

Abstract
RNA–RNA assembly governs key biological processes and is a powerful tool for engineering synthetic genetic circuits. Characterizing RNA assembly in living cells often involves monitoring fluorescent reporter proteins, which are at best indirect measures of underlying RNA–RNA hybridization events and are subject to additional temporal and load constraints associated with translation and activation of reporter proteins. In contrast, RNA aptamers that sequester small molecule dyes and activate their fluorescence are increasingly utilized in genetically encoded strategies to report on RNA-level events. Split-aptamer systems have been rationally designed to generate signal upon hybridization of two or more discrete RNA transcripts, but none directly function when expressed in vivo. We reasoned that the improved physiological properties of the Broccoli aptamer enable construction of a split-aptamer system that could function in living cells. Here we present the Split-Broccoli system, in which self-assembly is nucleated by a thermostable, three-way junction RNA architecture and fluorescence activation requires both strands. Functional assembly of the system approximately follows second-order kinetics in vitro and improves when cotranscribed, rather than when assembled from purified components. Split-Broccoli fluorescence is digital in vivo and retains functional modularity when fused to RNAs that regulate circuit function through RNA–RNA hybridization, as demonstrated with an RNA Toehold switch. Split-Broccoli represents the first functional split-aptamer system to operate in vivo. It offers a genetically encoded and nondestructive platform to monitor and exploit RNA–RNA hybridization, whether as an all-RNA, stand-alone AND gate or as a tool for monitoring assembly of RNA–RNA hybrids.
Keywords: binary aptamer, fluorescence complementation, RNA interaction, RNA AND gate, RNA synthetic biology, cotranscriptional RNA assembly
RNA–RNA interactions drive key processes in biology, such as RNA interference,1 retroviral genome dimerization,2 CRISPR-Cas activation3 and targeting,4 and post-transcriptional regulation of RNA by long noncoding RNAs.5 RNA is also increasingly utilized as a programmable element for the control of gene expression and cellular function within the field of synthetic biology.6,7 Riboregulators,8 Toehold switches,9 Small Transcription Activating RNAs (STARs),10 and other ribodevices exploit the hybridization of a trans-acting RNA with a target mRNA to control gene expression at either the transcriptional or translational level. Nevertheless, the toolkit for monitoring RNA–RNA hybridization events as they occur in real time is limited, relying primarily on fluorescent reporter proteins. However, this approach is indirect, as the downstream measurements are subject to additional variables related to translation, time delays related to fluorophore maturation, and additional resource burdens on the system. Monitoring RNA–RNA hybridization directly through an RNA-based system is therefore highly desirable. RNA-based systems have small genetic footprints and they offer improved temporal sensitivity, as signal amplification and decay are governed by transcription, degradation and assembly rates of the RNA. Furthermore, genetic circuits with a directly reportable output driven by RNA–RNA hybridization may provide an alternative detection strategy for cell-based or cell-free diagnostics based entirely on transcription.11
Intracellular RNA visualization strategies roughly fall into two categories: those that are genetically encoded and those that rely on chemically labeled probes.12 Genetically encoded strategies allow for nondestructive analysis in real time, unlike chemical probes that require exogenous introduction and may thereby perturb the system. Several encoded systems for RNA visualization have been developed that utilize RNA-protein interactions, such as those found in the bacteriophages MS2 and PP7. RNA hairpin motifs in these phage genomes serve as binding sites for phage coat proteins. When these motifs are inserted into an RNA of interest they can recruit coat proteins that have been fused to fluorescent reporter proteins, allowing for indirect visualization of the RNA.13,14 However, these systems require the insertion of up to 24 copies (∼1200 nucleotides) of the binding motif and the use of export and localization signals to overcome diffuse fluorescence arising from unbound fusion proteins. While some of the limitations of protein-based RNA visualization approaches can be mitigated by fluorescence complementation assays—which utilize split-fluorescent proteins that only fluoresce when brought into proximity through paired RNA binding motifs—these approaches still require proteins.
Genetically encoded RNA aptamers that bind and activate the fluorescence of small molecule dyes have seen increasing use in monitoring RNA directly without involving reporter proteins. Aptamers are single-stranded, functional nucleic acids that fold into three-dimensional structures to bind defined molecular targets.15 Several RNA aptamers have been selected against small molecule dyes for which fluorescence is activated upon binding and sequestration within the aptamer.16−18 While the malachite green aptamer (MGA)16 was the first to demonstrate such fluorescence activation, the recent selection of the Spinach aptamer,17 which binds to a freely diffusible and nontoxic dye (DFHBI), has resulted in a surge of publications reporting its use in a wide range of contexts, such as serving as the output for engineered genetic circuits,19,20 as a tool to monitor RNA transcription,21,22 and as a fluorescent sensor for metabolites.23,24 Several improvements to the original Spinach aptamer have been developed through directed evolution strategies,25−27 including the Broccoli aptamer, which was selected in vivo for improved physiological properties.28 The utility of these aptamers was further expanded by the introduction of DFHBI-1T, a brighter aptamer-activatable fluorophore compatible with the Spinach family of aptamers and optimized for conventional green fluorescence excitation and emission spectra used for GFP detection.29 Nonetheless, while several direct visualization strategies for RNA exist, including those that operate exclusively as RNA, none is designed specifically for visualization of RNA–RNA interactions.
We reasoned that RNA assembly processes could be monitored directly through the use of a split-aptamer system analogous to the split fluorescent proteins that have been used extensively to monitor cosynthesis, colocalization, and assembly of proteins. Split fluorescent aptamers could provide similar information for RNA, as they would only generate signal upon cosynthesis, colocalization, and hybridization of the component nucleic acids to form the functional aptamer. Indeed, split versions of MGA30 and Spinach31−33 aptamers have been developed for use in vitro. However, these systems have not been directly observed to function when transcribed in vivo. The triphenylmethane dye that binds to MGA is toxic, therefore limiting its use in living systems, and the Spinach aptamers are limited by weak signal in vivo. Multimerization can increase signal strength, but can also result in interference between the different aptamer modules. Several approaches to multimerizing or splitting functional nucleic acids utilize three-way junction (3WJ) architectures,34−38 which are replete throughout biology.39 Because the Broccoli aptamer exhibits improved physiological properties, such as lowered dependence on magnesium and higher folding efficiency at 37 °C,28 we reasoned that it would serve as an ideal platform for an improved split-aptamer system. We further reasoned that multimerization of the aptamer would overcome potential issues of low signal and that a self-assembling and thermodynamically stable RNA motif, such as a 3WJ, would allow for robust fluorescence complementation.
Here, we present the binary “Split-Broccoli” system as a stand-alone RNA logic gate and as a device for monitoring RNA–RNA hybridization in vitro and in vivo. The design–build–test cycle iterates from in silico design to in vitro implementation and finally to in vivo functionality. The structure-based strategy begins with an unsplit dimeric aptamer within a stabilizing RNA architecture and continues with its bisection into a two-component system. The system assembles reliably in vitro when purified components are thermally renatured, and the addition of transcription terminator structures improves the “OFF” level of the system. At physiological temperature, in vitro assembly approximately follows second-order kinetics but appears to be limited by kinetic traps that prevent fast refolding into the functional hybrid. However, when individual RNAs of the system are cotranscribed in vitro, fluorescence signal strength roughly approximates the unsplit variant over a 4-h time course. The Split-Broccoli system also assembles and activates fluorescence when expressed in vivo, whether as a stand-alone AND gate or as a tool to monitor an RNA–RNA hybridization event that drives translation of a red fluorescent protein. We anticipate that the Split-Broccoli system is an enabling addition to the toolkit for RNA biologists.
Results
Designs for Stabilized Broccoli Aptamers and the Split-Broccoli System
The published monomeric and dimeric forms of the Broccoli aptamer (mBroccoli and dBroccoli) served as starting points for engineering three dimeric forms of Broccoli aptamer.28 First, while the Spinach series of aptamers17,25 are typically embedded within a tRNALys3 scaffold40 to stabilize folding and in vivo accumulation, there are divergent reports as to whether Broccoli’s fluorescence activation requires transcription within a tRNA context.27,28,41 We therefore compared the performance of mBroccoli to dBroccoli without additional sequence and also to the Spinach2 aptamer embedded within a tRNALys3 scaffold (tSpinach2). Fluorescence from mBroccoli was equivalent to that of tSpinach2, but dBroccoli alone, without additional stabilizing base pairs, yielded approximately 25% of signal relative to mBroccoli (Figure S1). This represents an 8-fold reduction from the expected doubling, consistent with its predicted suboptimal folding (Figure S2). Alternative designs for stabilizing Broccoli dimer without greatly increasing transcript size were guided by RNA secondary structure predictions using the web-based NUPACK software suite.42 Adding 3 terminal G:C base pairs to the terminal stem—but not 1 or 2 G:C base pairs or 3 pairs that included A:U pairs—provided adequate stabilization to favor the expected structure, but stabilization by a mixed 4 base pair stem (GAGG:CCTC) exceeded that of the other designs. When this 4 base pair stem was appended to dBroccoli to form the 100-nucleotide “stabilized dimeric Broccoli” (SdB) (Figure S2d), fluorescence was more than 2-fold enhanced relative to mBroccoli (Figure S3). SdB was therefore used as a reference point for further engineering of the aptamers.
The second dimeric Broccoli aptamer design exploits the 3WJ motif derived from the packaging RNA (pRNA) component of the DNA packaging motor from bacteriophage Φ29. This stable element is a proven architecture for multimerization of functional nucleic acids35,43,44 and self-assembly from oligonucleotides.45−47 We therefore inserted Broccoli monomers into arms 2 and 3 of the 3WJ to form 3WJdB (Figure 1a). Because these arms extend in opposite directions from the 3WJ structural core in crystal structures of the Φ29 3WJ pRNA,48 this design is expected to keep the monomer units spatially distant and to minimize intersubunit misfolding or potential quenching that can arise from chromophore–chromophore interactions. While the work reported here was under way, Filonov et al. reported a similar design (“F30–2xBroccoli”) utilizing the same 3WJ element from Φ29 pRNA, but with Broccoli monomers inserted into arms 1 and 2.49 Our 3WJdB design incorporates a single nucleotide substitution (C → U) and conversion of a U:A base pair to an A:U pair, as Filonov et al. observed that these changes improved transcriptional yield of full-length RNA by interrupting a poly uridine tract that may serve as a cryptic transcription terminator.49 However, we also note that these substitutions include an adenosine whose phosphate is implicated in coordinating one of the core metal ions of the 3WJ,48 potentially affecting thermostability of the motif.
Figure 1.
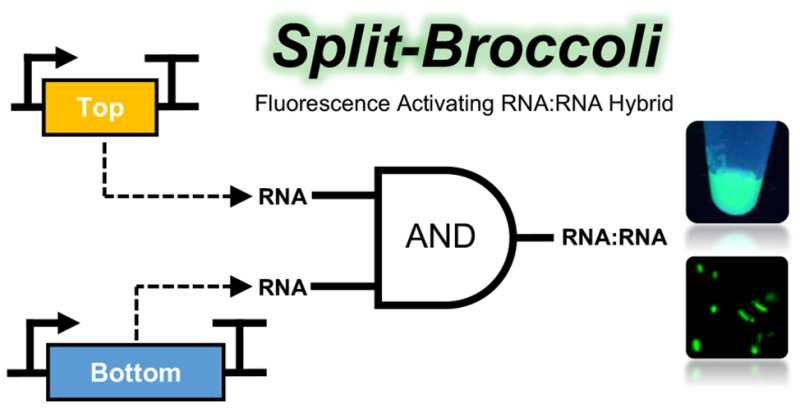
Design and NUPACK predicted secondary structures of 3WJdB and Split-Broccoli. (a) Monomers of Broccoli aptamer (green) were inserted into Arms 2 and 3 of the three-way junction (3WJ) RNA motif (black) to create the unimolecular, unsplit three-way junction dimeric Broccoli (3WJdB). (b) Design of the Split-Broccoli system required inversion of the Broccoli monomer present in Arm 2 to ensure that neither Top (yellow) nor Bottom (blue) strand alone contained the full sequence required to independently form a fully functional monomer of Broccoli (i.e., xn:xn′). Predicted hybridization of the two strands was strengthened with the addition of terminal stems. (c) The Split-Broccoli system illustrated as an RNA AND gate (left) and its corresponding truth table (right). Output from the system should be true (1) only when both inputs are also true.
The third dimeric Broccoli aptamer design bisects 3WJdB after first inverting the monomer in Arm 2 to ensure that neither strand alone would contain the full sequence required to form a functional aptamer. In addition, 8 or 12 base pairs were appended on the two distal stems to favor complete hybridization of the two strands and formation of the desired secondary structure (Figure 1b). The two autonomous RNA strands of the Split-Broccoli system are designated Top and Bottom. Neither strand contains the full sequence required to form a functional Broccoli monomer, and neither is predicted to fold into a secondary structure that resembles a functional monomer (Figure S4a, b). Engineering the Split-Broccoli system in this manner, wherein fluorescence activation (output) is only expected in the presence of both Top and Bottom strands (inputs), creates an AND gate capable of performing and reporting on logical operations entirely as RNA (Figure 1c).
Split-Broccoli Assembles In Vitro to Reconstitute Fluorescence Activation
To evaluate performance of the Split-Broccoli system, we compared two approaches for assembling Split-Broccoli from purified Top and Bottom strands. In the first approach, equimolar amounts of Top and Bottom were mixed and incubated with dye in buffer, then unfolded at 90 °C and renatured by slow cooling to 37 °C prior to measuring fluorescence. The thermally renatured Split-Broccoli exhibited approximately 82% of the signal relative to the unsplit, unimolecular 3WJdB, while neither Top nor Bottom strand alone generated robust fluorescence signal (Figure 2a). Fluorescence signal for the renatured Split-Broccoli was 22-fold greater than Bottom strand alone, which gave less than 4% of signal compared to 3WJdB. Additionally, the 3WJdB design exhibited higher signal when compared to SdB, suggesting that the 3WJ framework reinforces the productive fold better than the minimal stem that stabilizes SdB (Figure 2a). In the second approach, the two strands were first refolded separately and then mixed at 37 °C without renaturation to mimic a more biologically relevant assembly process. Fluorescence signal for the complex was again strongly above background, and little or no fluorescence was observed for either Top or Bottom strand alone (Figure 2b). These patterns were clearly visible when these samples were excited with a standard UV-light source (Figure 2c). For this simple mixing approach, fluorescence from the assembled complex was approximately 60% of the signal from unsplit 3WJdB under the same conditions and was roughly 15-fold over Bottom strand alone, which again generated ∼4% signal over the No RNA background sample (Figure 2b). The data from these two approaches show effective reconstitution of aptamer function from the binary Split-Broccoli system in vitro, with switch-like behavior that serves as a two-input AND gate and fluorescence signal strength that is slightly reduced relative to the unsplit 3WJdB and roughly equivalent to SdB.
Figure 2.

In vitro assembly of the Split-Broccoli system in the absence and presence of transcription terminator structures. Assembly of equimolar amounts of purified Split-Broccoli RNA components (Top + Bottom) demonstrate robust function comparable to fluorescence of the stabilized dimeric Broccoli (SdB) and 3WJdB, (a) when thermally renatured or (b) when simply incubated together at physiological temperature. Background signal from either Top or Bottom alone remains minimal for both assembly methods. (c) Fluorescence of 3WJdB and the Split-System (Top + Bottom, thermally renatured) is easily observed when excited with longwave ultraviolet light, whereas signal from Bottom alone is only slightly discernible. When transcribed with transcription terminator structures (denoted by appending “-T” to the names of the individual RNAs) and assembled in vitro (d, e), the Split-Broccoli system exhibits a decrease in relative fluorescence, but demonstrates a larger fold-change in fluorescence activation over either Top-T or Bottom-T alone. (f) Nondenaturing gel electrophoresis and dual staining with ethidium bromide and DFHBI-1T of the Split-Broccoli system with transcription terminator structures suggests that decreased fluorescence of the system is a result of incomplete hybridization between Top-T and Bottom-T, rather than nonfunctional assembly. (g) Functional assembly of Top-T and Bottom-T approximately follows second-order kinetics (y = A[Top][Bottom] = A[Top]2, for equimolar mixture). Mean values are shown with error bars to indicate standard deviations (n = 5 for panels a, b, d, e; n = 4 for panel g).
Transcription Terminator Structures Enhance Activation Ratio
The RNA species described above were generated by runoff transcription of linear dsDNA templates. In contrast, bacterial Rho-independent transcription termination utilizes a G:C rich, stem-loop structure followed by a U-rich tract that promotes dissociation of the polymerase from the DNA template.50 To characterize the performance of Split-Broccoli in a more biological sequence context, T7 transcription terminator structures were appended to both unsplit and split aptamer transcripts, denoted by appending “-T” to the names of the individual RNAs. Neither Top-T nor Bottom-T was predicted to contain a Broccoli-like secondary structure (Figure S4c,d), whereas hybridization of these two strands was predicted to form the two Broccoli domains flanking the 3WJ motif (Figure S5). Fluorescence from thermally renatured Top-T and Bottom-T strands was 486-fold above signal for either strand alone, representing switch-like, digital behavior for the Split-Broccoli system. In comparison with runoff transcripts, this improvement in activation ratio can be ascribed to the reduction in signal from Bottom-T alone to background No RNA levels, in spite of a decrease in signal from the assembled complex relative to the unsplit 3WJdB-T (∼43%, Figure 2d). Fluorescence activation remained high (186-fold) when the complex was assembled by incubating Top-T and Bottom-T strands at physiological temperature, even though signal strength for the complex dropped to approximately 19% of the unimolecular 3WJdB-T (Figure 2e). Fluorescence signal strength for unsplit 3WJdB and 3WJdB-T were roughly equivalent under both conditions (Figure S6), thus justifying comparison of relative fluorescence values of terminated transcripts to runoff transcripts.
Split-Broccoli Assembly In Vitro Approximately Follows Second-Order Kinetics
To address the molecular basis for limitations to the in vitro assembly of Top-T and Bottom-T strands at physiological temperature, assembly of Split-Broccoli was evaluated using a native gel stained with ethidium bromide, to locate all molecular species, and stained with DFHBI-1T to identify functionally assembled complexes.49 When incubated at 37 °C, each individual strand of the Split-Broccoli system migrated as a single nonfunctional band, and a new functional band appeared when the two strands were assembled together (Figures 2f, S7). However, a substantial amount of input material remains unassembled under these conditions, consistent with the observed decrease in relative fluorescence for the annealed complex upon adding transcription terminator structures (Figure 2e). Annealing efficiency and fluorescence intensity of the system improved when the system was thermally renatured. Although each strand remained independently nonfunctional when renatured, a second, non-functional isoform is evident for thermally renatured Bottom-T that is not evident when Bottom-T was incubated alone at 37 °C, indicating that the terminator hairpins may be cross-annealing to form a dimer of Bottom-T. Together, these results suggest that incomplete, rather than nonfunctional, assembly is responsible for the decrease in signal when purified components of the system are incubated at 37 °C. Robust assembly in vitro may thus be limited by kinetic folding traps within one or both strands that prevent fast refolding into the functional hybrid.
In principle, RNA-based genetic control elements can provide rapid responses in vitro or in vivo because signal development does not require translation. To better understand assembly kinetics, we measured the rate of functional assembly of Split-Broccoli at physiological temperature. Top-T and Bottom-T strands were separately folded, mixed with dye, and mixed with each other at equimolar concentrations. Development of fluorescence signal was monitored as a function of time (Figure S8), and the rates were plotted against input RNA concentrations. Split-Broccoli assembly approximately followed second-order kinetics (Figure 2g). The exponent (1.73) falls slightly below the expected value of 2.00, potentially as a consequence of competition from internal structure within the individual strands, as suggested by the native gel mobility shift assay described above.
Cotranscriptional Assembly of Split-Broccoli Improves Signal
Gel purification of RNA under non-native conditions can introduce conformational traps.51 In contrast, cotranscriptional folding in vivo is a sequential process in which the RNA folds from the 5′ end as synthesis occurs. Furthermore, folding is concurrent with, and can be influenced by, other RNA species that are present. Therefore, to better characterize in vitro the in vivo potential of the Split-Broccoli system, we evaluated cotranscriptional assembly by incubating equimolar amounts of linear dsDNA templates corresponding to 3WJdB-T, Top-T, Bottom-T, or both Top-T and Bottom-T together, and assaying for function in transcription reaction conditions optimized for time resolution (Figure 3). The unimolecular 3WJdB-T exhibited measurable fluorescence within a few minutes of incubation, and Split-Broccoli required approximately ten more minutes as the two strands accumulated to a sufficient concentration to associate with each other at a detectable level. Although the rate of signal development for the unimolecular 3WJdB-T is substantially faster than Split-Broccoli for the first 45 min, the rates are approximately equal for both the unsplit and split aptamers afterward. Additionally, when compared to SdB-T in transcription conditions optimized for RNA production, 3WJdB-T function is slightly improved and goes to completion, suggesting that the inclusion of a 3WJ scaffold does not delay aptamer folding (Figure S9). Cotranscription of the split-aptamer system reconstituted nearly all of the function of 3WJdB (∼88%) and demonstrated a remarkable 124-fold increase in signal relative to Bottom-T strand alone after a 4-h reaction. Cotranscription of the Split-Broccoli system with terminator structures therefore allows for robust functional assembly without the requirement for additional in vitro manipulations, such as thermal renaturation or coincubation of independently transcribed RNA that may be kinetically trapped in alternative, nonfunctional structures.
Figure 3.
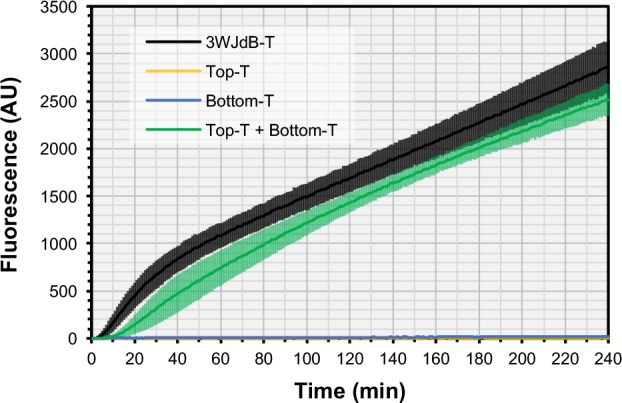
Cotranscriptional assembly of Split-Broccoli with transcription terminator structures improves signal relative to independently transcribed and assembled RNA. When cotranscribed with equimolar amounts of template DNA in a one-pot in vitro transcription reaction designed to maximize time resolution, Split-Broccoli (Top-T + Bottom-T, green) exhibits approximately 88% of the signal generated by the unimolecular, unsplit variant (3WJdB-T, black) after a 4-h reaction. By the final time point at 240 min, Split-Broccoli demonstrates a 124-fold increase over either Top-T (yellow) or Bottom-T (blue) alone. Mean values are shown (n = 4) with error bars to indicate standard deviations.
Split-Broccoli Functions In Vivo
To determine whether the Split-Broccoli system would function in vivo, we constructed expression plasmids for evaluation in Escherichia coli. Transcripts originate from the constitutive P70a promoter52 and were terminated with Rho-independent transcription terminators. Single-insert plasmids direct the transcription of either 3WJdB-T (pUC19-P70a-3WJdB-T), Top-T (pUC19-P70a-Top-T) or Bottom-T (pUC19-P70a-Bottom-T), while a dual-insert plasmid (pUC19-P70a-Top-T∼P70a-Bottom-T) directs transcription of Top-T and Bottom-T, with a 270-nucleotide spacer (∼) between transcription units to reduce topological tension arising from simultaneous transcription (Figure 4a and Supporting Information). As a control, we also included a variant of the dual-insert plasmid that was missing the second P70a promoter upstream of Bottom-T (pUC19-P70a-Top-T∼Bottom-T) to ensure that signal generated by the dual-promoted Split-Broccoli system is not a product of a single runon transcript folding onto itself.
Figure 4.

The Split-Broccoli system functions when expressed in vivo. (a) DNA templates corresponding to 3WJdB-T (black), Top-T (yellow), Bottom-T (blue) were individually cloned into the pUC19 plasmid. A single plasmid expressing both Top-T and Bottom-T was created (pUC19-P70a-Top-T∼P70a-Bottom-T), as was a control plasmid for runon transcription which lacked a promoter immediately upstream of Bottom-T (pUC19-P70a-Top-T∼Bottom-T). (b) A representative flow cytometry histogram of 5 × 104 events per population illustrates a shift in fluorescence for the plasmid containing the Split-Broccoli expression plasmid (green). Bacterial populations transformed with plasmids containing either Top-T or Bottom-T alone, or lacking a promoter upstream of Bottom-T, demonstrate background levels of fluorescence equivalent to the unmodified pUC19 plasmid control. (c) Relative mean fluorescence intensities for flow cytometric analyses of transformed populations, normalized to the pUC19 plasmid (set to 0) and 3WJdB-T (set to 1), are shown with error bars to indicate standard deviations (n ≥ 4). (d) Fluorescence microscopy imaging further validates the in vivo functionality of the Split-Broccoli system, as green fluorescence is only observed for E. coli transformed with either the unimolecular 3WJdB-T encoding plasmid or bimolecular Split-Broccoli encoding plasmid.
Transformed bacteria were grown to mid log phase and their fluorescence was measured by flow cytometry (Figure 4b,c). For the cell population containing the 3WJdB-T insert, mean fluorescence intensity was shifted approximately 25-fold (Figure 4b) relative to pUC19 transformants alone. Importantly, transcribing both Top-T and Bottom-T together generated a notable shift (approximately 6-fold above background) in mean fluorescence intensity. Fluorescence signal from populations containing only Top-T or Bottom-T inserts exhibited background levels of signal equivalent to the unmodified pUC19 plasmid containing no insert, representing a true OFF state, as did populations transformed with a plasmid that lacked the second promoter for transcribing Bottom-T, indicating that transcription of Top-T strand was effectively terminated (Figure 4b,c). These results were further confirmed with fluorescence microscopy (Figure 4d). The Split-Broccoli system therefore acts as a stand-alone, all-RNA AND gate in vivo, with a true OFF state in the absence of both strands and an ON state in their presence.
Split-Broccoli Is Functionally Modular and Can Be Used to Monitor RNA–RNA Assembly In Vivo
RNA molecules assemble with each other to drive many processes in biology. To evaluate whether the Split-Broccoli system could report on such assembly events, we chose to monitor activation of an RNA Toehold switch. Toehold switches are two-component RNA systems for gene regulation in which a trans-acting RNA (“Trigger”) relieves translational repression from a cis-acting hairpin structure (“Toehold”) that lies just upstream of a gene of interest.9 In the absence of a Trigger RNA, the Toehold structure effectively sequesters ribosomal access to the 5′ UTR. Base pairing of Trigger to Toehold relaxes the 5′ UTR hairpin structure, thereby exposing the ribosome binding site and start codon to allow for efficient translation initiation. We fused Top with a well-characterized Toehold that regulates an mCherry reporter protein to generate Top-Toehold-mCherry and we fused Bottom with the corresponding Trigger RNA to generate Trigger-Bottom (Figure 5a,b), and then determined fluorescence for bacteria carrying these constitutively expressing plasmids. Red fluorescence for bacteria encoding mCherry in the OFF state (Top-Toehold-mCherry only) showed minimal red fluorescence, potentially due to incomplete suppression of translation by the Toehold (Figures 5c, S10). Both Top-Toehold-mCherry alone and Trigger-Bottom alone exhibited less than 2% of green fluorescence of the functional Top+Bottom populations. Finally, when the complete Split-Broccoli system was fused with the complete Toehold switch system and expressed as two discrete transcripts (Top-Toehold-mCherry + Trigger-Bottom), both green and red fluorescence were activated in the cell population and observed with flow cytometry (Figures 5c, S10) and fluorescence microscopy (Figure 5d). Split-Broccoli is therefore modular and can be used to monitor RNA–RNA interactions in vivo.
Figure 5.

Split-Broccoli is modular and can be used to monitor RNA–RNA hybridization events in vivo. (a) A Split-Broccoli Toehold Switch plasmid was constructed to include two constitutively expressed transcription units. The first transcription unit encodes Top (yellow) and Toehold (gray) sequences within the 5′ UTR of the mCherry mRNA (red). Translation of the Top-Toehold-mCherry mRNA is suppressed due to sequestration of the ribosome binding site (orange) and start codon within the toehold structure (boxed). The second transcription unit encodes Trigger (gray) and Bottom (blue) sequences, which can base pair with Top-Toehold-mCherry. (b) Hybridization of Top-Toehold-mCherry with Trigger-Bottom allows fluorescence activation of the Split-Broccoli system and translation of mCherry. (c) Green fluorescence (left columns) and red fluorescence (right columns) from flow cytometric analysis of populations show background levels of fluorescence for plasmids encoding a single transcription unit only (Top-Toehold-mCherry or Trigger-Bottom). Top + Bottom, which transcribes the Split-Broccoli system, exhibits only green fluorescence, while the Split-Broccoli Toehold Switch plasmid (Top-Toehold-mCherry + Trigger-Bottom) exhibits both red and green fluorescence, indicating both hybridization of Split-Broccoli and translation of mCherry. Grand mean fluorescence intensity (n = 4) is shown with error bars to indicate standard deviations. (d) Fluorescence microscopy imaging of E. coli harboring the Split-Broccoli Toehold Switch plasmid confirms hybridization of the Top and Bottom components of Split-Broccoli (green fluorescence) and activation of mCherry translation (red fluorescence). (e) An E. coli cell-free system (TX-TL) was used to monitor transcription and translation of the Split-Broccoli Toehold Switch plasmid and demonstrates the increased temporal sensitivity of Split-Broccoli (green fluorescence, left axis) over mCherry (red fluorescence, right axis). Mean values (n = 3) are shown with error bars to indicate standard deviations.
An advantage of RNA-based fluorescent reporters over proteins such as GFP and mCherry is improved temporal sensitivity, as translation and fluorophore maturation would not be required for function. To investigate how quickly Split-Broccoli could be observed relative to a translated gene of interest, we utilized an E. coli cell-free transcription and translation system (TX-TL)52 with the Split-Broccoli Toehold Switch plasmid (Top-Toehold-mCherry + Trigger-Bottom). The TX-TL system is prepared from a crude E. coli cytoplasmic extract and contains the endogenous transcription and translation machinery, allowing for rapid in vitro characterization of synthetic gene circuits with in vivo-like conditions. Green fluorescence from Split-Broccoli was detectable in TX-TL almost immediately and increased rapidly before peaking around 90 min (Figure 5e). In contrast, red fluorescence from mCherry took approximately 45 min to detect and slowly increased over the remaining time course. While the fluorophore maturation time can be improved through the use of alternative fluorescent proteins, the nearly instantaneous response of the Split-Broccoli system demonstrates the speed at which an RNA-based reporter can act.
In this application of the Split-Broccoli system, the RNA–RNA hybridization event self-reports through green fluorescence of the assembled aptamer, while translational activation of the downstream mCherry gene is subsequently self-reported through red fluorescence. Most proteins do not self-report their synthesis and are not so easily monitored in real time. The Top-Toehold-mCherry and Trigger-Bottom design can be readily adapted to those proteins by replacing mCherry with another gene of interest for which translation is less readily monitored. The appearance of green fluorescence upon induction of Trigger-Bottom could then be taken as visual evidence that the gene of interest was being translated. Although we fused Split-Broccoli to an RNA Toehold switch, alternative systems for regulating gene expression could also be used, such as those regulated by Small Transcription Activating RNAs (STARs).10 The hybridization of an antisense STAR to its cognate sense RNA results in the formation of an antiterminator structure that allows transcription of an mRNA to continue. The Split-Broccoli system could therefore be used to monitor the STAR hybridization event upstream of a gene of interest.
Discussion
The 3WJ RNA architecture is a robust platform for dimerization of the Broccoli aptamer and allows for a straightforward bisection of aptamers into two-stranded systems that can restore function when hybridized. We have shown that the Split-Broccoli system functions when assembled from purified RNA in vitro, in both the presence and absence of transcription terminator structures and when thermally renatured or assembled at physiological temperature. In vitro performance appeared to be limited by an incomplete assembly process that was only partially restored through thermal renaturation, suggesting a high kinetic barrier for refolding of the RNA components into the hybridized complex. This barrier was inconsequential if the components of the system were cotranscribed, allowing for the more native RNA structures to hybridize efficiently and productively. These findings motivated us to explore the use of the system in vivo. Although Split-Broccoli signal in vivo was observable, it was not as robust as the unimolecular 3WJdB construct. We speculate that this reduction in signal could have arisen from instability of one or both of the strands against degradative cellular forces, such as RNases, rather than kinetic barriers to refolding. The decrease in relative signal could potentially be remediated by transcribing the split aptamer system with highly structured 5′ ends, as the 3′ end terminator structures likely offer some protection against nuclease activity targeting unstructured, linear RNA. Furthermore, when the Split-Broccoli system was fused to an RNA Toehold switch upstream of a fluorescent protein, Split-Broccoli fluorescence was observed tens of minutes before the fluorescent protein, demonstrating the speed and utility of an RNA-based split fluorescent aptamer in monitoring an RNA–RNA hybridization event.
To the best of our knowledge, Split-Broccoli is the first split-aptamer system directly shown to function when transcribed in vivo and is the first example of an in vivo logic gate with RNA inputs and a directly observable output composed of RNA. We posit that Split-Broccoli, or an analogously composed system, can be extended to detect RNA–RNA hybridization events nondestructively in real time as an alternative to chemically synthesized probes and without the requirement of a protein reporter. For RNA-based computation strategies that require a measurable reporter in living systems, Split-Broccoli may offer an approach that can operate without introducing an additional time delay or resource burden required by translating traditional protein-based reporters. Additionally, as transcription of the individual strands of the Split-Broccoli system can be regulated by independent transcription factors, the Split-Broccoli system can be used as an AND gate in vivo, further expanding the functional capabilities of RNA as a programmable and self-reporting molecule for RNA–RNA assembly.
Methods
Broccoli Designs and NUPACK Analysis
Sequences for the Spinach2 aptamer, Broccoli aptamer and the core domain of the Φ29 pRNA 3WJ motif were used as previously published.25,28,45SdB, 3WJdB and Split-Broccoli designs, with and without transcription terminators, were analyzed for predicted structures with the NUPACK web application at default settings for RNA at 37 °C.42 For Split-Broccoli hybridization, the two input strands were constrained to a maximum complex size of 2 and at a concentration of 1 μM each. For details of DNA sequences refer to Supporting Information.
DNA Templates and RNA Transcription
Oligonucleotides for generating DNA templates were ordered from Integrated DNA Technologies and ligated to generate PCR amplification templates. In short, oligonucleotides corresponding to the left and right halves of each sequence, and a reverse complement sequence bridging both halves, were designed. The oligonucleotides corresponding to the right half of each sequence were phosphorylated for ligation using T4 polynucleotide kinase (New England Biolabs). All three oligonucleotides per complete sequence were then incubated in equimolar amounts and annealed through a heat–cool step prior to ligation with T4 DNA ligase (New England Biolabs). Ligated oligonucleotides were PCR amplified with Pfu DNA polymerase, a forward primer to add the 5′ XbaI restriction site and T7 promoter, and a reverse primer to append the T7 terminator and 3′ XmaI restriction site. Amplification products were cloned into the pUC19 plasmid and inserts were confirmed through sequencing. Plasmids encoding the T7 RNA polymerase promoted templates were deposited to Addgene (IDs 87307–87310). For details of DNA sequences refer to Supporting Information.
Transcription templates were generated by PCR amplification of sequence-verified plasmids. The pET28c-Spinach2 plasmid (a gift from Samie Jaffrey) was used for PCR amplification and transcription of tSpinach2. Runoff transcription reactions were performed using T7 RNA polymerase, in vitro transcription buffer (50 mM Tris-HCl pH 7.5, 15 mM MgCl2, 5 mM DTT, and 2 mM spermidine), and 2 mM of each ATP, CTP, GTP and UTP. Reactions were incubated at 37 °C for a minimum of 4 h and halted with the addition of 2× RNA loading buffer (95% formamide and 5 mM EDTA, with trace amounts of Xylene Cyanol FF and Bromophenol Blue). RNAs were purified through denaturing polyacrylamide gel electrophoresis (0.75 mm 6% TBE-PAGE, 8 M urea) and bands corresponding to the expected product size were gel extracted and eluted while tumbling overnight in 300 mM Sodium Acetate pH 5.4. Eluates were ethanol precipitated, resuspended in buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA), and stored at −20 °C until further use. RNA concentrations were determined on a NanoDrop 1000 spectrophotometer (Thermo Scientific).
In Vitro Assembly and Fluorescence Assays
In vitro assembly reactions were prepared on ice with 25 picomoles of each RNA and 250 picomoles of DFHBI-1T dye (Lucerna Technologies) in buffer (40 mM HEPES pH 7.5, 100 mM KCl, 1 mM MgCl2) at a final volume of 50 μL. For thermal renaturation, samples were transferred into a preheated aluminum insert within a dry heat block set to 90 °C. Following addition of samples, the aluminum insert was immediately removed from the block heater and placed on the lab bench to cool to 37 °C before measurement. Assembly at 37 °C was performed by placing samples in a dry heat block at 37 °C for 15 min before measurement. Samples were then transferred into a clear, flat-bottom 96-well plate and measured for fluorescence (λex = 472 nm, λem = 507 nm) on an EnSpire Mulitmode plate reader (PerkinElmer) at room temperature. Measurements from No RNA samples, which contained buffer and dye, were averaged to establish background signal. Background signal was subtracted from all measurements and individual readings were normalized for fluorescence relative to 3WJdB. Normalized values were used to calculate mean and standard deviation.
Dual Staining Native Gel Shift Assay
Ten picomoles of each purified RNA (Top-T, Bottom-T, 3WJdB-T) were incubated at 37 °C for 15 min before being loaded onto a 0.75 mm 6% native TBE polyacrylamide gel in a final volume of 25 μL with 20% glycerol. Approximately 1 h after electrophoresis at 20 W in 4 °C, the gel was stained according to an in-gel imaging protocol.49 In brief, the gel was stained with 5 μM of DFHBI-1T at RT for 15 min and then imaged using a Typhoon FLA 9000 (GE Healthcare) with Alexa Fluor 488 settings (473 nm laser excitation, Y520 emission filter). A destaining step was then performed on the gel with 2 washes in ultrapure Milli-Q water (EMD Millipore) for 5 min each, followed by a 5 min incubation in ethidium bromide at 0.5 μg/mL. The gel was then reimaged on a Typhoon FLA 9000 (GE Healthcare) using the ethidum bromide setting (532 nm laser excitation, O580 emission filter). Densitometry analysis was performed in Fiji.53 Following a slight linear contrast adjustment, the intensity value of each band was estimated. The hybridization yield for the annealed complex was calculated according to the following formula: [(annealed complex)/(free Top-T + free Bottom-T + annealed complex)].
Split-Broccoli Assembly Kinetics
Purified samples of Top-T and Bottom-T RNA at various concentrations (0, 0.05, 0.1, 0.15, 0.2, 0.25, 0.3, 0.35, 0.4, 0.45, 0.5, and 0.6 μM) were independently prepared on ice, in buffer (40 mM HEPES pH 7.5, 100 mM KCl, 1 mM MgCl2) containing DFHBI-1T at 4 μM. Samples were aliquoted into a clear, flat-bottom 96-well plate and preincubated at 37 °C for 15 min. Assembly was initiated by using a multichannel pipet to transfer 50 μL of Top-T samples into 50 μL of Bottom-T samples at equimolar concentrations. Fluorescence measurements (λex = 485/25 nm, λem = 535/25 nm) were taken on an Infinite F200 Pro plate reader (Tecan) every 10 s for 3600 s. The linear region between 500 and 1000 s was used to fit a line and determine the slope (i.e., change in fluorescence as a function of time). Slope values from replicate experiments were averaged and then plotted against their concentration to obtain a value for the rate of assembly.
Cotranscription Fluorescence Assay
Cotranscription assays were performed with PCR amplification products purified through agarose gel electrophoresis (GeneJET Gel Extraction Kit, Thermo Scientific) to isolate bands corresponding to the desired DNA template size. Templates were quantified on a NanoDrop 1000 spectrophotometer (Thermo Scientific). To ensure equivalent transcriptional burden on the RNA polymerase across samples, each sample contained a total of 10 picomoles of T7-promoted template DNA. For 3WJdB-T, Top-T alone and Bottom-T alone, 5 picomoles of each DNA template was added to 5 picomoles of DNA template for an unrelated RNA aptamer (80N 433min2) with no observed fluorescence activation of DFHBI-1T. Split-Broccoli sample was composed of 5 picomoles of each Top-T and Bottom-T DNA templates, while background sample contained 10 picomoles of the 80N 433min2 control aptamer template. Samples were incubated with 2.5 nanomoles of each nucleotide triphosphate (ATP, CTP, GTP, UTP), 1 unit of inorganic pyrophosphatase (New England Biolabs), 1 nanomole of DFHBI-1T, and 40 U of T7 RNA polymerase with its supplied buffer (New England Biolabs) in a total volume of 50 μL. Samples were prepared and kept on ice until transfer to a flat-bottom 384-well plate and measurement on a Synergy HT plate reader (BioTek) prewarmed to 37 °C. Plate reader was configured to take readings at 1 min intervals over a period of 4 h (λex = 485/20 nm, λem = 528/20 nm). For Supporting Information Figure S9, samples volumes were reduced to 20 μL and T7 polymerase was used at 100 U, while all other amounts remained the same. Background measurement values at each time point were subtracted from sample values before calculating mean and standard deviation.
In Vivo Fluorescence
Expression plasmids for in vivo assays were constructed to contain constitutive E. coli P70a promoters52 and either a T7 transcription terminator or a derivative of the T500 transcription terminator from bacteriophage Φ82.54 Plasmids for the single insert corresponding to 3WJdB-T, Top-T, and Bottom-T were cloned using standard restriction digest cloning to insert the sequence between the XbaI and XmaI sites in the pUC19 plasmid and were terminated with the T7 terminator. Dual expression plasmids contained the T7 terminator immediately downstream of Top and the T500 terminator derivative immediately downstream of Bottom. DNA assembly (NEBuilder HiFi DNA Assembly, New England Biolabs) was used to insert the two inserts, along with a 270-nucleotide spacer sequence, between the NdeI and HindIII sites of the pUC19 plasmid. All plasmid inserts were confirmed through sequencing. Plasmids for in vivo transcription by native E. coli RNA polymerase were deposited to Addgene (IDs 87311–87315). For details of DNA sequences refer to Supporting Information.
The protocol for in vivo assessment in E. coli was adapted from the Spinach and Broccoli publications.17,28 In short, 100 ng of each plasmid was transformed into E. coli BL21(DE3) competent cells using a standard heat shock transformation protocol. Cells were plated on 2xYT-agar plates containing 100 μg/mL of ampicillin and incubated overnight at 37 °C. Single colonies from each plate were cultured overnight (∼16 h) at 37 °C with 250 rpm shaking in 3 mL of 2xYT broth containing 100 μg/mL of ampicillin. Following overnight culture, 100 μL of samples were added to 2.9 mL of fresh liquid media until reaching OD600 = 0.4, at which point 500 μL of sample was pelleted at 2000 RCF and resuspended in 1 mL Dulbecco’s phosphate-buffered saline (DPBS) with calcium and magnesium (Thermo Fisher). 200 μL aliquots were then incubated with 200 μM DFHBI-1T at 37 °C for 45 min prior to assessment on an Accuri C6 flow cytometer (BD Biosciences) configured with a 488 nm excitation laser and 533/30 nm filter. For each population, 5 × 104 events were analyzed and processed using FlowJo software.
Samples for fluorescence microscopy were prepared as described above. Following the centrifugation of freshly diluted overnight culture, samples were resuspended in 200 μL of DPBS and plated on poly-d-lysine-coated 8-chamber glass chamber slides (Lab-Tek). After a 45 min incubation at 37 °C, cells were washed twice in DPBS and incubated with 200 μM DFHBI-1T in DPBS for an additional 45 min. Fluorescence images were obtained at 100× oil objective magnification using QCapture Suite Plus software and Rolera-XR camera (QImaging) mounted on an Olympus 1 × 70 inverted fluorescent microscope. Green fluorescence (λex = 470/40 nm, λem = 525/50, 33 ms exposure) and bright field images were captured and exported into Fiji53 for linear adjustment of the brightness and contrast.
Split-Broccoli Toehold Switch Assays
Toehold (TS2_KS01) and Trigger (TS2_AT01) RNA sequences were used as published.9 Inserts for Top-Toehold-mCherry and Trigger-Bottom were synthesized as gBlocks gene fragments from Integrated DNA Technologies and cloned into pUC19 between the XbaI and XmaI restriction sites. For the dual-insert plasmid containing Top-Toehold-mCherry and Trigger-Bottom (“Split-Broccoli Toehold Switch”), the pUC19-Trigger-Bottom plasmid was linearized with NdeI and the insert for Top-Toehold-mCherry was inserted using Gibson assembly. Each transcription unit contained a constitutive E. coli P70a promoter52 and a T7 transcription terminator. In vivo analysis was performed as described above for in vivo fluorescence, with additional detection for red fluorescence using a 670 nm low-pass filter. For each population, 5 × 104 events were analyzed and processed using FlowJo software. Fluorescence microscopy was performed as described for In Vivo Fluorescence, with the addition of a red fluorescence filter set (λex = 562/40 nm, λem = 624/40, 100 ms exposure).
TX-TL transcription and translation reactions (a gift of Julius B. Lucks) were prepared and performed as described.52 In short, E. coli cytoplasmic extract, reaction buffer with metabolites and 20 μM DFHBI-1T were added to either 8 nM plasmid or H2O (blank) on ice, in a total volume of 10 μL. Samples were immediately transferred to a flat-bottom 384-well plate and measured on a Synergy H1 plate reader (BioTek) configured with filter sets for green fluorescence (λex = 472, λem = 507) and red fluorescence (λex = 587, λem = 615 nm). Readings were taken at 3 min intervals over the course of 12 h. Average blank values at each time point were subtracted from sample values before calculating mean and standard deviation. Plasmids and their sequences were deposited to Addgene (IDs 87314, 87316–87318). For details of DNA sequences refer to Supporting Information.
Acknowledgments
We acknowledge support for this work from the University of Missouri Department of Biochemistry, the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01AI074389) and the National Aeronautics and Space Administration (NNX12AD66G). We thank members of the Burke lab, Julius B. Lucks (Northwestern University), Maureen McKeague (ETH Zurich) and Raghav R. Poudyal (Pennsylvania State University) for providing feedback on the manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.7b00059.
Table S1, Figures S1–S10, Plasmid DNA Sequences and Annotated Maps (PDF)
Author Present Address
# Department of Chemical and Biological Engineering, Northwestern University, Evanston, Illinois 60208, United States.
Author Contributions
K.K.A. and D.H.B. conceived the Split-Broccoli system. K.K.A., K.D.T., M.F.L., D.P. and D.H.B. designed the experiments. K.K.A., K.D.T., M.F.L. and D.P. performed the experiments. K.K.A. and D.H.B. wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Cech T. R.; Steitz J. A. (2014) The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157, 77–94. 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Johnson S. F.; Telesnitsky A. (2010) Retroviral RNA dimerization and packaging: the what, how, when, where, and why. PLoS Pathog. 6, e1001007. 10.1371/journal.ppat.1001007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M.; Chylinski K.; Fonfara I.; Hauer M.; Doudna J. A.; Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudayyeh O. O.; Gootenberg J. S.; Konermann S.; Joung J.; Slaymaker I. M.; Cox D. B. T.; Shmakov S.; Makarova K. S.; Semenova E.; Minakhin L.; Severinov K.; Regev A.; Lander E. S.; Koonin E. V.; Zhang F. (2016) C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573–aaf5573. 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S.; Coller J. (2013) RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 14, 699–712. 10.1038/nrm3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeague M.; Wong R. S.; Smolke C. D. (2016) Opportunities in the design and application of RNA for gene expression control. Nucleic Acids Res. 44, 2987–2999. 10.1093/nar/gkw151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha M.; Rostain W.; Prakash S.; Duncan J. N.; Jaramillo A. (2016) Using RNA as Molecular Code for Programming Cellular Function. ACS Synth. Biol. 5, 795–809. 10.1021/acssynbio.5b00297. [DOI] [PubMed] [Google Scholar]
- Isaacs F. J.; Dwyer D. J.; Ding C.; Pervouchine D. D.; Cantor C. R.; Collins J. J. (2004) Engineered riboregulators enable post-transcriptional control of gene expression. Nat. Biotechnol. 22, 841–847. 10.1038/nbt986. [DOI] [PubMed] [Google Scholar]
- Green A. A.; Silver P. A.; Collins J. J.; Yin P. (2014) Toehold switches: de-novo-designed regulators of gene expression. Cell 159, 925–939. 10.1016/j.cell.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell J.; Takahashi M. K.; Lucks J. B. (2015) Creating small transcription activating RNAs. Nat. Chem. Biol. 11, 214–220. 10.1038/nchembio.1737. [DOI] [PubMed] [Google Scholar]
- Slomovic S.; Pardee K.; Collins J. J. (2015) Synthetic biology devices for in vitro and in vivo diagnostics. Proc. Natl. Acad. Sci. U. S. A. 112, 14429–14435. 10.1073/pnas.1508521112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath A. K.; Rentmeister A. (2015) Genetically encoded tools for RNA imaging in living cells. Curr. Opin. Biotechnol. 31, 42–49. 10.1016/j.copbio.2014.07.012. [DOI] [PubMed] [Google Scholar]
- Bertrand E.; Chartrand P.; Schaefer M.; Shenoy S. M.; Singer R. H.; Long R. M. (1998) Localization of ASH1 mRNA particles in living yeast. Mol. Cell 2, 437–445. 10.1016/S1097-2765(00)80143-4. [DOI] [PubMed] [Google Scholar]
- Larson D. R.; Zenklusen D.; Wu B.; Chao J. A.; Singer R. H. (2011) Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science 332, 475–478. 10.1126/science.1202142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozer A.; Pagano J. M.; Lis J. T. (2014) New Technologies Provide Quantum Changes in the Scale, Speed, and Success of SELEX Methods and Aptamer Characterization. Mol. Ther.--Nucleic Acids 3, e183. 10.1038/mtna.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grate D.; Wilson C. (1999) Laser-mediated, site-specific inactivation of RNA transcripts. Proc. Natl. Acad. Sci. U. S. A. 96, 6131–6136. 10.1073/pnas.96.11.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige J. S.; Wu K. Y.; Jaffrey S. R. (2011) RNA mimics of green fluorescent protein. Science 333, 642–646. 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgosheina E. V.; Jeng S. C. Y.; Panchapakesan S. S. S.; Cojocaru R.; Chen P. S. K.; Wilson P. D.; Hawkins N.; Wiggins P. A.; Unrau P. J. (2014) RNA mango aptamer-fluorophore: a bright, high-affinity complex for RNA labeling and tracking. ACS Chem. Biol. 9, 2412–2420. 10.1021/cb500499x. [DOI] [PubMed] [Google Scholar]
- Bhadra S.; Ellington A. D. (2014) Design and application of cotranscriptional non-enzymatic RNA circuits and signal transducers. Nucleic Acids Res. 42, e58–e58. 10.1093/nar/gku074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akter F.; Yokobayashi Y. (2015) RNA signal amplifier circuit with integrated fluorescence output. ACS Synth. Biol. 4, 655–658. 10.1021/sb500314r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höfer K.; Langejürgen L. V.; Jäschke A. (2013) Universal aptamer-based real-time monitoring of enzymatic RNA synthesis. J. Am. Chem. Soc. 135, 13692–13694. 10.1021/ja407142f. [DOI] [PubMed] [Google Scholar]
- Pothoulakis G.; Ceroni F.; Reeve B.; Ellis T. (2014) The spinach RNA aptamer as a characterization tool for synthetic biology. ACS Synth. Biol. 3, 182–187. 10.1021/sb400089c. [DOI] [PubMed] [Google Scholar]
- You M.; Litke J. L.; Jaffrey S. R. (2015) Imaging metabolite dynamics in living cells using a Spinach-based riboswitch. Proc. Natl. Acad. Sci. U. S. A. 112, E2756–65. 10.1073/pnas.1504354112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. C.; Wilson S. C.; Hammond M. C. (2016) Next-generation RNA-based fluorescent biosensors enable anaerobic detection of cyclic di-GMP. Nucleic Acids Res. 44, e139. 10.1093/nar/gkw580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack R. L.; Disney M. D.; Jaffrey S. R. (2013) A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat-containing RNA. Nat. Methods 10, 1219–1224. 10.1038/nmeth.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketterer S.; Fuchs D.; Weber W.; Meier M. (2015) Systematic reconstruction of binding and stability landscapes of the fluorogenic aptamer spinach. Nucleic Acids Res. 43, 9564–9572. 10.1093/nar/gkv944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autour A.; Westhof E.; Ryckelynck M. (2016) iSpinach: a fluorogenic RNA aptamer optimized for in vitro applications. Nucleic Acids Res. 44, 2491–2500. 10.1093/nar/gkw083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filonov G. S.; Moon J. D.; Svensen N.; Jaffrey S. R. (2014) Broccoli: rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J. Am. Chem. Soc. 136, 16299–16308. 10.1021/ja508478x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.; Strack R. L.; Svensen N.; Jaffrey S. R. (2014) Plug-and-play fluorophores extend the spectral properties of Spinach. J. Am. Chem. Soc. 136, 1198–1201. 10.1021/ja410819x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpashchikov D. M. (2005) Binary malachite green aptamer for fluorescent detection of nucleic acids. J. Am. Chem. Soc. 127, 12442–12443. 10.1021/ja0529788. [DOI] [PubMed] [Google Scholar]
- Rogers T. A.; Andrews G. E.; Jaeger L.; Grabow W. W. (2015) Fluorescent monitoring of RNA assembly and processing using the split-spinach aptamer. ACS Synth. Biol. 4, 162–166. 10.1021/sb5000725. [DOI] [PubMed] [Google Scholar]
- Ausländer S.; Fuchs D.; Hürlemann S.; Ausländer D.; Fussenegger M. (2016) Engineering a ribozyme cleavage-induced split fluorescent aptamer complementation assay. Nucleic Acids Res. 44, e94. 10.1093/nar/gkw117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi N.; Kolpashchikov D. M. (2016) Split Spinach Aptamer for Highly Selective Recognition of DNA and RNA at Ambient Temperatures. ChemBioChem 17, 1589–1592. 10.1002/cbic.201600323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Potty A. S. R.; Jackson G. W.; Stepanov V.; Tang A.; Liu Y.; Kourentzi K.; Strych U.; Fox G. E.; Willson R. C. (2009) Engineered 5S ribosomal RNAs displaying aptamers recognizing vascular endothelial growth factor and malachite green. J. Mol. Recognit. 22, 154–161. 10.1002/jmr.917. [DOI] [PubMed] [Google Scholar]
- Reif R.; Haque F.; Guo P. (2012) Fluorogenic RNA nanoparticles for monitoring RNA folding and degradation in real time in living cells. Nucleic Acid Ther. 22, 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent A. D.; Spiropulos N. G.; Heemstra J. M. (2013) General approach for engineering small-molecule-binding DNA split aptamers. Anal. Chem. 85, 9916–9923. 10.1021/ac402500n. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Zhang L.; Zhou Z.; Dong S.; Wang E. (2014) Aptamer-based sensing platform using three-way DNA junction-driven strand displacement and its application in DNA logic circuit. Anal. Chem. 86, 312–316. 10.1021/ac403235y. [DOI] [PubMed] [Google Scholar]
- Hou T.; Li W.; Zhang L.; Li F. (2015) A versatile and highly sensitive homogeneous electrochemical strategy based on the split aptamer binding-induced DNA three-way junction and exonuclease III-assisted target recycling. Analyst 140, 5748–5753. 10.1039/C5AN01176K. [DOI] [PubMed] [Google Scholar]
- Lescoute A.; Westhof E. (2006) Topology of three-way junctions in folded RNAs. RNA 12, 83–93. 10.1261/rna.2208106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponchon L.; Dardel F. (2007) Recombinant RNA technology: the tRNA scaffold. Nat. Methods 4, 571–576. 10.1038/nmeth1058. [DOI] [PubMed] [Google Scholar]
- Ageely E. A.; Kartje Z. J.; Rohilla K. J.; Barkau C. L.; Gagnon K. T. (2016) Quadruplex-Flanking Stem Structures Modulate the Stability and Metal Ion Preferences of RNA Mimics of GFP. ACS Chem. Biol. 11, 2398–2406. 10.1021/acschembio.6b00047. [DOI] [PubMed] [Google Scholar]
- Zadeh J. N.; Steenberg C. D.; Bois J. S.; Wolfe B. R.; Pierce M. B.; Khan A. R.; Dirks R. M.; Pierce N. A. (2011) NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 32, 170–173. 10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
- Haque F.; Shu D.; Shu Y.; Shlyakhtenko L. S.; Rychahou P. G.; Evers B. M.; Guo P. (2012) Ultrastable synergistic tetravalent RNA nanoparticles for targeting to cancers. Nano Today 7, 245–257. 10.1016/j.nantod.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu D.; Khisamutdinov E. F.; Zhang L.; Guo P. (2014) Programmable folding of fusion RNA in vivo and in vitro driven by pRNA 3WJ motif of phi29 DNA packaging motor. Nucleic Acids Res. 42, e10. 10.1093/nar/gkt885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu D.; Shu Y.; Haque F.; Abdelmawla S.; Guo P. (2011) Thermodynamically stable RNA three-way junction for constructing multifunctional nanoparticles for delivery of therapeutics. Nat. Nanotechnol. 6, 658–667. 10.1038/nnano.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binzel D. W.; Khisamutdinov E. F.; Guo P. (2014) Entropy-driven one-step formation of Phi29 pRNA 3WJ from three RNA fragments. Biochemistry 53, 2221–2231. 10.1021/bi4017022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binzel D. W.; Khisamutdinov E.; Vieweger M.; Ortega J.; Li J.; Guo P. (2016) Mechanism of three-component collision to produce ultrastable pRNA three-way junction of Phi29 DNA-packaging motor by kinetic assessment. RNA 22, 1710–1718. 10.1261/rna.057646.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Endrizzi J. A.; Shu Y.; Haque F.; Sauter C.; Shlyakhtenko L. S.; Lyubchenko Y.; Guo P.; Chi Y.-I. (2013) Crystal structure of 3WJ core revealing divalent ion-promoted thermostability and assembly of the Phi29 hexameric motor pRNA. RNA 19, 1226–1237. 10.1261/rna.037077.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filonov G. S.; Kam C. W.; Song W.; Jaffrey S. R. (2015) In-gel imaging of RNA processing using broccoli reveals optimal aptamer expression strategies. Chem. Biol. 22, 649–660. 10.1016/j.chembiol.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson M. H.; Greenleaf W. J.; Landick R.; Block S. M. (2008) Applied force reveals mechanistic and energetic details of transcription termination. Cell 132, 971–982. 10.1016/j.cell.2008.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlenbeck O. C. (1995) Keeping RNA happy. RNA 1, 4–6. [PMC free article] [PubMed] [Google Scholar]
- Garamella J.; Marshall R.; Rustad M.; Noireaux V. (2016) The All E. coli TX-TL Toolbox 2.0: A Platform for Cell-Free Synthetic Biology. ACS Synth. Biol. 5, 344–355. 10.1021/acssynbio.5b00296. [DOI] [PubMed] [Google Scholar]
- Schindelin J.; Arganda-Carreras I.; Frise E.; Kaynig V.; Longair M.; Pietzsch T.; Preibisch S.; Rueden C.; Saalfeld S.; Schmid B.; Tinevez J.-Y.; White D. J.; Hartenstein V.; Eliceiri K.; Tomancak P.; Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarnell W. S.; Roberts J. W. (1999) Mechanism of intrinsic transcription termination and antitermination. Science 284, 611–615. 10.1126/science.284.5414.611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.