

Abstract
Bicyclic peptides have greater conformational rigidity and metabolic stability than linear and monocyclic peptides and are capable of binding to challenging drug targets with antibody-like affinity and specificity. Powerful combinatorial library technologies have recently been developed to rapidly synthesize and screen large bicyclic peptide libraries for ligands against enzymes, receptors, and protein-protein interaction targets. Bicyclic peptides have been developed as potential therapeutics against a wide range of diseases, drug targeting agents, imaging/diagnostic probes, and research tools. In this minireview, we provide a summary of the recent progresses on the synthesis and applications of bicyclic peptides.
Keywords: combinatorial chemistry, high-throughput screening, macrocycles, protein-protein interactions, peptides, OBTC, phage display, mRNA display, click chemistry, Imaging agents
Graphical Abstract
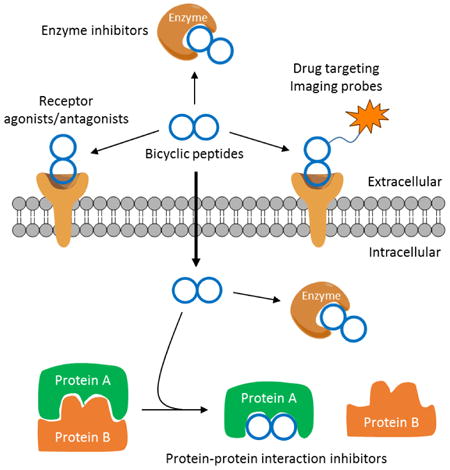
Bicyclic peptides can now be rapidly synthesized and screened to identify potent, specific ligands against enzymes, receptors, and protein-protein interactions, thus providing a new class of drug modalities and powerful research tools.
1. Introduction
The vast majority of FDA-approved drugs are small molecules (MW<500), which have the ability to modulate either extra- or intracellular targets. However, small molecules are generally limited to targeting proteins with deep binding pockets/grooves (e.g., enzymes, receptors, and ion channels), which represent ∼10% of all disease relevant human proteins.[1] Proteins that do not possess well defined binding pockets, such as those involved in protein-protein interactions (PPIs), are largely out of reach of small molecules. Since 1980's, an increasing number of macromolecules (MW>5000) have arrived into the clinic. While macromolecular drugs such as antibodies are capable of binding to essentially any target, they are too large to cross the cell membrane and thus limited to extracellular targets, which represent another ∼10% of disease relevant proteins[1]. This leaves the remaining ∼80% protein targets undruggable, including most of the proteins involved in intracellular PPIs.
The urgent demand for novel drug modalities to target the undruggable proteins, coupled with the clinical success of several macrocyclic peptide natural products (e.g., cyclosporine A, vancomycin, and daptomycin), has inspired researchers to explore synthetic macrocyclic peptides as the next-generation therapeutics. These efforts have resulted in numerous potent, selective monocyclic peptide inhibitors against both extracellular and intracellular proteins including many PPI targets.[2] In general, monocyclic peptides of small ring sizes (<10 amino acids) are relatively resistant to proteases and some have demonstrated oral bioavailability.[3] As the ring size increases, however, monocyclic peptides become progressively more conformationally flexible and susceptible to proteolytic degradation, especially when they consist of predominantly proteinogenic amino acids. One way to improve the proteolytic stability of macrocyclic peptides is to convert them into bicyclic structures, thus reducing the size of each ring to approximately half of the original size. The increased conformational rigidity of bicyclic peptides also further improves their target-binding affinity and selectivity. Additionally, the bicyclic system permits the two rings to function independently of each other, e.g., one ring for target binding while the other for cellular entry (vide infra). Consequently, generation of synthetic bicyclic peptides with novel biological activities has been an active area of research for several decades. While earlier studies were limited to the development of bicyclization methods and de novo design of bicyclic peptides, the field was greatly energized by the recent development of several powerful technologies for combinatorial synthesis of very large bicyclic peptide libraries in formats compatible with high-throughput screening for biological activity. These technologies have enabled researchers to quickly discover lead compounds that can either be used directly as probes of important biological functions or further improved into therapeutic agents. These compounds have also been conjugated to other drugs or biomolecules for drug targeting, diagnostic imaging or simply to enhance the properties of other therapeutic entities with specific pharmacokinetic deficiencies. This review will highlight the key developments in the preparation and application of bicyclic peptide ligands as chemical probes and therapeutics.
2. Bicyclic Peptides in Nature
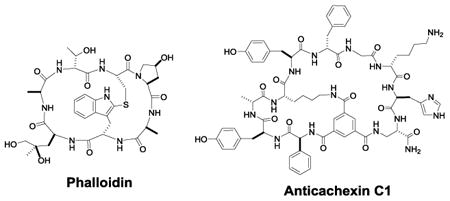
Like monocyclic peptides, bicyclic peptides are widely distributed in nature, exhibiting diverse biological activities.[4] For example, Moroidin (1, Figure 1), is a bicyclic octapeptide isolated from the seeds of Celosia argentea, which prevents eukaryotic cell division by inhibiting tubulin polymerization (IC50 = 3 μM).[5] Celogentins A-K are a related class of bicyclic octapeptides, which are also derived from C. argentea and possess anti-mitotic activity.[6] Among these compounds, Celogentin C (2, Figure 1) is most potent and inhibits tubulin polymerization with an IC50 value of 0.8 μM.[7] Other naturally occurring bicyclic peptides include phallotoxins and amatoxins, which are isolated from poisonous mushrooms of the genus amanita. Phalloidin (3, Figure 1), the first of seven bicyclic heptapeptidyl phallotoxins isolated from the death cap mushroom (Amanita phalloides), inhibits cytokinesis and cytotaxis by preventing the depolarization of actin filaments.[8] Phalloidin binds tightly and selectively to filamentous actin (F-actin), and fluorescently labeled phalloidin has been widely used as a tracer of F-actin in cells.[9] Amatoxins are bicyclic octapeptides, which exert their cytotoxicity by selectively inhibiting mammalian RNA polymerase II [LD50 = 0.1 mg/kg in rats for α-amanitin (4, Figure 1)].[10] The potent cytotoxicity of α-amanitin makes it an excellent “warhead” for selective tumor targeting approaches (vide infra). Finally, theonellamides A-G are a class of sterol binding bicyclic peptides isolated from marine sponge, which display both cytotoxic and antifungal activities.[11] Theonellamide F (5, Figure 1) induces damage to cellular membranes and stimulates Rho1 mediated 1,3-β-D-glucan synthesis.[12] This compound also showed promising cytotoxicity against P388 and L1210 leukemia cells.[11]
Figure 1.

Structures of naturally occurring bicyclic peptides moroidin (1), Celogentin C (2), phalloidin (3), α-amanitin (4), and theonellamide F (5).
3. Synthesis of Bicyclic Peptides
3.1 Rational Design
The diverse biological activities of naturally occurring bicyclic peptides suggest that synthetic bicyclic peptides may be generated to have desirable biological activities. In 1978, Zanotti et al. reported the first synthesis of a bicyclic heptapeptide (6, Figure 2a), by first forming a thioether bond between a cysteine residue and an oxidized tryptophan derivative and subsequently head to tail cyclization.[13] The CD spectrum of the resulting heptapeptide suggested a structural similarity to phallotoxins. Maggi et al. synthesized a bicyclic peptidyl inhibitor of the tachykinin NK2 receptor, MEN 10,627 (7, Figure 2b).[14] The linear peptide sequence was synthesized on hydroxymethylbenzoic acid (HMBA) resin, with the side chains of diaminopropionic acid (Dap) and aspartate protected with Boc and t-butyl groups, respectively. After removal of the Dap and Asp sidechain protecting groups with trifluoroacetic acid (TFA), the peptide was first cyclized on resin by forming an amide between the Dap and Asp side chains. The peptide was then released from the resin by base hydrolysis of the ester linkage and the second cyclization was effected in dilute DMF. Another bicyclization strategy was later reported by Tam and co-workers (Figure 3a).[15] Linear peptides containing two cysteine residues, one at the N-terminus and the other at an internal position, and a C-terminal thioester linkage was prepared by solid-phase Boc/BOP chemistry. Following side-chain deprotection with HF, the peptides underwent spontaneous N-to-C cyclization through an on-resin intramolecular thioester ligation reaction and concomitant release of the peptides from the resin. Subsequent incubation in DMSO resulted in the formation of an intramolecular disulfide bond and bicyclization of the peptides.
Figure 2.

Examples of rationally designed bicyclic peptides. Bicyclization of peptides 8 and 10 resulted in more potent inhibitors against uPA (9) and Grb7 SH2 domain (11), respectively. iBCCi, isobutyloxycarbonylchloride; HMBA, hydroxymethylbenzoic acid.
Figure 3.

Various strategies for chemical synthesis of bicyclic peptides. RCM, ring-close metathesis; RCAM, ring-close alkyne metathesis.
Because disulfide and thioether bonds have limited metabolic stability, Liskamp and co-workers replaced them with alkene staples by ring-closing metathesis (RCM)-mediated cyclization in preparation of a bicyclic mimic of the DE-ring system of the lantibiotic nisin (Figure 3b).[16] Lavilla and colleagues later expanded on this stapling approach via application of a palladium catalyzed C-C bond formation between tryptophan and iodinated tyrosine or phenylalanine (Figure 3c).[17] Very recently, Waldmann and Grossman[18] employed orthogonal ring-closing olefin and alkyne metathesis schemes to form either manacle or butterfly shaped bicyclic peptides (Figure 3d). In an alternative approach, Grandas et al. prepared metabolically stable bicyclic peptides via successive Michael addition reactions between thiols and maleimides.[19] To ensure formation of the correct bonds, one of the reactive maleimides was initially masked with 2,5-dimethylfuran. Structurally diverse bicyclic peptides can be accessed with this approach by varying the location of the reactive groups in the linear sequence. To further expand the accessible chemical space, Reymond and colleagues[20] obtained bridged bicyclic peptides by first crosslinking the side chains of lysine and glutamate residues followed by thioether formation between an N-terminal chloroacetyl group and the side chain of an internal cysteine (Figure 3e). This method resulted in globularly shaped compounds that form intramolecular hydrogen bond networks, mimicking β-turn and α-helical structures. Bicyclic peptides have also been synthesized by simply connecting two monocyclic peptides with various linkers.[21]
The above bicyclization methods have been used to optimize hit/lead peptides isolated from combinatorial libraries. For example, starting with a head-to-tail monocyclic peptide hit (8) isolated from a phage display library, Roodbeen et al. designed bicyclic peptide 9 as an inhibitor against urokinase-type plasminogen activator (uPA) (Figure 2c).[22] They replaced the original disulfide bond with an N- to C- amide bond for the first cyclization and then introduced a new intramolecular disulfide bond to achieve bicyclization. Similarly, Gunzberg et al. showed that addition of a carbon staple to G7-18NATE (10), a selective monocyclic peptide inhibitor of the Grb7 SH2 domain, increased the inhibitor potency by 2-3 fold [e.g., KD = 1.5 μM for G7-B1 (11, Figure 2d)].[23] Bicyclization has also been used to impose conformational constraints on linear binding motifs. Based on a crystal structure of vascular endothelial growth factor (VEGF) in complex with VEGF receptor-1 (VEGFR1), Inguimbert et al.[24] rationally designed a bicyclic 25-mer VEGF antagonist, to mimic three of the VEGF strands involved in the interaction with the receptor. In another example, rigidification of a citrulline-containing peptide with a pair of disulfides resulted in inhibitors with improved affinity for anti-citrullinated peptide antibodies.[25]
Cell-permeable bicyclic peptides have been designed to target intracellular proteins. By screening a combinatorial library, Lian et al. discovered a potent monocyclic peptide inhibitor (12, Figure 4a) against protein-tyrosine phosphatase 1B (PTP1B).[26] Inhibitor 12 had poor membrane permeability, despite containing a cell-penetrating motif, Arg-Arg-Arg-Arg-Nal-Phe (where Nal is L-naphthylalanine), in its sequence. Interestingly, conversion of peptide 12 into a bicyclic structure (13), in which the PTP1B-binding sequence was confined into one ring and the cell-penetrating motif in the second ring, greatly improved the cell permeability while retaining almost full PTP1B inhibitory activity (KD = 37 nM). Peptide 13 potentiated insulin receptor signaling in HepG2 cells at nanomolar concentrations. Qian et al. recently extended this strategy to develop a general method for intracellular delivery of peptide ligands.[27] Briefly, a linear peptide cargo is fused to a short CPP motif (e.g., Arg-Arg-Arg-Arg-Nal-Phe) and the fusion peptide is bicyclized around a small-molecule scaffold, 3,5-bis(mercaptomethyl)benzoic acid (BMB), by forming an amide and two disulfide bonds (Figure 4b). Upon cytosolic entry, the disulfide bonds are reduced by intracellular glutathione to release the biologically active linear peptide for binding to a target of interest. As a proof of principle, the researchers designed a bicyclic peptide (14) as an inhibitor against the NEMO-IκB kinase interaction, which blocked NF-κB activation in HEK293 cells via the canonical pathway. Inhibitor 14 exhibited greatly increased cellular uptake as well as serum stability compared to the linear peptide counterpart.
Figure 4.

Design of cell-permeable bicyclic peptide inhibitors against PTP1B (13) and NEMO-IKK interaction (14). The CPP motifs are shown in red. CPP, cell-penetrating peptide; GSH, glutathione.
3.2 Chemical Synthesis of Bicyclic Peptide Libraries
Tam and co-workers synthesized the first bicyclic peptide library of 9 different compounds by native chemical ligation and intramolecular disulfide formation.[15] To increase the odds of identifying bicyclic peptide ligands against a target of interest, it was clear that much larger and more structurally diverse bicyclic peptide libraries must be prepared in a format compatible with high-throughput screening protocols. Large libraries of bicyclic peptides can be readily synthesized in the one bead-one compound (OBOC) format with the split-and-pool method of Lam et al.[28] However, following screening of OBOC libraries, structural determination of bicyclic peptide hits was not possible. To overcome this challenge, Pei and co-workers synthesized bicyclic peptide libraries in the one bead-two compound (OBTC) format, in which each resin bead contains a unique bicyclic peptide in the surface layer but a linear peptide of identical sequence inside the bead as an encoding tag.[29,30] Peptide cyclization was mediated by the formation of three amide bonds between a rigid small-molecule scaffold, trimesic acid (TMA), and the N-terminal amine and the side chains of an internal lysine residue and a C-terminal Dap residue of the peptide (Figure 5a). During library screening against a protein target of interest, the protein only has access to the bicyclic peptides on the bead surface and the linear peptides inside the beads do not interfere with library screening. Once an active hit(s) is identified, its structure is readily determined by sequencing the linear peptide tags inside the bead(s) by using Edman degradation and/or mass spectrometric methods.[31] This method is highly versatile, capable of generating bicyclic peptides of different ring sizes and peptide sequences, and is compatible with both natural and unnatural amino acids (e.g., D-amino acids) as well as nonpeptidic moieties as building blocks. For example, Lian et al.[30] synthesized a bicyclic peptide library containing 3-5 random residues in each ring and 24 different L- and D-amino acids (Figure 5a), which has a theoretical diversity of 1.0 × 1014, although in practice the size of OBTC libraries is limited to 108 different compounds. Screening of the library identified a bicyclic peptide antagonist of TNF-α (15, Figure 5b) which inhibited TNF-α from binding to its cognate receptor in a concentration-dependent manner (IC50 = 3.1 μM).[30]
Figure 5.

Chemically synthesized bicyclic peptide libraries. a) Design of a bicyclic peptide library in the OBTC format; b) Structure of TNFα inhibitor (15) discovered from the OBTC library in a); and c) the structure a cell-permeable bicyclic peptide inhibitor against K-Ras G12V (16).
To obtain cell-permeable macrocyclic peptide ligands against intracellular targets directly from combinatorial libraries, Trinh et al. modified the OBTC bicycle library design so that one ring consisted of different peptide sequences for potential binding to a target of interest, while the second ring featured a CPP motif (Phe-Nal-Arg-Arg-Arg-Arg or Arg-Arg-Arg-Arg-Nal-Phe) for cellular entry.[32] This library was screened for in vitro binding to an oncogenic K-Ras mutant (G12V), a driver for many human cancers. Remarkably, all of the library hits were cell-permeable, suggesting that the bicyclic design offers a general approach to developing macrocyclic peptides against intracellular targets. One of the bicyclic peptidyl Ras inhibitors (16, Figure 5c) physically blocked the Ras-Raf interaction with an IC50 of 3.4 μM, inhibited Ras signaling in cell culture, and induced apoptosis of human cancer cells at low μM concentrations.[32]
3.3 Biological Synthesis of Bicyclic Peptide Libraries
Bicyclic peptides have also been produced biologically via ribosomal synthesis, which is capable of generating libraries of enormous diversity (up to 1014 different compounds). Heinis and Winter pioneered the biological synthesis of bicyclic peptides by phage display.[33] They first generated a linear peptide library of the sequence C-X6-C-X6-C (where C is cysteine and X is any of the 20 proteinogenic amino acids) on the surface of filamentous phage, using the method of Smith.[34] The linear peptides were then converted into bicyclic peptides by reacting the peptides with a small-molecule scaffold, tris(bromomethyl)benzene (TBMB), which selectively alkylated the three cysteine residues in the sequence (Figure 6a). The effectiveness of this approach was demonstrated by isolation of potent bicyclic peptide inhibitors against proteases plasma kallikrein and cathepsin [KI values of 2.9 and 100 nM, respectively]. In addition to TBMB, other small-molecule scaffolds such as 1,3,5-triacryoyl-1,3,5-triazinane (TATA), N,N',N”-(benzene-1,3,5-triyl)tris(2-bromoacetamide) (TBAB), and N,N',N”-benzene-1,3,5-triyltrisprop-2-enamide have also been used to generate bicyclic peptides of different ring sizes and flexibility.[35] Alternatively, phage displayed bicyclic peptides can be formed via two disulfide bonds. To prevent the formation of a mixture of different disulfide bridges, Heinis and co-workers designed di-thiol amino acids (DTaas).[36] Incorporation of one of these unnatural amino acids into a sequence containing two cysteine residues followed by oxidation produced a single bis-disulfide bicyclic species (Figure 6b). This method has not yet been widely adopted, presumably because of the relative lability of the disulfide bonds.
Figure 6.
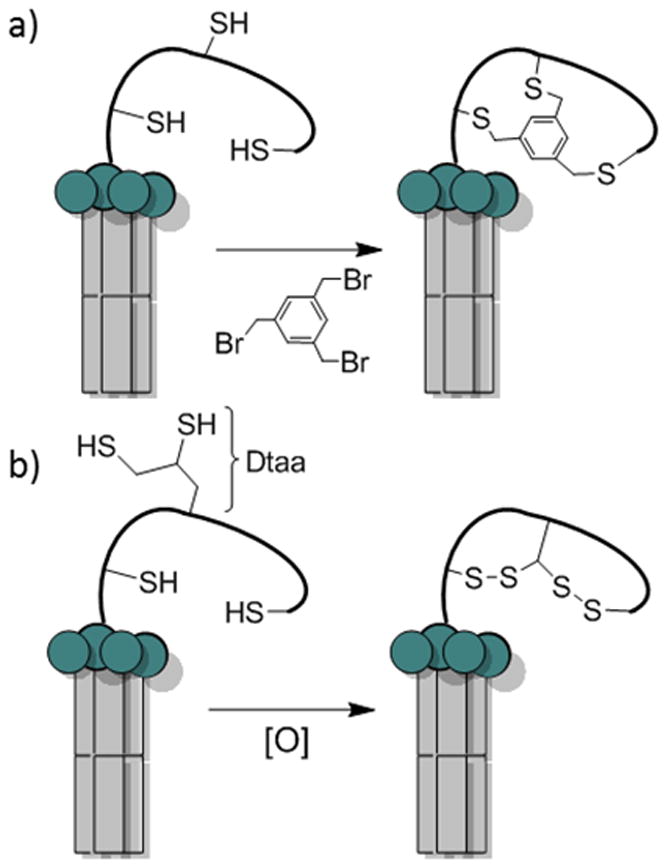
Phage displayed bicyclic peptide libraries. a) TBMB bicyclization of a phage display library with three cysteine residues; b) Oxidative bis-disulfide bicyclization of phage library with two cysteines and one di-thiol amino acid gives only one bicyclic isomer.
Another method for ribosomal synthesis of bicyclic peptides is mRNA display. In conventional mRNA display, each linear peptide is covalently linked to its encoding mRNA through the use of antibiotic puromycin and the identity of a hit peptide is determined by reverse transcription of the mRNA and sequencing the resulting cDNA (Figure 7a).[37] Suga and co-workers modified the mRNA display protocol by reprogramming the genetic code to incorporate unnatural amino acids. For example, they incorporated a fixed cysteine at the N-terminus of a peptide sequence and re-assigned the Phe codon (UUC) at an internal position to 4-(2-chloroacetyl)aminobutyric acid (Cab); the resulting newly translated peptide underwent spontaneous cyclization by forming a thioether linkage between the N-terminal cysteine and the Cab residue (Figure 7b).[38] Bicyclization was achieved by additionally incorporating azidohomoalanine and propargyl glycine residues into the peptide sequence, followed by Cu(I)-mediated azide-alkyne cycloaddition. More recently, Hacker et al. exploited the orthogonality between alkylation of cysteine side chains with bis(bromomethyl)benzene and click chemistry to produce bicyclic peptides that are either mannacle shaped or theta-bridged (Figure 7c).[39] Unlike previous bicyclic peptide libraries, in which the conformation of bicyclic peptides was pre-determined by the small-molecule scaffold, the bicyclization method of Hacker et al. is scaffold free, therefore allowing for different loop attachment points and access to multiple bicyclic topologies.
Figure 7.

Bicyclic peptide libraries synthesized by mRNA display and SICLOPPS. a) Scheme showing the key features of the mRNA display technology; b) bicyclization of mRNA displayed peptides by the method of Sako et al.;[36] c) bicyclization of mRNA displayed peptide library by the method of Hacker et al.;[37] and d) bicyclization of SICLOPPS-synthesized peptides by the method of Bionda and Fusan.[39]
Fasan and co-workers have adapted an intein splicing strategy, termed “split-intein circular ligation of peptides and proteins (SICLOPPS)”,[40] to generate bicyclic peptide libraries.[41] Inteins are self-excising protein domains that process to link their flanking sequences by a native peptide bond.[42] A SICLOPPS construct consists of the C-terminal fragment of an intein (INTC), a peptide sequence to be cyclized, and the N-terminal fragment of the intein (INTN) (Figure 7d). Upon transcription and translation, the N- and C-terminal intein fragments reassemble to form an active intein, which subsequently cyclizes the peptide sequence of interest through peptide splicing.[43] By integrating the SICLOPPS method with genetic incorporation of unnatural amino acids, Bionda and Fasan prepared natural product-like bicyclic peptides of varying ring sizes and intramolecular connectivity patterns.[41] The initial N-to-C macrocyclization was achieved by protein splicing using the split intein of DnaE, while further cyclization to form the bicycle was effected by thioether formation between the side chains of a genetically encoded O-(2-bromoethyl)-tyrosine and a cysteine residue. Future integration of this method with one of the peptide display techniques may allow access to combinatorial libraries of natural product-like bicyclic peptides that can be screened against a target of interest.
Among the methods described for bicyclic peptide library synthesis, the OBTC method is most versatile with respect to the nature of building blocks and the chemical reactions employed to connect the building blocks, producing structurally diverse, metabolically stable, and natural product-like bicyclic peptides. Its main disadvantage is that the library size is currently limited to ∼108 different compounds, although this size limit can be increased by ∼1000-fold by synthesizing DNA-encoded OBTC libraries.[44] So far, bicyclic PPI inhibitors derived from naïve OBTC libraries have typically exhibited high nM to low μM binding affinities to the intended targets and usually require further optimization by either conventional medicinal chemistry approaches or synthesis and screening of second-generation libraries (biased libraries). On the other hand, mRNA display libraries can reach 1014 in diversity and, as a result, highly potent ligands (with pM to low nM KD values) can often be directly isolated from such libraries. mRNA display libraries are, of course, limited to building blocks that are recognizable by the ribosome, namely proteinogenic amino acids and a subset of unnatural amino acids (e.g., Nα-methylated L-amino acids).[45] Phage-display and SICLOPPS libraries have an intermediate level of diversity (∼109, which is limited by the efficiency of bacterial transformation) and are generally limited to the 20 proteinogenic amino acids as building blocks.
4. Applications of Bicyclic Peptides
4.1 Therapeutics and Chemical Probes
4.1.1 Enzyme Inhibitors
A number of naturally occurring bicyclic peptides have been used as therapeutics and/or research tools. For example, Romidepsin (17, Figure 8) is a small bicyclic depsipeptide derived from the bacterium Chromobacterium violaceum and a potent histone deacetylase inhibitor. It has received FDA approval for treatment of cutaneous T-cell lymphoma and other peripheral T-cell lymphomas.[46] Another naturally occurring bicyclic peptide, bouvardin, is a translation inhibitor which has been patented for use as an anticancer agent, in combination with radiation therapy or other chemotherapeutic agents.[47] As mentioned previously, α-Amanitin (4, Figure 1) is a bicyclic peptidyl toxin that potently induces apoptosis of both rapidly dividing and slowly growing cells. To reduce its toxicity, scientists at Heidelberg Pharmaceuticals conjugated it to a tumor targeting antibody.[48] The resulting antibody-drug conjugate (ADC) has shown promise for the treatment of drug-resistant tumors. For example, conjugation of α-amanitin to an antibody against prostate-specific membrane antigen (PSMA) resulted in complete tumor remission in a prostate cancer mice model at a 150 μg/kg dose. In addition, because α-amanitin is highly water soluble, even at a relatively high drug loading, the ADC did not suffer from aggregation problems that are frequently encountered with small-molecule ADC's.
Figure 8.

Examples of bicyclic peptide inhibitors against histone deacetylase (17), chymotrypsin (18), factor XII (19), and peptidyl-prolyl isomerase Pin1 (20).
The success of biologically active bicyclic peptides from nature has inspired chemists to explore synthetic bicyclic peptides as therapeutic agents and research tools. Bicyclic peptides have been found to be highly effective enzyme inhibitors, especially against extracellular enzymes, since in the latter case, membrane permeation is not required. Bowman-Birk inhibitors (BBI) are small, disulfide-rich protein inhibitors of serine proteases which have shown promise for treatment of muscular atrophy, cancer and neurodegenerative, inflammatory, and cardiovascular diseases.[49] Ando et al. synthesized a bicyclic hexadecapeptide corresponding to the chymotrypsin-binding site of the soybean BBI (18, Figure 8), as a proteolytically stable and effective inhibitor of chymotrypsin.[50] Heinis and others screened phage displayed bicyclic peptide libraries against several proteases and discovered highly potent and specific inhibitors against both plasma kallikrein (PK) and urokinase-type plasminogen activator (uPA).[51] For example, UK18 showed >2000-fold selectivity for uPA relative to closely related proteases. Other efforts produced a bicyclic peptide inhibitor with a KI value of 8.1 nM against the coagulation factor XII.[52] Synthesis and testing of several dozen analogs led to bicyclic peptide 19 (Figure 8), which showed a 10-fold higher target-binding affinity and a ∼5-fold increase in blood plasma half-life.[53] As discussed previously, Lian et al. engineered cell-permeable bicyclic peptidyl inhibitors against intracellular enzymes PTP1B and peptidyl-prolyl cis-trans isomerase Pin1, by fusing monocyclic peptide inhibitors previously identified from OBTC libraries with a cyclic CPP.[26] The bicyclic peptidyl PTP1B inhibitor (13, Figure 4) showed both high potency (KD = 37 nM) and selectivity for PTP1B (17-fold lower potency for the closely related TCPTP and no inhibition of any of the other PTPs tested). The bicyclic PTP1B inhibitor provides a potential lead for further development of treatment for type II diabetes. Pin1 is dispensable in normal cells but is overexpressed in many cancers and required for cancer cell proliferation, suggesting that Pin1 inhibitors may have applications as anticancer agents.[54] The bicyclic peptidyl Pin1 inhibitor of Lian et al. (20, Figure 8) inhibited the intracellular function of Pin1 and the proliferation of HeLa cells.[26] The group later reported an improved analog of peptide 20, which is non-phosphorylated and metabolically more stable.[55] Bicyclic peptides are capable of achieving exquisite specificity for a given enzyme, because their large sizes allow them to interact with not only the active site, but also the less conserved regions surrounding the active site. In addition to therapeutic applications, highly selective enzyme inhibitors also provide powerful tools for chemical biology, allowing researchers to specifically “knockout” the activity of a given enzyme in a complex system with temporal control and assess the biological function of the enzyme in an otherwise native system.
4.1.2 PPI Inhibitors
One of the main impetuses for developing macrocyclic peptides is to inhibit PPIs, which are challenging targets for conventional drug modalities. Several bicyclic peptide PPI inhibitors have been rationally designed from their linear or monocyclic peptide precursors. For example, the Bcl2 homology (BH) proteins are well validated anticancer drug targets that regulate apoptosis by influencing mitochondrial outer membrane permeabilization (MOMP). Fairlie and colleagues showed that bicyclization of a previously reported BAD BH3 peptide resulted in shorter (8-14 AA's), more potent, and more drug-like peptidomimetics against Bcl-xL (21, Figure 9).[56] Grb2 is a key adaptor protein involved in receptor tyrosine kinase signaling pathways, by binding to specific phosphotyrosyl residues on the receptors via its SH2 domain.[57] Grb2 inhibitors thus have therapeutic applications as anticancer agents. Quartararo et al. designed a bicyclic peptide inhibitor of the Grb2 SH2 domain.[58] They found that conversion of a monocyclic undecapeptide, G1, into a bicyclic peptide, BC1 (22, Figure 9), resulted in 60-fold higher potency and 200-fold improved selectivity for the Grb2 SH2 domain. In another application, dimerization of a potent monocyclic peptide inhibitor produced an effective bicyclic peptide therapeutic against vascular endothelial growth factor (VEGF).[59]
Figure 9.

Examples of bicyclic peptide PPI inhibitors against Bcl-xL (21), Grb2 SH2 domain (22), K-Ras (23), and Smurf HECT domain (24).
Other investigators have discovered bicyclic peptide PPI inhibitors against several notable therapeutic targets from combinatorial libraries. Lian et al. screened an OBTC library of 3 × 105 bicyclic peptides against human TNFα and discovered two hits which prevented TNFα from binding to its cognate receptors and protected cells against TNFα-induced death [IC50 = 3.1 μM for anticachexin C1 (15, Figure 5b)].[30] Protein inhibitors against TNFα (e.g., Humira) are among the top-selling drugs for treatment of a range of inflammatory diseases. Metabolically stable bicyclic peptide TNFα inhibitors may provide a lower-cost, less immunogenic, and potentially orally active replacement for the protein inhibitors. Screening of the same library against oncogenic K-Ras G12V identified two different types of K-Ras ligands.[60] The first type [e.g., cyclorasin B2 (23, Figure 9); KD = 500 nM] apparently bind to K-Ras at or near the effector-binding site and inhibited the Ras-Raf interaction in vitro, while the second type of ligands bind at an yet undetermined site and do not affect effector binding. Unfortunately, these hits showed no significant anticancer activity because of poor membrane permeability. Trinh et al. later modified the library design by incorporating a CPP sequence into one of the rings.[32] Screening of this library against K-Ras G12V followed by hit optimization produced bicyclic peptide 16 (Figure 5c), which was cell-permeable, inhibited K-Ras signaling in cell culture, and induced apoptosis in lung cancer cells (LD50 ∼17 μM). Mund et al. identified several bicyclic peptides that prevented ubiquitination of the Smurf2 HECT domain, by screening a phage-displayed library.[61] The most potent compound (24, Figure 9) had an IC50 of 2.5 μM and excellent selectivity for the Smurf2 HECT domain, enabling its use as a probe during subsequent screening of small-molecule inhibitors. As described previously, a disulfide-mediated bicyclic peptide inhibitor against the NEMO-IKK interaction has been developed for potential treatment of inflammation and cancer.[27]
4.1.3 Receptor Agonists and Antagonists
A few bicyclic peptides have been reported as agonists or antagonists of cell surface receptors, most of which are natural products or derived from rational design efforts. BI-32169 (25, Figure 10), a 19-aa bicyclic peptide isolated from Streptomyces sp. (DSM 14996), is a moderately potent antagonist against the human glucagon receptor (IC50 440 nM) in a functional cell-based assay.[62] In addition to the synthetic NK2 receptor antagonist (7) described previously (Figure 2b), Hruby and co-workers synthesized a bicyclic peptide analog of oxytocin, 4,8-cyclo[Mpa1,Glu4,Cys6,Lys 8]oxytocin.[63] This bicyclic peptide acts as a very potent antagonist of oxytocin in the rat uterus assay with an in vitro pA2 value of 8.2 and a pA2 value of 6.45 in vivo. Discovery of agonists or antagonists from combinatorial libraries has been challenging, because library screening requires the availability of the receptor protein in purified forms, which is generally difficult for integral membrane proteins. Heinis and colleagues screened phage displayed bicyclic peptide libraries against the soluble extracellular domains (ECDs) of the epidermal growth factor receptor Her2 and the Notch receptor.[64,65] Although relatively potent and selective ligands were identified for both ECDs (KD of 304 and 150 nM, respectively), neither ligand showed agonist or antagonist activity against the respective receptor signaling. Presumably, the bicyclic peptides bound to the ECDs at functionally unimportant sites. These peptide ligands may, however, offer valuable starting points for the development of high-affinity receptor binders with potential application for tumor imaging and therapy (vide infra).
Figure 10.

Structure of hGR antagonist, BI-32169.
4.2 Drug Targeting
Antibody-drug conjugates (ADC's) have become a popular approach to improving the therapeutic window of small-molecule drugs.[66] Typically, a highly potent small-molecule drug is covalently attached to a monoclonal antibody specific for an antigen unique to or over-expressed on a target tissue (e.g., a tumor). Binding of the antibody to the antigen results in concentration of the ADC to the target tissue. However, ADC's suffer from several drawbacks, including low drug payload per unit mass of antibody, high cost of production, and most significantly, poor tissue penetration (e.g., solid tumors) due to the large size of antibodies. Since bicyclic peptides are capable of binding to diverse protein targets with antibody-like binding affinity and specificity but are much smaller is size, they are ideally suited as drug-targeting agents. In fact, bicyclic peptide-drug conjugates (BDC's) have become the platform technology at Bicycle Therapeutics (Cambridge, MA), for selective delivery of chemotherapeutic agents or other drugs to target cell populations. The small size of BDC's allows them to penetrate deeply into solid tumors, while retaining high renal clearance to reduce drug-induced toxicity. In collaboration with AstraZeneca, Bicycle Therapeutics is developing BDCs for the treatment of cancer as well as respiratory, cardiovascular and metabolic diseases. Their most promising lead compound, BT1718, a conjugate targeting membrane type-1 matrix metalloproteinase (MT1-MMP) for solid tumors, has passed GLP toxicology testing and is expected to enter clinical trials within the year.[67] BDC's utilizing the tumor targeting ability of RGD peptides have also been developed. For example, a prodrug intended to specifically target tumors over-expressing integrins avB3 and avB5 was synthesized by covalently attaching doxorubicin to a bis(disulfide)-mediated bicyclic RGD peptide (26, Figure 11).[68] A peptide sequence recognized by tumor specific plasmin protease was used as a linker to further improve tumor selectivity. The BDC showed plasmin-dependent cytotoxicity to cancer cells in vitro, although diminished binding affinity and poor solubility of the conjugate have limited its effectiveness in vivo. In another example, conjugation of the microtubulin inhibitor paclitaxel to a bicyclic RGD peptide allowed its selective delivery to integrin avB3-expressing tumors.[69] These initial successes clearly demonstrate that BDC's can be developed as viable (and superior) replacements of ADC's.
Figure 11.

Structure of a BDC between a RGD bicycle and doxorubicin.
4.3 Imaging and Diagnostics
Bicyclic peptides are very attractive as PET tracers and other diagnostic imaging agents. Compared to monoclonal antibodies and other protein binders, bicyclic peptides have much faster clearance, due to their smaller sizes, resulting in excellent signal-to-background ratios and shorter delays between dosing and imaging. The discovery of five somatostatin receptor subtypes (sst1-sst5) that are differentially over-expressed on the surface of different cancer cells led researchers to explore radiolabeled somatostatin derived bicyclic peptides for imaging applications. Researchers at Merck first developed bicyclic somatostatin analogs to improve the metabolic stability of the natural hormone, somatostatin-14 (SRIF-14).[70] Further investigation by Falb et al. revealed that conformationally constrained bicyclic analogs of somatostatin could target specific sst receptor subtypes.[71] Based on these findings, Fani et al. designed a bicyclic somatostatin analog conjugated to 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) doped with either 177Lu or 68Ga for potential use for sst receptor subtype targeting in diagnostic imaging.[72] The high clearance rate of the most promising compound, 68Ga-AM3 (27, Figure 12), from non-targeted tissues resulted in high tumor-to-blood and tumor-to-muscle ratios after only 1 h, which may permit its application in PET or CT studies of sst-positive tumors. However, poor internalization led to rapid washout from sst positive tumors, preventing their use with more long-lived radionuclides for therapeutic purposes. In another example, a monocyclic RGD peptide was further rigidified by incorporation of aminocyclopentane (ACP) or aminocyclohexane (ACH) as residue side chains.[73] Conjugation of the resulting bicyclic peptide to DOTA loaded with 64Cu generated an effective PET imaging agent. The conjugate (28, Figure 12) showed higher affinity for U87MG glioblastoma cells than their monocyclic counterparts and was able to efficiently localize to integrin positive tumors. Lastly, as mentioned previously, fluorescently labeled phalloidin (3, Figure 1) has also been widely used as a tool to visualize actin filaments in cellular studies.[9]
Figure 12.

Examples of bicyclic peptides as diagnostic imaging agents.
5. Summary and Outlook
Since 2009, there has been a rapid growth in the discovery and applications of bicyclic peptide ligands against enzymes, receptors, and PPI targets, thanks to the advent of several powerful combinatorial library technologies. It is now feasible to develop bicyclic peptides with antibody-like affinity and specificity for many (if not all) proteins. In the coming years, we anticipate that many of the traditional roles of antibodies (e.g., therapeutics, diagnostics, and research tools) will be played by bicyclic peptides (or other macrocyclic compounds), because the latter have better cell and tissue penetration, greater metabolic stability, and lower cost of production. Perhaps the most exciting application of bicyclic peptides will likely be their use as inhibitors of intracellular PPIs which are, as of date, largely out of reach of small molecules or antibodies. The remaining challenges will be how to selectively deliver the bicyclic peptides to diseased tissues and/or effectively deliver them into the cytosol (and the nucleus) of mammalian cells.
Acknowledgments
Macrocyclic peptide related research in the Pei laboratory was supported by NIH grants (GM062820, GM110208, and GM122459).
Biographies

Curran Rhodes is currently a graduate student pursuing his PhD degree in organic chemistry from The Ohio State University under the direction of Dr. Dehua Pei. He received his B. S. degree in biochemistry in 2013 from Ohio University. As a PhD candidate, he is also a member of the Chemistry and Biology Interface Program (CBIP) and his research efforts have been directed towards the development of novel bicyclic PPI inhibitors for challenging intracellular targets.

Dehua Pei is the Charles H. Kimberly Professor of Chemistry and Biochemistry at The Ohio State University. He received his B.S. degree in chemistry from Wuhan University, China and a PhD degree in organic chemistry from University of California, Berkeley. He was a Damon Runyon-Winchell Walter Cancer Fund postdoctoral fellow at Harvard Medical School before joining the faculty at The Ohio State University. His research group is currently developing new methodologies for combinatorial synthesis and screening of macrocyclic peptides/peptidomimetcis, cyclic cell-penetrating peptides for drug delivery, and macrocyclic inhibitors against previously undruggable targets, such as intracellular protein-protein interactions.
References
- 1.Verdine GL, Hilinski GJ. Methods Enzymol. 2012;503:3–30. doi: 10.1016/B978-0-12-396962-0.00001-X. [DOI] [PubMed] [Google Scholar]
- 2.a) Cardote TAF, Ciulli A. ChemMedChem. 2016;11:787–794. doi: 10.1002/cmdc.201500450. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dougherty P, Qian Z, Pei D. Biochem J. 2017;474:1109–1125. doi: 10.1042/BCJ20160619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Bock JE, Gavenonis J, Kritzer JA. ACS Chem Biol. 2013;8:488–499. doi: 10.1021/cb300515u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Giordanetto F, Kihlberg J. J Med Chem. 2014;57:278–295. doi: 10.1021/jm400887j. [DOI] [PubMed] [Google Scholar]
- 4.For recent reviews see: Chung BKW, Yudin AK. Org Biomol Chem. 2015;13:8768–8779. doi: 10.1039/c5ob01115a.Baeriswyl V, Heinis C. ChemMedChem. 2013;8:377–384. doi: 10.1002/cmdc.201200513.
- 5.Morita H, Shimbo K, Shigemori H, Kobayashi J. Bioorg Med Chem Lett. 2000;10:469–471. doi: 10.1016/s0960-894x(00)00029-9. [DOI] [PubMed] [Google Scholar]
- 6.a) Kobayashi J, Suzuki H, Shimbo K, Takeya K, Morita H. J Org Chem. 2001;66:6626–6633. doi: 10.1021/jo0103423. [DOI] [PubMed] [Google Scholar]; b) Suzuki H, Morita H, Iwasaki S, Kobayashi J. Tetrahedron. 2003;59:5307. [Google Scholar]; c) Suzuki H, Morita H, Shiro M, Kobayashi J. Tetrahedron. 2004;60:2489. [Google Scholar]
- 7.a) Harrison JR, Moody CJ. Tetrahedron Lett. 2003;44:5189. [Google Scholar]; b) Yuen AKL, Joliffe KA, Hutton CA. Austr J Chem. 2006;59:819. [Google Scholar]
- 8.Lynen F, Weiland U. Justus Liebigs Ann Chem. 1937;533:93–117. [Google Scholar]; b) Lengsfeld AM, Löw I, Wieland T, Dancker P, Hasselbach W. Proc Natl Acad Sci USA. 1974;71:2803–2807. doi: 10.1073/pnas.71.7.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Capani F, Deerinck TJ, Ellisman MH, Bushong E, Bobik M, Martone ME. J Histochem Cytochem. 2001;49:1351–1361. doi: 10.1177/002215540104901103. [DOI] [PubMed] [Google Scholar]
- 10.a) Lindell TJ, Weinberg F, Morris PW, Roeder RG, Rutter WJ. Science. 1970;170:447–449. doi: 10.1126/science.170.3956.447. [DOI] [PubMed] [Google Scholar]; b) Hallen HE, Luo H, Scott-Craig JS, Walton JD. Proc Natl Acad Sci USA. 2007;104:19097–19101. doi: 10.1073/pnas.0707340104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Matsunaga S, Fusetani N, Hashimoto K, Walchli M. J Am Chem Soc. 1989;111:2582–2588. [Google Scholar]; b) Matsunaga S, Fusetani N. J Org Chem. 1995;60:1177–1181. [Google Scholar]
- 12.Nishimura S, Arita Y, Honda M, Iwamoto K, Matsuyama A, Sharai A, Kawasaki H, Kakeya H, Kobayashi T, Matsunaga S, Yoshida M. Nat Chem Biol. 2010;6:519–526. doi: 10.1038/nchembio.387. [DOI] [PubMed] [Google Scholar]
- 13.Zanotti G, Birr C, Wieland T. Int J Pept Protein Res. 1978;12:204–216. doi: 10.1111/j.1399-3011.1978.tb02888.x. [DOI] [PubMed] [Google Scholar]
- 14.Maggi CA, Astolfi M, Guiliani S, Goso C, Manzini S, Meini S, Patacchini R, Pavone V, Pedone C, Quartara L. J Pharm Exp Ther. 1994;271:1489–1500. [PubMed] [Google Scholar]
- 15.Sun Y, Lu G, Tam JP. Org Lett. 2001;3:1681–1684. doi: 10.1021/ol015889i. [DOI] [PubMed] [Google Scholar]
- 16.Ghalit N, Reichwein JF, Hilbers HW, Breukink E, Rijkers DTS, Liskamp RMJ. ChemBioChem. 2007;8:1540–1554. doi: 10.1002/cbic.200700244. [DOI] [PubMed] [Google Scholar]
- 17.Mendive-Tapia L, Preciado S, Garcia J, Ramón R, Kielland N, Albericio F, Lavilla R. Nat Commun. 2015;6:7160. doi: 10.1038/ncomms8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cromm PM, Schaubach S, Spiegel J, Fürstner A, Grossmann TN, Waldmann H. Nat Commun. 2016;7:11300. doi: 10.1038/ncomms11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elduque X, Pedroso E, Grandas A. Org Lett. 2013;15:2038–2041. doi: 10.1021/ol400726y. [DOI] [PubMed] [Google Scholar]
- 20.Bartoloni M, Jin X, Marcaida MJ, Banha J, Dibonaventura I, Bongoni S, Bartho K, Gräbner O, Sefkow M, Darbre T, Reymond JL. Chem Sci. 2015;6:5473–5490. doi: 10.1039/c5sc01699a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oh D, Darwish SA, Shirazi AN, Tiwari RK, Parang K K. ChemMedChem. 2014;9:2449–2453. doi: 10.1002/cmdc.201402230. [DOI] [PubMed] [Google Scholar]
- 22.Roodbeen R, Paaske B, Jiang L, Christensen A, Neilsen JT, Huang M, Mulder FA, Nielsen NC, Andreasen PA, Jensen KJ. ChemBioChem. 2013;14:2179–2188. doi: 10.1002/cbic.201300335. [DOI] [PubMed] [Google Scholar]
- 23.Gunzberg MJ, Kulkarni K, Watson GM, Ambaye ND, Del Borgo MP, Brandt R, Pero SC, Perimutter P, Wilce MCJ, Wilce JA. Sci Rep. 2016;6:27060. doi: 10.1038/srep27060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goncalves V, Gautier B, Garbay C, Vidal M, Inguimbert N. J Pept Sci. 2008;14:767–772. doi: 10.1002/psc.965. [DOI] [PubMed] [Google Scholar]
- 25.Fischer S, Lamping M, Gold M, Röttger Y, Brödje D, Dodel R, Frantz R, Mraheil MA, Chakraborty T, Geyer A. Bioorg Med Chem. 2017;25:603–608. doi: 10.1016/j.bmc.2016.11.022. [DOI] [PubMed] [Google Scholar]
- 26.Lian W, Jiang B, Qian Z, Pei D. J Am Chem Soc. 2014;136:9830–9833. doi: 10.1021/ja503710n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian Z, Rhodes CA, McCroskey LC, Wen J, Appiah-Kubi G, Wang DJ, Guttridge DC, Pei D. Angew Chem Int Ed. 2017;56:1525–1529. doi: 10.1002/anie.201610888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 29.Joo SH, Xiao Q, Ling Y, Gopishetty B, Pei D. J Am Chem Soc. 2006;128:13000–13009. doi: 10.1021/ja063722k. [DOI] [PubMed] [Google Scholar]
- 30.Lian W, Upadhyaya P, Rhodes CA, Liu Y, Pei D. J Am Chem Soc. 2013;135:11990–11995. doi: 10.1021/ja405106u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thakkar A, Wavreille AS, Pei D. Anal Chem. 2006;78:5935–5939. doi: 10.1021/ac0607414. [DOI] [PubMed] [Google Scholar]
- 32.Trinh TB, Upadhyaya P, Qian Z, Pei D. ACS Comb Sci. 2016;18:75–85. doi: 10.1021/acscombsci.5b00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heinis C, Rutherford T, Freund S, Winter G. Nat Chem Biol. 2009;5:502–507. doi: 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- 34.Smith GP. Science. 1985;288:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 35.a) Rim C, Lahey LJ, Patel VG, Zhang H, Son DY. Tetrahedron Lett. 2009;50:745–747. [Google Scholar]; b) Angelini A, Diderich P, Morales-Sanfrutos J, Thurnheer S, Hacker D, Menin L, Heinis C. Bioconjug Chem. 2012;23:1856–1863. doi: 10.1021/bc300184m. [DOI] [PubMed] [Google Scholar]
- 36.Chen S, Gopalakrishnan R, Schaer T, Marger F, Hovius R, Bertrand D, Pojer F, Heinis C. Nat Chem. 2014;6:1009–1016. doi: 10.1038/nchem.2043. [DOI] [PubMed] [Google Scholar]
- 37.Roberts RW, Szostak JW. Proc Natl Acad Sci USA. 1997;94:12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sako Y, Morimoto J, Murakami H, Suga H. J Am Chem Soc. 2008;130:7232–7234. doi: 10.1021/ja800953c. [DOI] [PubMed] [Google Scholar]
- 39.Hacker DE, Hoinka J, Iqbal ES, Przytycka TM, Hartman MCT. ACS Chem Biol. 2017;12:795–804. doi: 10.1021/acschembio.6b01006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scott CP, Abel-Santos E, Wall M, Wahnon DC, Benkovic SJ. Proc Natl Acad Sci. 1999;96:13638–13643. doi: 10.1073/pnas.96.24.13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bionda N, Fasan R. ChemBioChem. 2015;16:2011–2016. doi: 10.1002/cbic.201500179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kane PM, Yamashiro CT, Wolczyk DF, Neff N, Goebl M, Stevens TH. Science. 1990;250:651–657. doi: 10.1126/science.2146742. [DOI] [PubMed] [Google Scholar]
- 43.Wu H, Hu Z, Liu XQ. Proc Natl Acad Sci USA. 1998;95:9226–9231. doi: 10.1073/pnas.95.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacConnell AB, McEnaney PJ, Cavett VJ, Paegel BM. ACS Comb Sci. 2015;17:518–534. doi: 10.1021/acscombsci.5b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ito K, Passioura T, Suga H. Molecules. 2013;18:3502–3528. doi: 10.3390/molecules18033502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.a) Masuoka Y, Shindoh N, Inamura N. Prog Drug Res. 2008;66:337–359. doi: 10.1007/978-3-7643-8595-8_7. [DOI] [PubMed] [Google Scholar]; b) Ueda H, Manda T, Matsumoto S, Mukumoto S, Nishigaki F, Kawamura I, Shimomura K. J Antibiot. 1994;47:315–323. doi: 10.7164/antibiotics.47.315. [DOI] [PubMed] [Google Scholar]; c) Sandor V, Bakke S, Robey RW, Kang MH, Blagosklonny MV, Bender J, Brooks R, Peikarz RL, Tucker E, Figg WD, Chan KK, Goldspiel B, Fojo AT, Balcerzak SP, Bates SE. Clin Cancer Res. 2002;8:718–728. [PubMed] [Google Scholar]
- 47.a) Su TT, Gladstone MN, Zhang G, Sammakia T. 2015 US0238562A1. [Google Scholar]; b) Su TT, Gladstone MN, Zhang G, Sammakia T. 2016 US9452215B2. [Google Scholar]
- 48.Parmley S. SciBx. 2014;7 doi: 10.1038/scibx.2014.1397. [DOI] [Google Scholar]
- 49.Clemente A, del Carmen Arques M. World J Gastroenterol. 2014;20:10305–10315. doi: 10.3748/wjg.v20.i30.10305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ando S, Yasutake A, Waki M, Nishino N, Kato T, Izumiya N. Biochim Biophys Acta. 1987;916:527–531. doi: 10.1016/0167-4838(87)90200-7. [DOI] [PubMed] [Google Scholar]
- 51.Pollaro L, Diderich P, Angelini A, Bellotto S, Wegner H, Heinis C. Anal Biochem. 2012;427:18–20. doi: 10.1016/j.ab.2012.04.025. [DOI] [PubMed] [Google Scholar]
- 52.Baeriswyl V, Calzavarini S, Chen S, Zorzi A, Bologna L, Angelillo-Scherrer A, Heinis C. ACS Chem Biol. 2015;10:1861–1870. doi: 10.1021/acschembio.5b00103. [DOI] [PubMed] [Google Scholar]
- 53.Middendorp SJ, Wilbs J, Quarroz C, Calzavarini S, Angelillo-Scherrer A, Heinis C. J Med Chem. 2017;60:1151–1158. doi: 10.1021/acs.jmedchem.6b01548. [DOI] [PubMed] [Google Scholar]
- 54.Lu KP, Zhou XZ. Nat Rev Mol Cell Biol. 2007;8:904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 55.Jiang B, Pei D. J Med Chem. 2015;58:6306–6312. doi: 10.1021/acs.jmedchem.5b00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shepherd NE, Harrison RS, Ruiz-Gomez G, Abbenante G, Mason JM, Fairlie DP. Org Biomol Chem. 2016;14:10939–10945. doi: 10.1039/c6ob02185a. [DOI] [PubMed] [Google Scholar]
- 57.Oligino L, Lung FD, Sastry L, Bigelow J, Cao T, Curran M, Burke TR, Jr, Wang S, Krag D, Roller PP, King CR. J Biol Chem. 1997;272:29046–29052. doi: 10.1074/jbc.272.46.29046. [DOI] [PubMed] [Google Scholar]
- 58.Quartararo JS, Eshelman MR, Peraro L, Yu H, Baleja JD, Lin YS, Kritzer JA. Bioorg Med Chem. 2014;22:6387–6391. doi: 10.1016/j.bmc.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Achen M, Hughes R, Stacker S, Cendron A. 2002 US20020065218A1. [Google Scholar]
- 60.Upadhyaya P, Qian Z, Habir NAA, Pei D. Tetrahedron. 2014;70:7714–7720. doi: 10.1016/j.tet.2014.05.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mund T, Lewis MJ, Masten S, Pelham HR. Proc Natl Acad Sci. 2014;111:16736–16741. doi: 10.1073/pnas.1412152111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Potterat O, Wagner K, Gemmecker G, Mack J, Puder C, Vettermann R, Streicher R. J Nat Prod. 2004;67:1528–1531. doi: 10.1021/np040093o. [DOI] [PubMed] [Google Scholar]
- 63.Hill PS, Smith DD, Slaninova J, Hruby VJ. J Am Chem Soc. 1990;112:3110–3113. [Google Scholar]
- 64.Diderich P, Heinis C. Tetrahedron. 2014;70:7733–7739. [Google Scholar]
- 65.Urech-Varenne C, Radtke F, Heinis C. ChemMedChem. 2015;10:1754–1761. doi: 10.1002/cmdc.201500261. [DOI] [PubMed] [Google Scholar]
- 66.Beck A, Goetsch L, Dumontet C, Covaïa N. Nat Rev Drug Discov. 2017;16:315–337. doi: 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 67.Bennet G, Harrison H, Campbell S, Teufel D, Langford G, Watt A, Bonny C. Eur J Cancer. 2016;69:S21. [Google Scholar]
- 68.de Groot FM, Broxterman HJ, Adams HP, van Vliet A, Tesser GI, Elderkamp YW, Schraa AJ, Kok RJ, Molema G, Pinedo HM, Scheeren HW. Mol Cancer Ther. 2002;1:901–911. [PubMed] [Google Scholar]
- 69.Chen X, Plasencia C, Hou Y, Neamati N. J Med Chem. 2005;48:1098–1106. doi: 10.1021/jm049165z. [DOI] [PubMed] [Google Scholar]
- 70.Veber DF, Holly FW, Paleveda WJ, Nutt RF, Bergstrand SJ, Torchiana M, Glitzer MS, Saperstein R, Hirschmann R. Proc Natl Acad Sci USA. 1978;75:2636–2640. doi: 10.1073/pnas.75.6.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Falb E, Salitra Y, Yechezkel T, Bracha M, Litman P, Olender R, Rosenfeld R, Senderowitz H, Jiang S, Goodman M. Bioorg Med Chem. 2001;9:3255–3264. doi: 10.1016/s0968-0896(01)00234-6. [DOI] [PubMed] [Google Scholar]
- 72.Fani M, Mueller A, Tamma ML, Nicolas G, Rink HR, Cescato R, Reubi JC, Maecke HR. J Nucl Med. 2010;51:1771–1779. doi: 10.2967/jnumed.110.076695. [DOI] [PubMed] [Google Scholar]
- 73.Park JA, Lee YJ, Lee JW, Lee KC, An GI, Kim KM, Kim BI, Kim TJ, Kim JY. ACS Med Chem Lett. 2014;5:979–982. doi: 10.1021/ml500135t. [DOI] [PMC free article] [PubMed] [Google Scholar]