

Abstract
Gas-perfused microchannels generated a linear oxygen gradient via diffusion across a 100 μm polydimethylsiloxane (PDMS) membrane. The device enabled exposure of a single monolayer of cells sharing culture media to a heterogeneous oxygen landscape, thus reflecting the oxygen gradients found at the microscale in the physiological setting and allowing for the real-time exchange of paracrine factors and metabolites between cells exposed to varying oxygen levels. By tuning the distance between two gas supply channels, the slope of the oxygen gradient was controlled. We studied the hypoxic activation of the transcription factors HIF-1α and HIF-2α in human endothelial cells within a spatial linear gradient of oxygen. Quantification of the nuclear to cytosolic ratio of HIF immunofluorescent staining demonstrated that the threshold for HIF-1α activation was below 2.5% O2 while HIF-2α was activated throughout the entire linear gradient. We show for the first time HIF-2α is subject to hyproxya, hypoxia by proxy, wherein hypoxic cells activate HIF in close proximity normoxic cells. These results underscore the differences between HIF-1α and HIF-2α regulation and suggest that a microfluidic oxygen gradient is a novel tool for identifying distinct hypoxic signaling activation and interactions between differentially oxygenated cells.
Keywords: hypoxia, oxygen, gradient, microfluidic, HIF-1α, HIF-2α, endothelial cells
Graphical Abstract
INTRODUCTION
Cells adapt to environmental oxygen levels by shifting metabolic processes and initiating transcriptional programs. When oxygen demand exceeds the supply, the intracellular oxygen levels decrease, and cells experience hypoxia. Hypoxia signaling pathways regulate a broad array of biological processes including angiogenesis1,2, embryonic development3,4, stem cell differentiation5,6, extracellular matrix remodeling7, tumor growth and progression8, and metastasis9. Hypoxia-inducible factors (HIFs) are a family of transcription factors that are the central regulators of the transcription of hypoxia-response genes, including growth factors, receptors, and metabolic enzymes. The key α-subunits, HIF-1α and HIF-2α, are subject to rapid turnover under normal physiological oxygen (normoxic) conditions via the action of prolyl-hydroxylases (PHDs) which hydroxylate proline residues within the oxygen-dependent degradation domain (ODD) of HIFs and thus promote ubiquitination and proteasomal degradation. Hypoxia suppresses PHD activity, thus allowing HIF-1α and HIF-2α to accumulate and translocate to the nucleus where they bind to the ubiquitously expressed HIF-1β (also known as ARNT) and initiate hypoxia-activated transcription.
The threshold for HIF activation has been reported to be cell type specific. Oxygen levels <1% O2 are considered particularly hypoxic and activate HIF-1α in several cell types10–12. However, there are notable exceptions such as papillary tip cells in rodent kidneys which do not demonstrate any significant HIF-1α activation even though these cells are physiologically exposed to <1% O2, but HIF-1α is strongly induced in those papillary cells during systemic in vivo hypoxic injury13. Especially at moderate hypoxia (O2 levels ≈1–10%), thresholds for hypoxic HIF-activation can vary widely between cell types and this is in part due to varying PHD isoform expression levels across tissues and cell types14. Dynamic studies showed transient 3 hour pulses of HIF-1α and HIF2α expression during a constant hypoxic condition15. Additionally, a temporal gradient was reported to demonstrate that HIF-1α responds to decrements in oxygen and not absolute oxygen levels16. Together, these studies underscore the complexities involved in understanding HIF activation. Importantly, factors that may be important in oxygen sensing and HIF activation may have not yet been defined. One such factor is a spatial oxygen gradient to examine the effect differentially oxygenated cells may have on each other. To date, no study has investigated HIF activation as a function of position within a spatial oxygen gradient, primarily from a lack of adequate tools.
Even though previous studies of the threshold of HIF activation examined a range of oxygen levels, these studies were conducted in homogeneous oxygen environments where each experiment exposes a whole monolayer of cells to a single homogeneous oxygen level 17–20. However, homogeneous oxygenation does not reflect in vivo hypoxia where oxygen gradients are present both radially and longitudinally in the microvasculature and tissue oxygenation decreases rapidly with increased distance from a microvessel21.
Microfluidic devices enable the exposure of cells to precisely controlled oxygen environments22,23 but have also predominantly been used to generate single-condition, homogeneous environments24–28. Some devices have demonstrated generation of oxygen gradients, but these have relied on complex internal or external architecture, including many gas inputs, off-chip gas mixers, and on-chip dilution trees29,30. Other microfluidic devices require flow of media perfusate or oxygen scavenging chemicals31–34. There has been limited control of gradient shape with most gradients having non-monotonic, sigmoidal, or exponentially decreasing profiles29,30,35,36. The few linear oxygen gradients reported have had significant drawbacks including requiring many gas inputs, not being characterized with respect to time required to reach equilibrium, and not considering the ability to tune the slope by altering the diffusion gap between microchannels29,35,36.
Here we report the generation of a stable, linear oxygen gradient with a tunable slope that depends on the size of the diffusion gap between two gas-perfused microchannels. We validate the gradient with oxygen measurements and show the equilibration of a linear profile over time. Importantly, we quantify the expression of HIF-1α and HIF-2α in human brain endothelial cells as a function of position via immunofluorescent staining and observe important differences in HIF-activation thresholds when cells are exposed to oxygen gradients as compared to homogeneous oxygen levels.
EXPERIMENTAL
Microfluidic device design and fabrication
The microchannel layer was fabricated using standard soft lithography techniques. Briefly, polydimethylsiloxane (PDMS) (Sylgard 184 Silicone Elastomer, Dow Corning) base was added to the curing agent at a weight ratio of 10:1. The mixture was mixed and degassed in a planetary centrifugal mixer (Thinky; Laguna Hills, CA) and cast on a silicon master containing SU-8 (MicroChem) microchannel features. The microchannel dimensions were 500 μm (width) × 20 mm (length) × 100μm (height). The PDMS channel layer was cured at 85 °C on a hot plate for 2 h and inlet/outlet ports were punched with a 15 gage (1.37 mm ID, 1.83 OD) blunt needle.
The PDMS membrane was fabricated by spinning uncured, degassed PDMS (10:1 weight ratio of base to curing agent) on a 100 mm diameter silicon wafer (University Wafer, Boston, MA) at 1000 rpm for 30 s using a spin coater (Laurell Technologies Corporation, North Wales, PA). After curing at 60 °C for 40 min, the membrane was bonded to the microchannel layer using surface treatment from a handheld corona discharge device (Electro Technic Products, Chicago, IL). The bonded membrane was punched with inlet/outlet ports, and the PDMS construct was then bonded on a 75 mm × 50 mm glass slide (Fisher Scientific).
Oxygen Modulation
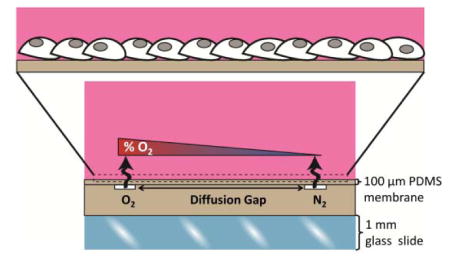
Two microfluidic perfusion channels (500 μm width and 100 μm depth) were separated by varying widths of a diffusion gap (Fig. 1A). One channel was continuously perfused with 5% CO2, balanced air and the second channel was continuously perfused with 5% CO2, balanced nitrogen. Diffusion of the gases across the 100 μm membrane and into the bulk created a linear gradient surface oxygen profile to which the cells cultured on the membrane were exposed. The experimental setup for gas perfusion flowed compressed gas from the regulator on the gas tank to a glass tube rotometer and then through a mini gas regulator and into a microchannel of the device (Fig. 1B). The connection of a manometer by a three-way valve allowed for real-time monitoring of the pressure within the microchannel. The pressure in the two microchannels was kept equal at 5 psi to establish a stable oxygen gradient and minimize variation within and between experiments.
Figure 1.

Constant perfusion of oxygen and nitrogen compressed gasses into microchannels separated by a diffusion gap exposes cells to a spatial linear oxygen gradient. (A) A schematic cross-sectional view of the microchannel device demonstrates diffusion from an oxygen channel and a nitrogen channel across a thin PDMS membrane establishes an oxygen gradient to which cells cultured on the membrane are exposed. (B) The experimental setup schematic for gas perfusion shows flow from a compressed gas tank to a glass tube rotometer and then through a mini gas regulator and into a microchannel of the device. The connection of a manometer by a three-way valve is used to monitor that pressure remains equal within each microchannel.
Oxygen gradient validation
The surface oxygen profile was characterized using oxygen sensors constructed from a 100 μm gas-permeable PDMS membrane impregnated with platinum(II) octaethylporphyrinketone (PtOEPK). The sensor was fabricated by first making a 35% w/w toluene/polystyrene mixture and then adding 0.5 mg mL−1 of PtOEPK to the solution of polystyrene dissolved in toluene. The PtOEPK-containing mixture was spin-coated on a cured 100 μm PDMS membrane on a silicon wafer. The dried polystyrene was washed away with isopropanol, leaving behind a PDMS membrane containing PtOEPK that can easily be cut to convenient sizes for desired sensors. The fluorescence of the PtEOPK fluorophore in the sensor is quenched in the presence of oxygen. Prior to beginning perfusion of compressed gas, the PDMS oxygen sensor was placed on top of the device’s PDMS membrane and the open well was filled with 5 mL of PBS. Scans across the sensor were used to determine the surface percent oxygen profile. Scans were taken over a 4 h period after introducing gas flow. Percent oxygen was plotted from the fluorescent intensity by solving the Stern–Volmer equation. The hypoxic channel was perfused with 5% CO2, balanced nitrogen, and the normoxic channel was perfused with 5% CO2, balanced air. The fluorescent intensity of the sensor at 4 h within the region directly above the hypoxic channel when perfused with 5% CO2, balanced nitrogen while nitrogen gas was injected in the environment surrounding the device was used as the 0% O2 calibration. The fluorescent intensity of the sensor directly above the normoxic channel under ambient conditions prior to gas perfusion was used as the 21% O2 calibration. The fabrication and oxygen validation of the homogeneous control devices is reported elsewhere36.
Oxygen conditions were modulated by introducing desired gas compositions from compressed gas tanks. Plastic connectors (McMaster-Carr) interfaced between the access ports of the device and the tubing (Tygon 1.59 mm (1/16 inch) ID and 3.18 mm(1/8 inch) OD; Cole-Parmer)supplying the compressed gas. The gas pressure was stabilized by running polyurethane tubing (6.35 mm (1/4 inch) OD, McMaster-Carr) from the gas regulator on the compressed gas tank to a glass tube rotameter (Omega Engineering, Inc., Stamford, CT) and then to a mini gas regulator (Marsh Bellofram, Newell, WV). Each gas line was adjusted to 5 psi in the device as monitored by a manometer (Dwyer Instruments, Michigan City, IN).
Endothelial cell culture
Fabricated devices were autoclaved and then filled with 2 mL of 0.1% gelatin (porcine skin type A; Sigma-Aldrich) in water. Gelatin was incubated at 37 °C overnight on the PDMS membrane. Gelatin was then aspirated, and endothelial cells were seeded at 250 000 cells per device. The human cerebral microvessel endothelial cell line hCMEC/D3 was cultured in EGM-2 MV (Lonza). Devices with cells were placed in Petri dishes and kept in a standard incubator at 37 °C in 5% CO2, balanced air. Media was changed every 24 h until cells reached >80% confluency.
In-device immunofluorescent staining
Cell culture media was aspirated, and the cells were washed twice with PBS, fixed with 4% paraformaldehyde for 10 min at room temperature, and washed two additional times with PBS. Cells were permeabilized in 0.2% Triton X-100 in PBS for 10 min at room temperature, washed three times with PBS, and blocked in 3% bovine serum albumin (BSA) in phosphate-buffered saline with Tween 20 (PBST) for 1 h at room temperature. Cells were subsequently incubated with anti-HIF-1α antibody or anti-HIF2α antibody (Novus Biologicals, Littleton, CO) with a 1:200 dilution in 3% BSA in PBST overnight at 4 °C. After three washes with PBST, cells were incubated with fluorescent secondary antibody (Alexa Fluor 633; Invitrogen, Carlsbad, CA) in 3% BSA in PBST with a 1:300 dilution for 2 h at room temperature. Cells were washed three times with PBST and nuclei were stained (Hoechst 33342; Invitrogen) with a 1:5000 dilution in PBST for 10 min at room temperature. Then a drop of antifade mounting solution (ProLong Gold AntifadeMountant; Thermofisher) was added directly to the immunostained cells on the membrane, a glass coverslip was placed on top, and left to cure in the dark for 24 h before imaging.
Microscopy and image analysis
Images were acquired using a confocal laser scanning microscope (Zeiss LSM 710). Devices were placed upside down in the slide holding stage insert to be imaged on the inverted confocal microscope. The entire gradient was imaged using the tiling function in the Zen imaging software. A63X Plan-Apochromat (1.46NA) objective was used during acquisition. Images were processed using FIJI open source software. Nuclear/cytosolic ratios were determined as follows: a nuclear mask was made from the Hoechst staining channel. Images acquired of the HIF staining channel were background subtracted and filtered (median). The nuclear mask was applied to quantify the mean pixel value of the 12-bit image (intensity min, max: 0, 4095) within the nuclear regions of the HIF staining channel per field of view frame. The mask was also used to subtract the area of the nuclei, leaving behind the cytosolic staining. The cytoplasmic signal was determined as the mean pixel value of the area in a field of view frame thresholded above a set background value (100 was used for HIF-1α and 200 was used for HIF-2α). The nuclear/cytoplasmic ratio was measured by dividing the mean nuclear intensity by the mean cytoplasmic intensity.
Statistical analysis
Oxygen validation experiments were repeated three independent times. The data are expressed as the mean ± SD. The slope was determined from a linear regression of the data across the spatial gradient after 4 h of oxygen modulation. Cell culture experiments were repeated three independent times. During each independent experiment, three scans of the cells across the gradient were acquired. The data are expressed as the mean ± SEM. Significance was determined from a two-way ANOVA with Bonferroni’s multiple comparison post hoc test. Analyses were performed using Prism 5 by Graphpad.
RESULTS
The microfluidic cell culture platform generates linear oxygen gradients
We developed a micro-engineered system to generate a stable, linear oxygen gradient and study HIF activation in a monolayer of human endothelial cells exposed to a range of oxygenation from 0% to ambient 21% (Fig. 1A), thus better mimicking the physiologic oxygen microenvironment in vivo where gradients of oxygen exist in tissues and within the vasculature. Gas perfusion of buried microchannels covered in a gas-permeable PDMS membrane allowed for diffusion of gas across the membrane to expose cells to a spatial gradient. The linear gradient allowed us to precisely assign a known level of oxygenation to which cells are exposed with a specific position. The linear gradient also allowed equal contribution of all the oxygen levels within the range of established oxygenation for the spatial profile. The experimental setup for gas perfusion integrated real-time monitoring of the pressure in the two microchannels via connection of each microchannel to a manometer (Fig. 1B).
The size of the diffusion gap tunes the slope of the linear oxygen gradient
We first determined the oxygen gradient in the device as a function of the position along the x-axis (Fig. 2A–D). Devices with a diffusion gap of 3 mm (Fig. 2A), 5 mm (Fig. 2C), and 7 mm (Fig 2B) were fully characterized with gradients from 21% to 0% O2 over a 4-hour duration of oxygen modulation. The 5 mm diffusion gap device was further characterized in the range from 7.5% to 0% O2 by substituting a 5% CO2, 7.5% O2, and balanced nitrogen tank for perfusion into the first microchannel (Fig. 2D) to demonstrate the usage of the device in a narrower range of oxygenation. The linear gradient developed over time, and by 2 hours of oxygen modulation, the established gradients were near equilibrium for all tested diffusion gaps. The slope of the gradient could be tuned by altering the width of the diffusion gap (Fig. 2E). The slope did not vary linearly with the diffusion gap width (Fig. 2F). However, a reduction in the diffusion gap resulted in a steepening of the oxygen gradient. The slopes characterized from the 21% to 0% range of the 3 mm, 5 mm, and 7 mm gaps were −5.5% ± 0.3%, −3.4% ± 0.1%, and −2.7% ± 0.1% O2/mm, respectively.
Figure 2.

Sensor measurements characterized the oxygen gradient. (A–C) The surface percent oxygen over a 4 h duration is shown as a function of position with microchannel inputs of 21% O2 and 0% O2 for a 3 mm, 7 mm, and 5 mm diffusion gap, respectively. (D) Microchannel inputs of 7.5% O2 and 0% O2 for a 5 mm diffusion gap demonstrates that choice of inputs determines the range of the linear gradient. (E) The 21%-0% range for 3 mm, 5 mm, and 7 mm diffusion gaps are compared, demonstrating that the slope is tunable by altering the diffusion gap parameter. The middle of each diffusion gap is centered at the 0 mm position. (F) The slope is plotted as a function of the size of the diffusion gap.
The oxygen gradient is maintained in the presence of cells
To determine whether the oxygen gradient was affected by cells cultured on the membrane, oxygen levels were measured in the presence of the cell monolayer to assess whether the linear gradient was preserved. Cells were cultured directly on a PDMS membrane impregnated with PtOEPK. Tiled scans of the PtOEPK signal on the membrane were acquired after 4 h of oxygen modulation with cells (Fig. 3A) and without cells (Fig. 3B) on the membrane of the device. The post Stern–Volmer analysis demonstrated that the spatial gradient was not affected by the presence of cells (Fig. 3C). As expected for such a device with constant perfusion of gas at constant, high pressure (5 psi) through the microfluidic channels, the cells do not consume oxygen at a rate fast enough to significantly alter the surface oxygen profile to which the cells were exposed. The slope of the gradient with cells and without cells was equal to −3.8 ± 0.2% O2/mm.
Figure 3.
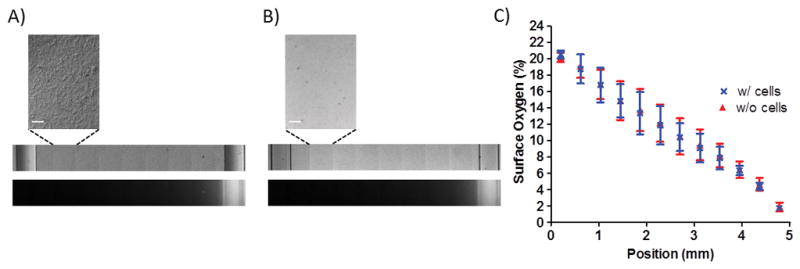
Cells cultured on the membrane do not affect the oxygen gradient to which the cells are exposed. Tiled scans of brightfield images of the membrane (top) and fluorescent images of the PtEOPK signal (bottom) demonstrate the presence of the oxygen gradient (A) with cells and (B) without cells. Scale bar: 100 μm. (C) The surface percent oxygen level as a function of position does not vary between the cell-occupied and cell-vacant condition.
HIF-1α requires low oxygen levels for activation
We next evaluated the activation of HIF-1α and HIF-2α in human endothelial cells within the oxygen gradient. The 5 mm gap device was used for all subsequent cell studies. D3 endothelial cells were cultured in the device until approximately 80% confluency. The cells were then subjected to 8 hours of a linear oxygen gradient. Immunofluorescence imaging of HIF-1α in the spatial gradient clearly showed nuclear translocation of HIF-1α in the cells cultured directly above the device’s hypoxic channel (Fig. 4D) and homogeneous hypoxic control devices (Fig. 4B) as compared to cells cultured above the normoxic channel (Fig. 4C) and homogeneous normoxic control devices (Fig. 4A). A tiled image scan of the entire gradient allowed us to determine the nuclear to cytosolic ratio for each field of view and these were plotted as the x-coordinate of the position at the center of the field of view. The nuclear to cytosolic ratios of cells within the oxygen gradient provided a measure of HIF-1α activation because active HIFs are translocated to the nucleus and were compared to the ratios found in cells exposed to homogeneous oxygen levels. The nuclear to cytosolic ratio of a homogeneous normoxic control device was normalized to 1. Nuclear to cytosolic ratio quantification of HIF-1α showed that the two closest fields of view to the hypoxic channel were statistically significantly different from the homogeneous normoxic control (Fig. 4E). Oxygen levels in the linear gradient below 2.5% resulted in statistically higher HIF activation. Importantly, all the positions with O2 levels higher than 2.5% (x-axis coordinate lower than 4.8 mm) did not show any significant HIF-1α activation. Importantly, the degree of HIF-1α activation at the 0–1% level approximated that of cells placed in homogeneous hypoxia (blue range, Fig 4E).
Figure 4.
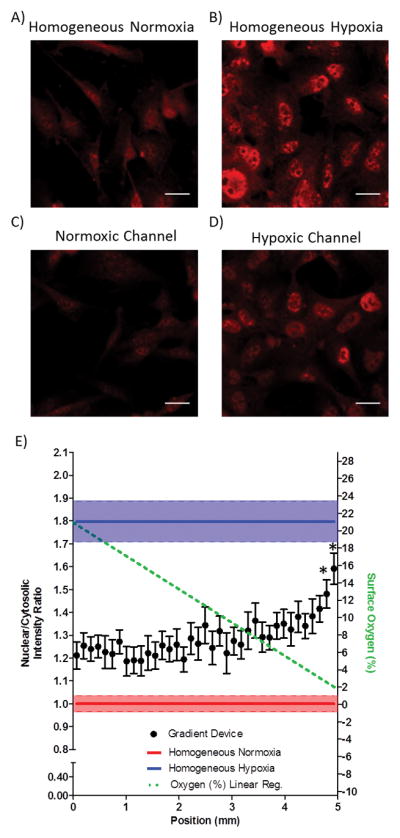
HIF-1α activation in endothelial cells occurs at low oxygen levels within a linear oxygen gradient. (A–B) Panels of HIF-1α immunofluorescent staining for normoxic and hypoxic homogeneous control devices. (C–D) HIF-1α immunofluorescent staining in the oxygen gradient device of cells directly above the normoxic gas supply microchannel and hypoxic gas supply microchannel, respectively. Scale bar: 20 μm. (E) Quantification of the nuclear to cytosolic ratio as a function of the position in the oxygen gradient. * p < 0.05 as compared to homogeneous normoxia.
HIF-2α is activated throughout the spatial oxygen gradient
In contrast to the narrowly confined range of HIF-1α activation at the extremely low O2 levels (0–2.5%), HIF-2α activation was seen throughout the entire spatial gradient. Even cells exposed to normoxia (by diffusion of 5% CO2, balanced air and cultured directly above the normoxic supply channel in the gradient device) had observable HIF-2α activation (Fig. 5C) when compared to cells cultured in homogeneous normoxic conditions (Fig. 5A), suggesting that normoxia within an oxygen gradient elicits HIF-2α activation whereas homogeneous normoxia does not. Quantification of images from a tiled scan of the gradient demonstrated widespread HIF-2α activation throughout the gradient when compared to cells exposed to homogeneous normoxia (Figure 5E).
Figure 5.
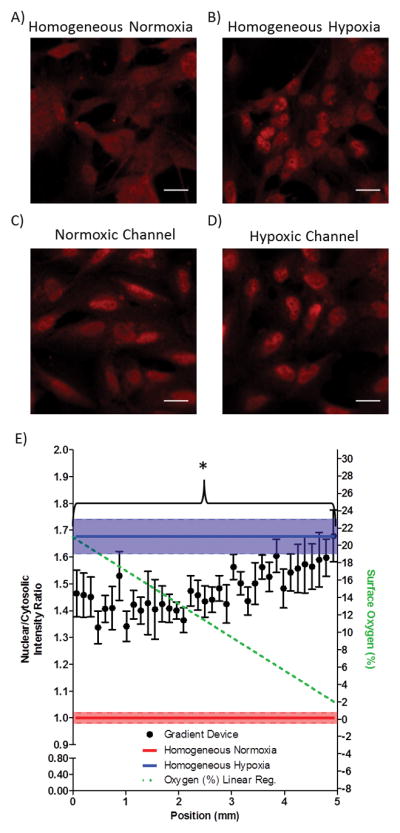
HIF-2α is activated throughout the entire linear gradient. (A–B) Panels of HIF-2α immunofluorescent staining for normoxic and hypoxic homogeneous control devices. (C–D) HIF-2α immunofluorescent staining in the oxygen gradient device of cells directly above the normoxic gas supply microchannel and hypoxic gas supply microchannel, respectively. Scale bar: 20 μm. (E) Quantification of the nuclear to cytosolic ratio as a function of the position in the oxygen gradient. * p < 0.05 as compared to homogeneous normoxia.
DISCUSSION
In our gas perfusion-based linear oxygen gradient cell culture platform, we showed the spatial gradient develops over time, becoming completely developed after several hours due to the bulk PDMS in the diffusion gap on the scale of several millimeters. Although this time to equilibrate may be seen as a limitation of the device, cells could easily be exposed specifically to the fully developed spatial gradient if, for example, the cells were cultured on a gas-permeable PDMS membrane that was placed on top of the base PDMS membrane after several hours of oxygen modulation. Oxygen diffuses across an additional 100 μm PDMS membrane on the scale of tens of seconds36,37.
Although our primary focus was demonstrating a linear spatial gradient, we acknowledge that certain oxygen control applications may also call for control of a temporal gradient. Computer-controlled microdispensing nozzles have been successfully used to deliver intermittent hypoxia to a PDMS-based gas perfusion device38 and could be applied to our platform to temporally control the oxygen levels in the device or change the direction of the gradient. Furthermore, not all physiological gradients are necessarily linear, however, for the purpose of defining clear activation thresholds, linear gradients are suitable for the experimental design.
Differences between the oxygen-dependent stabilities of HIF-1α and HIF-2α have been reported by others, although it is not known whether these two transcription factors respond differentially to oxygen gradients. HIF-2α has a different sub-nuclear distribution than HIF-1α, lending to its increased stability and slower mobility as compared to HIF-1α39. Higher levels of oxygen have been shown to inhibit HIF-2α degradation and allow for rapid accumulation in human endothelial cells as compared to oxygen levels required to inhibit HIF-1α degradation (3.5% O2 vs. 1.0% O2, respectively)40. The vast majority of studies which have established oxygen level thresholds have been performed in cells exposed to a single, homogeneous oxygen environment (e.g. one cell culture plate is exposed to 3% while a separate plate is exposed to 1%). Using microfluidic devices, we studied whether the threshold of activation differs if cells are cultured in an open-well gradient which allows for biological interactions between cells that are exposed to varying oxygen levels. Here, we show in endothelial cells exposed to an oxygen gradient from 0% to 21%, HIF-2α is activated throughout the entire gradient when compared to cells exposed to homogeneous normoxia, suggesting a “contagion” of HIF-2α activation. Even cells within the spatial gradient that are exposed to continuous normoxia exhibit significant accumulation and nuclear translocation of the HIF-2α transcription factor, likely due to the close proximity of cells exposed to hypoxia within the same gradient.
The activation of HIF-2α demonstrates hyproxya (hypoxia by proxy), which describes the activation of hypoxic signaling in normoxic cells by hypoxic neighbors which may constitute an adaptive signal within a tissue in which localized hypoxia can elicit a broader systemic response. It is noteworthy that this was only true for HIF-2α activation but not for HIF-1α activation, suggesting that HIF-1α is only activated in hypoxic cells and not when neighboring cells experience hypoxia.
The mechanism by which hyproxya of HIF-2α activation occurs is not yet known. It likely involves a multitude of potential signals by which hypoxic cells can act as sentinels for normoxic neighbors and could include paracrine signals such as the secretion of exosomes, metabolites, or growth factors. It has been previously reported that exosomal miRNAs may activate HIF independent of hypoxia2,41. Therefore, one can envision a scenario in which the exosomes from the hypoxia-exposed cells contain miRNAs that activate HIF in the nearby normoxia-exposed cells. Additionally, hypoxia alters cellular metabolism, increasing a cell’s dependence on glycolysis over oxidative phosphorylation to produce ATP. As a result, metabolites such as lactate, succinate, and fumarate have been shown to accumulate intracellularly42,43. These metabolites may then be secreted into the extracellular space and affect surrounding cells. A specific cell surface receptor for lactate and succinate has been identified44–46.
The source of the reported biological effects in hypoxia-conditioned media experiments is thought to be the secreted molecules or microvesicles. The disadvantage of traditional conditioned media experiments is that there exists a single, isolated time point when cell culture media is harvested and transferred to other cells. Oxygen gradient devices in which differentially oxygenated cells share media allow for continuous exposure of normoxic cells to the hypoxia-induced secretome. Interestingly, HIF-2α transactivation is reported as being sensitive to the composition of cell culture media. HIF-2α specifically acts as a response factor to glucose concentration in media47,48. Media supplemented with acetate, which can be released by tumors, results in acetylation of HIF-2α and nuclear localization of an acetate-dependent acetyl CoA synthetase ACSS2 required for induction49. Hypoxia-conditioned media from cells secreting acetate can induce this HIF-2α acetylation48.
Understanding how HIF-1α and HIF-2α are differentially regulated in oxygen gradients is critical because HIF-1α and HIF-2α can have opposing roles based on their downstream targets50–55. Our findings demonstrate a novel aspect of differential HIF-1α and HIF-2α activation: their upstream activation by low oxygen. HIF-2α activation does not require hypoxia in all cells but can be activated when only selective cells are exposed to low oxygen levels. These results highlight the importance of understanding the effect of cell-cell interactions in an oxygen gradient environment. Further mechanistic studies using the microfluidic device we engineered could allow for the isolation of the intercellular signals which mediate HIF-2α hyproxya –hypoxia by proxy– and allow for therapeutic modulation of HIF-2α levels even in the absence of direct hypoxia.
Insight Box.
Gas control microfluidics generated linear spatial oxygen gradients to study the regulation of HIF-1α and HIF-2α, the transcription factors mediating cellular adaptation to hypoxic microenvironments. Prior studies have identified HIF activation thresholds in uniform oxygen levels but little is known about activation thresholds in oxygen gradients. We found that HIF-1α was activated in low levels of oxygen (below 2.5%) in the gradient. In contrast, HIF-2α activation was markedly influenced by the oxygenation of surrounding cells. The presence of hypoxic cells in close proximity to normoxic cells activated HIF-2α throughout the entire spatial gradient including cells exposed to ambient oxygen levels. Assessing hypoxic activation within oxygen gradients better reflects conditions in vivowhere metabolic consumption and oxygen transport generate physiological gradients.
Acknowledgments
This work was supported by National Science Foundation 1253060, NIH T32-HL007829, and NIH P01-HL60678.
References
- 1.Pugh CW, Ratcliffe PJ. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh G, Subramanian IV, Adhikari N, Zhang X, Joshi HP, Basi D, Chandrashekhar YS, Hall JL, Roy S, Zeng Y, Ramakrishnan S. J Clin Invest. 2010;120:4141–4154. doi: 10.1172/JCI42980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunwoodie SL. Dev Cell. 2009;17:755–773. doi: 10.1016/j.devcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 4.Simon MC, Keith B. Nat Rev Mol Cell Biol. 2008;9:285–96. doi: 10.1038/nrm2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohyeldin A, Garzón-Muvdi T, Quiñones-Hinojosa A. Cell Stem Cell. 2010;7:150–161. doi: 10.1016/j.stem.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Mathieu J, Zhang Z, Nelson A, Lamba DA, Reh TA, Ware C, Ruohola-Baker H. Stem Cells. 2013;31:1737–1748. doi: 10.1002/stem.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilkes DM, Semenza GL, Wirtz D. Nat Rev Cancer. 2014;14:430–9. doi: 10.1038/nrc3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oncologist T. 2004;9:10–17. [Google Scholar]
- 9.Semenza GL. Oncogene. 2010;29:625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chacko SM, Ahmed S, Selvendiran K, Kuppusamy ML, Khan M, Kuppusamy P. AJP Cell Physiol. 2010;299:C1562–C1570. doi: 10.1152/ajpcell.00221.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uchida T, Rossignol F, Matthay MA, Mounier R, Couette S, Clottes E, Clerici C. J Biol Chem. 2004;279:14871–14878. doi: 10.1074/jbc.M400461200. [DOI] [PubMed] [Google Scholar]
- 12.Stolze IP, Tian YM, Appelhoff RJ, Turley H, Wykoff CC, Gleadle JM, Ratcliffe PJ. J Biol Chem. 2004;279:42719–42725. doi: 10.1074/jbc.M406713200. [DOI] [PubMed] [Google Scholar]
- 13.Schofield CJ, Ratcliffe PJ. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 14.Heyman SN, Rosen S, Rosenberger C. Crit Care. 2011;15:209. doi: 10.1186/cc9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bagnall J, Leedale J, Taylor SE, Spiller DG, White MRH, Sharkey KJ, Bearon RN, Sée V. J Biol Chem. 2014;289:5549–5564. doi: 10.1074/jbc.M113.500405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Millonig G, Hegedüsch S, Becker L, Seitz HK, Schuppan D, Mueller S. Free Radic Biol Med. 2009;46:182–191. doi: 10.1016/j.freeradbiomed.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 17.Bracken CP, Fedele AO, Linke S, Balrak W, Lisy K, Whitelaw ML, Peet DJ. J Biol Chem. 2006;281:22575–22585. doi: 10.1074/jbc.M600288200. [DOI] [PubMed] [Google Scholar]
- 18.Bruning U, Cerone L, Neufeld Z, Susan F, Cheong A, Scholz CC, David a, Leonard MO, Tambuwala MM, Cummins P, Taylor CT, Bruning U, Cerone L, Neufeld Z, Fitzpatrick SF, Cheong A, Scholz CC, Simpson Da, Leonard MO, Tambuwala MM, Cummins EP, Taylor CT. 2011;31:4087–4096. doi: 10.1128/MCB.01276-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen LK, Cavadas MAS, Scholz CC, Fitzpatrick SF, Bruning U, Cummins EP, Tambuwala MM, Manresa MC, Kholodenko BN, Taylor CT, Cheong A. J Cell Sci. 2013;126:1454–63. doi: 10.1242/jcs.119974. [DOI] [PubMed] [Google Scholar]
- 20.Jiang BH, Semenza GL, Bauer C, Marti HH. Am J Physiol. 1996;271:C1172–C1180. doi: 10.1152/ajpcell.1996.271.4.C1172. [DOI] [PubMed] [Google Scholar]
- 21.TSAI AG, JOHNSON PC, INTAGLIETTA M. Physiol Rev. 2003;83:933–963. doi: 10.1152/physrev.00034.2002. [DOI] [PubMed] [Google Scholar]
- 22.Brennan MD, Rexius-Hall ML, Elgass LJ, Eddington DT. Lab Chip. 2014;14:4305–18. doi: 10.1039/c4lc00853g. [DOI] [PubMed] [Google Scholar]
- 23.Oomen PE, Skolimowski M, Verpoorte S. Lab Chip. 2016;16:3394–3414. doi: 10.1039/c6lc00772d. [DOI] [PubMed] [Google Scholar]
- 24.Oppegard SC, Nam KH, Carr JR, Skaalure SC, Eddington DT. PLoS One. doi: 10.1371/journal.pone.0006891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oppegard SC, Blake AJ, Williams JC, Eddington DT. Lab Chip. 2010;10:2366. doi: 10.1039/c004856a. [DOI] [PubMed] [Google Scholar]
- 26.Di Caprio G, Stokes C, Higgins JM, Schonbrun E. Proc Natl Acad Sci. 2015;112:9984–9989. doi: 10.1073/pnas.1509252112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brennan MD, Rexius-Hall ML, Eddington DT. PLoS One. 2015;10:1–9. doi: 10.1371/journal.pone.0137631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morshed A, Dutta P. Biochim Biophys Acta - Gen Subj. 2017;1861:759–771. doi: 10.1016/j.bbagen.2017.01.017. [DOI] [PubMed] [Google Scholar]
- 29.Adler M, Polinkovsky M, Gutierrez E, Groisman A. Lab Chip. 2010;10:388–91. doi: 10.1039/b920401f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lo JF, Sinkala E, Eddington DT. Lab Chip. 2010;10:2394–401. doi: 10.1039/c004660d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehta G, Mehta K, Sud D, Song JW, Bersano-Begey T, Futai N, Heo YS, Mycek MA, Linderman JJ, Takayama S. Biomed Microdevices. 2007;9:123–134. doi: 10.1007/s10544-006-9005-7. [DOI] [PubMed] [Google Scholar]
- 32.Chen YH, Peng CC, Cheng YJ, Wu JG, Tung YC. Biomicrofluidics. 2013;7:3626–3633. doi: 10.1063/1.4829775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng CC, Liao WH, Chen YH, Wu CY, Tung YC. Lab Chip. 2013;13:3239–45. doi: 10.1039/c3lc50388g. [DOI] [PubMed] [Google Scholar]
- 34.Chang CW, Cheng YJ, Tu M, Chen YH, Peng CC, Liao WH, Tung YC. Lab Chip. 2014;14:3762–72. doi: 10.1039/c4lc00732h. [DOI] [PubMed] [Google Scholar]
- 35.Shiwa T, Uchida H, Tsukada K. Am J Biomed Eng. 2012;2:175–180. [Google Scholar]
- 36.Rexius-Hall ML, Mauleon G, Malik AB, Rehman J, Eddington DT. Lab Chip. 2014;14:4688–95. doi: 10.1039/c4lc01168f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lo JF, Sinkala E, Eddington DT, Guihot AL, Toutain B, Loufrani L, Henrion D, Langer R, Linderman JJ, Takayama S. Lab Chip. 2010;10:2394. doi: 10.1039/c004660d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lo JF, Wang Y, Blake A, Yu G, Harvat TA, Jeon H, Oberholzer J, Eddington DT. Anal Chem. 2012;84:1987–93. doi: 10.1021/ac2030909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor SE, Bagnall J, Mason D, Levy R, Fernig DG, See V. Open Biol. 2016;6:160195. doi: 10.1098/rsob.160195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gong H, Rehman J, Tang H, Wary K, Mittal M, Chatturvedi P, Zhao Y, Komorova YA, Vogel SM, Malik AB. J Clin Invest. 2015;125:652–664. doi: 10.1172/JCI77701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu CJ, Tsai MM, Hung PS, Kao SY, Liu TY, Wu KJ, Chiou SH, Lin SC, Chang KW. Cancer Res. 2010;70:1635–1644. doi: 10.1158/0008-5472.CAN-09-2291. [DOI] [PubMed] [Google Scholar]
- 42.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LAJ. Nature. 2013;496:238–42. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arbiser JL. J Invest Dermatol. 2011;131:1189–91. doi: 10.1038/jid.2011.45. [DOI] [PubMed] [Google Scholar]
- 44.Ge H, Weiszmann J, Reagan JD, Gupte J, Baribault H, Gyuris T, Chen J, Tian H, Li Y. J Lipid Res. 2008;49:797–803. doi: 10.1194/jlr.M700513-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Roland CL, Arumugam T, Deng D, Liu SH, Philip B, Gomez S, Burns WR, Ramachandran V, Wang H, Cruz-Monserrate Z, Logsdon CD. Cancer Res. 2014;74:5301–5310. doi: 10.1158/0008-5472.CAN-14-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He W, Miao FJP, Lin DCH, Schwandner RT, Wang Z, Gao J, Chen JL, Tian H, Ling L. Nature. 2004;429:188–193. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 47.Brusselmans K, Bono F, Maxwell P, Dor Y, Dewerchin M, Collen D, Herbert JM, Carmeliet P. J Biol Chem. 2001;276:39192–39196. doi: 10.1074/jbc.C100428200. [DOI] [PubMed] [Google Scholar]
- 48.Chen R, Xu M, Nagati JS, Hogg RT, Das A, Gerard RD, Garcia JA. PLoS One. doi: 10.1371/journal.pone.0116515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu M, Nagati JS, Xie J, Li J, Walters H, Moon Y-A, Gerard RD, Huang C-L, Comerford SA, Hammer RE, Horton JD, Chen R, Garcia JA. Nat Med. 2014;20:1018–26. doi: 10.1038/nm.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chae KS, Kang MJ, Lee JH, Ryu BK, Lee MG, Her NG, Ha TK, Han J, Kim YK, Chi SG. Oncogene. 2011;30:1213–1228. doi: 10.1038/onc.2010.498. [DOI] [PubMed] [Google Scholar]
- 51.Raval RR, Lau KW, Tran MGB, Sowter HM, Mandriota SJ, Li J-L, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Mol Cell Biol. 2005;25:5675–86. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Imamura T, Kikuchi H, Herraiz M, Park D, Mino-kenduson M, Lynch MP, Rueda BR, Benita Y, Xavier J, Chung DC. Int J. 2009;124:617–643. doi: 10.1002/ijc.24032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Florczyk U, Czauderna S, Stachurska A, Tertil M, Nowak W, Kozakowska M, Poellinger L, Jozkowicz A, Loboda A, Dulak J. Free Radic Biol Med. 2011;51:1882–1892. doi: 10.1016/j.freeradbiomed.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szendrői A, Szősz AM, Kardos M, Tőkés A. Oncotarget. doi: 10.18632/oncotarget.9669. [DOI] [Google Scholar]
- 55.Eubank TD, Roda JM, Liu H, O’Neil T, Marsh CB. Blood. 2011;117:323–332. doi: 10.1182/blood-2010-01-261792. [DOI] [PMC free article] [PubMed] [Google Scholar]