

Abstract
Cells infected by viruses can exhibit diverse patterns of viral and cellular gene expression. The patterns arise in part from the stochastic or noisy reaction kinetics associated with the small number of genomes, enzymes, and other molecules that typically initiate virus replication and activate cellular anti-viral defenses. It is not known what features, if any, of the early viral or cellular gene expression correlate with later processes of viral replication or cell survival. Here we used two fluorescent reporters to visualize innate immune activation of human prostate cancer (PC3) cells against infection by vesicular stomatitis virus. The cells were engineered to express green-fluorescent protein under control of the promoter for IFIT2, an interferon-sensitive component of the anti-viral response, while red-fluorescent protein was expressed as a byproduct of virus infection. To isolate and quantitatively analyze single-cells, we used a unique microwell array device and open-source image processing software. Kinetic analysis of viral and cellular reporter profiles from hundreds of cells revealed novel relationships between gene expression and the outcome of infection. Specifically, the relative timing rather than the magnitude of the viral gene expression and innate immune activation correlated with the infection outcome. Earlier viral or anti-viral gene expression favored or hindered virus growth, respectively. Further, analysis of kinetic parameters estimated from these data suggests a trade-off between robust antiviral signaling and cell death, as indicated by a higher rate of detectable cell lysis in infected cells with a detectable immune response. In short, cells that activate an immune response lyse at a higher rate. More broadly, we demonstrate how the intrinsic heterogeneity of individual cell behaviors can be exploited to discover features of viral and host gene expression that correlate with single-cell outcomes, which will ultimately impact whether or not infections spread.
Graphical Abstract
We have identified critical aspects of the competition between a virus and its host’s immune-response, within single-cells using fluorescent reporters.
Introduction
Hosts and viruses have co-evolved and developed multiple competing mechanisms to either detect and shut down infection progression or evade and suppress host immune response pathways. The early steps in these processes are often mediated by very few molecules or complexes (e.g., a few viral genomes or cellular toll-like receptors), and they lead to a dramatic amplification of other biological responses. The dynamics of this amplification are often variable, leading to stochastic behaviors [1–4]. Furthermore, variability in the local environment of a cell, such as differences in cell-cell contact and local paracrine signaling, affect both cellular gene expression [5] and the ability of a virus to infect a cell [6–9]. Infections are further complicated by the extraordinary genetic heterogeneity that exists in virus populations [10–12]. Thus, viruses interact with their hosts by integrating multiple noisy factors and processes, ultimately producing a diversity of potential outcomes. A thorough study of these interactions is challenging because most molecular and cellular assays provide measures of average behaviors drawn from large populations of cells. Such measures often mask the diversity of viral and cellular behaviors. In contrast, data from high-throughput single-cell techniques can reveal the intrinsic heterogeneity of the viral and cellular processes. While such datasets can be initially overwhelming, their careful analysis can provide a significant opportunity to gain new insights into virus-host interactions [13].
Approaches to quantitative, single-cell studies in virology began more than half a century ago with investigations of single-cell bacteriophage production [14], an endpoint measure which has remained time-consuming and laborious but nicely illustrates the magnitude of cell-to-cell variability that exists during infections [8,14–16]. More recently, myriad single-cell measures have been used in combination to elucidate viral and cellular mechanisms [2,7,17–25]. Many of these studies have been aided by the development of live-cell imaging of fluorescent reporters. Within-well cytometry methods, for example, use fluorescent microscopy to isolate the reporter signal from individual cells in order to obtain flow-cytometry like readouts [19,26–28]. Imaging cells in populations provides a more natural context; however acquiring kinetic measures from individual cells within a population can be challenging owing in part to the cell-tracking problem [29–31]. Methods such as micro-patterning and cell-isolation in microwells can be used to eliminate imaging issues by physically isolating cells [32–36]. Further, they can be adapted for other applications such as the detection or quantification of cell secretions [37–39].
Continuing such efforts, we recently developed a platform for the streamlined analysis of single-cell infections. This platform combines live-cell imaging with a unique microwell array design that can be used to physically isolate cells, and also uses open-source databasing and image processing software to quantify the number of cells in each microwell and measure the fluorescent signals emitted from those cells [27]. We have recently applied this platform to the study of viral interference, using a viral reporter system to analyze how defective interfering particles (DIPs) impact VSV protein production after co-infection with different ratios of DIPs to infectious particles [40]. Here, we use this platform to study the interaction between vesicular stomatitis virus (VSV) and prostate cancer (PC3) cells. This specific virus-host system is a relatively well-characterized model of the competition between virus replication and the innate immune response to virus infection [41,42]. Although VSV easily replicates to high titers in many cancer cell lines, PC3 cells carry an intact interferon pathway that can resist infection. A variant of VSV that has an M51R mutation in its matrix protein is impaired in its normal ability to shut down host responses, making it highly susceptible to shutdown by PC3 cells [42–45]. Dynamic readouts of immune activation in PC3 cells infected by the wild-type (VSV-rWT) or the immune-sensitive variant (VSV-M51R) are feasible using a dual-color reporter system [44]. The virus is engineered to produce a red fluorescent protein (RFP) in proportion to other viral proteins while the engineered cell line (PC3-IFIT2) produces a green fluorescent protein (GFP) in response to activation of an interferon stimulated response element (ISRE) that is associated with the cellular innate immunity [46–48]. Both fluorescent proteins (DS-RedExpress and ZS-Green) express with similar kinetics enabling direct comparisons of certain viral and innate immune events within single-cells [44]. During infection we acquired real-time measures of the viral and host fluorescent reporters and extracted descriptive parameters of each kinetic trajectory. Correlation analysis of the virus and host parameters revealed aspects of the viral replication and innate immune processes that were most predictive of infection outcomes at the single-cell level.
Materials and Methods
Cell and virus culture and dual-color reporter system
A reporter cell line created from the human prostate cancer cell line PC3 was used in this work. The reporter cell line, PC3-IFIT2-ZSGreen1-DR (PC3-IFIT2), was created as previously described [44]. Briefly, the engineered gene shares the promoter sequence of the human IFIT2 gene, a downstream reporter of interferon stimulation, and encodes for the Zs-Green fluorescent protein. Thus, upon promotion of the IFIT2 gene, PC3-IFIT2 cells produce both GFP and IFIT2. PC3-IFIT2 cells were passaged every 3–4 days and grown in RPMI 1640 (Gibco) supplemented with 10% FBS (Atlanta Biologicals) and 2 mM Glutamax (Gibco). Infections of these cells were done in the same media, but with the FBS content reduced to 2%. Experiments were performed with cells within 10 passages from stocking.
VSV is a negative-sense, non-segmented RNA virus that is known for its broad cellular tropism, exceptionally fast replication kinetics in permissive cell types, and its sensitivity to interferon-stimulated genes. Versions of the wild-type strain and M51R strain were engineered that contain the DsRed-Express DR gene in the 5th position (VSV-rWT and VSV-M51R, respectively) to enable observation and comparison of viral protein production [41]. These two strains are equivalent except for the M51R point mutation in the VSV-M51R strain.
Infections and microwell device seeding
PC3-IFIT2 cells were infected in solution with stock concentrations of either the VSV-M51R or the VSV-rWT strains as previously described [16]. Following in-solution adsorption of virus while on ice, the temperature of the virus-cell solution was increased to 37°C to allow for internalization of attached viral particles [49]. After 7 minutes at 37°C, the cell suspension was repeatedly (3×) centrifuged and resuspended in fresh media to remove excess virus. Although the concentration of virus added was very high, inefficient virus adsorption led to a detectable infection in approximately one-third of cells. The resuspension media contained Hoechst 33342 (AnaSpec 1μM) and HEPES (Sigma-Aldrich, 25mM).
The microwell array (MA) device used here is shown in Fig 1 and the dimensions and methods for developing the array have been previously described [27]. This device design allows reagents and excess fluid to be passively isolated into different regions of the array, enabling multiplexed experiments to be run on the same device in parallel, a unique capability of the platform. The isolated regions resemble multiple bull’s-eyes on the surface of the device (Fig 1a). A ‘moat’ or recessed ring is used to isolate a small droplet-sized region of microwells from surrounding microwells. Here, this feature was used to study isolated PC3-IFIT2 cells infected with either VSV-rWT or VSV-M51R on the same chip.
Fig. 1. Microwell array device design and infected cell seeding.

(a) Diagram of a PDMS microwell device showing 10 bull’s-eyes in a 2 × 5 array arranged to fit on a glass slide footprint. (b) Each bull’s-eye contains 2500 sub-nanoliter volume wells. (c) Cells within microwells were imaged by taking a 4×3 array of 4× magnification images centered around each bull’s-eye. A stitched image overlay of phase contrast, GFP, RFP, and DAPI fluorescent channels of one bull’s-eye is shown. (d) A small section of (c) is magnified in order to visualize individual microwells and the cells contained within those wells. (e) From the kinetic imaging data, fluorescent trajectories from isolated single cells can be plotted, and kinetic parameters describing those trajectories extracted. The first data point above the limit of detection (LOD) for this sample virus trajectory is defined as the delay-time (Delayv). The rise-time is defined as the period between the delay-time and the time at which 85% of the maximum signal is reached (Risev). The first four data-points above the LOD can be fit with an exponential curve to estimate the production-rate, approximated as alpha (α) in the equation y = Aeαt (Ratev). We can also record a maximum signal for each trajectory that reaches a maximum during imaging (Maxv) and note cell lysis events if they occur.
After removal of excess virus from the infected cell suspension, the cells were added in droplets at a concentration of ~1×105cells/ml to each bull’s-eye. The cells were allowed to settle for ~30 sec before the droplets were removed and fresh media was added and removed 2–3 times to remove cells from the surface of the microwell device. The microwells were then sealed with the addition of a glass slide that was secured onto the PDMS device with an aluminum slide holder.
Live-cell imaging and image processing
Cell health within the microwell device was maintained during imaging using a humidified (85% RH), stage-top incubator at a temperature of 37°C and 5% CO2 concentration (Pathology Devices, Westminster, MD). In addition to the stage-top incubator, a larger heated incubator was used that encased the majority of the Nikon Eclipse TE300 microscope used in these experiments. Images were taken in a 4 × 3 array at 4× magnification around the center of each bulls-eye in the following order: brightfield (5ms exposure), blue channel (20ms exposure), red channel (1500ms exposure) and green channel (1500ms exposure) using a Sedat Quad cube (Chroma Technology, Bellows Falls, VT). Imaging the entire array required approximately 18 minutes, and was looped at a 30 minute interval for 22 hours beginning 1.42 hpi, using Metamorph software controlling an EXI Aqua Q-imaging CCD camera and a Pryor automated stage. After completion of the experiment, the image set was imported into an image processing and databasing software called Je’Xperiment (JEX) (http://github.com/jaywarrick/JEX). Details on data acquisition using this software can be found in Warrick et al [27]. Briefly, the images were sorted in JEX according to location, time, and color and then registered. Following image registration, microwell locations were identified and numbered. The maxima in the blue channel (representing nuclei) were located and the number in each microwell determined. Finally, a cell diameter (13 pixels) was chosen and measurements were taken in each fluorescent color at all times. The measurement and cell count data tables were exported as ARFF files (Weka software, University of Waikato) for subsequent data analysis.
High-throughput data analysis
In this work, the image data analysis was performed in Matlab using scripts that could also be run in the open-source software Octave. Cell count and location data exported from JEX was used to identify data from microwells containing either 0 or 1 cell. This data was organized by image location, and then a series of background corrections and thresholding steps were performed. First, a local background subtraction was performed for all empty-well and single-cell data. Next, the signal from all empty-wells was used to correct for background noise. Finally, illumination correction was performed using images of fluorescent standard slides (Ted Pella, Redding, CA), by multiplying the measured signal for each cell by the normalized signal intensity at the same x and y location on the correction image. The final step requires the determination of a limit of detection (LOD), which was based on the corrected empty well measurements. The median plus 3 standard deviations of the corrected empty well signals at all times and at all locations (entire PDMS device) was calculated and the highest of those values was chosen as the LOD. Any measurement below the LOD was set to zero. Wells containing fluorescent debris artificially raise the LOD and were removed from the analysis. Once the data was cleaned and thresholded, several parameters were extracted from the fluorescent kinetics (Fig 1e). To extract a production rate estimate, we fit an exponential function to the first 4 data points detected above the LOD and used the exponential as the production rate estimate. The delay-time was defined as the time from infection to initial detection of a fluorescent signal and the rise-time was the delay-time subtracted from the time to 85% of the maximum signal.
Statistical Analysis
Correlations between many parameter sets were determined by calculating the Pearson correlation coefficient. The significance of each Pearson correlation was determined through calculation of 95% confidence intervals using bootstrapping. The bootstrapping was performed by sampling 100% of the sample size with replacement, and confidence intervals were determined based on 10,000 simulated data sets.
Results
Single-cell analysis enables identification and quantification of sub-populations and rare events
PC3-IFIT2 cells were infected with two recombinant strains of vesicular stomatitis virus encoding the gene for DSRed-Express, sealed in microwells, and imaged over the course of the infection. The two VSV strains used here are equivalent except the VSV-M51R strain has a point mutation in the matrix protein that eliminates its ability to block nuclear export of cellular mRNA, effectively neutralizing the primary mechanism for suppressing the innate immune response by VSV. Fig 2 shows the number of VSV-rWT or VSV-M51R infected cells that were positive for the viral RFP reporter, the innate immune GFP reporter, or both reporters at some point in the experiment. Generally, VSV-M51R infected cells were positive for both reporters and VSV-rWT infected cells were only RFP+, but there were rare exceptions such as three VSV-rWT cells that were positive for both RFP and GFP. Moreover, 6.6 percent of signaling VSV-M51R infected cells exhibited no detectable viral gene expression.
Fig. 2. Composition of virus and host reporter activation in single cell populations infected by wildtype (WT) or mutant (M51R) virus.
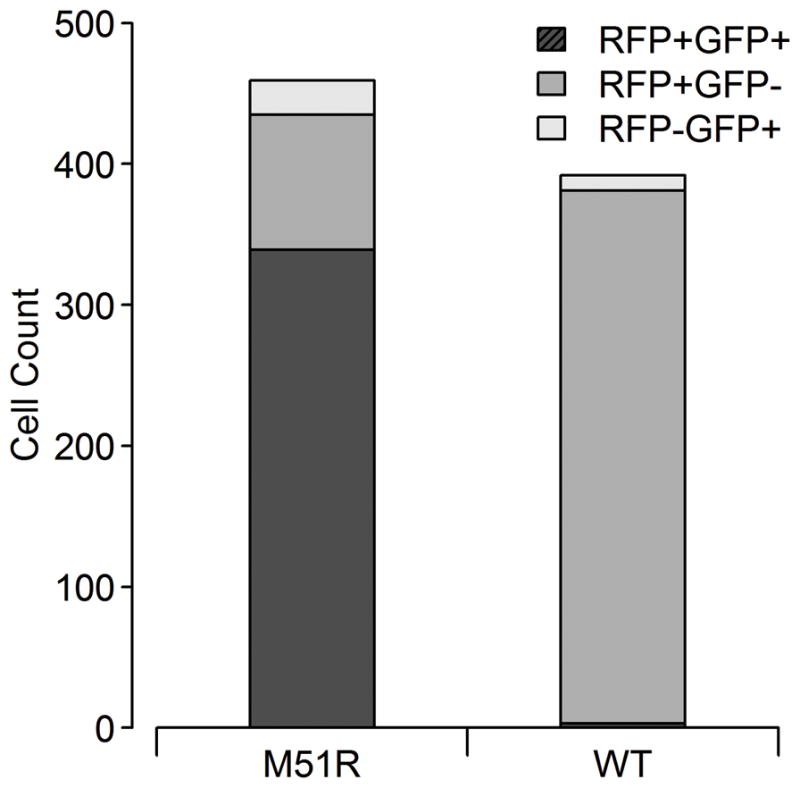
The majority of VSV-M51R infected cells expressing a fluorescent signal were positive for both the viral and innate immune reporters, while the majority of VSV-rWT cells expressed only the viral reporter at some point during imaging of cells (~1.4 – 23.4 hpi).
Parametrization of single-cell viral and host fluorescent trajectories reveals highly variable kinetics
Each infected host cell has the possibility of expressing two unique reporters, reflecting a potentially unique profile of viral and host gene expression over time. The fluorescence intensity for each reporter over time defines a trajectory from which one can extract descriptive parameters (Fig 1e). Delay-time, rise-time, maximum signal, presence or absence of cell lysis, and production-rate (α) parameters were extracted from each reporter trajectory. With five parameters from each reporter trajectory, and two reporters, we have the potential to obtain up to 10 quantitative characteristic parameters from each cell. The distributions for several parameters shown in Fig 3 illustrate the wide-ranging heterogeneity in virus protein production and host-response IFIT2 trajectories. The parameter distributions show that the viral and host rise-times (Risev and Riseh), which scale with the duration of the detectable reporter, varied greatly from less than 30 minutes to 19 hours while the production-rate (Ratev and Rateh) and maximum signal (Maxv and Maxh) varied 10- and 1000-fold, respectively. This 1000-fold range in Maxv is consistent with our previous observations that VSV infectious particle yields from single-cells can vary over three orders of magnitude [16]. Interestingly, although infections of PC3-IFIT2 cells were synchronized, delay-times to detectable viral reporter expression (Delayv) range from 1.4 hpi (the first time-point imaged) to over 22 hpi. These observations illustrate the extent of cell-to-cell variability that is available for exploring and identifying new quantitative relationships.
Fig. 3. Distributions of parameters for viral and host reporter protein trajectories.

These distributions represent trajectories from the VSV-M51R infected cells that were positive for one or both reporter signals, with virus parameters red and host parameters green. The value of σ was obtained using the median absolute deviation function in R (‘mad’), which applies a correction factor to approximate the standard deviation of the sample in a manner that is robust to outliers. Mean: x̄, Median: x̃, Std. Dev. Est.: σ, Range: min – max. Total cell counts for each histogram of virus/host parameters are (a) 350/308, (b) 435/363, (c) 373/295, and (d) 373/295 respectively.
Because of the extensive heterogeneity observed in these trajectories, analysis was in some cases limited to subsets of cells. For example, in some cases the RFP signal never reached a true maximum during imaging, and therefore analyses using either the rise-time or maximum signal parameters were limited to cells that reached a plateau in RFP intensity or lysed during imaging represented by 86% of all RFP+ cells infected by VSV-M51R, and 82% of all RFP+ cells infected by VSV-rWT. Further, analysis using the production-rate parameter was limited to trajectories from cells that were well fit with an exponential function (R2 value ≥ 0.9), represented by 72% of RFP trajectories and 82% of GFP trajectories. This parameter was sensitive to the limit of detection; bad fits were indicative of either slow rates of early reporter protein production, which may have delayed initial detection, or very noisy low-yield signals that reached a plateau within a few time-points of initial detection.
Innate-immune response activation correlates with higher viral protein production
In 22 percent of VSV-M51R infected cells (RFP+), the IFIT2 reporter was not detected (GFP−) (Fig 2). The analysis of parameters extracted from GFP+(immune positive) and GFP-(immune negative) VSV-M51R infected cell sub-populations (Table 1) shows a statistically significant difference in each metric (p ≤ 0.046), indicating that viral protein production was actually faster and produced at higher total amounts in innate immune activated cells (GFP+). Moreover, rates of viral protein production and overall yields were also significantly lower in VSV-rWT infected cells (Table 2), in which an immune response was rarely activated, and cell lysis rates were lower in both GFP− VSV-M51R infected cells (vs GFP+, Table 1) and VSV-rWT infected cells (vs GFP+ M51R, Table 2).
Table 1.
Comparison of viral RFP kinetic parameters in M51R-infected cell subpopulations.
| Parameter | M51R RFP+GFP+ | M51R RFP+GFP− | P-value |
|---|---|---|---|
| Delayv (hpi) | 7.10 | 9.64 | 1e-05 |
| Risev (hr) | 6.84 | 5.50 | 5e-04 |
| αv (1/hr) | 1.25 | 1.09 | 2e-04 |
| Maxv (au) | 854 | 420 | 6e-12 |
| % Lysed | 14.7 | 6.7 | 0.046 |
The mean of each parameter is given. A Mann-Whitney test was used for all p-values except lysed percentage. The z-test for comparison of two sample proportions was calculated to determine if the percentage of lysed cells was statistically different in the GFP+ and GFP− subpopulations.
Table 2.
Comparison of viral RFP kinetic parameters in subpopulations of M51R and WT-VSV infected cells.
| Parameter | M51R RFP+GFP+ | WT RFP+ | P-value |
|---|---|---|---|
| Delayv (hpi) | 7.10 | 7.40 | 0.014 |
| Risev (hr) | 6.84 | 6.46 | 0.079 |
| αv (1/hr) | 1.25 | 1.11 | 1e-11 |
| Maxv (au) | 854 | 370 | 2e-16 |
| % Lysed | 14.7 | 5 | 2e-5 |
Means of each parameter are given. A Mann-Whitney test was used for all p-values except lysed percentage. The z-test for comparison of two sample proportions was calculated to determine if the percentage of lysed cells was statistically different in the M51R RFP+/GFP+ and WT RFP+ subpopulations.
Viral and host gene expression is correlated in time, but not in magnitude
To further investigate the relationship between the innate immune response and viral protein production the Pearson’s correlation coefficient for each pair of parameters extracted from viral and cellular reporter trajectories was determined (Fig 4). Some correlations were as expected, such as the strong association between increasing viral protein expression (Risev) or viral production-rates (Ratev) and higher viral maximum signals (calculations based on the log10 of the Maxv) (R = 0.64 and 0.49, respectively). Others, such as the inverse relationship between Delayv and Maxv (R = −0.46) were less intuitive. Similar results were found for analogous host response measures. Conversely, there was no correlation between Maxv and Maxh (R = 0.1) or any viral parameters and the host maximum signal. There were, however, strong correlations between viral and host rise-times (R=0.54) and delay-times (R=0.59). These results indicate that the host response, represented by the presence of detectable IFIT2 reporter production, depends on the detection of infection processes that are strongly correlated with viral protein production; however, the degree of IFIT2 amplification is independent of downstream virus replication activity. Bootstrapping was used to calculate 95% confidence intervals for each parameter pair.
Fig. 4. Characteristic kinetic parameters of virus and host reporters correlate to different extents.

Results from RFP+GFP+ cells produced by reporter mutant virus (VSV-M51R) infections of reporter cells are shown. Each cell reported both a virus and host signal, which were used to estimate four virus and four host kinetic parameters, respectively. Then extents of correlation (R) were determined for all 28 unique pairs of virus-virus, host-host, or virus-host parameters. 95% confidence intervals were calculated through bootstrapping and insignificant correlations are marked with an X. The inset shows the extent of correlation between two host parameters: Maxh and Rateh.
Host reaction-time influences infection outcome
Fig 4 shows moderately negative correlations between Maxv and Delayv and also between Maxh and Delayh. To examine these further, we plotted all Max versus Delay pairs in Fig 5. However, given the large variability in the absolute timing of events across different cells (Fig 3), we explored the relative timing of events within each cell to clarify the behavior underlying these negative correlations. Plotting Delayh vs. Delayv shows that the host-response usually follows viral protein production, suggesting once again that the host responds to detection of some virus replication process (Figs 3 and 6a). Thus, the effect of the host response may be better appreciated by examining whether the magnitude of the virus and host responses, correlate with how rapidly the host reacts to infection. Here, we define the host reaction-time as (Delayh − Delayv), where a negative value means that the cell was detectably active prior to detection of viral gene expression, as previously observed [21]. Short or negative host-reaction times are associated with lower extents of viral gene expression reflected in Maxv (R = 0.38), as shown in Fig 6b. In contrast, lower extents of host gene expression reflected in Maxh are generally associated with longer host reaction-times (R = −0.49), as shown in Fig 6c. Likewise, patterns become apparent when considering how the duration of gene expression, described by the viral or host parameters Risev or Riseh, respectively, correlate with host reaction-time (Figs 6d and e). Notably, a relatively strong correlation is revealed when the difference in duration of viral and host gene expression (Risev − Riseh) is plotted versus the host reaction-time (Fig 6f). The positive slope indicates that the dynamics of viral and host protein production are influenced by early events, and the timing of the host-response is critical; faster host responses (shorter host-reaction times) correlate with shorter periods of viral protein production.
Fig. 5. Extents of viral and host reporter expression versus delay-times.

(a) Maxv appears to decrease with longer Delayv but (b) Maxv has no obvious relationship to Delayh. (c) Maxh is not affected by Delayv, but (d) Maxh may decrease with longer Delayh.
Fig. 6. Effect of host reaction-time on infection timing and outcome.

Comparison of (a) delay-time for virus and host reporter protein expression in single-cells. The legend shows the number of cells that have the same behavior. The extent of (b) viral gene expression (Maxv) and (c) host gene expression (Maxh) are plotted against the host reaction-time (Delayh − Delayv). Likewise, the duration of (d) viral gene expression (Risev) and (e) host gene expression (Riseh) are plotted against the host reaction-time. When (f) differences in the duration of reporter gene expression (Risev − Riseh) are plotted versus host reaction-time, a relatively strong linear correlation (R=0.57) is revealed. The host expresses earlier or has the time advantage when host reaction-time has a negative value. Conversely, the virus has the time advantage when host reaction-time has a positive value. All data refers to VSV-M51R infections.
Discussion
The complexity of virus-host interactions is reflected in the myriad strategies viruses have evolved to infect cells and evade innate immunity as well as the ways host cells have evolved to detect infection and prevent its spread. The development of single-cell techniques has provided new avenues to study such virus-host interactions and revealed significant heterogeneity in these interactions. Increasingly, single-cell cytometry is being used to leverage this heterogeneity for additional insight into virus-host interactions. In this work, we employed a unique microwell array device to monitor the kinetics of viral and host-response reporter trajectories from hundreds of isolated single-cell infections. From each trajectory, we extracted kinetic parameters to explore and characterize the heterogeneity in VSV-rWT (wild-type) and VSV-M51R (mutant) infections of PC3 cells.
The primary advantage of our particular single-cell method is the ability to unambiguously track the fluorescence expression from a single-cell through an entire infection cycle. This ability allowed us to look at paired viral and host reporter trajectories over time. We were also able to detect and quantify rare behaviors that would be difficult or impossible to find in population-level studies by simply counting infected cells that were positive or negative for either reporter. For example, we found three VSV-rWT infected cells out of hundreds that were positive for both the viral and host-response reporters, and we measured the fraction of VSV-M51R infected cells that never initiated a detectable immune response. Specifically, we identified a sub-population of 6.6 percent cells that appeared to be activated in the absence of infection, perhaps representing a sub-population of first responder or sentinel cells [2,21]. Future developments of this approach that collect individual cells and their viral progeny for gene expression or sequencing studies may reveal to what extent genetic, environmental or other factors contribute to the emergence of each sub-group.
The PC3 cell reporter gene was controlled by the same promotor as the interferon stimulated gene, IFIT2. When either VSV-rWT or VSV-M51R infected cells begin expressing GFP, both the reporter protein and IFIT2 are produced. Two aspects of IFIT2 activation may relate to anti-viral activity and potentially affect virus reporter production. First, IFIT2 is known to be associated with a pro-apoptotic cell-state [50,51]. Consistent with this observation, the percentage of lysed cells was higher in VSV-M51R infected GFP+ vs. GFP− cells (Table 1), although we are currently unable to distinguish between apoptosis and other mechanisms or forms of cell death such as necrosis. The second potential role of IFIT2 is as a viral translation suppressor through an interaction with eIF3 [48,52]. Thus, we might expect higher IFIT2 reporter expression to be associated with lower Ratev or Maxv; however, we did not observe such correlations. In fact, when we compare GFP+ and GFP− populations, IFIT2 activation was associated with earlier detection of viral protein, higher Ratev, and higher Maxv (i.e., a generally more robust infection, Table 1). This result may seem surprising, but it supports a proposed role for IFIT2 as a viral stress inducible gene (VSIG), as previously suggested [44,52,53].
While most VSIGs can be activated through IFN signaling they can also be directly activated by detection of viral replication intermediates, such as dsRNA, leading to phosphorylation of IRF-3 and activation of its associated response. Therefore, our finding that cells generally begin expressing IFIT2 two hours after detection of viral RFP is consistent with the understanding that transcription of these genes is directly linked to levels of viral intermediates present in each cell [54–56]. In many studies, expression of viral-induced stress genes, such as the dsRNA-activated protein kinase (PKR), leads to a global reduction in translation [58,59], yet we observed that viral RFP levels were higher in GFP+ cells infected with VSV-M51R versus GFP− cells (Table 1), and we also found higher average RFP signals in cells infected with the VSV-M51R strain versus the VSV-rWT strain (Table 2). Both results indicate that the viral stress response may be enhancing viral protein production. The stress response of the cell may be creating an environment that enables a rapid response but has the side-effect of promoting viral protein production, resulting in both high IFIT2 production and viral protein yields. It is an open question how this balance of activation and viral protein production plays out within spreading infections. How the race between the viral progeny and defensive cytokines plays out will ultimately depend not only on their production and release kinetics, but also their physical transport to neighboring susceptible cells, reflecting an integration of biological and physical processes [43,44, 60–62].
Whether an infection spreads likely depends both on the levels of virus and host cytokine production, as well as the timing of these two competing processes. While we observed no direct connection between the maximum levels of viral (Maxv) and host (Maxh) gene expression, we did find a very striking relationship between the maximum expression of each reporter and the host reaction-time (Delayh − Delayv). Generally, the viral reporter was detected first (shorter delay) and followed by detection of the host reporter (longer delay), giving rise to positive host reaction-times. Longer host reaction-times allowed for higher levels of viral gene expression and lower levels of host gene expression (Fig 6b/c). Further, within single cells, longer durations of viral gene expression correlated with longer host reaction times (R=0.30), consistent with a mechanism where virus and host race for finite biosynthetic resources, which are distributed to expression of viral or host genes depending on their relative timing, often favoring viral over host gene expression (Fig 6f).
However, there were instances when the host appeared to be activated prior to infection. The resulting negative host reaction-times could be caused by a cell that was already activated at the start of imaging, perhaps representing the presence of a process (stochastic or otherwise) that ensures at least a small sub-population is at any time activated as part of an early warning system. Alternatively, negative host reaction-times may be due to activation by a slow growing virus, in which amplification of the IFIT2 reporter occurs much faster than the viral reporter. Previous validation of this two-color virus-host system has indicated that the basal level of GFP expression in normal cell culture is ~1% [44].
The observed single-cell host responses suggest how these infections might, over multiple cycles, spread or be contained. Although we focused on IFIT2 activation here, its activation may reflect the many similarly regulated factors that contribute to activation of a cellular innate immune response. Further, the ability to measure the relative timing of events through quantitative kinetic measurements allowed us to observe that a more rapid reaction-time by the host cells was associated with reduced viral protein production and increased activation of the IFIT2 reporter protein. Therefore, we hypothesize that as infections spread and anti-viral cytokines transport to and activate uninfected neighboring cells, the growing time advantage ‘observed’ or integrated by these cells would allow suppression of a subsequent infection. Thus, what might be a short-term loss from the perspective of a single cell at the initial site of infection (higher viral protein production), might be a long-term gain for the tissue, where an early warning of susceptible cells inhibits their expression of viral genes and overall suppression of infection spread.
INSIGHT
Among virus-infected cells, there are subpopulations that do and do not initiate an innate immune response. Here, we show that the subpopulation that activates an innate immune response makes viral protein earlier and to a greater extent than non-activated cells. Further, we find that the relative timing of viral and host signaling greatly affect the outcome of infections.
INNOVATION
This work brings together time-matched dual-color gene expression reporters, a microwell array to enable study of virus and immune reporters in single-cells, data acquisition protocols, and open-source image processing software to extract gene-expression trajectories from hundreds of individual infected cells.
INTEGRATION
Some infected cells robustly amplify protein production to secrete anti-viral cytokines, which coincidentally also enhances viral protein synthesis. But the cytokines then warn neighboring cells, granting them a ‘time advantage’ to suppress subsequent waves of infection spread.
Acknowledgments
We thank Nathan Sherer for valuable feedback and David Beebe for access to microscale device fabrication equipment and materials. This work was supported by the National Institutes of Health (R01-AI091646, U19-AI0104317) and the Graduate School of the University of Wisconsin-Madison. A.T and J.W were NIH trainees (T32-AI078985).
Footnotes
Author Contributions
Conceptualization: AT, JW, JY. Investigation: AT. Formal Analysis: AT, JW. Writing – Review & Editing: AT, JW, JY.
Conflicts of interest
There are no conflicts of interest to declare.
References
- 1.Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 2.Rand U, Rinas M, Schwerk J, Nöhren G, Linnes M, Kröger A, et al. Multi-layered stochasticity and paracrine signal propagation shape the type-I interferon response. Mol Syst Biol. 2012;8:1–13. doi: 10.1038/msb.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levin D, Harari D, Schreiber G. Stochastic receptor expression determines cell fate upon interferon treatment. Mol Cell Biol. 2011;31:3252–66. doi: 10.1128/MCB.05251-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao M, Zhang J, Phatnani H, Scheu S, Maniatis T. Stochastic expression of the interferon-β gene. PLoS Biol. 2012;10:1–16. doi: 10.1371/journal.pbio.1001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Volfson D, Marciniak J, Blake WJ, Ostroff N, Tsimring LS, Hasty J. Origins of extrinsic variability in eukaryotic gene expression. Nature. 2006;439:861–4. doi: 10.1038/nature04281. [DOI] [PubMed] [Google Scholar]
- 6.Oliere S, Arguello M, Mesplede T, Tumilasci V, Nakhaei P, Stojdl D, et al. Vesicular stomatitis virus oncolysis of T lymphocytes requires cell cycle entry and translation initiation. J Virol American Society for Microbiology (ASM) 2008;82:5735–5749. doi: 10.1128/JVI.02601-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snijder B, Sacher R, Rämö P, Damm E-M, Liberali P, Pelkmans L. Population context determines cell-to-cell variability in endocytosis and virus infection. Nature. 2009;461:520–3. doi: 10.1038/nature08282. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Y, Yongky A, Yin J. Virology. Vol. 385. Elsevier Inc; 2009. Growth of an RNA virus in single cells reveals a broad fitness distribution; pp. 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He Y, Xu K, Keiner B, Zhou J, Czudai V, Li T, et al. Influenza A Virus Replication Induces Cell Cycle Arrest in G0/G1 Phase. J Virol American Society for Microbiology (ASM) 2010;84:12832–12840. doi: 10.1128/JVI.01216-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Domingo E, Sheldon J, Perales C. Viral quasispecies evolution. Microbiol Mol Biol Rev MMBR. 2012;76:159–216. doi: 10.1128/MMBR.05023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature Nature Publishing Group. 2006;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McWilliam Leitch EC, McLauchlan J. Determining the cellular diversity of hepatitis C virus quasispecies by single-cell viral sequencing. J Virol. 2013;87:12648–55. doi: 10.1128/JVI.01602-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pelkmans L. Using cell-to-cell variability - A New Era in Molecular Biology. Science. 2012;336:425–426. doi: 10.1126/science.1222161. [DOI] [PubMed] [Google Scholar]
- 14.Delbruck M. The burst size distribution in the growth of bacterial viruses (bacteriophages) J Bacteriol. 1945;50:131–5. doi: 10.1128/JB.50.2.131-135.1945. Available: http://www.ncbi.nlm.nih.gov/pubmed/20989330. [DOI] [PubMed] [Google Scholar]
- 15.Lwoff a, Dulbecco R, Vogt M, Lwoff M. Kinetics of the release of poliomyelitis virus from single cells. Virology. 1955;1:801–805. doi: 10.1111/j.1749-6632.1955.tb42536.x. Available: http://dx.doi.org/10.1016/0042-6822(55)90010-6. [DOI] [PubMed] [Google Scholar]
- 16.Timm A, Yin J. Virology. Vol. 424. Elsevier Inc; 2012. Kinetics of virus production from single cells; pp. 11–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snijder B, Sacher R, Rämö P, Liberali P, Mench K, Wolfrum N, et al. Single-cell analysis of population context advances RNAi screening at multiple levels. Mol Syst Biol. 2012;8:579. doi: 10.1038/msb.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu D, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;509:363–9. doi: 10.1038/nature13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rand U, Hillebrand U, Sievers S, Willenberg S, Köster M, Hauser H, et al. Uncoupling of the dynamics of host-pathogen interaction uncovers new mechanisms of viral interferon antagonism at the single-cell level. Nucleic Acids Res. 2014;42:e109. doi: 10.1093/nar/gku492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heldt FS, Kupke SY, Dorl S, Reichl U, Frensing T. Single-cell analysis and stochastic modelling unveil large cell-to-cell variability in influenza A virus infection. Nat Commun. 2015:6. doi: 10.1038/ncomms9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patil S, Fribourg M, Ge Y, Batish M, Tyagi S, Hayot F, et al. Single-cell analysis shows that paracrine signaling by first responder cells shapes the interferon-β response to viral infection. 2015;8:1–13. doi: 10.1126/scisignal.2005728. [DOI] [PubMed] [Google Scholar]
- 22.Ramji R, Wong VC, Chavali AK, Gearhart LM, Miller-Jensen K. A passive-flow microfluidic device for imaging latent HIV activation dynamics in single T cells. Integr Biol. 2015;7:990–1010. doi: 10.1039/C5IB00094G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulte MB, Andino R. Single-cell analysis uncovers extensive biological noise in poliovirus replication. J Virol. 2014;88:6205–6212. doi: 10.1128/JVI.03539-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu Y, Xue Q, Eisele MR, Sulistijo ES, Brower K, Han L, et al. Highly multiplexed profiling of single-cell effector functions reveals deep functional heterogeneity in response to pathogenic ligands. Proc Natl Acad Sci U S A. 2015;17:E607–15. doi: 10.1073/pnas.1416756112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaudet S, Miller-Jensen K. Redefining signaling pathways with an expanding single-cell toolbox. Trends Biotechnol. 2016;34:458–69. doi: 10.1016/j.tibtech.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ecker R, Steiner G. Microscopy-based multicolor tissue cytometry at the single-cell level. Cytom Part A. 2004;59:182–190. doi: 10.1002/cyto.a.20052. [DOI] [PubMed] [Google Scholar]
- 27.Warrick JW, Timm A, Swick A, Yin J. Tools for single-cell kinetic analysis of virus-host interactions. PLoS One. 2016:1–17. doi: 10.1371/journal.pone.0145081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.González-Jara P, Fraile A, Canto T, García-Arenal F. The multiplicity of infection of a plant virus varies during colonization of its eukaryotic host. J Virol. 2009;83:7487–7494. doi: 10.1128/JVI.00636-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maska M, Ulman V, Svoboda D, Matula P, Matula P, Ederra C, et al. A benchmark for comparison of cell tracking algorithms. Bioinformatics. 2014;30:1609–1617. doi: 10.1093/bioinformatics/btu080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kan A, Chakravorty R, Bailey J, Leckie C, Markham J, Dowling MR. Automated and semi-automated cell tracking: addressing portability challenges. J Microsc. 2011;244:194–213. doi: 10.1111/j.1365-2818.2011.03529.x. [DOI] [PubMed] [Google Scholar]
- 31.Li K, Miller ED, Chen M, Kanade T, Weiss LE, Campbell PG. Cell population tracking and lineage construction with spatiotemporal context. Med Image Anal. 2008;12:546–566. doi: 10.1016/j.media.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science Aaas. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 33.Endler EE, Duca KA, Nealey PF, Whitesides GM, Yin J. Propagation of viruses on micropatterned host cell. Biotechnol Bioeng. 2003;20:719–25. doi: 10.1002/bit.10516. [DOI] [PubMed] [Google Scholar]
- 34.Guldevall K, Vanherberghen B, Frisk T, Hurtig J, Christakou AE, Manneberg O, et al. Imaging immune surveillance of individual natural killer cells confined in microwell arrays. PLoS One. 2010;5:e15453. doi: 10.1371/journal.pone.0015453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasuga Y, Iwasawa T, Terada K, Oe Y, Sorimachi H, Ohara O, et al. Single-cell chemical lysis method for analyses of intracellular molecules using an array of picoliter-scale microwells. Anal Chem. 2008;80:9141–9149. doi: 10.1021/ac8016423. Available: http://www.ncbi.nlm.nih.gov/pubmed/19551983. [DOI] [PubMed] [Google Scholar]
- 36.Han Q, Bradshaw EM, Nilsson B, Hafler Da, Love JC. Multidimensional analysis of the frequencies and rates of cytokine secretion from single cells by quantitative microengraving. Lab Chip. 2010;10:1391–400. doi: 10.1039/b926849a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi J, Love KR, Gong Y, Gierahn TM, Love JC. Immuno-hybridization chain reaction enhances detection of individual cytokine-secreting human peripheral mononuclear cells. Anal Chem. 2011;83:6890–6895. doi: 10.1021/ac2013916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Torres AJ, Hill AS, Love JC. Nanowell-Based Immunoassays for Measuring Single-Cell Secretion: Characterization of Transport and Surface Binding. Anal Chem. 2014;86:11562–11569. doi: 10.1021/ac4030297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma C, Fan R, Ahmad H, Shi Q, Comin-Anduix B, Chodon T, et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med. 2011;17:738–743. doi: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akpinar F, Timm A, Yin J. High-throughput single-cell kinetics of virus infections in the presence of defective interfering particles. J Virol. 2016:90. doi: 10.1128/JVI.02190-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carey BL, Ahmed M, Puckett S, Lyles DS. Early steps of the virus replication cycle are inhibited in prostate cancer cells resistant to oncolytic vesicular stomatitis virus. J Virol. 2008;82:12104–15. doi: 10.1128/JVI.01508-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahmed M, Cramer SD, Lyles DS. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology. 2004;330:34–49. doi: 10.1016/j.virol.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 43.Lam V, Duca KA, Yin J. Arrested spread of vesicular stomatitis virus infections in vitro depends on interferon-mediated antiviral activity. Biotechnol Bioeng. 2005;90:793–804. doi: 10.1002/bit.20467. [DOI] [PubMed] [Google Scholar]
- 44.Swick A, Baltes A, Yin J. Visualizing infection spread: Dual-color fluorescent reporting of virus-host interactions. Biotechnol Bioeng. 2014;111:1200–1209. doi: 10.1002/bit.25170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, Lyles DS. Ability of the Matrix Protein of Vesicular Stomatitis Virus To Suppress Beta Interferon Gene Expression Is Genetically Correlated with the Inhibition of Host RNA and Protein Synthesis. J Virol. 2003;77:4646–4657. doi: 10.1128/JVI.77.8.4646-4657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fensterl V, Sen GC. The ISG56/IFIT1 gene family. J Interferon Cytokine Res. 2011;31:71–8. doi: 10.1089/jir.2010.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou X, Michal JJ, Zhang L, Ding B, Lunney JK, Liu B, et al. Interferon induced IFIT family genes in host antiviral defense. Int J Biol Sci. 2013;9:200–8. doi: 10.7150/ijbs.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Terenzi F, Hui DJ, Merrick WC, Sen GC. Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. J Biol Chem. 2006;281:34064–71. doi: 10.1074/jbc.M605771200. [DOI] [PubMed] [Google Scholar]
- 49.Miller DK, Lenard J. Inhibition of vesicular stomatitis virus infection by spike glycoprotein. Evidence for an intracellular, G protein-requiring step. J Cell Biol. 1980;84:430–437. doi: 10.1083/jcb.84.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reich NC. A death-promoting role for ISG54/IFIT2. J Interferon Cytokine Res. 2013;33:199–205. doi: 10.1089/jir.2012.0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stawowczyk M, Van Scoy S, Kumar KP, Reich NC. The interferon stimulated gene 54 promotes apoptosis. J Biol Chem. 2011;286:7257–66. doi: 10.1074/jbc.M110.207068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Terenzi F, Pal S, Sen GC. Induction and mode of action of the viral stress-inducible murine proteins, P56 and P54. Virology. 2005;340:116–24. doi: 10.1016/j.virol.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 53.Fensterl V, Sen GC. Interferon-induced Ifit proteins: Their role in viral pathogenesis. J Virol. 2014;89:2462–2468. doi: 10.1128/JVI.02744-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakaya T, Sato M, Hata N, Asagiri M, Suemori H, Noguchi S, et al. Gene induction pathways mediated by distinct IRFs during viral infection. Biochem Biophys Res Commun. 2001;283:1150–6. doi: 10.1006/bbrc.2001.4913. [DOI] [PubMed] [Google Scholar]
- 55.Bandyopadhyay SK, Leonard GT, Bandyopadhyay T, Stark GR, Sen GC. Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J Biol Chem. 1995;270:19624–19629. doi: 10.1074/jbc.270.33.19624. [DOI] [PubMed] [Google Scholar]
- 56.Elco J, Guenther JM, Williams BRG, Sen GC. Analysis of genes nduced by Sendai Virus infection of mutant cell lines reveals essential roles of interferon regulatory factor 3, NF-κB, and interferon but not Toll-Like Receptor 3. J Virol. 2005;79:3920–3929. doi: 10.1128/JFI.79.73920-3929.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loo Y-M, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–45. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gale M, Katze MG. Molecular Mechanisms of Interferon Resistance Mediated by Viral-Directed Inhibition of PKR, the Interferon-Induced Protein Kinase. Pharmacol Ther. 1998;78:29–46. doi: 10.1016/S0163-7258(97)00165-4. [DOI] [PubMed] [Google Scholar]
- 59.Sen GC, Peters GA. Viral stress-inducible genes. Adv Virus Res. 2007;70:233–263. doi: 10.1016/S0065-3527(07)70006-4. [DOI] [PubMed] [Google Scholar]
- 60.Duca KA, Lam V, Keren I, Endler EE, Letchworth GJ, Novella IS, Yin J. Quantifying viral propagation in vitro: toward a method for characterization of complex phenotypes. Biotechnol Prog. 2001;17:1156–65. doi: 10.1021/bp010115m. [DOI] [PubMed] [Google Scholar]
- 61.Haseltine EL, Lam V, Yin J, Rawlings JB. Image-guided modeling of virus growth and spread. Bull Math Biol. 2008;70:1730–48. doi: 10.1007/s11538-008-9316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Voigt EA, Swick A, Yin J. Rapid induction and persistence of paracrine-induced cellular antiviral states arrest viral infection spread in A549 cells. Virology. 2016;496:59–66. doi: 10.1016/j.virol.2016.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]