatase – ethyl eosin co-loading experiments, alkaline phosphatase was loaded at a concentration of 1 mg/mL within the reservoir, while 10 μL of a 5 mg/mL ethyl eosin solution in ethanol was added to 490 μL of THF and 20 mg of polymer. For calcein and DiI co-loading experiments, the aqueous solvent consisted of a 0.4 mM calcein solution in 1xPBS, which was impinged against 490 μL THF with 10 μL DiI (as supplied) and 20 mg of polymer. For the experiments examining the relative loading of fluorescein-dextran and cascade blue-dextran, both were used from stock solutions of 1 mg/mL in 1xPBS. For the GFP loading experiments, 50 μL of a 200 μg/mL was dissolved in 450 μL of 1xPBS (for syringe loading) or 2.45 mL 1xPBS (for reservoir loading), while 20 mg of polymer was dissolved in 500 μL of THF.
Multiple Impingement Experiments
For the multiple impingement samples, the organic and aqueous impingement solutions were prepared as previously described for the calcein/ethyl eosin co-loading experiments with a few exceptions. In this case, rather than containing 2.5 mL of 1xPBS, the scintillation vial reservoir was initially empty. Following each impingement, the resulting solution containing 1:1 THF:1xPBS, 20 mg of polymer, calcein, and ethyl eosin, was split evenly into two 1 mL syringes, and reintroduced into the CIJ mixer. This process was repeated between one and four times, with the final impingement emptying into a reservoir containing 2.5 mL of 1 xPBS.
Spectrometric Fluorescence and Absorbance Measurements
Fluorescence and absorbance measurements were taken on a SpectraMax M3 microplate reader (Molecular Devices). All readings were taken in black-walled clear bottom 96-well plates (Corning 07-200-567), at 100 μL volumes. All readings were normalized to a 100 μL 1xPBS blank control. The following settings were used for fluorescence measurements for each fluorophore (excitation/emission, filter used): calcein: 470/509, 495 filter), ethyl eosin: 525/560, 550 filter, tetramethylrhodamine: 555/580, 570 filter, fluorescein: 494/524, 515 filter, cascade blue: 400/420, 420 filter, indocyanine green: 780/820, no filter, DiI: 549/565, 550 filter, DiD: 644/670, 665 filter, ovalbumin texas red: 594/615, 610 filter, and GFP: 485/535, 495 filter. NBT diformazan was detected via absorbance at 620 nm.
Rapamycin Loading Efficiency by HPLC UV Absorbance
50 μL aliquots of formulations containing rapamycin were frozen at −80 C for at least 3 hours, then were lyophilized overnight. Lyophilized samples were dissolved in HPLC grade DMF and were vortexed then briefly centrifuged to pellet salt from the formulation. Samples were run on a Thermo Fisher Dionex UltiMate 3000 system with an Agilent Polypore 7.5 × 300mm column with an Agilent Polypore 7.5 × 50 mm guard column, housed at 60 C. The organic mobile phase was HPLC grade DMF, run at 0.5 mL/min. Rapamycin was detected using UV absorbance at 270 nm. For loading efficiency, aliquots were taken from samples before and after column purification. Analysis was performed using the Thermo Scientific Chromeleon software.
Alkaline Phosphatase Enzyme Activity Assay
Alkaline phosphatase was dissolved at 1 mg/mL in 2.5 mL of 137 mM NaCl solution for the reservoir. Unloaded polymersomes were formed by similarly but without alkaline phosphatase. All polymersome samples were purified by size exclusion chromatography (sepharose 4B) to remove THF and unloaded cargo. A stock solution of premixed NBT and BCIP was used (560 μM BCIP and 480 μM NBT prepared in 10 mM Tris and 59.3 mM MgCl2). 40 μL of polymersomes was added to wells of a 96 well plate, along with 10 uL of NBT/BCIP solution. Cargo release was performed by the addition of 50 μL of Triton X-100 solution, with final concentrations of Triton X-100 of 0.1%, 0.05%, and 0.01%. As a control, free alkaline phosphatase was diluted to the final concentration loaded within polymersomes (170 μg/mL), and was used as described for the polymersomes samples. Absorbance measurements were taken every minute for 10 minutes.
Size Measurements
Hydrodynamic diameters of nanostructures were measured using nanoparticle tracking analysis (NTA) on a Nanosight NS300 (Malvern). Measurements were taken using samples at a 1:1000 dilution in 1xPBS, resulting in approximately 0.1 mg/mL polymer concentrations. Readings were performed using a 633 nm laser. Five 1 minute videos were recorded per sample, with results averaged across the five readings. For micellar samples, typically >20 nm in diameter, dynamic light scattering measurements were performed using a Zetasizer (Malvern) to more accurately measure the size distribution, due to the limitations of NTA for smaller diameter aggregates.
LogP Values
LogP values were pulled from the ZINC15 database (zinc15.docking.org) [87].
Cryogenic Transmission Electron Microscopy
Specimens for cryoTEM were prepared by applying 4 μL of 1 mg/ml sample on a pretreated, holey carbon 400 mesh TEM grids and were plunge-frozen with a Gatan Cryoplunge freezer. Images were collected in vitreous ice using a JEOL 3200FSC transmission electron microscope operating at 300 keV at 4,000 x nominal magnification. A total dose of ~10 e−/Å2 and a nominal defocus range of 2.0–5.0 μm were used. Micrographs were acquired using a Gatan 3.710 × 3,838 pixel K2 Summit direct electron detector operating in counting mode. Each micrograph was acquired as 20-frame movies during a 5 s exposure. After data acquisition, the individual frames of each micrograph were aligned using Digital Micrograph software (Gatan) to compensate for stage and beam-induced drift, and the aligned images were summed for further image processing.
Animals
C57BL/6 male mice, 6–8 weeks old, were purchased from Jackson Labs. All mice were housed and maintained in the Center for Comparative Medicine at Northwestern University. All animal experimental procedures were performed according to protocols approved by the Northwestern University Institutional Animal Care and Use Committee (IACUC).
Live-Cell Confocal Microscopy
Bone marrow derived dendritic cells were generated from bone marrow collected from the tibias and femurs of C57BL/6 mice, in a protocol slightly modified from those described previously [88]. Tibias and femurs of C57BL/6 mice were cleaned of tissue, cut, and flushed through with 10% FBS in HBSS. Cells were centrifuged, supernatant was removed, and they were then treated with ACK lysis buffer for 5 minutes. Lysis was stopped with the addition of excess HBSS, and cells were centrifuged again, resuspended in 10 mL 10% FBS 1x Penstrep RPMI, and were plated in 100 mm petri dishes. Every three days, 200 ng of GM-CSF and 100 ng of IL-4 were added to culture. On the 8th day of maturation, cells were collected and plated into a FluoroDish at 3×105 cells/mL, along with 15 uL of dual-loaded GFP ethyl eosin polymersomes. Lysotracker Blue (lysosome stain) and SYTO 61 (nuclear stain) were added at 1:10,000 dilutions. Plated cells were imaged within a humidified chamber using a 63x oil-immersion objective on a SP5 Leica Confocal Microscope using HyD detectors and four lasers: 415 nm diode laser for Lysotracker Blue, 488 nm argon laser for GFP, 514 nm argon laser for ethyl eosin, and 633 nm HeNe laser for SYTO 61.
In Vivo Rapamycin Delivery
Formulations of rapamycin polymersomes and blank polymersomes were formed by flash nanoprecipitation using 20 mg of PEG17-bl-PPS36-Thiol polymer, with or without 0.5 mg rapamycin, respectively, dissolved in THF. Sterile 1xPBS was used as the aqueous phase and reservoir solution. Fluorescently labeled polymersomes of both formulations were formed similarly, with the addition of 25 μg of DiD in the organic phase. A formulation of free rapamycin was made in a solution of 8% ethanol, 10% PEG300, and 10% Tween 80 in 1xPBS. Briefly, rapamycin was dissolved in ethanol (3 mg/mL), and 31.2 μL was added to 1 mL of a solution of 10% PEG300 and 10% Tween 80 in 1xPBS (3.1% ethanol final, ~125 μg/mL rapamycin concentration). Vehicle was also injected without rapamycin, in which case 31.2 μL of pure ethanol was added to the 10% PEG300 10% Tween 80 solution. Mice were injected subcutaneously, slightly anterior to the scapula, with 1 mg/kg doses of rapamycin, or equivalent injections of vehicle or blank polymersomes, N = 3 per treatment group. Injections were performed on days 1, 4 and 7, with the final set of injections containing fluorescently labeled polymersomes, when applicable. Mice were sacked on day 8, and the draining (brachial) lymph nodes were collected, along with the spleens. Organs were mechanically homogenized in RPMI media and passed through a 70 μm cell strainer before being stained for flow cytometry.
Flow Cytometry
Splenic cells were first treated with ACK lysis buffer for 5 minutes on ice before being spun down and resuspended in blocking buffer. Cells were stained with Zombie Aqua as a fixable live/dead stain and FcRs were blocked with anti-mouse CD16/CD32. Cells were stained with a cocktail of antibodies in three panels. Panel 1: anti-mouse CD45 FITC, anti-mouse CD3 APC-Cy7, anti-mouse CD4 PE-Cy5, anti-mouse CD8a PE-Cy7, anti-mouse CD19 Pacific Blue, anti-mouse CD49b PerCp-Cy5.5, and anti-mouse CD25 PE. Panel 2: anti-mouse CD11b PerCp-Cy5.5, anti-mouse CD11c Pacific Blue, anti-mouse I-A/I-E FITC, anti-mouse B220 PE, anti-mouse Gr-1 APC-Cy7, and anti-mouse CD8a PE-Cy7. Panel 3: anti-mouse CD11b PerCp-Cy5.5, anti-mouse CD11c Pacific Blue, anti-mouse F4/80 FITC, anti-mouse Ly-6C APC-Cy7, and anti-mouse Ly-6G PE-Cy7. After washes, cells were fixed by IC cell fixation buffer (Biosciences). Flow cytometry was performed with FACSDiva on a LSRII flow cytometer (BD Biosciences), with the APC channel used to detect the DiD loaded into polymersomes. Data was analyzed using CytoBank online software.
Supplementary Material
Acknowledgments
We would like to thank Devang Amin and Philip Messersmith for their generous aid in the initial stages of procuring a CIJ mixer. We acknowledge staff and instrumentation support from the Structural Biology Facility at Northwestern University. The support from the R.H. Lurie Comprehensive Cancer Center of Northwestern University and the Northwestern University Structural Biology Facilities is acknowledged. The Gatan K2 direct electron detector was purchased with funds provided by the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust. We also thank the following facilities at Northwestern University: the Keck Interdisciplinary Surface Science Facility, the Structural Biology Facility, the Biological Imaging Facility, the Center for Advanced Molecular Imaging, and the Analytical Bionanotechnology Equipment Core. We greatly appreciate the useful and extensive discussions of this work with Rikkert Nap, David Malaspina and Igal Szleifer. This research was supported by the National Science Foundation grant 1453576 and the 2014 McCormick Catalyst Award. SDA was supported in part by NIH predoctoral Biotechnology Training Grant T32GM008449.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributions
S.D.A. and E.A.S. contributed to the conception and study design. S.D.A. and O.A.O. synthesized and characterized materials. S.D.A. and O.A.O performed the experiments. S.D.A. and E.A.S. contributed to the data analysis. S.D.A. and E.A.S. wrote the manuscript.
References
- 1.Duan X, Li Y. Physicochemical Characteristics of Nanoparticles Affect Circulation, Biodistribution, Cellular Internalization, and Trafficking. Small. 2013;9(9–10):1521–1532. doi: 10.1002/smll.201201390. [DOI] [PubMed] [Google Scholar]
- 2.Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007;2(4):249–55. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stano A, Scott EA, Dane KY, Swartz MA, Hubbell JA. Tunable T cell immunity towards a protein antigen using polymersomes vs. solid-core nanoparticles. Biomaterials. 2013;34(17):4339–46. doi: 10.1016/j.biomaterials.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 4.Choi CH, Hao L, Narayan SP, Auyeung E, Mirkin CA. Mechanism for the endocytosis of spherical nucleic acid nanoparticle conjugates. Proc Natl Acad Sci U S A. 2013;110(19):7625–30. doi: 10.1073/pnas.1305804110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang T, Bai J, Jiang X, Nienhaus GU. Cellular uptake of nanoparticles by membrane penetration: a study combining confocal microscopy with FTIR spectroelectrochemistry. ACS Nano. 2012;6(2):1251–9. doi: 10.1021/nn203892h. [DOI] [PubMed] [Google Scholar]
- 6.Tang C, Amin D, Messersmith PB, Anthony JE, Prud’homme RK. Polymer directed self-assembly of pH-responsive antioxidant nanoparticles. Langmuir. 2015;31(12):3612–20. doi: 10.1021/acs.langmuir.5b00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saad WS, Prud’homme RK. Principles of nanoparticle formation by flash nanoprecipitation. Nano Today. 2016;11(2):212–227. [Google Scholar]
- 8.Johnson BK, Prud’homme RK. Flash NanoPrecipitation of organic actives and block copolymers using a confined impinging jets mixer. Aust J Chem. 2003;56(10):1021–1024. [Google Scholar]
- 9.Han J, Zhu Z, Qian H, Wohl AR, Beaman CJ, Hoye TR, Macosko CW. A simple confined impingement jets mixer for flash nanoprecipitation. J Pharm Sci. 2012;101(10):4018–23. doi: 10.1002/jps.23259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng JC, Vigil RD, Fox RO. A competitive aggregation model for Flash NanoPrecipitation. J Colloid Interface Sci. 2010;351(2):330–342. doi: 10.1016/j.jcis.2010.07.066. [DOI] [PubMed] [Google Scholar]
- 11.Chmielowski RA, Abdelhamid DS, Faig JJ, Petersen LK, Gardner CR, Uhrich KE, Joseph LB, Moghe PV. Athero-inflammatory nanotherapeutics: Ferulic acid-based poly(anhydride-ester) nanoparticles attenuate foam cell formation by regulating macrophage lipogenesis and reactive oxygen species generation. Acta Biomater ( 2017 doi: 10.1016/j.actbio.2017.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao L, Shen G, Ma G, Yan X. Engineering and delivery of nanocolloids of hydrophobic drugs. Adv Colloid Interface Sci. 2017 doi: 10.1016/j.cis.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Tang C, Prud’homme RK. Targeted Theragnostic Nanoparticles Via Flash Nanoprecipitation: Principles of Material Selection. In: Vauthier C, Ponchel G, editors. Polymer Nanoparticles for Nanomedicines: A Guide for their Design, Preparation and Development, Springer International Publishing, Cham. 2016. pp. 55–85. [Google Scholar]
- 14.Pinkerton NM, Behar L, Hadri K, Amouroux B, Mingotaud C, Talham DR, Chassaing S, Marty JD. Ionic Flash NanoPrecipitation (iFNP) for the facile, one-step synthesis of inorganic-organic hybrid nanoparticles in water. Nanoscale. 2017;9(4):1403–1408. doi: 10.1039/c6nr09364g. [DOI] [PubMed] [Google Scholar]
- 15.Johnson BK, Prud’homme RK. Mechanism for rapid self-assembly of block copolymer nanoparticles. Phys Rev Lett. 2003;91(11):118302. doi: 10.1103/PhysRevLett.91.118302. [DOI] [PubMed] [Google Scholar]
- 16.Shillcock JC. Spontaneous vesicle self-assembly: a mesoscopic view of membrane dynamics. Langmuir. 2012;28(1):541–7. doi: 10.1021/la2033803. [DOI] [PubMed] [Google Scholar]
- 17.Mai Y, Eisenberg A. Self-assembly of block copolymers. Chem Soc Rev. 2012;41(18):5969–85. doi: 10.1039/c2cs35115c. [DOI] [PubMed] [Google Scholar]
- 18.Rikken RS, Engelkamp H, Nolte RJ, Maan JC, van Hest JC, Wilson DA, Christianen PC. Shaping polymersomes into predictable morphologies via out-of-equilibrium self-assembly. Nat Commun. 2016;7:12606. doi: 10.1038/ncomms12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salva R, Le Meins JF, Sandre O, Brulet A, Schmutz M, Guenoun P, Lecommandoux S. Polymersome shape transformation at the nanoscale. ACS Nano. 2013;7(10):9298–311. doi: 10.1021/nn4039589. [DOI] [PubMed] [Google Scholar]
- 20.Lipowsky R. The morphology of lipid membranes. Curr Opin Struct Biol. 1995;5(4):531–40. doi: 10.1016/0959-440x(95)80040-9. [DOI] [PubMed] [Google Scholar]
- 21.Dionzou M, Morere A, Roux C, Lonetti B, Marty JD, Mingotaud C, Joseph P, Goudouneche D, Payre B, Leonetti M, Mingotaud AF. Comparison of methods for the fabrication and the characterization of polymer self-assemblies: what are the important parameters? Soft Matter. 2016;12(7):2166–76. doi: 10.1039/c5sm01863c. [DOI] [PubMed] [Google Scholar]
- 22.Bleul R, Thiermann R, Maskos M. Techniques To Control Polymersome Size. Macromolecules. 2015;48(20):7396–7409. [Google Scholar]
- 23.Kumar V, Prud’homme RK. Nanoparticle stability: Processing pathways for solvent removal. Chem Eng Sci. 2009;64(6):1358–1361. [Google Scholar]
- 24.Kumar V, Wang L, Riebe M, Tung HH, Prud’homme RK. Formulation and stability of itraconazole and odanacatib nanoparticles: governing physical parameters. Mol Pharm. 2009;6(4):1118–24. doi: 10.1021/mp900002t. [DOI] [PubMed] [Google Scholar]
- 25.Le Meins JF, Sandre O, Lecommandoux S. Recent trends in the tuning of polymersomes’ membrane properties. Eur Phys J E Soft Matter. 2011;34(2):14. doi: 10.1140/epje/i2011-11014-y. [DOI] [PubMed] [Google Scholar]
- 26.Discher BM, Won YY, Ege DS, Lee JC, Bates FS, Discher DE, Hammer DA. Polymersomes: tough vesicles made from diblock copolymers. Science. 1999;284(5417):1143–6. doi: 10.1126/science.284.5417.1143. [DOI] [PubMed] [Google Scholar]
- 27.Allen S, Liu YG, Scott E. Engineering nanomaterials to address cell-mediated inflammation in atherosclerosis. Regen Eng Transl Med. 2016;2(1):37–50. doi: 10.1007/s40883-016-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vasdekis AE, Scott EA, Roke S, Hubbell JA, Psaltis D. Vesicle Photonics. Annu Rev Mater Res. 2013;43:283–305. [Google Scholar]
- 29.Robertson JD, Yealland G, Avila-Olias M, Chierico L, Bandmann O, Renshaw SA, Battaglia G. pH-sensitive tubular polymersomes: formation and applications in cellular delivery. ACS Nano. 2014;8(5):4650–61. doi: 10.1021/nn5004088. [DOI] [PubMed] [Google Scholar]
- 30.Cui J, Xu J, Zhu Y, Jiang W. Shear flow controlled morphological polydispersity of amphiphilic ABA triblock copolymer vesicles. Langmuir. 2013;29(50):15704–10. doi: 10.1021/la404186u. [DOI] [PubMed] [Google Scholar]
- 31.Thiermann R, Mueller W, Montesinos-Castellanos A, Metzke D, Lob P, Hessel V, Maskos M. Size controlled polymersomes by continuous self-assembly in micromixers. Polymer. 2012;53(11):2205–2210. [Google Scholar]
- 32.La Y, Park C, Shin TJ, Joo SH, Kang S, Kim KT. Colloidal inverse bicontinuous cubic membranes of block copolymers with tunable surface functional groups. Nat Chem. 2014;6(6):534–41. doi: 10.1038/nchem.1946. [DOI] [PubMed] [Google Scholar]
- 33.van Oers MC, Rutjes FP, van Hest JC. Tubular polymersomes: a cross-linker-induced shape transformation. J Am Chem Soc. 2013;135(44):16308–11. doi: 10.1021/ja408754z. [DOI] [PubMed] [Google Scholar]
- 34.Rikken RS, Kerkenaar HH, Nolte RJ, Maan JC, van Hest JC, Christianen PC, Wilson DA. Probing morphological changes in polymersomes with magnetic birefringence. Chem Commun (Camb) 2014;50(40):5394–6. doi: 10.1039/c3cc47483f. [DOI] [PubMed] [Google Scholar]
- 35.Kim KT, Zhu J, Meeuwissen SA, Cornelissen JJ, Pochan DJ, Nolte RJ, van Hest JC. Polymersome stomatocytes: controlled shape transformation in polymer vesicles. J Am Chem Soc. 2010;132(36):12522–4. doi: 10.1021/ja104154t. [DOI] [PubMed] [Google Scholar]
- 36.Soo PL, Eisenberg A. Preparation of block copolymer vesicles in solution. J Polym Sci Pol Phys. 2004;42(6):923–938. [Google Scholar]
- 37.Yabu H. Creation of Functional and Structured Polymer Particles by Self-Organized Precipitation (SORP) Bull Chem Soc Jpn. 2012;85(3):265–274. [Google Scholar]
- 38.Shum HC, Kim JW, Weitz DA. Microfluidic fabrication of monodisperse biocompatible and biodegradable polymersomes with controlled permeability. J Am Chem Soc. 2008;130(29):9543–9. doi: 10.1021/ja802157y. [DOI] [PubMed] [Google Scholar]
- 39.Scott EA, Stano A, Gillard M, Maio-Liu AC, Swartz MA, Hubbell JA. Dendritic cell activation and T cell priming with adjuvant- and antigen-loaded oxidation-sensitive polymersomes. Biomaterials. 2012;33(26):6211–9. doi: 10.1016/j.biomaterials.2012.04.060. [DOI] [PubMed] [Google Scholar]
- 40.Hammer DA, Kamat NP. Towards an artificial cell. FEBS Lett. 2012;586(18):2882–2890. doi: 10.1016/j.febslet.2012.07.044. [DOI] [PubMed] [Google Scholar]
- 41.Pessi J, Santos HA, Miroshnyk I, Yliruusi J, Weitz DA, Mirza S. Microfluidics-assisted engineering of polymeric microcapsules with high encapsulation efficiency for protein drug delivery. Int J Pharm. 2014;472(1–2):82–87. doi: 10.1016/j.ijpharm.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 42.Napoli A, Valentini M, Tirelli N, Muller M, Hubbell JA. Oxidation-responsive polymeric vesicles. Nat Mater. 2004;3(3):183–9. doi: 10.1038/nmat1081. [DOI] [PubMed] [Google Scholar]
- 43.Nicol E, Nicolai T, Durand D. Dynamics of poly(propylene sulfide) studied by dynamic mechanical measurements and dielectric spectroscopy. Macromolecules. 1999;32(22):7530–7536. [Google Scholar]
- 44.Cerritelli S, O’Neil CP, Velluto D, Fontana A, Adrian M, Dubochet J, Hubbell JA. Aggregation Behavior of Poly(ethylene glycol-bl-propylene sulfide) Di- and Triblock Copolymers in Aqueous Solution. Langmuir. 2009;25(19):11328–11335. doi: 10.1021/la900649m. [DOI] [PubMed] [Google Scholar]
- 45.Cerritelli S, Fontana A, Velluto D, Adrian M, Dubochet J, De Maria P, Hubbell JA. Thermodynamic and kinetic effects in the aggregation behavior of a poly(ethylene glycol-b-propylene sulfide-b-ethylene glycol) ABA triblock copolymer. Macromolecules. 2005;38(18):7845–7851. [Google Scholar]
- 46.Vasdekis AE, Scott EA, O’Neil CP, Psaltis D, Hubbell JA. Precision intracellular delivery based on optofluidic polymersome rupture. ACS Nano. 2012;6(9):7850–7. doi: 10.1021/nn302122h. [DOI] [PubMed] [Google Scholar]
- 47.Segura T, Hubbell JA. Synthesis and in vitro characterization of an ABC triblock copolymer for siRNA delivery. Bioconjugate Chem. 2007;18(3):736–745. doi: 10.1021/bc060284y. [DOI] [PubMed] [Google Scholar]
- 48.O’Neil CP, Suzuki T, Demurtas D, Finka A, Hubbell JA. A novel method for the encapsulation of biomolecules into polymersomes via direct hydration. Langmuir. 2009;25(16):9025–9. doi: 10.1021/la900779t. [DOI] [PubMed] [Google Scholar]
- 49.Akbulut M, Ginart P, Gindy ME, Theriault C, Chin KH, Soboyejo W, Prud’homme RK. Generic Method of Preparing Multifunctional Fluorescent Nanoparticles Using Flash NanoPrecipitation. Adv Funct Mater. 2009;19(5):718–725. [Google Scholar]
- 50.Wohl AR, Michel AR, Kalscheuer S, Macosko CW, Panyam J, Hoye TR. Silicate esters of paclitaxel and docetaxel: synthesis, hydrophobicity, hydrolytic stability, cytotoxicity, and prodrug potential. J Med Chem. 2014;57(6):2368–79. doi: 10.1021/jm401708f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu Z. Flash nanoprecipitation: prediction and enhancement of particle stability via drug structure. Molecular pharmaceutics. 2014;11(3):776–86. doi: 10.1021/mp500025e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nichols MD, Scott EA, Elbert DL. Factors affecting size and swelling of poly(ethylene glycol) microspheres formed in aqueous sodium sulfate solutions without surfactants. Biomaterials. 2009;30(29):5283–91. doi: 10.1016/j.biomaterials.2009.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scott EA, Nichols MD, Kuntz-Willits R, Elbert DL. Modular scaffolds assembled around living cells using poly(ethylene glycol) microspheres with macroporation via a non-cytotoxic porogen. Acta Biomater. 2010;6(1):29–38. doi: 10.1016/j.actbio.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McKenzie BE, de Visser JF, Friedrich H, Wirix MJM, Bomans PHH, de With G, Holder SJ, Sommerdijk NAJM. Bicontinuous Nanospheres from Simple Amorphous Amphiphilic Diblock Copolymers. Macromolecules. 2013;46(24):9845–9848. [Google Scholar]
- 55.Lobling TI, Borisov O, Haataja JS, Ikkala O, Groschel AH, Muller AH. Rational design of ABC triblock terpolymer solution nanostructures with controlled patch morphology. Nat Commun. 2016;7:12097. doi: 10.1038/ncomms12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saito N, Liu C, Lodge TP, Hillmyer MA. Multicompartment Micelles from Polyester-Containing ABC Miktoarm Star Terpolymers. Macromolecules. 2008;41(22):8815–8822. [Google Scholar]
- 57.Jeon SI, Lee JH, Andrade JD, Degennes PG. Protein Surface Interactions in the Presence of Polyethylene Oxide .1. Simplified Theory. J Colloid Interface Sci. 1991;142(1):149–158. [Google Scholar]
- 58.Stewart S, Liu G. Block Copolymer Nanotubes. Angew Chem Int Ed Engl. 2000;39(2):340–344. [PubMed] [Google Scholar]
- 59.Grumelard J, Taubert A, Meier W. Soft nanotubes from amphiphilic ABA triblock macromonomers. Chem Commun (Camb) 2004;13:1462–3. doi: 10.1039/b405671j. [DOI] [PubMed] [Google Scholar]
- 60.Massignani M, Canton I, Sun T, Hearnden V, Macneil S, Blanazs A, Armes SP, Lewis A, Battaglia G. Enhanced fluorescence imaging of live cells by effective cytosolic delivery of probes. PLoS One. 2010;5(5):e10459. doi: 10.1371/journal.pone.0010459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pustulka KM, Wohl AR, Lee HS, Michel AR, Han J, Hoye TR, McCormick AV, Panyam J, Macosko CW. Flash nanoprecipitation: particle structure and stability. Molecular pharmaceutics. 2013;10(11):4367–77. doi: 10.1021/mp400337f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pinkerton NM, Grandeury A, Fisch A, Brozio J, Riebesehl BU, Prud’homme RK. Formation of stable nanocarriers by in situ ion pairing during block-copolymer-directed rapid precipitation. Mol Pharm. 2013;10(1):319–28. doi: 10.1021/mp300452g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maherani B, Arab-Tehrany E, Kheirolomoom A, Geny D, Linder M. Calcein release behavior from liposomal bilayer; influence of physicochemical/mechanical/structural properties of lipids. Biochimie. 2013;95(11):2018–33. doi: 10.1016/j.biochi.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 64.van Dongen SF, Nallani M, Cornelissen JJ, Nolte RJ, van Hest JC. A three-enzyme cascade reaction through positional assembly of enzymes in a polymersome nanoreactor. Chemistry. 2009;15(5):1107–14. doi: 10.1002/chem.200802114. [DOI] [PubMed] [Google Scholar]
- 65.Rameez S, Alosta H, Palmer AF. Biocompatible and biodegradable polymersome encapsulated hemoglobin: a potential oxygen carrier. Bioconjug Chem. 2008;19(5):1025–32. doi: 10.1021/bc700465v. [DOI] [PubMed] [Google Scholar]
- 66.Arifin DR, Palmer AF. Polymersome encapsulated hemoglobin: a novel type of oxygen carrier. Biomacromolecules. 2005;6(4):2172–81. doi: 10.1021/bm0501454. [DOI] [PubMed] [Google Scholar]
- 67.Lomas H, Massignani M, Abdullah KA, Canton I, Presti CLo, MacNeil S, Du J, Blanazs A, Madsen J, Armes SP, Lewis AL, Battaglia G. Non-cytotoxic polymer vesicles for rapid and efficient intracellular delivery. Faraday Discuss. 2008;139:143–59. doi: 10.1039/b717431d. discussion 213–28, 419–20. [DOI] [PubMed] [Google Scholar]
- 68.Wang L, Chierico L, Little D, Patikarnmonthon N, Yang Z, Azzouz M, Madsen J, Armes SP, Battaglia G. Encapsulation of biomacromolecules within polymersomes by electroporation. Angew Chem Int Ed Engl. 2012;51(44):11122–5. doi: 10.1002/anie.201204169. [DOI] [PubMed] [Google Scholar]
- 69.Horwitz JP, Chua J, Noel M, Donatti JT, Freisler J. Substrates for cytochemical demonstration of enzyme activity. II. Some dihalo-3-indolyl phosphates and sulfates. J Med Chem. 1966;9(3):447. doi: 10.1021/jm00321a059. [DOI] [PubMed] [Google Scholar]
- 70.Coenen JJ, Koenen HJ, van Rijssen E, Kasran A, Boon L, Hilbrands LB, Joosten I. Rapamycin, not cyclosporine, permits thymic generation and peripheral preservation of CD4+ CD25+ FoxP3+ T cells. Bone Marrow Transplant. 2007;39(9):537–45. doi: 10.1038/sj.bmt.1705628. [DOI] [PubMed] [Google Scholar]
- 71.Noris M, Casiraghi F, Todeschini M, Cravedi P, Cugini D, Monteferrante G, Aiello S, Cassis L, Gotti E, Gaspari F, Cattaneo D, Perico N, Remuzzi G. Regulatory T cells and T cell depletion: role of immunosuppressive drugs. J Am Soc Nephrol. 2007;18(3):1007–18. doi: 10.1681/ASN.2006101143. [DOI] [PubMed] [Google Scholar]
- 72.Cao W, Manicassamy S, Tang H, Kasturi SP, Pirani A, Murthy N, Pulendran B. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat Immunol. 2008;9(10):1157–64. doi: 10.1038/ni.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. 2009;9(5):324–37. doi: 10.1038/nri2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Turnquist HR, Raimondi G, Zahorchak AF, Fischer RT, Wang Z, Thomson AW. Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J Immunol. 2007;178(11):7018–31. doi: 10.4049/jimmunol.178.11.7018. [DOI] [PubMed] [Google Scholar]
- 75.Haddadi A, Elamanchili P, Lavasanifar A, Das S, Shapiro J, Samuel J. Delivery of rapamycin by PLGA nanoparticles enhances its suppressive activity on dendritic cells. J Biomed Mater Res A. 2008;84(4):885–98. doi: 10.1002/jbm.a.31373. [DOI] [PubMed] [Google Scholar]
- 76.Jhunjhunwala S, Raimondi G, Thomson AW, Little SR. Delivery of rapamycin to dendritic cells using degradable microparticles. J Control Release. 2009;133(3):191–7. doi: 10.1016/j.jconrel.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mercalli A, Calavita I, Dugnani E, Citro A, Cantarelli E, Nano R, Melzi R, Maffi P, Secchi A, Sordi V, Piemonti L. Rapamycin unbalances the polarization of human macrophages to M1. Immunology. 2013;140(2):179–90. doi: 10.1111/imm.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yi S, Allen SD, Liu YG, Ouyang BZ, Li X, Augsornworawat P, Thorp EB, Scott EA. Tailoring Nanostructure Morphology for Enhanced Targeting of Dendritic Cells in Atherosclerosis. ACS Nano. 2016;10(12):11290–11303. doi: 10.1021/acsnano.6b06451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dowling DJ, Scott EA, Scheid A, Bergelson I, Kats DI, Sanchez-Schmitz G, Van Haren SD, Ninkovic J, Bonner DK, Hirosue S, Hubbell JA, Levy O. TLR8 agonist encapsulating polymersomes mimic the immunomodulating effect of the live BCG vaccine on neonatal dendritic cells while dramatically enhancing IL-12p70 production. Journal of Clinical Investigation In Press. 2017 [Google Scholar]
- 80.Li X, Garcia K, Sun Z, Xiao Z. Temporal regulation of rapamycin on memory CTL programming by IL-12. PloS one. 2011;6(9):e25177. doi: 10.1371/journal.pone.0025177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu XL, Luo L, Mu RH, Liu BB, Geng D, Liu Q, Yi LT. Fluoxetine regulates mTOR signalling in a region-dependent manner in depression-like mice, Scientific reports 5 ( 2015:16024. doi: 10.1038/srep16024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stoica L, Zhu PJ, Huang W, Zhou H, Kozma SC, Costa-Mattioli M. Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks long-term synaptic plasticity and memory storage. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(9):3791–6. doi: 10.1073/pnas.1014715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ikonen TS, Gummert JF, Serkova N, Hayase M, Honda Y, Kobayase Y, Hausen B, Yock PG, Christians U, Morris RE. Efficacies of sirolimus (rapamycin) and cyclosporine in allograft vascular disease in non-human primates: trough levels of sirolimus correlate with inhibition of progression of arterial intimal thickening. Transpl Int. 2000;13(Suppl 1):S314–20. doi: 10.1007/s001470050351. [DOI] [PubMed] [Google Scholar]
- 84.Xiong A, Duan L, Chen J, Fan Z, Zheng F, Tan Z, Gong F, Fang M. Flt3L combined with rapamycin promotes cardiac allograft tolerance by inducing regulatory dendritic cells and allograft autophagy in mice. PloS one. 2012;7(10):e46230. doi: 10.1371/journal.pone.0046230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Poston RS, Billingham M, Hoyt EG, Pollard J, Shorthouse R, Morris RE, Robbins RC. Rapamycin reverses chronic graft vascular disease in a novel cardiac allograft model. Circulation. 1999;100(1):67–74. doi: 10.1161/01.cir.100.1.67. [DOI] [PubMed] [Google Scholar]
- 86.Kishimoto TK, Ferrari JD, LaMothe RA, Kolte PN, Griset AP, O’Neil C, Chan V, Browning E, Chalishazar A, Kuhlman W, Fu FN, Viseux N, Altreuter DH, Johnston L, Maldonado RA. Improving the efficacy and safety of biologic drugs with tolerogenic nanoparticles. Nat Nanotechnol. 2016;11(10):890–899. doi: 10.1038/nnano.2016.135. [DOI] [PubMed] [Google Scholar]
- 87.Sterling T, Irwin JJ. ZINC 15—Ligand Discovery for Everyone. J Chem Inf Model. 2015;55(11):2324–37. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223(1):77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.