

SUMMARY
Tuberous Sclerosis Complex (TSC) is a neurodevelopmental disorder characterized by mutations in either the TSC1 or TSC2 genes whose products form a critical inhibitor of the mechanistic target of rapamycin (mTOR). Loss of TSC1/2 gene function renders an mTOR-overactivated state. Clinically, TSC manifests with epilepsy, intellectual disability, autism, and sleep dysfunction. Here we report that mouse models of TSC have abnormal circadian rhythms. We show that mTOR regulates the proteostasis of the core clock protein BMAL1, affecting its translation, degradation, and subcellular localization. This results in elevated levels of BMAL1 and a dysfunctional clock that displays abnormal timekeeping in constant conditions and exaggerated responses to phase resetting. Genetically lowering the dose of BMAL1 rescues circadian behavioral phenotypes in TSC mouse models. These findings indicate that BMAL1 deregulation is a feature of the mTOR-activated state and suggest a molecular mechanism for mitigating circadian phenotypes in a neurodevelopmental disorder.
Keywords: Tuberous Sclerosis Complex, circadian rhythms, BMAL1, translation, neurodevelopmental disorder, mTOR, sleep disorders, autism
eTOC Blurb
Sleep disorders are common in neurodevelopmental disorders such as the Tuberous Sclerosis Complex (TSC). Lipton et al. demonstrate that increased translation and decreased degradation of the core clock protein BMAL1 underlie circadian rhythm abnormalities in TSC mouse and cellular models.
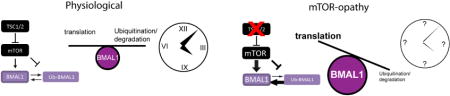
INTRODUCTION
Tuberous Sclerosis Complex (TSC) is a multisystem, autosomal dominant, neurodevelopmental disorder characterized by epilepsy, intellectual disability, autism, and sleep dysfunction(Bruni et al., 1995; Henske et al., 2016; van Eeghen et al., 2011). TSC is caused by mutation in either TSC1 or TSC2 whose products act as a tumor suppressor complex that inhibits the mechanistic target of rapamycin (mTOR) kinase. Without TSC1 or TSC2 gene function, mTOR is activated even in the absence of upstream growth regulatory cues. Thus, TSC is a paradigmatic mTOR-opathy and a monogenic cause for complex neurodevelopmental phenotypes(Lipton and Sahin, 2014).
mTOR is an atypical PIKK-family Ser/Thr kinase that integrates upstream environmental signals with cellular nutritive status to regulate protein synthesis, lipogenesis, and autophagy(Saxton and Sabatini, 2017). While many aspects of TSC have been intensively studied, sleep has not. Disrupted sleep has been linked to impaired daytime behavior, mood disorders, metabolic disease, and increased seizure risk in epilepsy and is estimated to occur in ~50% of TSC patients. Symptoms of sleep disruption in TSC include early arousals, sleep maintenance insomnia, and circadian clock disruption(Bruni et al., 1995; Hancock et al., 2005a; Hancock et al., 2005b; Hunt, 1993; Hunt and Stores, 1994). Almost nothing is known about the underlying mechanisms of sleep disturbance in TSC.
Sleep is regulated by both a homeostatic process and the circadian timing system(Borbely, 1982; Huang et al., 2011). The circadian clock is an evolutionarily conserved biological timing system that synchronizes organismal function with the light-dark cycle. Disruption of circadian rhythms has been linked to depression, schizophrenia, autism, obesity and diabetes(Jagannath et al., 2013b; Marcheva et al., 2013; Oliver et al., 2012; Wulff et al., 2010). Indeed, circadian disruption has been reported in several well established animal models of neurodevelopmental disorders including Fragile X, Angelman, Smith-Magenis and PTEN hamartoma tumor syndromes (De Leersnyder, 2006; De Leersnyder et al., 2006; Ebrahimi-Fakhari and Sahin, 2015; Gatto and Broadie, 2009; Ogawa et al., 2007). Importantly, treatment for circadian or sleep dysfunction in these disorders remains exclusively empirical because mechanistic insight into its pathophysiology is lacking.
On a molecular level, the circadian clock constitutes a negative feedback loop that has transcriptional, translational, and post-translational components. The bHLH-PAS domain containing transcription factors BMAL1 and CLOCK heterodimerize to activate the transcription of the Period and Cryptochrome genes whose products subsequently heterodimerize to suppress the transcriptional activity of BMAL1-CLOCK (Buhr and Takahashi, 2013; Takahashi, 2016). Phosphorylation and ubiquitination are among the post-translational modifications that regulate circadian timing and connect the clock to cellular signaling(Fang et al., 2007; Lee et al., 2001).
There is considerable evidence for a role for mTOR signaling in the regulation of the circadian clock. Activity of the mTOR pathway in the suprachiasmatic nucleus (SCN) is stimulated by light and mutations in the mTOR kinase itself or in the mTOR effector protein eukaryotic initiation factor 4E binding protein (4E-BP) regulate circadian timing(Cao et al., 2011; Cao et al., 2015; Cao et al., 2008; Cao et al., 2010; Cao et al., 2013). Interestingly, mTOR pathway activity demonstrates circadian oscillations(Cao et al., 2013; Cornu et al., 2014; Jouffe et al., 2013; Khapre et al., 2014; Lipton et al., 2015). Heterozygous mutations of either mTOR in mice or Tsc1/2 orthologs in Drosophila clock neurons can result in circadian arrhythmia, together suggesting that a delicate balance of mTOR signaling is required for optimizing circadian synchrony(Cao et al., 2013; Zheng and Sehgal, 2010). We have recently demonstrated that BMAL1 is phosphorylated by the mTOR effector kinase S6K1, and thereby promotes protein synthesis rhythms in cells by associating with the translational machinery in the cytoplasm. Whether this mechanism impacts circadian function in TSC remains unclear(Lipton et al., 2015).
To reciprocally delineate the mechanisms of sleep disruption in TSC and investigate the mechanisms by which mTOR signaling contributes to circadian timing, we tested mouse models of TSC for circadian behavior. We show that Tsc-deficient animals and cells demonstrate abnormal circadian rhythms and that BMAL1 translation is increased and its degradation decreased, together resulting in elevated levels of BMAL1 protein. Finally, tempering the expression of BMAL1 reverses abnormalities of circadian behavior in Tsc2 heterozygous mice. These findings contribute to an understanding of the translational regulation of the circadian clock and suggest novel therapeutic paradigms for targeting specific neurological phenotypes in TSC and other mTOR-opathies.
RESULTS
Mouse Models of TSC Have Abnormal Circadian Rhythms
We asked whether a Tsc1 loss-of-function model in mammals could recapitulate circadian anomalies seen with loss of the Drosophila ortholog of Tsc1. Homozygous loss of either Tsc1 or Tsc2 in mice results in embryonic lethality(Kobayashi et al., 1999; Kobayashi et al., 2001). We therefore tested a transgenic mutant in which the Tsc1 gene is deleted under the control of the SynapsinICre (SynICre) allele and results in the selective deletion of Tsc1 in post-mitotic neurons of the forebrain and brainstem(Meikle et al., 2008; Meikle et al., 2007) (Figure 1A,B). Tsc1flox/flox;SynICre mice develop seizures, ataxia, and die at about P30-40, phenotypes which can be abrogated with chronic rapamycin treatment(Meikle et al., 2008). To test the circadian function of adults with Tsc1 knockdown, we treated mutant and wildtype littermates with rapamycin until adulthood and then implanted them with data loggers to monitor core body temperature as a proxy for the output of the circadian clock (Figure 1A). This approach was used over running wheels because the ataxia of mutant mice limits their locomotion. After two weeks of recovery from implantation, rapamycin was discontinued to ‘uncover’ the effects of Tsc1 loss (Figure 1A). Tsc1flox/flox;SynICre mice demonstrated strikingly abnormal circadian rhythms in core body temperature compared to controls in constant darkness (DD) (Figure 1B–D). When placed in 12hour light:12 hour dark (LD) conditions, mutants appeared capable of re-entrainment. Fast Fourier transform analysis and examination of the mean core temperature as a function of circadian phase, demonstrated loss of a dominant circadian frequency in some Tsc1flox/flox;SynICre mice in DD conditions and corresponding reduction in mean circadian amplitude for the population (Figure 1D, F). Sufficient rhythmicity was preserved in some mutant animals, enabling the calculation of shortened free-running period (Figure 1E). Importantly, neither mean temperature nor respiratory physiology differed between mutants and controls, suggesting that the gross control of physiology was not altered by Tsc1 loss (Figure 1F, Figures S1 and S2).
Figure 1. Abnormal Circadian Rhythms in Brain-Specific Tsc1 Knockout Mice.
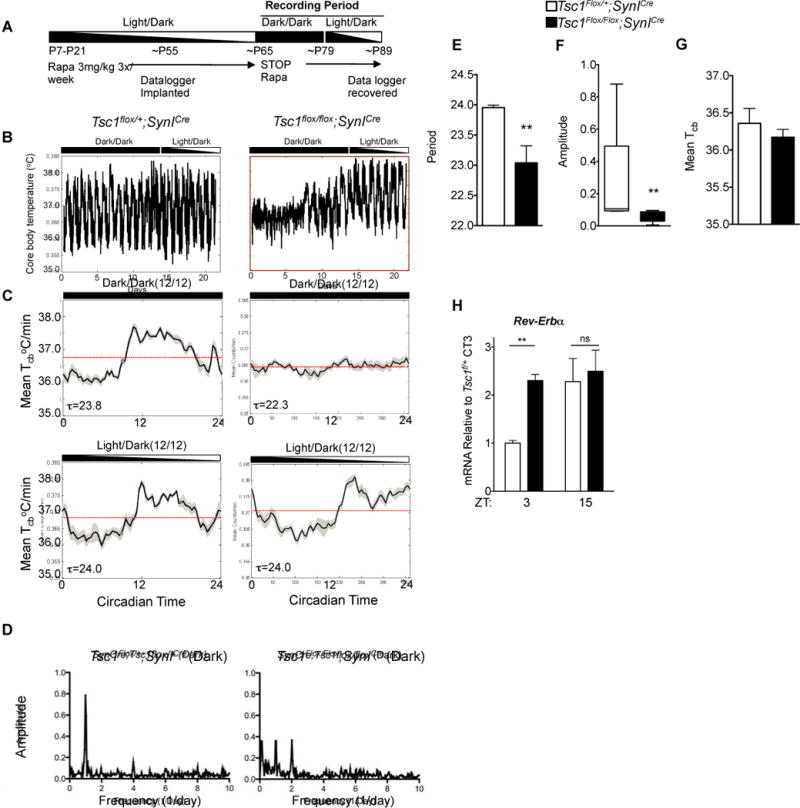
(A) Outline of experimental protocol for measurement of core body temperature in Tsc1 brain-specific knockout mice.
(B) Daily core body temperature measurements in representative SynICre;Tsc1flox/flox compared to controls. Note daily oscillations in temperature in DD are dysregulated in mutant compared to controls.
(C) Representative mean temperature recordings as a function of circadian time in L/D or D/D conditions for mice of indicated genotype.
(D) Fast Fourier Analysis demonstrating amplitude of power spectrum relative to period (1/frequency).
(E) Histogram of circadian period in DD conditions in SynICre;Tsc1flox/flox compared to controls. Student’s t test, *p<0.05 n=8 per genotype.
(F) Histogram of mean circadian amplitude from indicated genotypes. Student’s t test, **p<0.01.
(G) Mean core body temperature (Tcb) does not differ between SynICre;Tsc1flox/flox and controls.
(H) Histogram of diurnal variation of Rev-Erbα in the cortex of SynICre;Tsc1flox/flox mice. n=3 biological replicates each measured in duplicate, Student’s t test **p<0.01.
Because both the mTOR pathway and the circadian clock regulate metabolic outputs, we interrogated the diurnal expression of several previously defined clock-controlled metabolic genes. We found abnormal diurnal expression of several genes in Tsc1flox/flox;SynICre brain (Figure S1G). In support of a disruption in circadian rhythmicity in the brain of mutant mice, we found that the expression of the core clock gene Rev-Erbα was altered in Tsc1flox/flox;SynICre brains when compared to controls (Figure 1H). These data demonstrate that the Tsc pathway regulates the diurnal output of metabolic gene expression in the brain.
Rapamycin Corrects a Short Period Phenotype in Tsc2+/− Heterozygous Mice
While we observed abnormal circadian rhythms in neuron-specific Tsc1 knockouts, TSC is an autosomal dominant condition and patients are heterozygotes, prompting us to investigate the circadian phenotypes of Tsc2+/− constitutively heterozygous mice. Tsc2+/− have been shown to demonstrate abnormalities in axon guidance, synaptic plasticity, and cognitive function(Ehninger et al., 2008; Nie et al., 2010). We found that both Tsc2+/+ and Tsc2+/− mice efficiently entrained to LD conditions and showed strong circadian rhythms of locomotor and metabolic activity (Figure 2A and Figure S1). By contrast, under DD conditions, Tsc2+/− mutants, like Tsc1flox/flox;SynICre mutants, had a shorter free-running period when compared to wildtype littermates (Figure 2B); there was no change in circadian amplitude or overall activity (Figure 2C,D).
Figure 2. Shortened Free-running Period in Tsc2+/− Mice Is Responsive to Rapamycin.
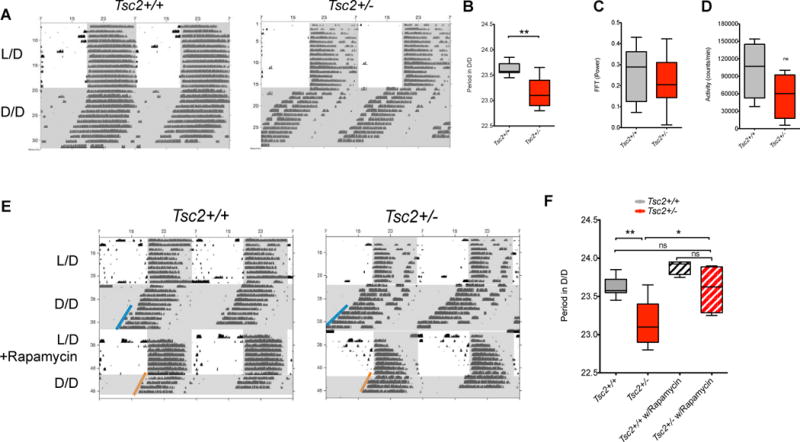
(A) Representative double-plotted actograms demonstrating wheel running activity in Tsc2+/− and Tsc2+/+ mice. Shading demonstrates timing of light-dark periods.
(B) Circadian period in DD conditions (error bars reflect minima and maxima of n=9 per genotype), Student’s t test, **p<0.003.
(C) Fast Fourier transform power in DD conditions (error bars reflect minima and maxima of n=9 per genotype). Student’s t test. ns = not significant
(D) Activity quantified as number of wheel rotations/minute in DD conditions (error bars reflect minima and maxima of n=9 per genotype), Student’s t test.
(E) Representative double-plotted actograms demonstrating wheel running activity in Tsc2+/− and Tsc2+/+ mice. Shading demonstrates timing of light-dark periods.
(F) Circadian period in DD conditions before and after rapamycin injection (error bars reflecting minima and maxima of n=9 per genotype without rapamycin, n=3 each genotype with rapamycin), 2-way ANOVA, *p<0.05, **p=0.02.
See Figure S1.
We predicted that the short period in DD conditions observed in Tsc2+/− mice was secondary to an upregulation of mTOR signaling in the brain of these mice relative to wildtype littermates. Thus, we injected light-entrained mice with intraperitoneal rapamycin prior to switching them to DD conditions. As predicted, treatment with the mTOR inhibitor rapamycin normalized the relative shortening of circadian free-running period in Tsc2+/− to wildtype levels while having no significant effect on wildtype mice (Figure 2E,F). These data suggest that the shortened period phenotype in Tsc2+/− mice results from aberrantly exuberant mTOR signaling.
Tsc2 knockout cells Mutant Cells Demonstrate Abnormal Circadian Phenotypes and Elevated BMAL1-CLOCK Transcriptional Targets
To gain mechanistic insight into a possible role for the TSC pathway in mammalian cells, we examined the expression of core circadian clock proteins in Tsc2 knockout mouse embryonic fibroblasts (MEFs). The expression of neither Bmal1 nor Clock differed in serum-starved Tsc2−/− cells compared to wildtype MEFs (from control littermates) (Figure 3A). In contrast, we observed marked elevations in Per1 and Per2 expression and more modest increase in Cry1 and Cry2 mRNA expression (Figure 3A). Thus, while the transcript levels of Bmal1 and Clock were not changed while that of several of their transcriptional targets did.
Figure 3. Abnormal Circadian Timing in Tsc2 Knockout Cells.
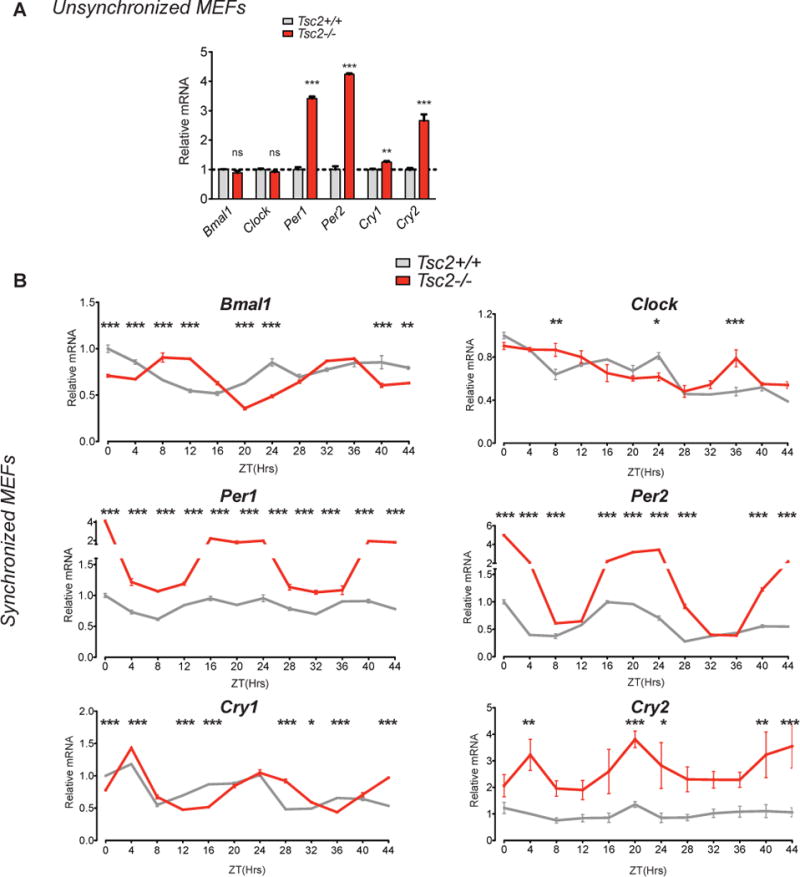
(A) Quantitative PCR of indicated core clock components in unsynchronized MEFs. Tsc2−/− compared to Tsc2+/+ cells, n=3 biological replicates per genotype run in duplicate and normalized to wildtype cells. Student’s t test, *p<0.05, **p<0.001, ***p<0.0001.
(B) qPCR of indicated core clock components in dexamethasone-synchronized Tsc2−/− compared to Tsc2+/+ cells demonstrate abnormal circadian timing of relative mRNA expression. 2-way ANOVA (zeitgeber time (ZT) and genotype) with Bonferonni post-hoc test of every data point compared to Tsc2+/+ cell at ZT0, normalized arbitrarily to wildtype cells at ZT0. *p<0.05, **p<0.001, ***p<0.0001, n=3 biological replicates per genotype each run in duplicate.
Because the expression of Per1 and Per2 were so markedly elevated, we wondered whether Tsc2−/− cells might demonstrate abnormal circadian dynamics. We synchronized these cells and examined gene expression every four hours over the circadian cycle. While the amplitude of Bmal1 did not differ in Tsc2−/− cells, we observed a near inversion in circadian phase compared to wildtype cells. We observed little difference in the transcription of Clock, which appeared to be non-rhythmic (Figure 3B). When we tested the transcriptional targets of BMAL1-CLOCK in synchronized cells, we observed striking abnormalities in the amplitude and period of Per1, Per2, Cry1, and Cry2 (Figure 3B). Thus, while the transcriptional amplitudes of Bmal1 and Clock are not changed by Tsc2 loss in unsynchronized cells, the circadian rhythms of core clock genes are markedly altered.
Increased BMAL1 Protein Expression in Tsc-deficient cells and animals
Because we observed no significant changes in Bmal1 or Clock transcription in Tsc2 knockout cells, but marked changes in their transcriptional targets and strong circadian timing phenotypes in both cells and animals, we hypothesized that Tsc-deficiency changes the translation and/or localization of BMAL1 protein. We observed increased BMAL1 expression in Tsc2−/− cells compared to wildtype contols and a trend toward increased expression of CLOCK that was not significant (Figure 4A). To examine the subcellular localization in MEFs, we examined BMAL1 in serum-starved wildtype MEFs by immunofluorescence and observed a preferential accumulation of BMAL1 in the cytoplasm (Figure 4B–C). In contrast, BMAL1 expression was notably higher in Tsc2−/−cells. Moreover, there was more BMAL1 in the nucleus of Tsc2−/− cells both before and after serum-stimulation for 2 hours (a commonly used method of circadian synchronization(Jagannath et al., 2013a)). This phenomenon was blocked by treatment with the mTOR inhibitor Torin 1 suggesting that it is mediated by mTOR over-activation in knockout cells (Figure 4C). Together these data suggest that mTOR promotes BMAL1 protein expression and localization to the nucleus.
Figure 4. Increased BMAL1 Protein Expression in Tsc-deficient Cells and Animals.

(A) Western blot of whole cell MEF lysates of indicated genotype, n=6 biological replicates, Student’s t test, ***p<0.001, ns=not signfiicant.
(B) Immunocytochemistry of Tsc2 MEFs of indicated genotype serum-starved overnight or maintained in serum-containing media. Quantification of nuclear/cytoplasmic ratio normalized to serum-starved wildtype cells; scale bar = 50μm, ***p<0.0001, 1-way ANOVA with Bonferonni post-hoc test.
(C) Representative western blot of nuclear and cytoplasmic fractions of Tsc2 MEFs of the indicated genotype treated as shown (n = 3 replicates). Nuclear and cytoplasmic fractions were run simultaneously and membranes were bisected prior to antibody application, incubated and processed together and then re-assembled for comparison.
(D) Western blots from brain lysates from Tsc2+/− and Tsc2+/+ littermates collected at indicated circadian times under constant conditions. (Below) Densitometry measurements of BMAL1 relative to actin normalized to Tsc2+/+ controls at CT3. 2-way ANOVA, p=0.02 for interaction, p=0.02 for genotype, p=0.02 for circadian time.
(E) Representative Western blots from SynICre;Tsc1flox/flox brain compared to controls with densitometry measurements of BMAL1 relative to tubulin, n=4 per genotype, Students t test, ***p<0.001.
(F) Representative BMAL1 immunohistochemistry of suprachiasmatic nuclei at ZT2 from indicated genotypes. See Figure S3.
To test these findings in vivo, we examined the brains of Tsc2+/− mice collected every 6 hours during the circadian cycle to test BMAL1 protein levels. We observed elevated BMAL1 protein levels in whole brain lysates with evidence for diminished circadian variability in BMAL1 levels in Tsc2+/− compared to lysates of wildtype littermates (Figure 4D). Similarly, we found that BMAL1 protein levels were also significantly elevated in Tsc1flox/flox;SynICre brain (Figure 4E). We also observed a marked increase in BMAL1 expression in the SCN at ZT2 in Tsc1flox/flox;SynICre compared to controls suggesting that the effect of Tsc-deficiency on BMAL1 could impact both the central clock in the SCN and peripheral clock (Figure 4F and Figure S3). Overall, our data demonstrate that BMAL1 protein expression is elevated in Tsc2 mutant cells and animals.
BMAL1 Translation Is Regulated by Tsc2
The mTOR pathway is an important translation regulatory pathway(Hsieh et al., 2012; Thoreen et al., 2012). Consistently, elevated protein synthesis has been reported in Tsc2−/− MEFs(Yu et al., 2014). The elevation of BMAL1 protein despite unchanged expression of its mRNA in Tsc2−/− cells led us to hypothesize that the mTOR pathway regulates BMAL1 translation. To test this directly, we performed bio-orthogonal non-canonical amino acid tagging (BONCAT) in Tsc2 knockout and wildtype MEFs (Figure 5A)(Dieterich et al., 2006). We labeled newly synthesized protein with the methionine analog L-azido-methionine (L-AHA) followed by copper-mediated cycloaddition of biotin (Click chemistry) and streptavidin-based affinity purification to isolate L-AHA-incorporated, de novo synthesized proteins (Figure 5B,C). We subsequently immunoblotted these proteins to assay the amount of newly synthesized BMAL1. We found an elevated level of de novo protein synthesis in Tsc2−/− MEFs compared to control cells consistent with previous reports (Figure 5B, D and Figure S5)(Zhang et al., 2014). When we immunoblotted biotinylated-L-AHA complexes for BMAL1, we found about a 50% increase in the amount of de novo synthesized BMAL1 in Tsc2−/− cells compared to control (Figure 5B,C). Importantly, we observed little biotinylation signal with either control methionine(Met)-labeled cells, cycloheximide treatment, or with cells labeled without L-AHA suggesting that the observed signal judiciously represents de novo protein (Figure S4). In a parallel approach, we immunoprecipitated 35S-labeled endogenous BMAL1 from Tsc2−/− or control cells and observed a similar increase in BMAL1 translation (Figure 5D). These data demonstrate that BMAL1 translation is increased in Tsc2−/− cells and offer an explanation for the elevated BMAL1 protein levels in the Tsc-deficient background.
Figure 5. BMAL1 Translation Is Increased in Tsc2 −/− MEFs.
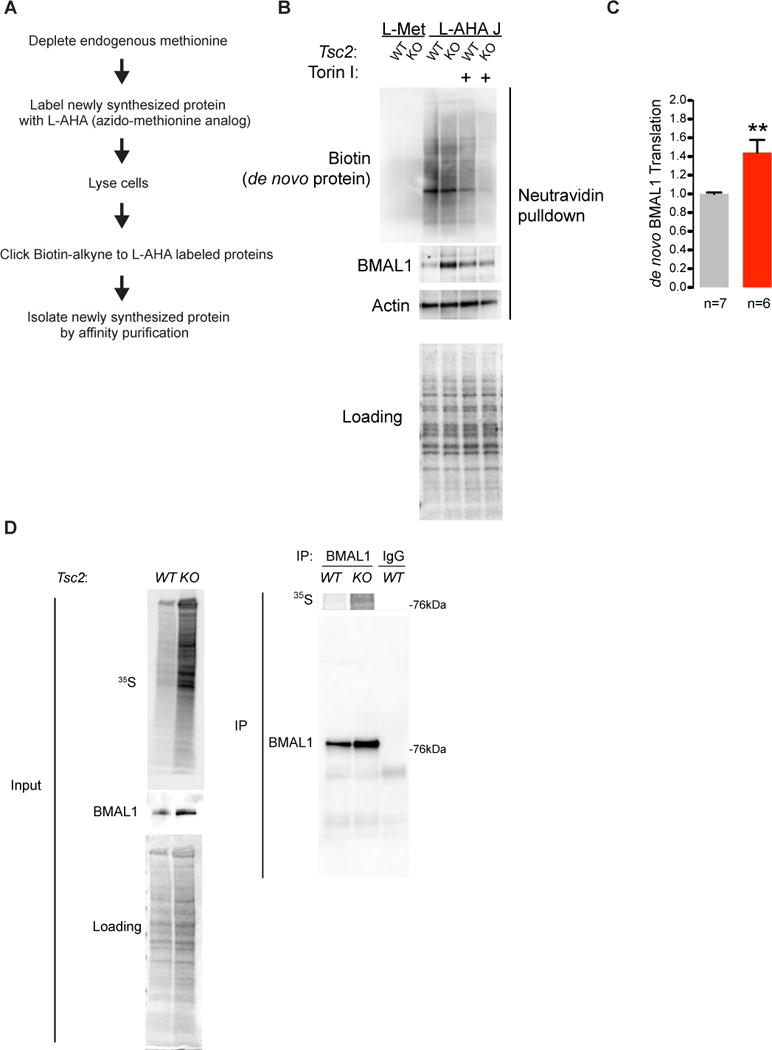
(A) Experimental outline of BONCAT and affinity purification of de novo-synthesized proteins.
(B) Representative immunoblots of cytoplasmic lysates and streptavidin-affinity purification of biotinylated-L-AHA-tagged de novo synthesized protein demonstrates increased BMAL1 in Tsc2−/− cells. See Figure S4.
(C) Quantification of total BMAL1 and de novo synthesized BMAL1 in MEFs of indicated genotype, n=3 biological replicates, Student’s t test, **p<0.001.
(D) Tsc2 MEFs of indicated genotype were pulse-labeled with 35S-Met/Cys for one hour followed by immunoprecipitation with BMAL1 monoclonal antibody and quantified by densitometry. See Figure S5.
mTOR Regulates BMAL1 Degradation, Ubiquitination, and UBE3A Association
Recent studies have suggested that the mTOR pathway coordinates the production and degradation of proteins and under growth conditions, suppresses the ubiquitination of relatively long-lived proteins(Zhao et al., 2015). To test whether changes in BMAL1 degradation contributed to increased BMAL1 levels in Tsc mutant backgrounds, we examined BMAL1 expression in Tsc2 MEFs treated with the protein synthesis inhibitor cycloheximide (CHX). In Tsc2 knockout cells, in which mTOR activity is elevated compared to wildtype controls, BMAL1 degraded more slowly than in control cells (Figure 6A, B).
Figure 6. Impaired BMAL1 Ubiquitination, Degradation, and UBE3A Association in Tsc2 −/− Cells.
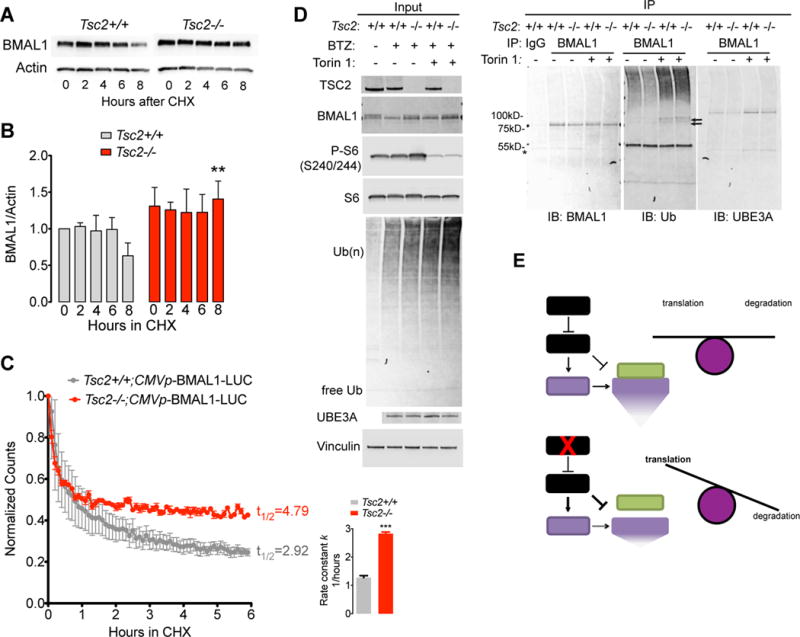
(A) Tsc2 MEFs were treated with 100μg/mL cycloheximide (CHX) and whole cell lysates were collected at indicated times for Western blot.
(B) Quantification of experiments in (A) analyzed by 2-way ANOVA for genotype and time, with Bonferonni correction, **p<0.01.
(C) Luminometry of CMV-BMAL1-Luciferase fusion reporter transfected into either Tsc2+/+ or Tsc2−/− MEFs and incubated with CHX. Shown is the average luminescence ± SEM of 6 samples per genotype per time point normalized to time 0 for each genotype. Histogram of exponential decay rate constant (1/hour) k ± SD,***p<0.0001, t test.
(D) Tsc2 MEFs were treated with 100nM bortezomib (BTZ) and either DMSO or 250nM Torin 1 for four hours followed by cell lysis and anti-BMAL1 immunoprecipitation. *indicates IgG heavy chain; black arrow indicates presumed low-molecular weight ubiquitinated BMAL1. All images represent samples run on the same membranes cut for visual clarity.
(E) Model of mTOR regulation of BMAL1 proteostasis in the presence or absence of TSC1/2 function.
To further validate these data, we constructed a constitutively expressed a fusion reporter in which the BMAL1 ORF was fused in frame with firefly Luciferase (BMAL1-Luc). We transiently transfected BMAL1-Luc into Tsc2+/+ and Tsc2−/− cells, treated cells with CHX and performed luminometry every 6 minutes in the presence of CHX. Consistent with previous findings, we observed a reduction in the overall half life of the BMAL1-Luc reporter and an increase in the rate constant in Tsc2−/− cells compared to controls (Figure 6C).
Because BMAL1 has been shown to be ubiquitinated by the E3 ligase UBE3A and undergo proteasomal degradation, we hypothesized that the changes in BMAL1 stability in Tsc2−/− cells could be explained by alterations in BMAL1 ubiquitination (Gossan et al., 2014; Sahar et al., 2010; Shi et al., 2015). We treated Tsc2 MEFs with the proteasome inhibitor bortezomib (BTZ) for 4 hours with or without Torin 1. Consistent with previous reports, BTZ treatment resulted in an increase in ubiquitinated protein compared to cells treated with DMSO in line with the model that inhibition of mTOR signaling promotes protein turnover(Zhao et al., 2015). We therefore hypothesized that in Tsc2−/− cells, the amount of ubiquitinated BMAL1 might be reduced. Indeed, we observed a reduction in BMAL1 associated ubiquitin in BTZ-treated Tsc2−/−cells compared to controls(Figure 6D). This effect was abrogated by Torin 1, suggesting that the effect is mediated through the relative increase in mTOR activity in Tsc2−/− cells. We next hypothesized that the reduction in BMAL1 ubiquitination in Tsc2−/− cells could result from reduced association of BMAL1 with UBE3A(Figure 6D). We observed less UBE3A associating with immunoprecipitated BMAL1 from Tsc2−/− cells whereas treatment with Torin 1 was again able to block this effect. Taken together, these data suggest that increased BMAL1 levels in Tsc2 knockout cells likely results from a combination of mTOR-mediated stimulation of translation and suppression of proteasome-mediated degradation (Figure 6E).
Tsc2-deficient Cells and Animals Demonstrate Abnormal Responses to a Resetting Stimulus
Because of the increase in BMAL1 expression and its aberrantly high expression in the nucleus in Tsc2 knockout cells, we surmised that circadian synchronization in these cells would be affected. We examined the transcriptional responses of Per1, a well-defined output of circadian clock phase resetting, during a two hour serum shock. Prior to synchronization, we again noted a large increase in Per1 expression (Figure 7A). After 120 minutes, Per1 levels rose in Tsc2+/+ cells whereas they plummeted in Tsc2−/−cells in response to serum shock. This phenotype was largely abrogated by co-treatment with the mTOR inhibitor Torin 1 suggesting that the transcriptional response to serum shock is, at least in part, mediated through the mTOR pathway (Figure 7A). Consistent with a role for mTOR pathway signaling in limiting the response to resetting(Cao et al., 2013), we also found that either rapamycin or Torin 1 treatment of human U2-OS:Bmal1-luciferase (Bmal1-luc) delayed the onset of the circadian nadir in response to phase resetting by dexamethasone, also consistent with other studies(Feeney et al., 2016) (Figure 6B–C). These data together suggest that the Tsc/mTOR pathway regulates the response to phase resetting in both mouse and human cells in response to either serum or dexamethasone, respectively.
Figure 7. Tsc-deficient Cells and Mice Demonstrate Rapid Responses to Phase Shifts Ameliorated by Lowering Bmal1 Dose.
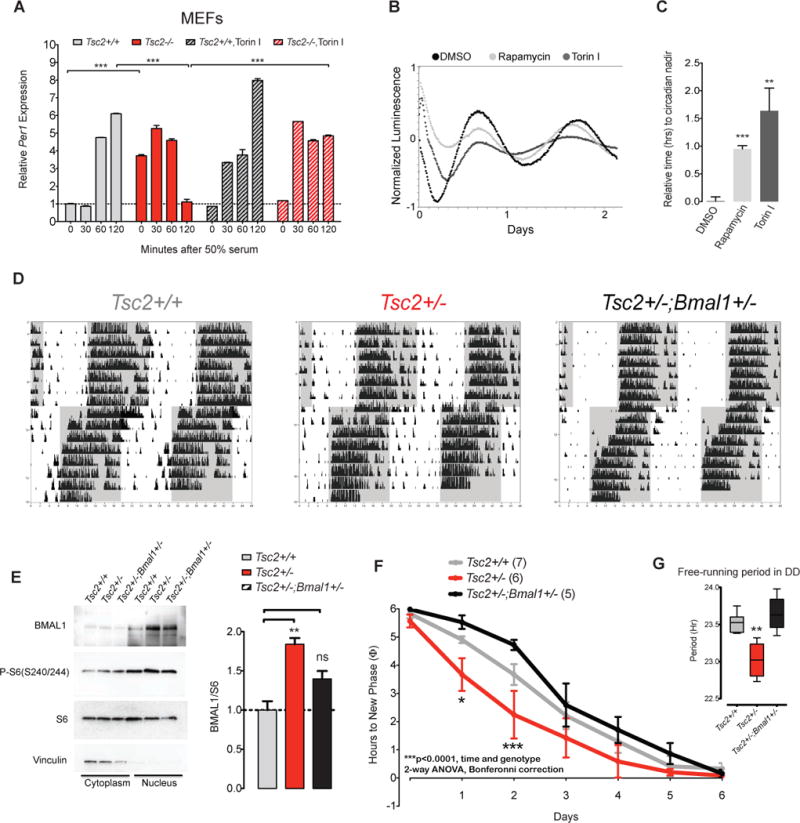
(A) Per1 expression during serum synchronization in Tsc2−/− and control MEFs treated with or without 250 nM Torin 1, n=3 biological replicate per genotype, each sample run in duplicate. 2-way ANOVA (time and genotype) with Bonferroni correction, ***p<0.0001.
(B) Detrended bioluminescence of U2-OS(Bmal1:luciferase) reporter cells treated with DMSO, 100μM rapamycin or 250nM Torin 1 after dexamethasone synchronization.
(C) Quantification of time to first nadir in Bmal1:luciferase in U2-OS(Bmal1:luciferase) cells, n=6 from two independent experiment, 1 way ANOVA, Bonferonni post-test, **p<0.01, ***p<0.001.
(D) Representative actograms of Tsc2+/+, Tsc2+/−, and Tsc2+/−;Bmal1+/− littermates in LD conditions after 6 hour phase advancement.
(E) Representative Western blot of cortical lysates separated into cytoplasmic and nuclear fractions from indicated genotypes (n=3 per genotype).
(F) Phase resetting of indicated genotypes quantified as time to reach new phase; number of animals per genotype noted in parenthesis (2-way ANOVA with Bonferroni post-tests, p=0.002 for interaction, p<0.0001 for genotype, p<0.0001 for time; for individual time point comparisons: *p<0.05, ***p<0.001).
(G) Free running period in DD conditions for indicated genotype (1-way ANOVA, **p<0.01).
Because Per1 expression is a crucial mediator of the phase-shifting response to zeitgebers, we asked whether the abnormal Per1 dynamics we observed in Tsc2−/− cells in response to serum-shock could be recapitulated in behavioral responses to zeitgebers such as light. We tested whether Tsc2+/− mice and wildtype littermates in a phase shifting paradigm in which the onset of the light-dark cycle was advanced by 6 hours. We maintained Tsc2+/− and wildtype littermates in LD for one week followed by a 6 hour phase advancement in the LD cycle. Tsc2+/− mutants re-phased significantly faster than wildtype controls, possibly related to the rapid loss of Per1-mediated repression observed in Tsc2−/− cells (Figure 6D).
Lowering Bmal1 Genetic Dose Rescues Circadian Phenotypes in Tsc2+/− Mice
We surmised that elevated Per1 levels in Tsc-deficient mice may result from aberrantly elevated BMAL1 protein levels. This led us to the logical hypothesis that a decrease in BMAL1 could potentially ameliorate the over-exuberant resetting phenotype seen in the Tsc2+/− mice. We tested double heterozygous Tsc2+/−;Bmal1+/− mice compared to Tsc2+/+ and Tsc2+/− littermates (and Bmal1+/−). Tsc2+/−;Bmal1+/− cortices expressed about half the level of BMAL1 with marked differences in the nuclear fraction compared to wildtype or Tsc2+/− littermates (Figure 7E). Remarkably, while Tsc2+/− mice again demonstrated a shortened free-running period and rapid shifting in response to advanced light schedules, Tsc2+/−;Bmal1+/− double heterozygotes did not (Figure 7D,F). Moreover, the average free-running period in DD conditions of double heterozygotes did not differ from that of wildtype mice (Figure 7G). We conclude that a lowered dose of Bmal1 normalizes the circadian phenotypes in Tsc2+/− mice implying that the Tsc2+/−phenotype results from excessive BMAL1 expression in these animals.
DISCUSSION
Here we have provided several lines of evidence that demonstrate that mouse models of the neurodevelopmental disorder Tuberous Sclerosis Complex – a paradigmatic mTORopathy – have circadian rhythm abnormalities attributable to deregulated proteostasis of the core circadian clock protein BMAL1. Previous studies in flies and mice have implicated Tsc1/2 and mTOR in clock regulation(Zheng and Sehgal, 2010) (Cao et al., 2013), however mechanisms linking deregulated mTOR signaling in disease models have not been reported. We have found that Tsc2+/− heterozygotes have shortened free-running circadian periods in constant conditions and brain-specific Tsc1 knockout mice demonstrate a loss of circadian temperature rhythms in constant conditions. Interestingly, in both animal models, circadian rhythms are restored by light/dark cycles suggesting that the presence of TSC1/2 activity is not essential for entrainment but rather regulates autonomous properties of the clock mechanism.
We have demonstrated that in Tsc2 knockout cells, BMAL1 translation is increased and its degradation decreased. Recent studies have implicated eIF4E phosphorylation in the translational control of mPER1 and mPER2 but not BMAL1(Cao et al., 2015). The mTOR pathway regulates the translation of thousands of genes, especially those with terminal 5′ oligopyrimidine (TOP) motifs(Thoreen et al., 2012). BMAL1 has not been previously reported among the targets of mTOR-mediated translation, possibly because target identification in large-scale profiling experiments are not geared toward targets of greatest functional relevance but rather those that undergo the greatest degree of change. Another study in liver suggested that BMAL1 protein expression is largely attributable to regulation of its cognate transcript(Janich et al., 2015). It is possible however, that the genomic-wide analysis of wildtype mice might belie the deregulation of BMAL1 translation we have observed in Tsc1/2 mutant backgrounds suggesting that the mechanism we have elucidated may be specific to a background of constitutive mTOR over-activation.
We find that the mTOR pathway regulates the ubiquitination of BMAL1 perhaps by regulating its association with UBE3A, a culprit gene for Angelman syndrome. Interestingly, maternally imprinted Ube3A mouse mutants appear to demonstrate subtle lengthening of circadian period and rapid phase resetting – the former is similar and the latter divergent from the phenotypes we observe in the TSC mouse models(Ehlen et al., 2015; Shi et al., 2015). Thus, while our findings highlight an unexpected potential molecular interaction between Angelman’s syndrome and TSC through the regulation of BMAL1 proteostasis, it is likely that these interactions are likely complex and are consistent with the only partial overlap (e.g. epilepsy and intellectual disability) in phenotypic similarities between these syndromes(Buiting et al., 2016; Robinson-Shelton and Malow, 2016).
Our study is consistent with emerging models of coordinated regulation of anabolic and catabolic aspects of proteostasis by the mTOR pathway and enables a potential framework for understanding how circadian clock control may integrate with nutrient signaling through BMAL1 proteostasis(Zhao and Goldberg, 2016). We do not yet understand how mTOR signaling regulates the ubiquitination of BMAL1. An attractive hypothesis is that mTOR signaling regulates various aspects of BMAL1 phosphorylation, which may in turn affect its recognition by ubiquitin-modifying enzymes such as UBE3A. Future work is required to further elucidate these mechanisms.
We have previously found that BMAL1 regulates translation by binding to the cap binding machinery in response to S6K1-mediated phosphorylation(Lipton et al., 2015). Taken together with our present findings, we speculate that the mTOR pathway regulates the proteostasis of BMAL1 as well as its phosphorylation, thereby controlling both the levels of BMAL1 and its function in the cell. These findings could support a feedforward model in which mTOR regulates the translation of BMAL1 and its subsequent function through phosphorylation. Indeed, we observe an increase in protein synthesis in Tsc2−/− cells with amplified peaks without a clear loss of rhythmicity in translation (Figure S5) suggesting the existence of a preserved, yet unidentified negative feedback mechanism which maintain translational oscillations even under conditions of mTOR over-activation.
Mutations in the mTOR pathway have been repeatedly implicated in neurodevelopmental diseases such as autism(Lipton and Sahin, 2014). Our study provides evidence implicating the same pathway in the control of circadian timekeeping and adds to a growing body of work suggests that circadian dysregulation may be a common feature of neurodevelopmental syndromes(Bourgeron, 2007; Ogawa et al., 2007; Tordjman et al., 2015). We show that the normalization of deregulated translation of a core clock protein BMAL1 heralds the circadian system as not only a source of potential liability in neurodevelopmental disease but also as a rational opportunity for treating it.
EXPERIMENTAL PROCEDURES
Cell Lines and Culture
All cells were maintained in basal media (DMEM/10%FBS/Pen-Strep/2mM Glutamine) unless otherwise specified at 37°C in 5% CO2. Tsc1 and Tsc2 MEFs were kindly provided by the laboratory of Dr. David Kwiatkowski (Dana-Farber Cancer Center, Brigham and Women’s Hospital, Harvard Medical School). Bmal1:Luciferase/U2OS and Per2:Luciferase cells were generously provided by Dr. John Hogenesch (University of Pennsylvania). Rapamycin (LC laboratories) or Torin 1 (Tocris) or Bortezomib (AG Scientific) dissolved in DMSO.
Animal Studies
All animal studies were performed with approval of the Children’s Hospital Boston Animal Care and Use Committee. All animals used were adult males under 6 months of age.
Synchronization
Synchronization was performed by treating cells with either DMEM/50% FBS or 100nM dexamethasone for 2 hours followed by replacement with basal media. The time of replacement media was designated clock time (or zeitgeber time (ZT)) 0.
BONCAT
For metabolic labeling of MEFs, 2 million cells were plated in basal media overnight in 10cm plates. Cells were washed once with warm pre-warmed PBS and then incubated in Met/Cys-free media (Life Technologies) supplemented with 10% dialyzed fetal calf serum for 30 minutes. 50μM L-AHA (Click Chemistry Tools) was added for 35 minutes to metabolically label cells. Cells were then washed twice with ice cold PBS and harvested with a cell lifter in 1mL of ice cold PBS and placed immediately on ice. Cells were then pelleted for 5 minutes at 1000rpm at 4°C. Cells were lysed in 20mM HEPES, 150mM NaCl, 1% NP-40, freshly supplemented with EDTA-free Complete protease inhibitor (Roche, 1 pill per 10mL buffer) and incubated on ice for 10 minutes. Lysates were cleared for 10 minutes at 16000rpm at 4°C, and supernatants retrieved and normalized for total protein.
To normalized lysates, copper-mediated cycloaddition was performed by incubating lysate with 20μM biotin-PEG4-alkyne (Click Chemistry Tools) was added with 50μM tris-hydroxypropyltriazolylmethylamine (THPTA) (Click Chemistry Tools) ligand, 50μM copper(II) sulfate, and 300μM sodium ascorbate (Sigma) as a reducing agent at room temperature protected from light for one hour. Reactions were placed over pre-washed high-affinity streptavidin coated agarose beads for 4 hours at 4°C. Beads were washed 5 times with lysis buffer and proteins were eluted by incubation with 2× Laemmli buffer and boiling for 5 minutes. Eluates were then analyzed by SDS-PAGE.
Immunoprecipitation and ubiquitination
MEFs were maintained in basal media overnight. Cells were treated with fresh media containing 100nM bortezomib (AG Scientific), 250 nM Torin I (Tocris), or DMSO for four hours. Cells were then washed twice with ice cold PBS. Cells were lifted from plates in 500μL of lysis buffer (50mM HEPES, 150mM sodium chloride, 10mg/mL sodium deoxycholate, 0.1% SDS, 1% NP-40) supplemented with freshly made 2mM N-ethylmaleimide (NEM) (Thermo) and EDTA-free protease inhibitor (Thermo), phosphatase inhibitor (Thermo), and 0.5μL Benzonase (Santa Cruz) and then incubated for 5 minutes on ice. Cells were then centrifuged at 16000 G for 5 minutes at 4°C. Lysates were normalized against a standard curve with bicinchonic acid assay (Bio-Rad). 2mg of total protein was used for immunoprecipitations. 10% was reserved as “input”. Lysates were pre-cleared for 10 minutes with 10μL of pre-rinsed DynaBead Protein G magnetic beads. Lysates were placed in a fresh tube with 3μg of either BMAL1 mouse monoclonal antibody (Santa-Cruz #365635) or normal mouse IgG (Santa Cruz) and placed in an end-over-end shaker overnight at 4°C. Immunocomplexes were immobilized on pre-rinsed 50μL of magnetic beads/sample for 1 hours. Beads were then washed four times with ice cold lysis buffer supplemented with 2mM NEM. Immunocomplexes were eluted by addition of 60μL of 2× Laemmli buffer and boiling for 5 minutes at 95°C and the unbound fraction was processed by immunoblotting.
Luminescence
Full length firefly luciferase was amplified from the pGL3-Control vector (Promega) and subcloned in frame at the EcoRV site of CMV-FLAG-BMAL1/pcDNA3.1(Lipton et al., 2015) by Gibson cloning. The construct was verified by sequencing. 2μg of DNA was transfected into Tsc2 wildtype or knockout MEFs with polyethylenimine (PEI) by incubating 3mg/mL of PEI per μg of DNA (Polysciences Inc.). MEFs were maintained in basal media until they were plated in 24 well plates at a density of 5 × 104 cells per well. The following day, basal media was replaced with “Lumicycle media” (phenol-free DMEM (Sigma D-2902), 4mM sodium bicarbonate, 20mM glucose, 10% FBS, freshly supplemented with 100μM beetle luciferin (Promega) and 100μg/mL of cycloheximide and placed in a Lumicycle 96 (Actimetrics) at 35°C. Half life and rate constant (k) were calculated with a first-order exponential decay equation: ln(Nt/N0)=−kt where k is the rate constant.
Preparation of Cytoplasmic Brain Lysates
Freshly harvested brain was washed twice in PBS and flash frozen in liquid nitrogen. ~100g of tissues was cut with an ice cold razor blade and placed in a Dounce homogenizer containing 1mL cold lysis buffer (50mM Tris-HCl, pH 7.4, 3mM calcium chloride, 1mM magnesium chloride, 0.25M sucrose, PhosStop and EDTA-free Complete inhibitors). Tissue was homogenized with 6–8 turns of a loose pestle a 6–8 turns of the tight pestle. The lysate was transferred to a 15mL conical tube containing another 3mL of lysis buffer and briefly vortexed at low setting. The sample was centrifuge at 4°C at 1000G. Supernatant was retrieved and saved as the cytoplasmic portion.
Statistical Methods
Statistical analysis was performed with GraphPad Prism 6.0 (GraphPad Software). Throughout the manuscript, the distribution of data points is expressed as mean ± SEM unless otherwise noted. Student t test for comparison of two conditions or ANOVAs were utilized with post hoc Bonferroni’s multiple-comparisons test for three or more conditions. p < 0.05 was considered significant.
Supplementary Material
HIGHLIGHTS.
TSC mouse models and cells demonstrate abnormal circadian behavior and physiology
Increased BMAL1 protein in Tsc knockout cells and animals results from defective proteostasis
Genetically lowering BMAL1 expression rescued circadian phenotypes in Tsc2+/− mice
Acknowledgments
We thank D. Kwiatkowski (Harvard Medical School) for providing critical reagents. This work was supported by the American Academy of Neurology Clinical Research Training Program, the Tuberous Sclerosis Alliance, the William Randolph Hearst Foundation, the American Sleep Medicine Foundation Physician Scientist Training Award, and the NICHD (K08 HD071026)(to J.O.L.) and the NINDS (R01 NS058956), John Merck Fund, Nancy Lurie Marks Family Foundation, and the Children’s Hospital Boston Translational Research Program (to M.S.). We thank S. Goldman and J. Leech for technical assistance and T. Archer and E. Mahoney for preparation of DNA clones.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conceptualization: J.O.L. and M.S. Methodology: J.O.L. and M.S. Software Programming: A.S.. Investigation: L.B, E.Y., K.H., F.C., P.T. and J.O.L.. Resources: J.O.L., F.D., M.S. Writing: J.O.L. and M.S. (original draft preparation). J.O.L., L.B., K.H., E.Y., P.T., F.D. (Review and Editing). Funding Acquisition: J.O.L. and M.S. Supervision: J.O.L. and M.S.
References
- Borbely AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204. [PubMed] [Google Scholar]
- Bourgeron T. The possible interplay of synaptic and clock genes in autism spectrum disorders. Cold Spring Harb Symp Quant Biol. 2007;72:645–654. doi: 10.1101/sqb.2007.72.020. [DOI] [PubMed] [Google Scholar]
- Bruni O, Cortesi F, Giannotti F, Curatolo P. Sleep disorders in tuberous sclerosis: a polysomnographic study. Brain Dev. 1995;17:52–56. doi: 10.1016/0387-7604(94)00118-h. [DOI] [PubMed] [Google Scholar]
- Buhr ED, Takahashi JS. Molecular components of the Mammalian circadian clock. Handbook of experimental pharmacology. 2013:3–27. doi: 10.1007/978-3-642-25950-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buiting K, Williams C, Horsthemke B. Angelman syndrome - insights into a rare neurogenetic disorder. Nat Rev Neurol. 2016;12:584–593. doi: 10.1038/nrneurol.2016.133. [DOI] [PubMed] [Google Scholar]
- Cao R, Anderson FE, Jung YJ, Dziema H, Obrietan K. Circadian regulation of mammalian target of rapamycin signaling in the mouse suprachiasmatic nucleus. Neuroscience. 2011;181:79–88. doi: 10.1016/j.neuroscience.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Gkogkas CG, de Zavalia N, Blum ID, Yanagiya A, Tsukumo Y, Xu H, Lee C, Storch KF, Liu AC, et al. Light-regulated translational control of circadian behavior by eIF4E phosphorylation. Nat Neurosci. 2015;18:855–862. doi: 10.1038/nn.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Lee B, Cho HY, Saklayen S, Obrietan K. Photic regulation of the mTOR signaling pathway in the suprachiasmatic circadian clock. Mol Cell Neurosci. 2008;38:312–324. doi: 10.1016/j.mcn.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Li A, Cho HY, Lee B, Obrietan K. Mammalian target of rapamycin signaling modulates photic entrainment of the suprachiasmatic circadian clock. J Neurosci. 2010;30:6302–6314. doi: 10.1523/JNEUROSCI.5482-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Robinson B, Xu H, Gkogkas C, Khoutorsky A, Alain T, Yanagiya A, Nevarko T, Liu AC, Amir S, Sonenberg N. Translational control of entrainment and synchrony of the suprachiasmatic circadian clock by mTOR/4E-BP1 signaling. Neuron. 2013;79:712–724. doi: 10.1016/j.neuron.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornu M, Oppliger W, Albert V, Robitaille AM, Trapani F, Quagliata L, Fuhrer T, Sauer U, Terracciano L, Hall MN. Hepatic mTORC1 controls locomotor activity, body temperature, and lipid metabolism through FGF21. Proc Natl Acad Sci U S A. 2014;111:11592–11599. doi: 10.1073/pnas.1412047111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leersnyder H. Inverted rhythm of melatonin secretion in Smith-Magenis syndrome: from symptoms to treatment. Trends Endocrinol Metab. 2006;17:291–298. doi: 10.1016/j.tem.2006.07.007. [DOI] [PubMed] [Google Scholar]
- De Leersnyder H, Claustrat B, Munnich A, Verloes A. Circadian rhythm disorder in a rare disease: Smith-Magenis syndrome. Mol Cell Endocrinol. 2006;252:88–91. doi: 10.1016/j.mce.2006.03.043. [DOI] [PubMed] [Google Scholar]
- Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT) Proc Natl Acad Sci U S A. 2006;103:9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Sahin M. Autism and the synapse: emerging mechanisms and mechanism-based therapies. Curr Opin Neurol. 2015;28:91–102. doi: 10.1097/WCO.0000000000000186. [DOI] [PubMed] [Google Scholar]
- Ehlen JC, Jones KA, Pinckney L, Gray CL, Burette S, Weinberg RJ, Evans JA, Brager AJ, Zylka MJ, Paul KN, et al. Maternal Ube3a Loss Disrupts Sleep Homeostasis But Leaves Circadian Rhythmicity Largely Intact. J Neurosci. 2015;35:13587–13598. doi: 10.1523/JNEUROSCI.2194-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Sathyanarayanan S, Sehgal A. Post-translational regulation of the Drosophila circadian clock requires protein phosphatase 1 (PP1) Genes Dev. 2007;21:1506–1518. doi: 10.1101/gad.1541607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeney KA, Hansen LL, Putker M, Olivares-Yanez C, Day J, Eades LJ, Larrondo LF, Hoyle NP, O’Neill JS, van Ooijen G. Daily magnesium fluxes regulate cellular timekeeping and energy balance. Nature. 2016;532:375–379. doi: 10.1038/nature17407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto CL, Broadie K. The fragile X mental retardation protein in circadian rhythmicity and memory consolidation. Mol Neurobiol. 2009;39:107–129. doi: 10.1007/s12035-009-8057-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossan NC, Zhang F, Guo B, Jin D, Yoshitane H, Yao A, Glossop N, Zhang YQ, Fukada Y, Meng QJ. The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic Acids Res. 2014;42:5765–5775. doi: 10.1093/nar/gku225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock E, O’Callaghan F, English J, Osborne JP. Melatonin excretion in normal children and in tuberous sclerosis complex with sleep disorder responsive to melatonin. J Child Neurol. 2005a;20:21–25. doi: 10.1177/08830738050200010301. [DOI] [PubMed] [Google Scholar]
- Hancock E, O’Callaghan F, Osborne JP. Effect of melatonin dosage on sleep disorder in tuberous sclerosis complex. J Child Neurol. 2005b;20:78–80. doi: 10.1177/08830738050200011302. [DOI] [PubMed] [Google Scholar]
- Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035. doi: 10.1038/nrdp.2016.35. [DOI] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Ramsey KM, Marcheva B, Bass J. Circadian rhythms, sleep, and metabolism. J Clin Invest. 2011;121:2133–2141. doi: 10.1172/JCI46043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt A. Development, behaviour and seizures in 300 cases of tuberous sclerosis. J Intellect Disabil Res. 1993;37(Pt 1):41–51. doi: 10.1111/j.1365-2788.1993.tb00868.x. [DOI] [PubMed] [Google Scholar]
- Hunt A, Stores G. Sleep disorder and epilepsy in children with tuberous sclerosis: a questionnaire-based study. Dev Med Child Neurol. 1994;36:108–115. doi: 10.1111/j.1469-8749.1994.tb11819.x. [DOI] [PubMed] [Google Scholar]
- Jagannath A, Butler R, Godinho SI, Couch Y, Brown LA, Vasudevan SR, Flanagan KC, Anthony D, Churchill GC, Wood MJ, et al. The CRTC1-SIK1 pathway regulates entrainment of the circadian clock. Cell. 2013a;154:1100–1111. doi: 10.1016/j.cell.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannath A, Peirson SN, Foster RG. Sleep and circadian rhythm disruption in neuropsychiatric illness. Curr Opin Neurobiol. 2013b;23:888–894. doi: 10.1016/j.conb.2013.03.008. [DOI] [PubMed] [Google Scholar]
- Janich P, Arpat AB, Castelo-Szekely V, Lopes M, Gatfield D. Ribosome profiling reveals the rhythmic liver translatome and circadian clock regulation by upstream open reading frames. Genome Res. 2015;25:1848–1859. doi: 10.1101/gr.195404.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouffe C, Cretenet G, Symul L, Martin E, Atger F, Naef F, Gachon F. The circadian clock coordinates ribosome biogenesis. PLoS Biol. 2013;11:e1001455. doi: 10.1371/journal.pbio.1001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khapre RV, Patel SA, Kondratova AA, Chaudhary A, Velingkaar N, Antoch MP, Kondratov RV. Metabolic clock generates nutrient anticipation rhythms in mTOR signaling. Aging (Albany NY) 2014;6:675–689. doi: 10.18632/aging.100686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Minowa O, Kuno J, Mitani H, Hino O, Noda T. Renal carcinogenesis, hepatic hemangiomatosis, and embryonic lethality caused by a germ-line Tsc2 mutation in mice. Cancer Res. 1999;59:1206–1211. [PubMed] [Google Scholar]
- Kobayashi T, Minowa O, Sugitani Y, Takai S, Mitani H, Kobayashi E, Noda T, Hino O. A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc Natl Acad Sci U S A. 2001;98:8762–8767. doi: 10.1073/pnas.151033798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- Lipton JO, Sahin M. The neurology of mTOR. Neuron. 2014;84:275–291. doi: 10.1016/j.neuron.2014.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton JO, Yuan ED, Boyle LM, Ebrahimi-Fakhari D, Kwiatkowski E, Nathan A, Guttler T, Davis F, Asara JM, Sahin M. The Circadian Protein BMAL1 Regulates Translation in Response to S6K1-Mediated Phosphorylation. Cell. 2015;161:1138–1151. doi: 10.1016/j.cell.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheva B, Ramsey KM, Peek CB, Affinati A, Maury E, Bass J. Circadian clocks and metabolism. Handbook of experimental pharmacology. 2013:127–155. doi: 10.1007/978-3-642-25950-0_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28:5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, Kwiatkowski DJ. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27:5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie D, Di Nardo A, Han JM, Baharanyi H, Kramvis I, Huynh T, Dabora S, Codeluppi S, Pandolfi PP, Pasquale EB, Sahin M. Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat Neurosci. 2010;13:163–172. doi: 10.1038/nn.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S, Kwon CH, Zhou J, Koovakkattu D, Parada LF, Sinton CM. A seizure-prone phenotype is associated with altered free-running rhythm in Pten mutant mice. Brain Res. 2007;1168:112–123. doi: 10.1016/j.brainres.2007.06.074. [DOI] [PubMed] [Google Scholar]
- Oliver PL, Sobczyk MV, Maywood ES, Edwards B, Lee S, Livieratos A, Oster H, Butler R, Godinho SI, Wulff K, et al. Disrupted circadian rhythms in a mouse model of schizophrenia. Curr Biol. 2012;22:314–319. doi: 10.1016/j.cub.2011.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson-Shelton A, Malow BA. Sleep Disturbances in Neurodevelopmental Disorders. Curr Psychiatry Rep. 2016;18:6. doi: 10.1007/s11920-015-0638-1. [DOI] [PubMed] [Google Scholar]
- Sahar S, Zocchi L, Kinoshita C, Borrelli E, Sassone-Corsi P. Regulation of BMAL1 protein stability and circadian function by GSK3beta-mediated phosphorylation. PloS one. 2010;5:e8561. doi: 10.1371/journal.pone.0008561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SQ, Bichell TJ, Ihrie RA, Johnson CH. Ube3a imprinting impairs circadian robustness in Angelman syndrome models. Curr Biol. 2015;25:537–545. doi: 10.1016/j.cub.2014.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 2016;18:164–179. doi: 10.1038/nrg.2016.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tordjman S, Davlantis KS, Georgieff N, Geoffray MM, Speranza M, Anderson GM, Xavier J, Botbol M, Oriol C, Bellissant E, et al. Autism as a disorder of biological and behavioral rhythms: toward new therapeutic perspectives. Front Pediatr. 2015;3:1. doi: 10.3389/fped.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eeghen AM, Numis AI, Staley BA, Therrien SE, Thibert RL, Thiele EA. Characterizing sleep disorders of adults with tuberous sclerosis complex: a questionnaire-based study and review. Epilepsy Behav. 2011;20:68–74. doi: 10.1016/j.yebeh.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff K, Gatti S, Wettstein JG, Foster RG. Sleep and circadian rhythm disruption in psychiatric and neurodegenerative disease. Nat Rev Neurosci. 2010;11:589–599. doi: 10.1038/nrn2868. [DOI] [PubMed] [Google Scholar]
- Yu X, Zecharia A, Zhang Z, Yang Q, Yustos R, Jager P, Vyssotski AL, Maywood ES, Chesham JE, Ma Y, et al. Circadian factor BMAL1 in histaminergic neurons regulates sleep architecture. Curr Biol. 2014;24:2838–2844. doi: 10.1016/j.cub.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nicholatos J, Dreier JR, Ricoult SJ, Widenmaier SB, Hotamisligil GS, Kwiatkowski DJ, Manning BD. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature. 2014;513:440–443. doi: 10.1038/nature13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Goldberg AL. Coordinate regulation of autophagy and the ubiquitin proteasome system by MTOR. Autophagy. 2016;12:1967–1970. doi: 10.1080/15548627.2016.1205770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhai B, Gygi SP, Goldberg AL. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc Natl Acad Sci U S A. 2015;112:15790–15797. doi: 10.1073/pnas.1521919112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Sehgal A. AKT and TOR signaling set the pace of the circadian pacemaker. Curr Biol. 2010;20:1203–1208. doi: 10.1016/j.cub.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.