

Abstract
Background
Interstitial pneumonia with autoimmune features (IPAF) identifies a recently recognized autoimmune syndrome characterized by interstitial lung disease and autoantibodies positivity, but absence of a specific connective tissue disease diagnosis or alternative etiology. We retrospectively reviewed the clinical presentation, diagnostic workup and management of seven critically ill patients who met diagnostic criteria for IPAF. We compared baseline characteristics and clinical outcome of IPAF patients with those of the population of ARDS patients admitted in the same period.
Results
Seven consecutive patients with IPAF admitted to intensive care unit for acute respiratory distress syndrome (ARDS) were compared with 78 patients with ARDS secondary to a known risk factor and with eight ARDS patients without recognized risk factors. Five IPAF patients (71%) survived and were discharged alive from ICU: Their survival rate was equal to that of patients with a known risk factor (71%), while the subgroup of patients without risk factors had a markedly lower survival (38%). According to the Berlin definition criteria, ARDS was severe in four IPAF patients and moderate in the remaining three. All had multiple organ dysfunction at presentation. The most frequent autoantibody detected was anti-SSA/Ro52. All patients required prolonged mechanical ventilation (median duration 49 days, range 10–88); four received extracorporeal membrane oxygenation and one received low-flow extracorporeal CO2 removal. All patients received immunosuppressive therapy.
Conclusions
This is the first description of a cohort of critical patients meeting the diagnostic criteria for IPAF presenting with ARDS. This diagnosis should be considered in any critically ill patient with interstitial lung disease of unknown origin. While management is challenging and level of support high, survival appears to be good and comparable to that of patients with ARDS associated with a known clinical insult
Electronic supplementary material
The online version of this article (doi:10.1186/s13613-017-0320-3) contains supplementary material, which is available to authorized users.
Keywords: Interstitial pneumonia with autoimmune features, Lung-dominant connective tissue disease, ARDS, ECMO
Background
The term “connective tissue disease” (CTD) refers to a heterogeneous group of disease that targets the connective tissues of the body. The autoimmune CTDs are caused by overactivity of the immune system, resulting in the production of autoantibodies. Their diagnosis is based on the combination of clinical history, physical examination and laboratory and radiologic findings, according to the established diagnostic criteria [1, 2].
Clinical presentation ranges from mild symptoms to life-threatening manifestations. Up to 30% of patients with CTD require intensive care unit (ICU) admission, and the CTD is frequently diagnosed during the ICU stay [3, 4]. The main reason for ICU admission is acute respiratory failure (ARF): type and frequency of lung involvement vary among the different CTDs [5], but a typical presentation is represented by interstitial lung disease (ILD) [6, 7].
The early recognition of the etiology of ILD (CTD, environmental exposures, drugs or idiopathic conditions) is crucial to choose the appropriate therapeutic strategy but can be really challenging. Nonetheless, a significant number of patients with ILD have clinical and/or serologic features suggesting an autoimmune etiology but do not fulfill established diagnostic criteria for a definite CTD: In these cases, different definitions have been proposed like undifferentiated CTD (UCTD) [8, 9], lung-dominant CTD (LD-CTD) [10, 11] and autoimmune-featured ILD (AIF-ILD) [12], but none of these is universally accepted. Recently, a consensus on the term “interstitial pneumonia with autoimmune features” (IPAF) has been reached by a joint European Respiratory Society (ERS)–American Thoracic Society (ATS) task force [13].
We present here for the first time a series of seven patients meeting the diagnostic criteria for IPAF requiring ICU admission for acute respiratory distress syndrome (ARDS) and multiple organ failure. Baseline characteristics and clinical outcome of IPAF patients were compared with those of the population of ARDS patients admitted in the same period.
Methods
Patient population
We retrospectively reviewed the electronic files of patients with a diagnosis of ARDS (according to the Berlin definition criteria) [14] admitted to the tertiary level ICUs of San Gerardo Hospital in Monza and of Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico in Milan (Italy) from May 2012 to October 2016. Based on the presence or absence of a recognized risk factor for ARDS among those listed in the Berlin definition criteria [14], patients were subdivided into two groups: (1) patients with a known risk factor; (2) patients without a known risk factor. The latter group was further subdivided into two subgroups according to the presence of diagnostic criteria for IPAF [13]: a. patients with IPAF (study group); b. patients without both a recognized risk factor and not fulfilling the criteria for IPAF.
The following data at ICU admission were recorded: demographics (sex, age), comorbidities, severity scores (SAPSII and SOFA), risk factors for ARDS, PaO2/FiO2 (P/F) ratio, intrapulmonary shunt (Qs/Qt), ventilator and respiratory mechanics parameters [positive end-expiratory pressure (PEEP), tidal volume (Vt), plateau pressure (P plat), driving pressure (ΔP), respiratory rate (RR), static respiratory system compliance (C rs)].
P plat was measured during an end-inspiratory pause of at least 2 s ; ΔP was defined as P plat − PEEPtot; static C rs was calculated as Vt/ΔP.
The following outcome data were collected: use of prone positioning, need for extracorporeal membrane oxygenation (ECMO) support, duration of invasive mechanical ventilation (IMV) and ECMO (if applicable), ICU length of stay (ICU-LOS) and ICU mortality.
IPAF was diagnosed according to the criteria described by the joint ATS–ERS task force (Table S1) [13].
In all patients with IPAF (study group) and with ARDS without known risk factors, computed tomography (CT) of the chest and bronchoalveolar lavage (BAL) were performed within 2 days from ICU admission, and their findings were reviewed according to the ATS Guidelines [15, 16]. Timing, dose and schedule of administration of steroid therapy and other eventual immunosuppressive treatments were recorded. Episodes of ventilator-associated pneumonia (VAP) occurring in IPAF patients were analyzed. Finally, in patients with IPAF we obtained long-term follow-up data on lung morphology (by CT scan), respiratory function (by spirometry) and immunological status.
Statistical analysis
Data are presented as absolute frequency (% of the included patients) or as median and interquartile range. To evaluate differences between patients’ groups, the Kruskal–Wallis test and Pearson’s Chi-squared test were utilized to compare continuous and nominal variables, respectively. Two-tailed values of p below 0.05 were considered statistically significant. The JMP 11 statistical program (SAS Institute Inc, Cary, NC) was used for statistical analysis.
Results
Data from 93 patients with ARDS were reviewed. As previously described, patient population was subdivided into three groups (Fig. 1): (1) patients with a known risk factor for ARDS (n = 78); (2) patients without risk factors but fulfilling the diagnostic criteria for IPAF (n = 7); (3) patients without risk factors and not meeting the criteria for IPAF (n = 8).
Fig. 1.
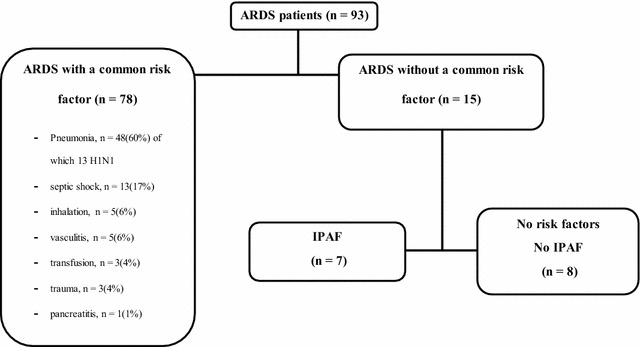
Patient population
Among the 78 patients with ARDS secondary to a known clinical insult, pneumonia was the most frequent risk factor (47 cases, 13 of viral origin), as shown in Fig. 1.
Description of IPAF patients
Seven patients (four males and three females) with ARF due to a newly diagnosed ILD meeting the diagnostic criteria for IPAF were identified. Median age was 61 years (IQR 55–64); median SAPS II and SOFA scores at admission were 32 (32–36) and 11 (11–12.5), respectively. Five patients (71%) survived: Their median ICU stay was 54 (23–78) days and they were all discharged alive from the hospital. Two patients died 72 and 23 days after ICU admission, respectively (Table 1).
Table 1.
Patient characteristics, ventilator and respiratory mechanics parameters (at admission) severity scores (at admission) and clinical outcomes of the seven IPAF patients included in the study
| PT | Age (years) | Sex | PEEP (cmH2O) | P plat (cmH2O) | ΔP (cmH2O) | C rs (mL/cmH2O) | P/F (mmHg) | Qs/Qt | SAPS II score | SOFA score | ICU stay (days) | MV duration (days) | ECMO duration (days) | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PT 1 | 62 | M | 16 | 25 | 9 | 42 | 80 | 51 | 37 | 11 | 84 | 81 | 64 | Alive |
| PT 2 | 45 | M | 12 | 30 | 18 | 22 | 97 | 54 | 37 | 10 | 72 | 72 | 63 | Dead |
| PT 3 | 50 | M | 14 | 26 | 12 | 30 | 141 | 35 | 24 | 11 | 95 | 88 | 53 | Alive |
| PT 4 | 66 | F | 10 | 31 | 21 | 12 | 120 | NA | 36 | 13 | 23 | 21 | 0 | Alive |
| PT 5 | 61 | M | 14 | 29 | 15 | 26 | 163 | NA | 32 | 14 | 10 | 10 | 0 | Alive |
| PT 6 | 74 | F | 12 | 26 | 14 | 27 | 77 | 25 | 32 | 12 | 23 | 23 | 0 (ECCO2R) | Dead |
| PT 7 | 61 | F | 12 | 24 | 14 | 14 | 122 | 26 | 32 | 11 | 54 | 49 | 17 | Alive |
P plat: end-inspiratory plateau pressure; ΔP: driving pressure; C rs: respiratory system compliance; P/F: PaO2/FiO2 ratio; Qs/Qt: intrapulmonary shunt; SAPS Simplified Acute Physiology Score, SOFA Sequential Organ Failure Assessment, ICU intensive care unit, MV mechanical ventilation, ECMO extracorporeal membrane oxygenation; ECCO2R: extracorporeal CO2 removal; NA not available
All patients were admitted to the hospital for acute dyspnoea and hypoxemia. Extrathoracic symptoms were few and nonspecific (fatigue, weight loss, fever), and none had skin lesions or arthritis. No patient had previous history of ILD and/or CTD, and detailed medication and occupational history were negative.
According to the oxygenation criteria established by the Berlin definition [14], three patients had moderate and four had severe acute respiratory distress syndrome (ARDS); however, in none of them a “known clinical insult” [14] could be identified as the cause of ARDS. At admission, median P/F ratio was 120 mmHg (range 80–163) and median respiratory system compliance 26 ml/cmH2O. All patients required intubation and invasive mechanical ventilation (MV). At first day of MV, median positive end-expiratory pressure (PEEP) was 12 cmH2O, plateau airway pressure 26 cmH2O and tidal volume 380 mL (Table 1). The time course of selected parameters during the ICU stay is depicted in Additional file 1: Figure S1. The following rescue therapies were applied: recruitment maneuvers (all patients), inhaled nitric oxide (three patients) and prone positioning (four patients). The median duration of MV was 49 days (21–81) and six subjects (85%) were tracheostomized. Five patients (71%) required an extracorporeal respiratory support with veno-venous extracorporeal membrane oxygenation (ECMO) in four cases and with low-flow extracorporeal CO2 removal (ECCO2R) with a dedicated device (Prolung™, Estor) in one case. Three of these patients survived and the median ECMO duration was 53 days (17–63).
All patients needed vasopressor support during the ICU stay and two presented with shock at admission. One patient required continuous renal replacement therapy.
Bacterial, viral or fungal infections were excluded in all patients by means of a complete microbiological workup, consisting of cultural, serological and molecular biology tests on blood, urine and BAL samples.
Other potentially reversible causes, such as pneumothorax, pulmonary embolism, left heart failure, acute eosinophilic pneumonia and hypersensitivity pneumonia, were ruled out.
Findings of BAL samples and CT scans are detailed in Table 2. Briefly, cytomorphological analysis of BAL showed mixed alveolitis with significant lymphocytosis (>30%) in three cases and significant neutrophilia (>50%) in two other patients, without signs of viral inclusion or diffuse alveolar hemorrhage.
Table 2.
Autoantibodies pattern, BAL and CT scan findings of IPAF patients
| PT | Autoantibodies | BAL | CT findings at presentation | ILD pattern at CT | CT findings at follow-up |
|---|---|---|---|---|---|
| PT 1 | ANA; SSA/Ro52; anti-centromere | Lymphocytic cellular pattern (MA 42%; L 35%; PMN 14%; E 2%) | Extensive GGO with crazy-paving mainly in dependent zones and with subpleural sparing; consolidations in costophrenic sulci; some cystic lesions mainly in lower areas | LIP | Minimal diffuse GGO and subpleural reticulations; rare traction bronchiolectasis; enlarged cystic lesions (20 months) |
| PT 2 | SSA/Ro52 | Neutrophilic cellular pattern (MA 9%; L 0%; PMN 91%; E 0%); Foam cells +++ | Focal subpleural and peribroncovascular consolidations in upper lobes and extensive consolidations of RLL, with air bronchogram | OP | Extensive parenchymal fibrosis with large bilateral pleural effusions (after 2 months of ICU stay) |
| PT3 | SSA/Ro52 | Physiological cellular pattern (MA 93%; L 2%; PMN 5%, E 0%); Foam cells +++ | Subpleural consolidations with air bronchogram, mainly in lower lobes, with perilobular consolidations in RUL | OP | Diffuse reticulations with architectural distortion and subpleural curvilinear lines; limited traction bronchiectasis (17 months) |
| PT 4 | ANA; SSA/Ro52 | Neutrophilic cellular pattern (MA 9%; L 9%; PMN 83%; E 5%) | Limited gravity-dependent consolidations with air bronchogram and GGO; some areas of perilobular consolidations | OP | Gradual development of extensive GGO with reticulations, mainly in lower lobes with traction bronchiectasis (24 months) |
| PT 5 | Anti-Jo1; SSA/Ro52 | Lymphocytic cellular pattern (MA 27%; L 57%; PMN 14%; E 2%) | Diffuse GGO with subpleural sparing and crazy paving; subpleural consolidations mainly dorsal; initial signs of fibrosis with corkscrew-like traction bronchiectasis in RUL consolidated areas | AIP/DAD | Minimal subpleural GGO with reticulations; traction bronchiolectasis in RUL (23 months) |
| PT 6 | ANA; SSA/Ro52 | Lymphocytic cellular pattern (MA 60%; L 30%; PMN 10%; E 0%) | Bilateral consolidations with air bronchogram in lower lobes and dorsal segments of upper lobes. Some GGO in lower lobes and anterior segments of upper lobes. Limited reticulations anteriorly | OP | NA |
| PT 7 | ANA (nucleolar pattern) | Physiological cellular pattern (MA 95%; L 1%; PMN 3%, E 1%) | Bilateral quite extensive GGO in upper and lower lobes, with subpleural and peribronchovascular distribution, associated with minimal reticulations in upper lobes and limited consolidations in lower lobes | OP/NSIP overlap | NA |
ANA antinuclear antibodies, RUL right upper lobe, GGO ground-glass opacities, RLL right lower lobe, MA macrophages, L lymphocytes, PMN polymorphonuclear cells, E eosinophils, LIP lymphocytic interstitial pneumonia, OP organizing pneumonia, AIP acute interstitial pneumonia, DAD diffuse alveolar damage, NA not available
At CT scans, consolidations were present in all cases and mainly found in subpleural caudal areas, often associated with perilobular opacities (Fig. 2a; Additional file 1: Figure S2). Ground-glass opacities, in some cases with subpleural sparing, were also quite common but more diffuse and predominant at follow-up scans (Fig. 2b; Additional file 1: Figure S2). In four patients, mediastinal or hilar lymphadenopathies (with a diameter greater than 10 mm) were observed, while pleural effusion was present in only one case. Of the five patients who survived, two had limited or widespread organizing pneumonia (OP) consolidations; one had typical features of AIP/DAD with ground-glass opacities, patchy consolidations and fibrotic changes; one had extensive ground-glass opacities in lower lobes with some cysts suggestive of lymphocytic interstitial pneumonia (LIP) but with atypical extensive superimposition of “crazy-paving” pattern; the last one had and OP/nonspecific interstitial pneumonia (NSIP) overlap pattern. Both patients who died had an OP pattern at presentation, but in one of them the initial OP pattern rapidly progressed to acute interstitial pneumonia/diffuse alveolar damage (AIP/DAD) pattern and he subsequently developed striking extensive fibrotic parenchymal involvement. Follow-up scans were obtained in the first four surviving patients after a median interval from hospital discharge of 20 months. Patients with OP pattern at presentation developed ground-glass opacities both in new parenchymal territories and in previously consolidated areas. Three patients developed mild to moderate signs of fibrosis (traction bronchiectasis and mild parenchymal distortion), but no patient developed honeycombing. Representative images from baseline and follow-up CT scans of the first five patients are presented in Additional file 1: Figure S2.
Fig. 2.
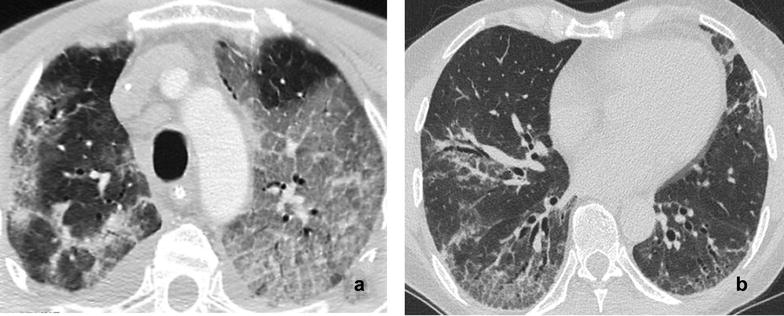
a Contrast-enhanced computed tomography scan of the thorax of Patient 5 at the level of the carina at ICU admission (slice thickness 2 mm). The picture shows bilateral, diffuse ground-glass opacities with partial subpleural sparing and crazy-paving pattern. b Follow-up high-resolution computed tomography scan of the thorax of Patient 4 at the level of the middle lobe (slice thickness 1 mm). The picture shows bilateral, mainly dorsal ground-glass opacities with reticulations and traction bronchiectasis
A complete autoantibody panel was obtained, consisting of antinuclear antibodies (ANA), anti-SSA/Ro52, anti-SSB/LA, anti-ribonucleoprotein, anti-Scl70, anti-Smith, anti-Jo1, anti-centromere, anti-double-stranded DNA, anti-neutrophil cytoplasmic antibody (ANCA), cyclic anti-citrullinated peptide and rheumatoid factor (RF). All patients had anti-SSA/Ro52; one patient had high titer ANA and anti-centromere autoantibodies and one was positive for anti-Jo1. Since no other causes of ILD were identified, extrathoracic symptoms were nonspecific and pulmonary manifestations were by far the most important, all patients met the criteria for IPAF.
All patients received immunosuppressive therapy while in ICU, after a median interval of 10 days from admission. Five of them received high doses of methylprednisolone (1000 mg/day for 3 days and then 1 mg/kg/day), followed by cyclophosphamide (10–15 mg/kg every 2–3 weeks); they also received iv immunoglobulin infusion, and two were treated with cyclosporine for inadequate response to cyclophosphamide. The remaining two patients were treated with methylprednisolone (1 mg/kg/day) without loading dose.
Five patients (71%) developed a ventilator-associated pneumonia, after a median time interval of 19 (10–31) days from admission: Pneumonia was caused by multidrug-resistant Gram-negative bacteria in three cases and by Aspergillus fumigatus in one case.
The median follow-up of the first four surviving patients was 21 months. At the last visit, forced vital capacity range was 77–112% and diffusing lung capacity for carbon monoxide 39–95% of predicted and no patient requires oxygen supplementation. Immunological follow-up examinations showed that anti-SSA/Ro52 became negative in three out of four patients. Maintenance immunosuppressive therapy with low-dose prednisone, azathioprine and mycophenolate is ongoing in three patients.
All IPAF patients, except from the two who died, gave their written informed consent to publication. Due to the retrospective nature of the study and since written consent was obtained from the study patients, ethics committee approval was waived according to local regulations.
Comparison between IPAF patients and control population
Table 3 shows the comparison of baseline characteristics and clinical outcome among the three patient groups. We did not find significant differences in regard to demographics, severity of hypoxemia (P/F ratio and Qs/Qt) and ventilator and respiratory mechanics parameters. IPAF patients tended to have a higher median SOFA score compared to both control groups (11 vs 8 vs 8.5; p = 0.068) and a higher frequency of use of prone positioning (86 vs 53 vs 43%; p = 0.197) and ECMO (71 vs 41 vs 25%; p = 0.176). ICU survival of IPAF patients was exactly equal to that of patients with a known risk factor (71%), while it was markedly higher than that of the patients without risk factors (38%); however, due to the small number of patients, this difference was not statistically significant. Compared to the other subgroups, IPAF patients had significantly longer median ICU-LOS (54 vs 19 vs 15.5 days; p = 0.0045) and duration of IMV (49 vs 17 vs 15.5 days; p = 0.031) and ECMO (53 vs 9.5 vs 28; p = 0.006).
Table 3.
Patient characteristics, severity scores at admission, ventilator and respiratory mechanics parameters at admission and clinical outcome data in the three groups of patients included in the study
| ARDS with known risk factors | No risk factors | IPAF | p | |
|---|---|---|---|---|
| Gender (male) | 54 (69%) | 1 (25%) | 4 (57%) | 0.763 |
| Age (years) | 55 (45–67.25) | 58.5 (38.5–72.25) | 61 (50–66) | 0.726 |
| SAPS II | 41.5 (31–50.5) | 42 (35–59) | 32 (32–37) | 0.133 |
| SOFA | 8 (5.75–12) | 8.5 (8–11) | 11 (11–13) | 0.068 |
| PaO2/FiO2 (mmHg) | 93 (67.75–125.25) | 103 (62.75–150.75) | 120 (80–141) | 0.515 |
| PEEP (mmHg) | 12.5 (10–15) | 10 (7.25–14.75) | 12 (10–14) | 0.303 |
| Compliance (mL/cmH2O) | 29.4 (24–37) | 26.5 (16.25–43.5) | 26 (14–30) | 0.336 |
| Intrapulmonary shunt (%) | 35 (25–46.5) | 33 (14.5–40.5) | 35 (25.5–52.5) | 0.588 |
| Plateau pressure (cmH2O) | 28 (26–30) | 27.5 (22.5–29.75) | 26 (25–30) | 0.796 |
| Driving pressure (cmH2O) | 14 (11–18) | 15.5 (10.25–21.25) | 14 (12–18) | 0.977 |
| ECMO | 32 (41%) | 2 (25%) | 5 (71%) | 0.176 |
| Tracheostomy | 45 (58%) | 3 (43%) | 5 (71%) | 0.557 |
| Pronation | 41 (53%) | 3 (43%) | 6 (86%) | 0.197 |
| Survival | 55 (71%) | 3 (38%) | 5 (71%) | 0.159 |
| ICU length of stay (days) | 19 (8.75–32.25)* | 15.5 (5.5–35.75)* | 54 (23–84) | 0.045 |
| ECMO duration (days) | 9.5 (6–13)* | 28 (8–48) | 53 (17–63) | 0.006 |
| IMV duration (days) | 17 (6–28.5)* | 15.5 (5.5–34.25)* | 49 (21–81) | 0.031 |
Data are presented as absolute frequency (% of the included patients) or as median and interquartile range
IPAF interstitial pneumonia with autoimmune features, SAPS II Simplified Acute Physiology Score, SOFA Sequential Organ Failure Assessment, PEEP positive end-expiratory pressure, ECMO extracorporeal membrane oxygenation, ICU intensive care unit, IMV invasive mechanical ventilation
* p < 0.05 versus IPAF group
In all the eight control patients without risk factors, microbiological and immunological screening resulted negative. Revision of chest CT scans and BAL performed in these patients did not allow the identification of a typical radiologic or cytomorphologic pattern, similarly to what observed IPAF patients. Of note, in all these patients a course of corticosteroid therapy was performed, mainly for “nonresolving ARDS.”
Discussion
In this paper, we reviewed the clinical presentation, diagnostic workup and management of seven critically ill patients with IPAF. Our patients had an extremely severe clinical picture, with ARDS and multiple organ failure; they required prolonged mechanical ventilation and, in three cases, prolonged ECMO support. To the best of our knowledge, this is the first description of a cohort of patients with IPAF requiring ICU admission and invasive ventilation. Compared to the control population of ARDS patients admitted in the same period, ICU survival of patients with IPAF was equal to that of patients with ARDS associated with a known risk factor, while it was markedly higher than that of the subgroup of patients without risk factors (71 vs 38%). Hence, in our case series, IPAF patients had similar baseline characteristics and outcome to patients with ARDS associated with a recognized clinical insult, while patients without a known risk factor had a worse outcome. Of note, despite similar severity of hypoxemia and impairment of respiratory mechanics, IPAF patients had a significantly higher ICU-LOS and duration of IMV and required more frequently prone positioning and ECMO. Our findings confirm the data of Gibelin et al. [17] on a large series of ARDS patients: they observed that patients lacking exposure to common risk factors had a significantly higher mortality (66%) than other ARDS patients and found that the absence of known risk factors was independently associated with mortality (adjusted OR 2.06). Of note, De Prost et al. [18] recently published a secondary analysis on the cohort of ARDS patients without risk factors included in the LUNG SAFE study: ARDS without risk factors accounted for 8.3% of all ARDS cases and in 80% of these patients the etiology of ARDS was not identified.
Identification of the etiology of ILDs and their management remain a clinical challenge. The low incidence of ILD-associated ARF requiring ICU admission is a major obstacle to the assessment of outcome predictors and treatment optimization. Moreover, available studies report a high mortality among patients requiring invasive MV [7, 19, 20], ranging from 47 to 89.7%. For these reasons, ICU physicians tend to be reluctant to admit patients with ILD of uncertain etiology and are even more reluctant to administer immunosuppressive drugs in the absence of a definite diagnosis of an autoimmune disease.
Many authors have underlined the need to categorize patients with prevalent pulmonary involvement in probable autoimmune origin but with insufficient diagnostic criteria for a definite CTD. The rheumatologist would classify these patients as UCTD [8]; however, UCTD usually identifies patients with milder disease, nonspecific clinical features and low incidence of pulmonary involvement [2, 9].
Thus, Vij proposed the definition AIF-ILD [12]: Diagnostic criteria are the presence of ILD associated with at least one symptom/sign and one abnormal serologic test. In Vij’s retrospective review of 200 patients with ILD, 63 were classified as AIF-ILD and had significantly lower survival rates than patients with ILD associated with a definite CTD.
An alternative classification has been presented by Fischer, who proposed the definition of LD-CTD for “cases where ILD has a rheumatologic flavor as supported by specific autoantibodies and yet does not meet criteria for a defined CTD based on the lack of adequate extrathoracic features” [11]. However, ICU physicians are still not familiar with these diagnostic categories: Neither AIF-LD nor LD-CTD was considered in the large survey published by Dumas on 363 critically ill patients with systemic rheumatic diseases [21].
In the attempt to create consensus, an ERS-ATS task force recently proposed the definition of IPAF [13], based on the combination of at least one feature from at least two of three diagnostic domains: clinical (specific extrathoracic manifestations), serologic (specific circulating autoantibodies) and morphologic (suggestive radiologic or histopathologic pattern). The main characteristics of the classification are summarized in Additional file 1: Table S1.
All our patients but one were positive for anti-SSA/Ro52: Ro52 is a protein implicated in the process of ubiquitination and is upregulated at sites of autoimmune inflammation [22]. Several studies have demonstrated that anti-Ro52 is associated with ILD in patients with CTD, particularly in anti-synthetases syndromes [23]. Anti-tRNA synthetases were detected only in one of our patients, who had anti-Jo1 positivity but did not meet the diagnostic criteria for the anti-synthetase syndrome. However, diagnostic assays for anti-tRNA synthetases other than anti-Jo1 were not routinely available in our hospitals before 2015, and this might have reduced our ability to diagnose the disease.
As noted above, the definition of IPAF requires the presence of morphologic features, namely nonspecific interstitial pneumonia (NSIP), organizing pneumonia (OP) or lymphoid interstitial pneumonia (LIP) patterns at HRTC or lung biopsy [13]. Histologic and radiologic presentation in these patients is unspecific and variable, as recently demonstrated by Omote in a series of 44 patients with LD-CTD who underwent open lung biopsy [24]: the major histologic patterns were usual interstitial pneumonia (UIP) followed by NSIP, and UIP pattern was associated with worse prognosis. Similar findings were described in two recent studies on larger cohorts of patients meeting the diagnostic criteria for IPAF: More than half of the patients had a high-confidence diagnosis of UIP pattern on CT that was associated with worse prognosis particularly when associated with honeycombing or pulmonary artery enlargement [25–27]. We did not identify any specific radiologic (CT scan) or cytologic (BAL) findings in IPAF patients; a significant lymphocytosis in BAL, which strongly suggests an autoimmune etiology, was indeed observed only in three patients. Interestingly, none of these patients had a UIP pattern on CT scan, and this may explain the good clinical outcome of our cohort. Interestingly, lack of a typical CT scan and BAL findings was observed also in control patients without common risk factors. However, we acknowledge that the correlation between lung histology and radiologic pattern at CT scan may be quite poor, especially in critically ill patients undergoing mechanical ventilation.
One important limitation of our study is the lack of histologic data, due to the high risk of serious complications when a lung biopsy is performed during mechanical ventilation and ECMO. We acknowledge that lung histology is extremely helpful in patients with ARDS of unknown origin or in cases of nonresolving ARDS, since it can provide important diagnostic and prognostic information and guide patient management [28, 29]. Another limitation resides in the very small number of patients, which limits statistical power of the analysis even in case of large differences among the subgroups; moreover, the limited sample size precluded the possibility of performing a multivariate analysis of risk factors associated with mortality.
To the best of our knowledge, this is the first report of a series of critical patients fulfilling the diagnostic criteria for IPAF requiring ICU admission for ARDS. The number of patients is small, but the patient population is very well characterized and the clinical condition is rare. We think that our patient series demonstrates that the possibility of an autoimmune etiology and in particular the diagnosis of IPAF must be considered in any critically ill patient with “ARDS” according to the Berlin definition criteria but without a known risk factor. Once the diagnosis is established, these patients should receive a “full code” treatment, including ECMO if necessary, especially if the CT scan does not show a UIP pattern with associated signs of fibrosis or pulmonary hypertension. Management of IPAF patients is really challenging, but they can have a very good outcome if the appropriate therapy is instituted: immunosuppressive treatment can lead to a significant improvement in pulmonary manifestations and should be initiated as soon as an infectious cause has been excluded.
However, selecting the appropriate diagnostic and therapeutic strategy is extremely complex and requires a multidisciplinary approach: the intensivist should become part of a team together with the rheumatologist, the pulmonologist, the radiologist and the pathologist.
Conclusions
Interstitial pneumonia with autoimmune features (IPAF) is a rare autoimmune form of interstitial lung disease that can present acutely with ARDS and multiple organ failure, requiring ICU admission and advanced life support measures (included ECMO, if needed). This diagnosis should be considered in any patient presenting with interstitial pneumonia and ARDS lacking exposure to common risk factors for ARDS. In our small cohort of patients, the clinical response to immunosuppressive therapy was good, with a survival rate equal to that of patients with ARDS associated with a known clinical insult. Findings of the present study need to be confirmed prospectively on larger patient series.
Additional file
Additional file 1. Electronic Supplementary Material
Authors’ contributions
GG takes responsibility for the content of the manuscript and contributed substantially to the study design, data analysis and interpretation, preparation, writing and submission of this manuscript. BV, VS, MM and VS contributed to data collection and interpretation, preparation and writing of this manuscript. MRP, AP contributed to study design, data interpretation, review and final approval of the manuscript. GD analyzed all the radiologic studies and contributed to writing, review and submission of the manuscript. AP contributed to study design, review and final approval of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We are indebted to Prof. Giacomo Bellani and Prof. Francesco Blasi for their critical reading of the manuscript and for their helpful comments and suggestions.
Competing interests
Giacomo Grasselli received payment for lectures from Thermo-Fisher and Pfizer Pharmaceuticals and travel–accommodation–congress registration support from Biotest. (All these relationships are unrelated with the present work) No conflict of interest has been declared by co-authors.
Ethics approval, consent to participate and consent for publication
The following statement is included at the end of “Results” section: “All patients, except from the two who died, gave their written informed consent to publication. Due to the retrospective nature of the study and since written consent was obtained from the patients, ethics committee approval was waived according to local regulations.”
Funding
No financial support was received for the study, including funding from NIH or HHMI and departmental or institutional funding
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations
Abbreviations
- AIF-ILD
Autoimmune-featured interstitial lung disease
- ANA
Antinuclear antibodies
- ARDS
Acute respiratory distress syndrome
- ARF
Acute respiratory Failure
- ATS
American Thoracic Society
- BAL
Bronchoalveolar lavage
- CT
Computed tomography
- CTD
Connective tissue disease
- ECMO
Extracorporeal membrane oxygenation
- ERS
European Respiratory Society
- ICU
Intensive care unit
- ILD
Interstitial lung disease
- IPAF
Interstitial pneumonia with autoimmune features
- LD-CTD
Lung-dominant connective tissue disease
- LIP
Lymphocytic interstitial pneumonia
- MV
Mechanical ventilation
- NSIP
Nonspecific interstitial pneumonia
- OP
Organizing pneumonia
- RF
Rheumatoid factor
- UCTD
Undifferentiated connective tissue disease
- UIP
Usual interstitial pneumonia
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13613-017-0320-3) contains supplementary material, which is available to authorized users.
Contributor Information
Giacomo Grasselli, Phone: +39 02 55033285, Email: giacomo.grasselli@unimi.it.
Beatrice Vergnano, Email: beavergnano79@gmail.com.
Maria Rosa Pozzi, Email: gattinielli@gmail.com.
Vittoria Sala, Email: sala.vittoria@gmail.com.
Gabriele D’Andrea, Email: dandrea.gabriele@gmail.com.
Vittorio Scaravilli, Email: vittorio.scaravilli@gmail.com.
Marco Mantero, Email: mantero.marco@gmail.com.
Alberto Pesci, Email: alberto.pesci@unimib.it.
Antonio Pesenti, Email: antonio.pesenti@unimi.it.
References
- 1.Goldblatt F, O’Neill SG. Clinical aspects of autoimmune rheumatic diseases. Lancet. 2013;382:797–808. doi: 10.1016/S0140-6736(13)61499-3. [DOI] [PubMed] [Google Scholar]
- 2.Vij R, Strek ME. Diagnosis and treatment of connective tissue disease-associated interstitial lung disease. Chest. 2013;143:814–824. doi: 10.1378/chest.12-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janssen NM, Karnad DR, Guntupalli KK. Rheumatologic diseases in the intensive care unit: epidemiology, clinical approach, management and outcome. Crit Care Clin. 2002;18:729–748. doi: 10.1016/S0749-0704(02)00025-8. [DOI] [PubMed] [Google Scholar]
- 4.Quintero OL, Rojas-Villarraga A, Mantilla RD, Anaya JM. Autoimmune diseases in the intensive care unit. An update. Autoimmun Rev. 2013;12:380–395. doi: 10.1016/j.autrev.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Cottin V. Idiopathic interstitial pneumonias with connective tissue diseases features: a review. Respirology. 2016;21:245–258. doi: 10.1111/resp.12588. [DOI] [PubMed] [Google Scholar]
- 6.Corte TJ, Copley SJ, Desai SR, Zappala CJ, Hansell DM, Nicholson AG, et al. Significance of connective tissue disease features in idiopathic interstitial pneumonia. Eur Respir J. 2012;39:661–668. doi: 10.1183/09031936.00174910. [DOI] [PubMed] [Google Scholar]
- 7.Zafrani L, Lemiale V, Lapidus N, Lorillon G, Schlemmer B, Azoulay E. Acute respiratory failure in critically ill patients with interstitial lung disease. PLoS ONE. 2014;9(8):e104897. doi: 10.1371/journal.pone.0104897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mosca M, Neri R, Bombardieri S. Undifferentiated connective tissue diseases (UCTD): a review of the literature and a proposal for preliminary classification criteria. Clin Exp Rheumatol. 1999;17:615–620. [PubMed] [Google Scholar]
- 9.Kinder BW, Collard HR, Koth L, Daikh DI, Wolters PJ, Elicker B. Idiopathic nonspecific interstitial pneumonia: lung manifestation of undifferentiated connective tissue disease? Am J Respir Crit Care Med. 2007;176:691–697. doi: 10.1164/rccm.200702-220OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer A, du Bois RM. Interstitial lung disease in connective tissue disorders. Lancet. 2012;380:689–698. doi: 10.1016/S0140-6736(12)61079-4. [DOI] [PubMed] [Google Scholar]
- 11.Fischer A, West SG, Swigris JJ, Brown KK, du Bois RM. Connective tissue disease-associated interstitial lung disease: a call for clarification. Chest. 2010;138:251–256. doi: 10.1378/chest.10-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vij R, Noth I, Strek ME. Autoimmune-featured interstitial lung disease: a distinct entity. Chest. 2011;140:1292–1299. doi: 10.1378/chest.10-2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. ERS/ATS Task Force on Undifferentiated Forms of CTD-ILD: an official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46:976–987. doi: 10.1183/13993003.00150-2015. [DOI] [PubMed] [Google Scholar]
- 14.ARDS Definition Task Force Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 15.Travis WD, Costabel U, Hansell DM, King TE, Jr, Lynch DA, Nicholson AG, et al. ATS/ERS Committee on Idiopathic Interstitial Pneumonias: an official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, Du Bois RM, et al. American Thoracic Society Committee on BAL in Interstitial Lung Disease: an official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185:1004–1014. doi: 10.1164/rccm.201202-0320ST. [DOI] [PubMed] [Google Scholar]
- 17.Gibelin A, Parrot A, Maitre B, Brun-Buisson C, Mekontso Dessap A, Fartoukh M, et al. Acute respiratory distress syndrome mimickers lacking common risk factors of the Berlin definition. Intensive Care Med. 2016;42:164–172. doi: 10.1007/s00134-015-4064-y. [DOI] [PubMed] [Google Scholar]
- 18.De Prost N, Pham T, Carteaux G, Mekontso Dessap A, Brun-Buisson C, Fan E, Bellani G, et al. Etiologies, diagnostic work-up and outcomes of acute respiratory distress syndrome with no common risk factor: a prospective multicenter study. Ann Intensive Care. 2017;7:69. doi: 10.1186/s13613-017-0281-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Perez ER, Yilmaz M, Jenad H, Daniels CE, Ryu JH, Hubmayr RD, et al. Ventilator settings and outcome of respiratory failure in chronic interstitial lung disease. Chest. 2008;133:1113–1119. doi: 10.1378/chest.07-1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gungor G, Tatar D, Salturk C, Cimen P, Karakurt Z, Kirakli C, et al. Why do patients with interstitial lung diseases fail in the ICU? A two-center cohort study. Respir Care. 2013;58:525–531. doi: 10.4187/respcare.01734. [DOI] [PubMed] [Google Scholar]
- 21.Dumas G, Géri G, Montlahuc C, Chemam S, Dangers L, Pichereau C, et al. Outcomes in critically ill patients with systemic rheumatic disease: a multicenter study. Chest. 2015;148(4):927–935. doi: 10.1378/chest.14-3098. [DOI] [PubMed] [Google Scholar]
- 22.Oke V, Wahren-Herlenius M. The immunobiology of Ro52 (TRIM21) in autoimmunity: a critical review. J Autoimmun. 2012;39:77–82. doi: 10.1016/j.jaut.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 23.La Corte R, Lo Monaco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity. 2006;39:249–253. doi: 10.1080/08916930600623791. [DOI] [PubMed] [Google Scholar]
- 24.Omote N, Taniguchi H, Kondoh Y, Watanabe N, Sakamoto K, Kimura T, et al. Lung-dominant connective tissue disease. Clinical, radiologic and histologic features. Chest. 2015;148:1438–1446. doi: 10.1378/chest.14-3174. [DOI] [PubMed] [Google Scholar]
- 25.Oldham JM, Adegunsoye A, Valenzi E, Lee C, Witt L, Chen L, et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J. 2016;47:1767–1775. doi: 10.1183/13993003.01565-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luppi F, Wells AU. Interstitial Pneumonia with Autoimmune Features (IPAF): a work in progress. Eur Respir J. 2016;47:1622–1624. doi: 10.1183/13993003.00690-2016. [DOI] [PubMed] [Google Scholar]
- 27.Chung JH, Montner SM, Adegunsoye A, Lee C, Oldham JM, Husain AN, et al. CT findings, radiologic-pathologic correlation, and imaging predictors of survival for patients with interstitial pneumonia with autoimmune features. Am J Roentgenol. 2017;208:1229–1236. doi: 10.2214/AJR.16.17121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guerin C, Bayle F, Leray V, Debord S, Stoian A, Yonis H, et al. Open lung biopsy in nonresolving ARDS frequently identifies diffuse alveolar damage regardless of the severity stage and may have implications for patient management. Intensive Care Med. 2015;41:222–230. doi: 10.1007/s00134-014-3583-2. [DOI] [PubMed] [Google Scholar]
- 29.Aublanc M, Perinel S, Guerin C. Acute respiratory distress syndrome: the role of lung biopsy. Curr Opin Crit Care. 2017;23:24–29. doi: 10.1097/MCC.0000000000000373. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Electronic Supplementary Material