

Abstract
Neurofibromatosis type-1 (NF1) is a common neurogenetic disorder and an important cause of intellectual disability. Brain-behaviour associations can be examined in vivo using morphometric magnetic resonance imaging (MRI) and diffusion tensor imaging (DTI) to study brain structure. Here, we studied structural and behavioural phenotypes in heterozygous Nf1 mice (Nf1+/−) using T2-weighted imaging MRI and DTI, with a focus on social recognition deficits. We found that Nf1+/− mice have larger volumes than wild-type (WT) mice in regions of interest involved in social cognition, the prefrontal cortex (PFC) and the caudate-putamen (CPu). Higher diffusivity was found across a distributed network of cortical and subcortical brain regions, within and beyond these regions. Significant differences were observed for the social recognition test. Most importantly, significant structure–function correlations were identified concerning social recognition performance and PFC volumes in Nf1+/− mice. Analyses of spatial learning corroborated the previously known deficits in the mutant mice, as corroborated by platform crossings, training quadrant time and average proximity measures. Moreover, linear discriminant analysis of spatial performance identified 2 separate sub-groups in Nf1+/− mice. A significant correlation between quadrant time and CPu volumes was found specifically for the sub-group of Nf1+/− mice with lower spatial learning performance, suggesting additional evidence for reorganization of this region. We found strong evidence that social and spatial cognition deficits can be associated with PFC/CPu structural changes and reorganization in NF1.
Keywords: Brain, magnetic resonance imaging, mice, neurofibromatosis type-1, social cognition, spatial learning
Neurofibromatosis type-1 (NF1) is a relatively frequent neurogenetic disorder with a prevalence of 1/3500 worldwide. Mutations in the NF1 gene lead to loss of function of neurofibromin which is a negative regulator of cellular proliferation and differentiation (Theos & Korf 2006). Its role on synaptic plasticity, learning and memory is also well documented (Costa & Silva 2003; Shilyansky et al. 2010a, b).
Intellectual disabilities occur in up to 50% of NF1 children; they may manifest as cognitive slowing, memory disturbances, difficulties in solving strategic problems, visuospatial impairments, deficits in motor coordination, mental flexibility and affective disorders (Diggs-Andrews & Gutmann 2013; Ribeiro et al. 2012; Silva et al. 1997; Violante et al. 2013; Zöller et al. 1997). Recent evidence suggests that social cognitive deficits are common in NF1 and that this condition qualifies as an autism spectrum disorder (Lehtonen et al. 2013; Loitfelder et al. 2015; Plasschaert et al. 2015; Pride et al. 2013, 2014; Walsh et al. 2013).
As in NF1 subjects, behavioural studies in mouse models of NF1 have identified spatial learning (Shilyansky et al. 2010a,b) and motor coordination (van der Vaart et al. 2011) deficits. Studies with NF1 mouse models showed that increased Ras/MAPK (mitogen-activated protein kinases) signalling result in higher GABA (gamma-aminobutyric acid) release during learning and consequently in deficits in hippocampal long-term potentiation (LTP) that could account for the spatial learning and memory deficits of these mutant mice (Costa et al. 2002; Cui et al. 2008). Interestingly, treatment with lovastatin reversed the molecular, electrophysiological and learning deficits of these mice. Transcranial magnetic stimulation studies in NF1 patients also identified cortical potentiation deficits that seemed to be caused by enhanced inhibition and that could be reversed by treatment with lovastatin (Mainberger et al. 2013).
Shilyansky et al. (2010a,b) identified similar working memory deficits in Nf1+/− mice that resemble the human phenotype. In mice, the working memory deficits seemed to be caused by increased GABA neurotransmission in prefrontal cortex (PFC) and striatum. Human studies revealed working memory deficits, and functional magnetic resonance imaging (fMRI) studies indicated that these deficits were correlated with hypoactivation of PFC circuits, a result consistent with the idea that enhanced inhibition accounts for working memory deficits in NF1.
In parallel with studies that identified social cognition deficits in NF1 (Lehtonen et al. 2013; Plasschaert et al. 2015; Pride et al. 2013, 2014; Walsh et al. 2013), selective social learning deficits have also been identified in mice heterozygous for the Nf1 gene (Nf1+/−), and related to changes in GABA neurotransmission (Molosh et al. 2014). Consistent with previous electrophysiological findings in the hippocampus (Costa et al. 2002; Cui et al. 2008), studies in the amygdala have indeed suggested that social cognition deficits in Nf1+/− mice are caused by enhanced GABA neuro-transmission that lead to deficits in LTP (Molosh et al. 2014). Deletion of the p21 protein-activated kinase (Pak1) gene reversed the amygdala molecular and electrophysiological deficits and rescued the social learning deficits of these mice (Molosh et al. 2014). However, these therapeutic approaches may only reverse functional deficits and not the structural changes induced by abnormal cell proliferation during development.
Other studies revealed that phenotypic changes are not confined to neurons and identified mild astrocytic abnormalities in Nf1+/− mice. Rizvi et al. (1999) observed an increased number of astrocytes in periaqueductal grey (PAG) and nucleus accumbens (Acc) of Nf1+/− mice, as well as increased astrocyte sizes in PAG. Gutmann et al. (2001) showed that astrocytes from Nf1+/− mice have reduced cell attachment, actin cytoskeletal abnormalities as well as increased cell motility.
Structural brain abnormalities have been documented in NF1 patients, although their relation to learning and social cognition deficits remains elusive. Increases in the volume of cortical and subcortical structures have been frequently observed and are summarized in Table 1 (Balestri et al. 2003; Cutting et al. 2000; Duarte et al. 2014; Greenwood et al. 2005; Margariti et al. 2007; Moore et al. 2000; Pride et al. 2014; Steen et al. 2001). Magnetic resonance imaging (MRI) and diffusion tensor imaging (DTI) studies pointed to a generalized change in mean diffusivity (MD) associated with volumetric changes (for a summary see also Table 1) (Filippi et al. 2013; Karlsgodt et al. 2012; Santos 2011; Wignall et al. 2010; Zamboni et al. 2007).
Table 1. Summary of human MRI/DTI studies in NF1 (ADC, apparent diffusion coefficient; λ1, longitudinal diffusivity; λ2 and λ3, transverse diffusivity).
| Authors | WM volume | GM volume | Brain volume | ADC | MD | FA | RD | λ2, λ3 | λ1 |
|---|---|---|---|---|---|---|---|---|---|
| Cutting et al. (2000) | H | ||||||||
| Moore et al. (2000) | H* | H | H | ||||||
| Steen et al. (2001) | H | ND | H | ||||||
| Cutting et al. (2002) | H | H/L | H | ||||||
| Greenwood et al. (2005) | H | H | H | ||||||
| Margariti et al. (2007) | H | H | |||||||
| Zamboni et al. (2007) | H | L | |||||||
| Wignall et al. (2010) | H* | H | L* | H* | ND* | ||||
| Santos (2011) | H* | L* | H* | ND* | |||||
| Filippi et al. (2013) | ND | H | L† | H† | ND† | ||||
| Karlsgodt et al. (2012) | H | H | H | L | H | H | |||
| Duarte et al. (2014) | H | H/L | |||||||
| Pride et al. (2014) | ND | H/L | H |
H, higher values in NF1 patients compared to control subjects; L, lower values in NF1 patients compared to control subjects; H/L, H and L were obtained for different brain regions; ND, no differences between NF1 patients and control subjects.
Observations restricted to visual-callosal WM pathways.
Observations restricted to the corpus callosum.
It has been difficult to relate these structural findings with the cognitive impairments observed in NF1. Accordingly, the relation of grey matter (GM) changes with attention or social-cognitive deficits remains controversial (Cutting et al. 2002; Greenwood et al. 2005; Moore et al. 2000; Pride et al. 2014).
Although some studies of Nf1 mice models have been conducted using MRI, they were focused just on high-intensity lesions in T2-weighted images (Robinson et al. 2010; Rosenbaum et al. 1999). Brain studies comparing MRI findings in Nf1 mouse models and NF1 subjects would be invaluable in validating these models. Additionally, structural deficits might emerge as key biomarkers for clinical investigation, and these deficits can also be studied in mouse models. In this study, we addressed brain morphometry in Nf1+/− mice, in key regions involved in social cognition, using T2-MRI as well as DTI, as well as the relation between the structural phenotypes of these mice and social cognition deficits.
Materials and methods
Mice models
Nf1+/− mice were F1 hybrids from a cross between C57BL/6NTac and 129T2/SvEmsJ. All experiments used littermates as controls and were done with the experimenter blinded to genotype. A total number of 13 wild-type (WT, 4 males and 9 females) and 15 Nf1+/− (8 males and 7 females) mice were randomly chosen for behavioural assessment and MRI/DTI studies (due to technical reasons, 1 Nf1+/− mouse and 4 WT mice were excluded from the structural studies). Animals were group housed (2–4) on a 12h light/dark cycle in vivarium at University of California Los Angeles (UCLA), CAU and CNC.IBILI. All included animals were healthy (discomfort score 0). Experimenters were blind for genotype. All mice completed successfully the behavioural component of this study at UCLA, USA, and were then transported to the University of Coimbra, Portugal, for structural studies.
All efforts were made to reduce animal stress along the experimental stages and transportation, and both groups were submitted exactly to the same procedure to minimize group effect. The phenotype for the animal model is stable up to 10 months of age, and all the measurements were comprised between this period: behavioura tests between 3 and 5 months of age and MRI/DTI acquisitions between 6 and 8 months of age.
All experimental protocols at the UCLA were approved by the Chancellor's Animal Research Committee of the University of California, Los Angeles, in accordance with National Institutes of Health (NIH) guidelines. At the University of Coimbra, animal experiments were conducted according to the European Council Directives on Anima Care (2010/63/UE) and to the National Authorities
MRI acquisition
In vivo image acquisitions were conducted with a 9.4T magnetic resonance small animal scanner (BioSpec 94/20, Bruker Corporation, Ettlingen, Germany), at the Institute for Nuclear Sciences Applied to Health (ICNAS), University of Coimbra. Animals were anesthetised with isoflurane (delivered through the system E-Z SA800, Euthanex, Palmer, PA, USA), with constant temperature monitoring (Haake SC 100, Thermo Scientific, Waltham, Massachusetts, USA) and assessment of cardiorespiratory function (1030, SA Instruments Inc., Stony Brook, NY, USA).
For volumetric analyses, T2-weighted images were acquired in coronal planes using a RARE sequence: repetition time (TR) = 3800 ms; echo time (TE) = 33 ms; 10 averages; pixel size of 0.0781 mm × 0.0781 mm and slice thickness of 0.5 mm without spacing between slices (total head 256 pixels × 256 pixels × 34 slices)
For diffusivity analyses, DTI images were acquired using an EPI (echo-planar imaging) sequence with 85 gradient directions; b-value = 1000 second/mm2; TR = 3000 milliseconds; TE = 18.5121 milliseconds; pixel size = 0.1563 mm × 0.1563 mm and slice thickness = 0.5 mm, without spacing between slices (total head 128 pixels × 128 pixels × 21 slices).
Volumetric analysis
For the analyses of brain structure volumes, images were pre-processed and segmented in Matlab R2012b, version number 8, including the following steps. (1) Correction of magnetic field inhomogeneity generated from the surface coil: the corrections were implemented using intensity curves from T2-weighted images obtained for an homogeneous phantom, and acquired with the same coil and system configuration. (2) Images were co-reregistered to the same space of a reference volume using rigid transformation (this reference was created from the mean of seven co-registered brain volumes, apart from this study). (3) The heads were skull-stripped using a binary mask. (4) Brain intensities were normalized to match the range of a custom made template. (5) Images were segmented (labelled) into 27 structures following the Markov Random Field approach (Fischl et al. 2002), and using a custom made template. (6) After automatic segmentation, manual corrections were performed in the structures of interest (i.e. PFC, caudate putamen -CPu- and the hippocampus) to further minimize potential errors resulting from the procedure inaccuracies. We implemented this procedure in Matlab, where the T2-weighted image and the corresponding segmentation are superimposed, and after visual inspection, the operator redefines the misaligned borders of the structures of interest using a cursor (7) The volume values were computed by multiplying the number of voxels labelled as belonging to each structure of interest, by the voxel size. An example of the images resulting from the T2-weighted sequence and the resulting segmentation is presented in Fig. 1. (8) Independent samples t-tests were implemented for each structure, to depict the existence of significant differences between volumes of WT and Nf1+/− mice (significance level = 5%). (9) The testing of a gender effect in the measured volumes was performed using analysis of variance (anova).
Figure 1.
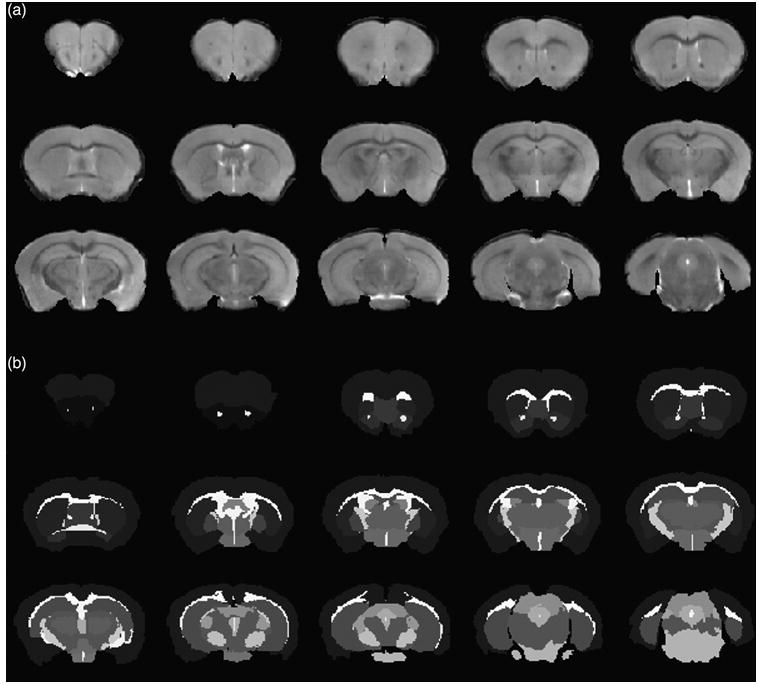
(a) Anatomical (T2-weighted) images and (b) the resulting segmentation, from a subset of slices including PFC, CPu and the hippocampus.
DTI analysis
We analysed fractional anisotropy (FA), radial diffusivity (RD) and the λ1 diffusion tensor eigenvalue, involving the following steps. (1) Eddy current correction was applied using the FDT diffusion toolbox of the FMRIB's Software Library (FSL). (2) Using the same software and toolbox, parametric maps were constructed, including images with inactive diffusion gradient (b0), MD, FA, λ1, λ2 and λ3 maps (see Figure S1, Supporting Information, for representative data). (3) Using the b0 maps, images from each mouse were co-registered to the same space. First, b0 images from WT mice were co-registered with the b0 map of one mouse; next, we computed the mean from b0 maps of all WT mice; finally, the b0 maps from all WT and Nf1+/− mice were co-registered to the mean-b0 map, and the resultant transformation matrix was applied to the remaining parametric maps. In this step, an affine transformation with nine parameters was applied, using Matlab R2012b. (4) The RD maps for each mouse were constructed as (λ2+ λ3)/2. (5) The maps were smoothed using the Statistical Parametric Mapping, version number 8 (SPM8) software, with an isotropic Gaussian kernel, FWHM (full width to half maximum) [mm] =[0.5 0.5 0.5]. (6) Statistical analyses for comparison between WT and Nf1+/− characteristics were first performed through independent samples Student's t-tests in a voxel-by-voxel wise.
The significance level was set at P = 5% followed by false discovery rate (FDR) correction for multiple comparisons (Q = 5%). (7) Also, the averages of diffusivity parameters were computed over the structures of interest using binary masks, and analysed by independent samples Student's t-tests (significance level = 5%).
Behavioural assessment
Morris water maze
In the hidden-platform version of the Morris water maze, mice were trained with two trials per day for five consecutive days [inter-trial interval (ITI) = 1 min]. This training regimen is ideal to not over train the mice, and better detect a difference between Nf1+/− and WT phenotypes. In each training trial, mice were released from a different starting position, and then were allowed to search for the escape platform for 60 seconds. The platform was submerged 1 cm under the surface of the water. Once a mouse found the platform, it was left there for 15 seconds. If a mouse did not find the platform within 60 seconds, it was guided to the platform and allowed to remain on the platform for 15 seconds before being removed from the pool. Memory was assessed in probe trials that were given after completion of training. During the probe trials, the platform was removed and the mice were allowed to search for it for 60 seconds. Data were acquired and analysed using WaterMaze software (Actimetrics, Wilmette, Illinois, USA).
Social recognition
The social recognition task included training and test sessions. Before training, mice were handled 2 min per day for 3 days, and then habituated to an experimental box for 10 min for another 2 days. During the 10 min training session, mice were allowed to explore the box for 5 min. Then they were exposed and interacted with an ovariectomized (OV) female mouse (3–6 month old, 129/SvJ – #000691 – purchased from Jackson Lab) that was placed under a wired cylinder for another 5 min. During the test session (24 h after training), mice were placed back into the experimental box and allowed to interact with two OV females one familiar and one novel.
Analyses of the behavioural data
For social recognition performance, we compared the time spent with a novel vs. a familiar mouse, for WT and Nf1+/− mice, (paired sample Student's t-tests, significance level of 5%). We also computed a two-way ANOVA to test for an interaction between genotype and social stimulus type. For the water maze probe tests, three parameters were analysed: the time spent in the training quadrant, the number of platform crossings and the proximity to the training platform. For quadrant time, we compared the time the mice spent in the training quadrant vs. the average time they spent in the other three quadrants. For platform crossings, we compared the number of crossings of the site where the training platform was during training vs. the average number of crossings in comparable sites in the other three quadrants. These analyses were implemented through two-way ANOVA tests. We also evaluated the average proximity to the training platform, with independent samples Student's t-test.
Correlation between volumetric and behavioural data
We assessed the correlation between brain structure volumes and social recognition performance (fraction of time spent with a novel mice), at a significance level of 5%, followed by FDR correction.
Spatial performance revealed a clustering pattern consistent with variable phenotypic penetrance: a cluster of Nf1+/− mice showing performances similar to WT mice (higher performance cluster), while another cluster of Nf1+/− mice showed considerably reduced performance levels, which were linearly separable (see below) from the higher performance cluster. This justified the calculation of correlations separately for each cluster.
To get an overall measure of spatial learning performance, we first combined the measurements from the spatial learning task into a unique combined index (Indexc, eqn 1).
| (1) |
The discriminant index for quadrant time (IndexQT, eqn 2) and the discriminant index for platform crossings (IndexPC, eqn 3) are shown below:
| (2) |
| (3) |
The normalised proximity (ProximityN, eqn 4) was computed as:
| (4) |
where Distancei is the mean distance to the platform for each mouse, Distancemax and Distancemin are the maximum and minimum average distances overall for the Nf1+/− group. Thus, the combined index will have values closer to zero for animals with lower performance, and values closer to one for animals with higher performance.
The combined index was then used to divide the Nf1+/− mice into two groups matched in size (high and low performance mice), and then we submitted it to 3D linear discriminant analysis (LDA) for testing accuracy and precision of this division (Fig. 2). The presence of maximum accuracy and precision encouraged us to proceed with the correlation analysis for each cluster separately.
Figure 2. Result from LDA (with maximum accuracy and precision) on dividing Nf1+/− mice in two groups based on water maze test performance.
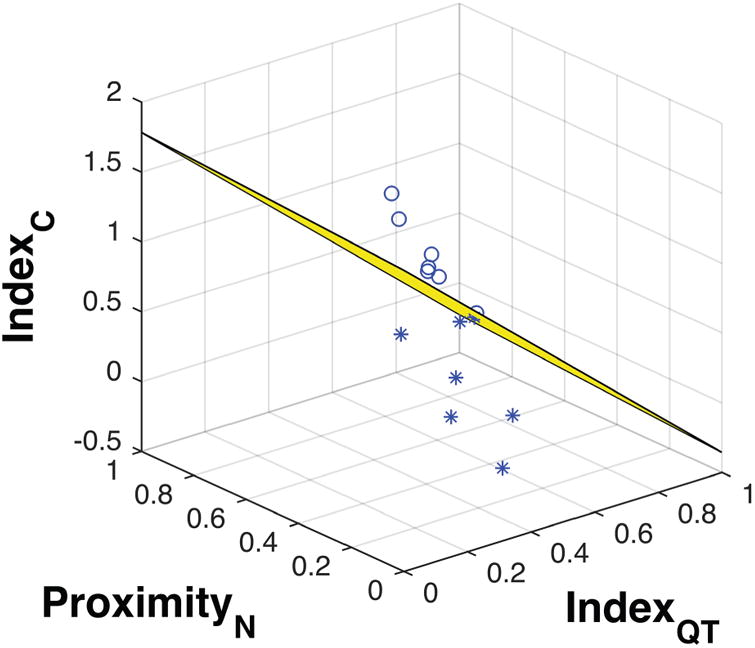
Spherical and star markers represent the high and low performance groups, respectively.
Results
Volumetric analysis
Significant group differences were found in the PFC, CPu and hippocampus, and volumes were higher for Nf1+/− mice in all these regions (Fig. 3). No volumetric correlation with animal sex (PFC: P = 0.5359, F = 0.4; CPu: P = 0.3028, F = 1.12; hip -pocampus: P =0.1286, F =2.52) and no sex–genotype interactions (PFC: P =0.1863, F = 1.88; CPu: P =0.9829, F =0; hippocampus: P =0.1678, F =2.06) were found, when analysed by ANOVA.
Figure 3. Volume measurements for WT and Nf1+/− mice on: PFC (P = 0.0268, t = −2.42, df = 17), hippocampus (P <0.0001, t = −5.77, df = 21), CPu (P = 0.0007, t = −4.16, df = 16) and accumbens (P = 0.8139, t = −0.24, df = 14).
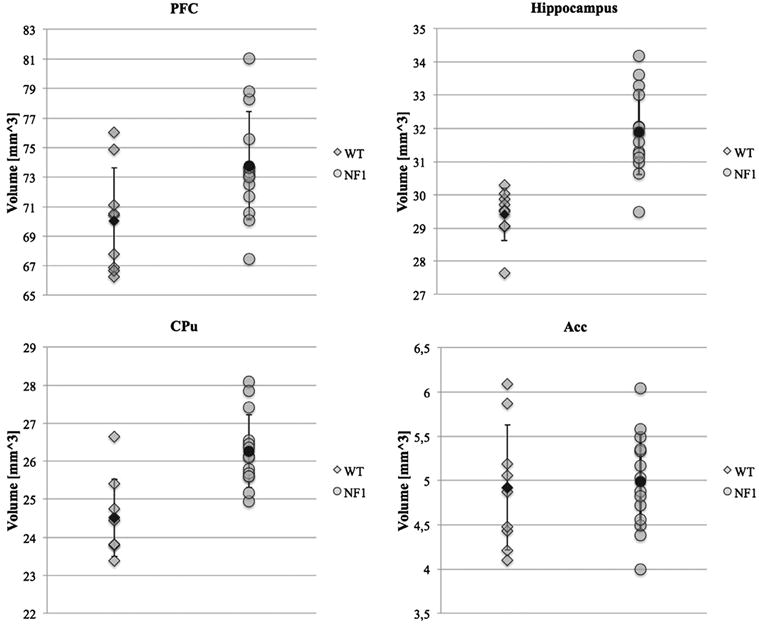
Statistical results were obtained from independent samples t-test. Mean values (black shapes) and standard deviation bars are represented.
DTI analysis
From the voxel-based analysis, and using a significance level of 5% followed by FDR correction, we found significant group differences concerning MD maps. MD was higher in Nf1+/− mice in cortical structures (including the PFC), septum, striatum, globus pallidus, hippocampus, thalamus, PAG and midbrain (Fig. 4). With a voxel-based analysis, no significant differences were found for FA maps.
Figure 4. Regions with significant statistical differences (green) concerning MD maps distributed along the cortex, striatum, globus pallidus, thalamus, hippocampus, midbrain and PAG.
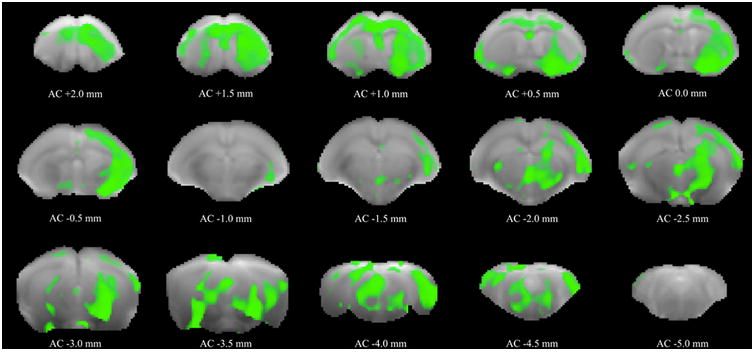
The results were obtained from independent samples t-tests with 5% of significance level, followed by FDR correction (Q = 5%). In the figure, statistical data are superimposed to b0 images, and the plane positions are indicated with reference to the anterior commissure (AC).
When assessing the average of diffusivity parameters over the main structures of interest, significant differences were seen for all of them, as presented in Table 2. For all the structures of interest and for all the parameters evaluated, higher diffusivity values were observed in Nf1+/− mice
Table 2. Average diffusivity parameters over PFC, CPu and hippocampus, and the respective statistical analysis.
| MD | FA | λ1 | RD | |||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||
| WT | Nf1 | WT | Nf1 | WT | Nf1 | WT | Nf1 | |
| PFC | 6.59e-4 ± 1.85e-5 | 7.05e-4 ± 2.97e-5 | 0.0919 ± 0.0196 | 0.1244 ± 0.0449 | 7.2e-4 ± 2.62e-5 | 7.97e-4 ± 5.02e-5 | 6.28e-4 ± 1.73e-5 | 6.59e-4 ± 2.58e-5 |
| P values | 1.53e-4 | 0.0096 | 1.12e-4 | 0.0023 | ||||
| t | −4.61 | −2.85 | −4.77 | −3,47 | ||||
| df | 21 | 21 | 20 | 21 | ||||
| CPu | 6.09e-4 ± 1.71e-5 | 6.55e-4 ± 3.44e-5 | 0.0903 ± 0.0281 | 0.128 ± 0.0382 | 6.65e-4 ± 3.08e-5 | 7.44e-4 ± 5.66e-5 | 5.81e-4 ± 1.37e-5 | 6.11e-4 ± 2.84e-5 |
| P values | 3.77e-4 | 0.013 | 3.28e-4 | 0.003 | ||||
| t | −4.26 | −2.72 | −4.3 | −3.37 | ||||
| df | 20 | 20 | 21 | 20 | ||||
| Hippocamp | us 6.61e-4 ± 2.31e-5 | 7.04e-4 ± 3.37e-5 | 0.0928 ± 0.0269 | 0.123 ± 0.0327 | 7.24e-4 ± 4.27e-5 | 7.93e-4 ± 5.07e-5 | 6.3e-4 ± 1.55e-5 | 6.6e-4 ± 2.98e-5 |
| P values | 0.0014 | 0.0259 | 0.0022 | 0.0042 | ||||
| t | −3.67 | −2.41 | −3.53 | −3.22 | ||||
| df | 21 | 20 | 19 | 20 | ||||
Moreover, comparisons between right and left hemispheres revealed significantly different MD values in the structures of interest, as shown in the Table 3. No differences were observed for FA maps
Table 3. Comparison of MD values between right and left hemispheres in the PFC, CPu and hippocampus.
| PFC | CPu | Hippocampus | ||
|---|---|---|---|---|
| WT | P values | 0.4825 | 0.1592 | 0.0082 |
| t | −0.74 | 1.55 | 3.49 | |
| df | 8 | 8 | 8 | |
| Nf1+/- | P values | 0.015 7 | 0.0083 | 0.00005 |
| t | −2.78 | 3.11 | 5.95 | |
| df | 13 | 13 | 13 |
Behavioural assessment
We replicated previous findings (Costa et al. 2002; Cui et al. 2008; Shilyansky et al. 2010a, b) of impaired spatial learning performance in Nf1+/− mice. As expected, WT mice but not Nf1+/− mice crossed the site where the platform was during training more often than comparable sites in the other three quadrants (Fig. 5a). Unlike Platform crossings, which require that mice cross the exact site where the platform was during training, quadrant time simply measures the time the mice spent in the quadrant where the platform was during training With this less spatially stringent measure, we found evidence that some Nf1+/− mice acquired some spatial information Although the fraction of time spent in quadrant did not show a genotype × place interaction, the average time the WT mice spent in the training quadrant compared to the other three quadrants is significantly higher, while analysis of the performance of Nf1+/− mice showed similar searching time in the target quadrant vs. other quadrants (Fig. 5b).
Figure 5. Results from the water maze and social recognition tests (mean values and standard deviation).
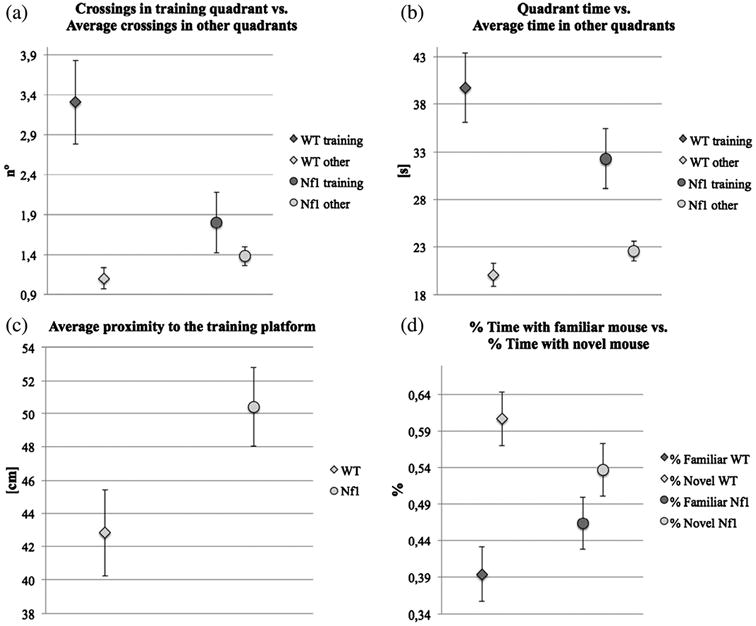
(a) Platform crossings vs. average crossings in other quadrants, two-way ANOVA, P = 0.0156, F1,26 = 6.693; multiple comparisons: WT P = 0.0004 and Nf1+/− P = 0.6117. (b) Time in training quadrant vs. average time in other quadrants, two-way ANOVA, P = 0.1317, F1,26 = 2.422; multiple comparison: WT P = 0.0006 and Nf1+/− P = 0.0683. (c) Average proximity, for WT vs. Nf1+/− mice (P = 0.0448, t = −2.15, df = 19). (d) Fractions of time with familiar mouse vs. fraction of time with novel mouse, for WT (P = 0.0156, t = −2.85, df = 11) and Nf1+/− (P = 0.3328, t = −1.01, d f = 13) mice; two-way ANOVA, interaction term P = 0.06, F = 3.57.
However, as previously reported, their performance is not uniform (Silva et al. 1997) and LDA of spatial performance identified at least two clusters (see below). Finally, average proximity was significantly different between the WT and the Nf1+/− mice used for MRI analysis (Fig. 5c).
In the platform crossing measure during probe trials, Nf1+/− mutants crossed the training platform site significantly less frequently than WT mice (this is a more sensitive measure than latency, see Figures S2 and S3; two-way anova, P = 0.0156, F126 = 6.693; multiple comparison: WT, n=13, P = 0.0004; Nf1+/−, n = 15, P = 0.6117). In % time spend in quadrant, WT mice showed significantly higher searching time in target quadrant than other quadrants; Nf1 +/− mutants showed similar searching time in target quadrant vs. other quadrants (Figure S4; two-way anova, P = 0.1317 F 1,26 = 2.422; multiple comparison: WT, n=13, P = 0.0006; Nf1+/−, n = 15, P = 0.0683).
Previous studies had shown that Nf1+/− mice display mild deficits in water maze task in comparison to WT mice. In this experiment, we used three common measures to quantify learning. Two of the measures, platform crossing and proximity, showed significant genotype-related interaction between WT and NF1+/−. The other measure, % of time spent in quadrant, did not show genotypes × place interaction. Nevertheless, a planned comparison showed that WT mice exhibit significant learning (more time in platform quadrant than other quadrants) while NF1+/− mice do not, as previous studies showed. All together, these results demonstrate that that NF1+/− display learning deficits in water maze task.
From the test for social cognition assessment, we found that unlike WT mice, which spent significantly more time with the novel mouse, Nf1+/− mice did not discriminate between novel and familiar mice (Fig. 5d), a result that confirms their social cognition deficits (Molosh et al. 2014). A two-way ANOVA also suggested an interaction between genotype and type of social stimulus (F = 3.57, P = 0.06) although at a marginal significance level.
Correlation between volumetric and behavioural data
We found a significant correlation between PFC volumes in the Nf1+/− group and social discrimination performance as measured by the fraction of time spent with novel mice (P = 0.0206; r = −0.601; Fig. 6) and this correlation survived to FDR corrections. In contrast, no such correlation was found for the WT mice (P = 0.853, r = −0.0787; Fig. 6). Although the nature of this analysis is not causal, it agrees with the notion that the PFC has a critical role in social functioning (Adolphs 2009).
Figure 6.
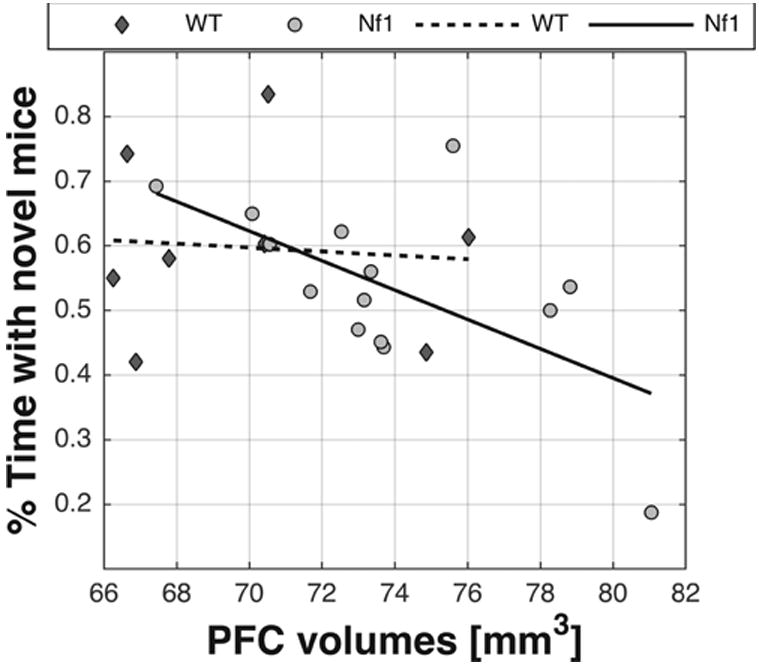
Linear correlation between PFC volumes and % time with novel mice: WT (diamond markers; r = −0.0787; P = 0.853) and Nf1+/− mice (round markers; r = −0.601; P = 0.0206).
Analyses of spatial learning in the Nf1+/− group with lower performance uncovered a significant correlation between the IndexQT and CPu volumes (P = 0.0447; r = 0.7658). We did not found significant correlation between parameters related to spatial learning and the other investigated structures (i.e. PFC and hippocampus). Despite the correlational nature of observations, they are in line with the notion of a dampened contribution to learning.
Discussion
In this study, featuring a structure–function correlation approach, we found indications for an important role of the PFC in the social cognition deficits in the Nf1+/− mouse model. The PFC and CPu were chosen as regions of interest for analyses because previous human studies had already shown impairments in these structures (Cutting et al. 2002; Duarte et al. 2014; Greenwood et al. 2005; Karlsgodt et al. 2012; Loitfelder et al. 2015; Shilyansky et al. 2010a, b; Steen et al. 2001; Violante et al. 2013). Despite their correlational (non-causal) nature, these results are interesting because they point to an important role of high-level structures such as the PFC in social cognition, in addition to the amygdala (Molosh et al. 2014).
Our studies also suggest that compensatory mechanisms in the CPu may be associated with spatial learning impairments in underperforming Nf1+/− mice. Although not the main focus of this article, these results are relevant because of the known interaction, often of competitive nature, between the hippocampus and basal ganglia structures during learning (Poldrack & Rodriguez 2004).
The volumetric analyses developed in this study, revealed higher volumes in key brain regions of interest in Nf1+/− mice, including the PFC, CPu and hippocampus. These results are largely consistent with structural studies in NF1 patients, where increases in white matter (WM) and GM volumes were frequently observed (Duarte et al. 2014; Karlsgodt et al. 2012; Margariti et al. 2007; Pride et al. 2014), as opposed to GM reductions in a few other regions (Cutting et al. 2002; Duarte et al. 2014; Pride et al. 2014; see also, Table 1).
Concerning brain diffusivity we found a widespread, beyond our regions of interest, cortical and subcortical increase in MD, as shown from MD maps, which is also consistent with studies in NF1 patients (Filippi et al. 2013; Santos 2011; Table 1). Importantly, the average diffusivities over the structures of interest also showed increases on MD and FA, due to increases in both RD and λ1. No major axonal disorganization or disruption seems to be present in Nf1+/− mice brains, and the MD and FA differences could be a consequence of increased spacing between axons. This notion is also consistent with a study in NF1 patients by Violante et al. (2013). Moreover, inter-hemispheric differences in MD were also observed, which a pattern often found in neurodevelopmental disorders due to their slightly distinct developmental trajectory (Table 3).
The PFC, CPu and hippocampus showed differences in both volumetric and DTI analyses. These brain structures are known to be involved in a wide range of cognitive functions. Concerning the hippocampus, a clear relationship has been previously established with impairments in spatial learning and working memory which have been related to abnormalities in gabaergic mediated inhibition identified in the hippocampus (Costa et al. 2002; Cui et al. 2008). These seem to also be present in the PFC and CPu (Shilyansky et al. 2010a,b).
Social discrimination deficits had been previously recognized in Nf1+/− mice (Molosh et al. 2014). Compared to WT mice, the Nf1+/− mice showed reduced preference for novel conspecifics during a social memory test given 24 h after the initial social interaction. These findings are consistent with evidence in human behavioural and structural studies indicating that NF1 patients have social cognition impairments consistent with the autistic spectrum (Lehtonen et al. 2013; Plasschaert et al. 2015; Pride et al. 2013, 2014; Walsh et al. 2013). Importantly, a significant linear correlation was observed between PFC volumes and social discrimination ability in the Nf1+/− group, as measured by the fraction of time spent with novel mice. This result does not allow for causal inference but supports the hypothesis that in addition to the amygdala (Molosh et al. 2014), alterations in PFC in Nf1+/− mice (and also in NF1 patients) contribute to the social deficits of these mice. This finding is consistent with a recent human study in NF1 patients, suggesting an important role for the frontal cortex in social cognition performance (Loitfelder et al. 2015).
Interestingly, a significant correlation was observed between the IndexQT and CPu volumes in underperforming Nf1+/− mice. CPu volumes were also found to be increased in NF1 patients (Violante et al. 2013). This finding is consistent with that of striatal networks being largely prone to functional reorganization (Poldrack & Rodriguez 2004).
The findings reported here suggest an avenue for future research. For example, some of the authors have previously observed that lovastatin reverses the behavioural phenotype of Nf1+/− mice (Li et al. 2005). It remains an outstanding question whether structural and neurochemical impairments could be reversed. Nevertheless, the former might only be partially reversible, given the nature of neural proliferation during development.
To conclude, in this structure–function phenotyping study, we found evidence that suggests a role for the PFC and the CPu in the social cognition deficits found in the Nf1+/− mouse model. Importantly, changed PFC volumes were related to social deficits observed in Nf1+/− mice. The structural and functional findings reported here may be relevant for longitudinal studies of cognitive function in NF1, and they may also instruct current and future efforts to find a treatment for cognitive and social deficits in this population.
Supplementary Material
Figure S1: Example of MD and FA maps.
Figure S2: Latency to the platform in Nf1+/− and WT mice in the hidden-platform version of the water maze, two-way ANOVA, P = 0.39, F252 = 0.959.
Figure S3: Number of platform crossings during a 60s probe trial given after 3 days of training, two-way anova, P = 0.0156, F1 26 = 6.693; multiple comparisons: (WT, n = 13, P = 0.0004; Nf1+/−, n = 15, P = 0.6117).
Figure S4: Percentage time spent in quadrant during a 60s probe trial given after 3 days of training, two-way anova, P = 0.1317 F1 26 = 2.422; multiple comparisons: (WT, n = 13, P = 0.0006; Nf1+/−, n = 15, P = 0.0683)
Table S1: Results from anova test on evaluating sex × genotype interaction.
Acknowledgments
This work was supported by MH084315 to A.J.S. and CENTRO-07-ST24-FEDER-00205, PTDC/SAU-ORG/118380/2010, Grant FCT UID/NEU/04539/2013-2020, COMPETE, POCI-01-0145-FEDER-007440 to M.C.B. and FCT fellowship SFRH/BPD/ 112863/2015 to L.I.P.
Footnotes
No conflicts of interests are to be reported by any of the authors.
Supporting Information: Additional supporting information may be found in the online version of this article at the publisher's web-site
References
- Adolphs R. The social brain: neural basis of social knowledge. Annu Rev Psychol. 2009;60:693–716. doi: 10.1146/annurev.psych.60.110707.163514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestri P, Vivarelli R, Grosso S, Santori L, Farnetani MA, Galluzzi P, Vatti GP, Calabrese F, Morgese G. Malformations of cortical development in neurofibromatosis type 1. Neurology. 2003;61:1799–1801. doi: 10.1212/01.wnl.0000099080.90726.ba. [DOI] [PubMed] [Google Scholar]
- Costa RM, Silva AJ. Mouse models of neuro bromatosis type I: bridging the GAP. Trends Mol Med. 2003;9:19–23. doi: 10.1016/s1471-4914(02)00008-4. [DOI] [PubMed] [Google Scholar]
- Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Kucherlapati R, Jacks T, Silva AJ. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415:526–530. doi: 10.1038/nature711. [DOI] [PubMed] [Google Scholar]
- Cui Y, Costa RM, Murphy GG, Elgersma Y, Zhu Y, Gutmann DH, Parada LF, Mody I, Silva AJ. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell. 2008;135:549–560. doi: 10.1016/j.cell.2008.09.060.Neurofibromin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutting LE, Koth CW, Burnette CP, Abrams MT, Kaufmann WE, Bridge Denckla M. Relationship of cognitive functioning, whole brain volumes, and T2-weighted hyperintensities in neurofibromatosis-1. J Child Neurol. 2000;15:157–160. doi: 10.1177/088307380001500303. [DOI] [PubMed] [Google Scholar]
- Cutting LE, Cooper KL, Koth CW, Mostofsky SH, Kates WR, Denckla MB, Kaufmann WE. Megalencephaly in NF1: predominantly white matter contribution and mitigation by ADHD. Neurology. 2002;59:1388–1394. doi: 10.1212/01.WNL.0000032370.68306.8A. [DOI] [PubMed] [Google Scholar]
- Diggs-Andrews KA, Gutmann DH. Modeling cognitive dysfunction in neurofibromatosis-1. Trends Neurosci. 2013;36:237–247. doi: 10.1016/j.tins.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte JV, Ribeiro MJ, Violante IR, Cunha G, Silva E, Castelo-Branco M. Multivariate pattern analysis reveals subtle brain anomalies relevant to the cognitive phenotype in neurofibromatosis type 1. Hum Brain Mapp. 2014;35:89–106. doi: 10.1002/hbm.22161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippi CG, Watts R, Duy LAN, Cauley KA. Diffusion-tensor imaging derived metrics of the corpus callosum in children with neurofibromatosis type I. Am J Roentgenol. 2013;200:44–49. doi: 10.2214/AJR.12.9590. [DOI] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, Van Der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/S0896-6273(02)00569-X. [DOI] [PubMed] [Google Scholar]
- Greenwood RS, Tupler LA, Whitt JK, Buu A, Dombeck CB, Harp AG, Payne ME, Eastwood JD, Krishnan KRR, Mac-Fall JR. Brain morphometry, T2-weighted hyperintensities, and IQ in children with neurofibromatosis type 1. Arch Neurol. 2005;62:1904–1908. doi: 10.1001/archneur.62.12.1904. [DOI] [PubMed] [Google Scholar]
- Gutmann DH, Wu YL, Hedrick NM, Zhu Y, Guha A, Parada LF. Heterozygosity for the neurofibromatosis 1 (NF1) tumor suppressor results in abnormalities in cell attachment, spreading and motility in astrocytes. Hum Mol Genet. 2001;10:3009–3016. doi: 10.1093/hmg/10.26.3009. [DOI] [PubMed] [Google Scholar]
- Karlsgodt KH, Rosser T, Lutkenhoff ES, Cannon TD, Silva AJ, Bearden CE. Alterations in white matter microstructure in neurofibromatosis-1. PLoS One. 2012;7:1–11. doi: 10.1371/journal.pone.0047854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtonen A, Howie E, Trump D, Huson SM. Behaviour in children with neurofibromatosis type 1: cognition, executive function, attention, emotion, and social competence. Dev Med Child Neurol. 2013;55:111–125. doi: 10.1111/j.1469-8749.2012.04399.x. doi:10. 1111/j.1469-8749.2012.04399.x. [DOI] [PubMed] [Google Scholar]
- Li W, Cui Y, Kushner SA, Brown RAM, Jentsch JD, Frankland PW, Cannon TD, Silva AJ. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol. 2005;15:1961–1967. doi: 10.1016/j.cub.2005.09.043. [DOI] [PubMed] [Google Scholar]
- Loitfelder M, Huijbregts SCJ, Veer IM, Swaab HS, Van Buchem MA, Schmidt R, Rombouts SA. Functional connectivity changes and executive and social problems in neurofi-bromatosis type I. Brain Connect. 2015;5:312–320. doi: 10.1089/brain.2014.0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainberger F, Jung NH, Zenker M, Wahlländer U, Freudenberg L, Langer S, Berweck S, Winkler T, Straube A, Heinen F, Granström S, Mautner VF, Lidzba K, Mall V. Lovas-tatin improves impaired synaptic plasticity and phasic alertness in patients with neurofibromatosis type 1. BMC Neurol. 2013;13:131–142. doi: 10.1186/1471-2377-13-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margariti PN, Blekas K, Katzioti FG, Zikou AK, Tzoufi M, Argyropoulou MI. Magnetization transfer ratio and volumetric analysis of the brain in macrocephalic patients with neurofibromatosis type 1. Eur Radiol. 2007;17:433–438. doi: 10.1007/s00330-006-0323-1. [DOI] [PubMed] [Google Scholar]
- Molosh AI, Johnson PL, Spence JP, Arendt D, Federici LM, Bernabe C, Janasik SP, Segu ZM, Khanna R, Goswami C, Zhu W, Park SJ, Li L, Mechref YS, Clapp DW, Shekhar A. Social learning and amygdala disruptions in Nf1 mice are rescued by blocking p21-activated kinase. Nat Neurosci. 2014;17:1583–1590. doi: 10.1038/nn.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore BD, Slopis JM, Jackson EF, De Winter AE, Leeds NE. Brain volume in children with neurofibromatosis type 1: relation to neuropsychological status. Neurology. 2000;54:914–920. doi: 10.1212/WNL.54.4.914. [DOI] [PubMed] [Google Scholar]
- Plasschaert E, Descheemaeker MJ, Van Eylen L, Noens I, Steyaert J, Legius E. Prevalence of autism spectrum disorder symptoms in children with neurofibromatosis type 1. Am J Med Genet B Neuropsychiatr Genet. 2015;168B:72–80. doi: 10.1002/ajmg.b.32280. [DOI] [PubMed] [Google Scholar]
- Poldrack RA, Rodriguez P. How do memory systems interact? Evidence from human classification learning. Neurobiol Learn Mem. 2004;82:324–332. doi: 10.1016/j.nlm.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Pride NA, Crawford H, Payne JM, North KN. Social functioning in adults with neurofibromatosis type 1. Res Dev Disabil. 2013;34:3393–3399. doi: 10.1016/j.ridd.2013.07.011. [DOI] [PubMed] [Google Scholar]
- Pride NA, Korgaonkar MS, Barton B, Payne JM, Vucic S, North KN. The genetic and neuroanatomical basis of social dysfunction: lessons from neurofibromatosis type 1. Hum Brain Mapp. 2014;35:2372–2382. doi: 10.1002/hbm.22334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro MJ, Violante IR, Bernardino I, Ramos F, Saraiva J, Reviriego P, Upadhyaya M, Silva ED, Castelo-Branco M. Abnormal achromatic and chromatic contrast sensitivity in neurofibromatosis type 1. Invest Ophthalmol Vis Sci. 2012;53:287–293. doi: 10.1167/iovs.11-8225. [DOI] [PubMed] [Google Scholar]
- Rizvi TA, Akunuru S, Courten-Myers G, Switzer RC, Nordlund ML, Ratner N. Region-specific astrogliosis in brains of mice heterozygous for mutations in the neurofibromatosis type 1 (Nf1) tumor suppressor. Brain Res. 1999;816:111–123. doi: 10.1016/S0006-8993(98)01133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson A, Kloog Y, Stein R, Assaf Y. Motor deficits and neurofibromatosis type 1 (NF1)-associated MRI impairments in a mouse model of NF1. NMR Biomed. 2010;23:1173–1180. doi: 10.1002/nbm.1546. [DOI] [PubMed] [Google Scholar]
- Rosenbaum T, Engelbrecht V, Krölls W, Van Dorsten FA, Hoehn-Berlage M, Lenard HG. MRI abnormalities in neurofibromatosis type 1 (NF1): a study of men and mice. Brain Dev. 1999;21:268–273. doi: 10.1016/S0387-7604(99)00024-8. [DOI] [PubMed] [Google Scholar]
- Santos A. Fac Sci Technol Univ Coimbra. University of Coimbra; 2011. Visual-callosal white matter pathways in neurofi-bromatosis type 1: a Diffusion Tensor Imaging – Fiber Tracking (DTI-FT) study. [Google Scholar]
- Shilyansky C, Karlsgodt KH, Cummings DM, Sidiropoulou K, Hardt M, James AS, Ehninger D, Bearden CE, Poirazi P, Jentsch JD, Cannon TD, Levine MS, Silva AJ. Neurofibromin regulates corticostriatal inhibitory networks during working memory performance. Proc Natl Acad Sci USA. 2010a;107:13141–13146. doi: 10.1073/pnas.1004829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilyansky C, Lee YS, Silva AJ. Molecular and cellular mechanisms of learning disabilities: a focus on NF1. Annu Rev Neurosci. 2010b;33:221–243. doi: 10.1146/annurev-neuro-060909-153215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Frankland PW, Marowitz Z, Friedman E, Laszlo GS, Cioffi D, Jacks T, Bourtchuladze R. A mouse model for the learning and memory deficits associated with neurofibromatosis type I. Nat Genet. 1997;15:281–284. doi: 10.1038/ng0397-281. [DOI] [PubMed] [Google Scholar]
- Steen RG, Taylor JS, Langston JW, Glass JO, Brewer VR, Reddick WE, Mages R, Pivnick EK. Prospective evaluation of the brain in asymptomatic children with neurofibromatosis type 1: relationship of macrocephaly to T1 relaxation changes and structural brain abnormalities. AJNR Am J Neuroradiol. 2001;22:810–817. [PMC free article] [PubMed] [Google Scholar]
- Theos A, Korf BR. Pathophysiology of neurofibromatosis type 1. Ann Intern Med. 2006;144:842–849. doi: 10.7326/0003-4819-144-11-200606060-00010. [DOI] [PubMed] [Google Scholar]
- van der Vaart T, van Woerden GM, Elgersma Y, de Zeeuw CI, Schonewille M. Motor deficits in neurofibromatosis type 1 mice: the role of the cerebellum. Genes Brain Behav. 2011;10:404–409. doi: 10.1111/j.1601-183X.2011.00685.x. doi:101111/j1601-183X.201100685.x. [DOI] [PubMed] [Google Scholar]
- Violante IR, Ribeiro MJ, Silva ED, Castelo-Branco M. Gyrification, cortical and subcortical morphometry in neurofibromatosis type 1: an uneven profile of developmental abnormalities. J Neurodev Disord. 2013;5:1–13. doi: 10.1186/1866-1955-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh KS, Vélez JI, Kardel PG, Imas DM, Muenke M, Packer RJ, Castellanos FX, Acosta MT. Symptomatology of autism spectrum disorder in a population with neurofibromatosis type 1. Dev Med Child Neurol. 2013;55:131–138. doi: 10.1111/dmcn.12038. doi:101111/dmcn.12038. [DOI] [PubMed] [Google Scholar]
- Wignall EL, Griffiths PD, Papadakis NG, Wilkinson ID, Wallis LI, Bandmann O, Cowell PEE, Hoggard N. Corpus callosum morphology and microstructure assessed using structural MR imaging and diffusion tensor imaging: initial findings in adults with neurofibromatosis type 1. AJNR Am J Neuroradiol. 2010;31:856–861. doi: 10.3174/ajnr.A2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamboni SL, Loenneker T, Boltshauser E, Martin E, Il'yasov KA. Contribution of diffusion tensor MR imaging in detecting cerebral microstructural changes in adults with neurofibromatosis type 1. AJNR Am J Neuroradiol. 2007;28:773–776. doi:28/4/773[pii] [PMC free article] [PubMed] [Google Scholar]
- Zöller ME, Rembeck B, Bäckman L. Neuropsychologica deficits in adults with neurofibromatosis type 1. Acta Neurol Scand. 1997;95:225–232. doi: 10.1111/j.1600-0404.1997.tb00103.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Example of MD and FA maps.
Figure S2: Latency to the platform in Nf1+/− and WT mice in the hidden-platform version of the water maze, two-way ANOVA, P = 0.39, F252 = 0.959.
Figure S3: Number of platform crossings during a 60s probe trial given after 3 days of training, two-way anova, P = 0.0156, F1 26 = 6.693; multiple comparisons: (WT, n = 13, P = 0.0004; Nf1+/−, n = 15, P = 0.6117).
Figure S4: Percentage time spent in quadrant during a 60s probe trial given after 3 days of training, two-way anova, P = 0.1317 F1 26 = 2.422; multiple comparisons: (WT, n = 13, P = 0.0006; Nf1+/−, n = 15, P = 0.0683)
Table S1: Results from anova test on evaluating sex × genotype interaction.