

Abstract
Tryptophan-rich peptides, being short and suitable for large-scale chemical synthesis, are attractive candidates for developing a new generation of antimicrobials to combat antibiotic-resistant bacteria (superbugs). Although there are numerous pictures for membrane-bound structure of a single tryptophan (W), how multiple Trp amino acids assemble themselves and interact with bacterial membranes are poorly understood. This communication presents the three-dimensional structure for an eight-residue Trp-rich peptide (WWWLRKIW-NH2 with 50% W) determined by the improved 2D NMR method, which includes the measurements of 13C and 15N chemical shifts at natural abundance. This peptide forms the shortest two-turn helix with a distinct amphipathic feature. A unique structural arrangement is identified for the Trp triplet, WWW, that forms a π configuration with W2 as the horizontal bar and W1/W3 forming the two legs. Arginine scan reveals that the WWW motif is essential for killing methicillin-resistant Staphylococcus aureus USA300 and disrupting preformed bacterial biofilms. This unique π configuration for the WWW motif is stabilized by aromatic-aromatic interactions as evidenced by ring current shifts as well as nuclear Overhauser effects. By maintaining the WWW motif, a change of I7 to R led to a potent antimicrobial and antibiofilm peptide with four-fold improvement in cell selectivity. Collectively, this study elucidated the structural basis of antibiofilm activity of the peptide, identified a better peptide candidate via structure-activity relationship studies, and laid the foundation for engineering future antibiotics based on the WWW motif.
Graphical Abstract
Naturally occurring antimicrobial peptides (AMPs) are endogenous defense molecules of living organisms.1 They remain potent for millions of years, making them appealing templates for developing the next generation of antimicrobials to combat superbugs and difficult-to-kill viruses. The amino acid use in such peptides is biased so that they have the desired sequence feature to recognize invading pathogens. In α-helical amphibian AMPs, glycine, leucine, alanine, and lysine are rich, whereas glycine, arginine, and cysteine are abundant in β-sheet peptides. Such abundant amino acids can determine the peptide scaffold.2 In contrast, certain amino acids, such as histidine (His) and tryptophan (Trp), are less frequently deployed on average in natural AMPs (http://aps.unmc.edu/AP; Figure S1 in Supporting Information). However, Trp and His can be abundant in some peptides. Interest in Trp-rich peptides remains high because they are relatively short yet potent against superbugs. Usually, these Trp-rich peptides are also accompanied by arginine (Arg).3 The combination of hydrophobic Trp and cationic Arg are sufficient to generate amphipathic sequences. Such amphipathic sequences lay the foundation for AMPs to target bacterial membranes. While cationic residues can recognize anionic surfaces, hydrophobic Trp amino acids can anchor the entire peptide to the membrane. Structural determination of membrane-bound AMPs enables us to view the active conformation and provides a basis for structure-based peptide design. Most of the structures of small AMPs are determined by the classic 2D NMR method,4a which typically requires the recording of TOCSY, DQF-COSY, and NOESY spectra. Due to the complex nature of biological membranes, most of the structural characterization of Trp-containing AMPs was conducted in membrane-mimetic environments such as micelles. The structures of indolicidin and tritrpticin are extended with multiple turns.3 How the multiple aromatic rings bind to membranes is not always obvious because they are not all located on the same hydrophobic surface. The story is further complicated since those Trp-rich peptides can also interact with bacterial DNA.5
Here we present a unique structure for a newly designed Trp-rich peptide TetraF2W-RK (WWWLRKIW-amide),6 which contains 50% Trp, one of the highest percentages known to date. This peptide appears to act on bacterial membranes. First, Staphylococcus aureus USA300 can be rapidly killed. Second, the peptides synthesized using L- or D-amino acids give an identical minimal inhibitory concentration (MIC) against S. aureus, suggesting a chiral protein-like interface is not involved. Third, after peptide treatment, a non-permeable dye fluorescein isothiocyanate (FITC) can enter S. aureus in 30 sec, suggesting membrane damage by the peptide.6 These results laid a solid basis for us to determine the 3D structure of TetraF2W-RK bound to membrane-mimetic micelles. We chose to utilize the improved 2D NMR method7a for structural determination because recent studies show the importance of this method for peptides rich in certain amino acids (e.g., leucine and cysteine).7b,7c In this method, five 2D NMR spectra are recorded (1H-1H TOCSY, DQF-COSY, NOESY, 1H-13C HSQC, and 1H-15N HSQC). The 15N and 13C resonances have a broader range of chemical shifts, enabling the validation of proton assignments. In addition, these heteronuclear chemical shifts also contain structural information and can be used to refine the nuclear Overhauser effect (NOE)-derived structure to achieve high quality.7c Different from indolicidin and tritrpticin, this membrane-targeting Trp-rich peptide, TetraF2W-RK, forms a regular two-turn α-helix with a clear amphipathic nature, enabling us to decipher the role of each residue via single residue substitutions. Our results reveal that the π configuration of the N-terminal Trp triplet (WWW) of this short cationic peptide plays a critical role in disrupting the preformed biofilms of S. aureus USA300 based on quantification of both biomass and live bacteria.
The NMR spectra of TetraF2W-RK bound to perdeuterated dodecylphosphocholine (DPC-d38) are well dispersed (Figure 1, A&B), leading to complete assignments. Based on these 1H chemical shifts (Table S1), a total of 225 NOEs were assigned and converted to distance restraints for structural calculations. The number of NOEs per residue is 28, which is high for a micelle-bound peptide.3 Multiple (i, i+3) and (i, i+4) types of NOEs indicate a helical conformation, consistent with the secondary 1Hα chemical shift analysis (Table S2). Moreover, peptide backbone dihedral angles derived from a set of heteronuclear chemical shifts also indicate a helical structure.4e All the distance and angle restraints were utilized for structural calculations. Figure 1C presents an ensemble of 20 energy minimized 3D structures with backbone atoms superimposed. The backbone structure of TetraF2W-RK was well defined (rmsd = 0.06 Å). The 3D structure of the peptide consists of a two-turn helix. This can be seen in the Ramachandran plot, where nearly all the backbone angles (ϕ and φ) are clustered into the helical region (Figure S2). Remarkably, the side chains of the peptide also superimpose well and each Trp side chain is clearly visible (Figure 1, D & E). The aromatic rings of three Trp residues (W1, W3, and W8), as well as L4 and I7, occupy the same side, constituting the hydrophobic surface (Figure 1, D–F). W2, however, is located between the hydrophilic and hydrophobic interface where it is adjacent to the side chain of K6 (Figure 1, D–F). R5 is the only side chain that is clearly on the hydrophilic surface. Hence, TetraF2W-RK forms one of the shortest amphipathic helices after binding to membrane-mimetic micelles.
Figure 1.

NMR spectra (A–B) and 3D structure (C–F) of TetraF2W-RK bound to perdeuterated dodecylphosphocholine micelles at 25°C and pH 6.2. Portions of natural abundance 1H-15N HSQC (A) and 1H-1H NOESY (B) spectra illustrate data quality. Also shown are (C) superimposed backbones of an ensemble of 20 structures that form the shortest two-turn helix as indicated by the dihedral angles on the Ramachandran plot (Figure S2), side (D) and end-on (E) views of the amphipathic helix with superimposed side chains, and a side-view of the amphipathic structure featuring the π configuration of the WWW triplet in red (H). Color code: L4, I7, W8 in grey, R5 and K6 in multiple colors: nitrogen, blue; oxygen, red; carbon, grey; and hydrogen, white. NMR data were collected, processed, and assigned as described in the supporting document.4,7a Additional sample conditions and chemical shifts are provided in Table S1.
To provide evidence for the binding of the amphipathic helix to the micelles, we also compared the water exchange cross peaks of the side chain NH of the four Trp residues (Figure S3). The exchange peak for W2 is strongest, indicating that it was most exposed to water. The exchange cross peaks for W1 and W3 side chains reached 50% of the peak intensity of W2, implying that these two aromatic rings inserted deeper into the micelles. There is no clear cross peak with water for the side chain of W8, implying an even deeper penetration into the micelles. Such a Trp side chain proton exchange picture of the peptide with water is fully consistent with the amphipathic structure of TetraF2W-RK with the hydrophobic side chains buried into the micelles (Figure 1D).
There are multiple NOE cross peaks between the aliphatic portion of K6 and the aromatic ring of W2, suggesting a possible aromatic-charge interaction. However, it did not appear to matter much whether it is lysine or arginine (Arg) at position 6, because a swap of the positions between R5 and K6, leading to TetraF2W-KR, did not influence bacterial killing efficiency.6 When we compared all possible dibasic pairs in the same peptide context, the order of S. aureus USA300 killing efficiency is RR > RK ~ KR > KK. This means that arginines at both positions of this peptide template are more effective than lysines in bacterial killing (based on both colony count and membrane permeation experiments). Indeed, results in the literature reveal the preference of Trp-rich peptides to bind anionic membranes, indicating that these arginines are involved in interaction with anionic phosphatidylglycerols (PGs).3 We have observed direct Arg-PGs contacts between a human cathelicidin LL-37 peptide and dioctanoylphosphatidylglycerol by intermolecular NOE spectroscopy.8 Interestingly, the double arginine variant, TetraF2W-RR, is less cytotoxic to human HEK293 and HaCaT cells.6 It appears that the Arg-Trp combination, frequently observed in Trp-rich peptides, offers an evolutional advantage by optimizing the desired antimicrobial effect and minimizing toxicity to the host.
We then asked whether insertion of additional arginines to the peptide would be helpful to the peptide in terms of potency and selectivity. For this purpose, we have made six single arginine variants based on the above double arginine variant TetraF2W-RR template (wild type WT). The sequences of these peptides are given in Table 1. The quality of these synthetic peptides can be seen from the HPLC chromatograms (Figure S4) as well as correct masses. Antimicrobial activities of these peptides were evaluated by the microdilution method as described6 using both Gram-negative Escherichia coli and Gram-positive S. aureus (Table 1). In the case of E. coli ATCC 25922, there was a four-fold increase in the minimal inhibitory concentration (MIC) for all the variants, except for the I7R peptide (viz, I7 is substituted by R), which retained the same MIC. When methicillin-resistant S. aureus (MRSA) USA300 was tested, we also observed a four-fold loss in activity for the W1R, W3R, L4R, and W8R variants compared to the WT (Table 1). However, the anti-MRSA activity of W2R was less reduced, suggesting that W2 is less important here than other Trps in the peptide. Interestingly, like the case of E. coli, the activity of I7R against S. aureus USA300 also remained the same (MIC 3.1 μM).
Table 1.
Antibacterial and hemolytic effects of TetraF2W-RR (WT) and its single residue variantsa
| Name | Peptide Sequence | S. aureus (μM) | E. coli (μM) | HL50 (μM) | tR (min) |
|---|---|---|---|---|---|
| WT | WWWLRRIW | 3.1 | 6.2 | 35 | 12.10 |
| W1R | RWWLRRIW | 12.5 | 25 | >200 | 11.48 |
| W2R | WRWLRRIW | 6.2 | 25 | 100 | 10.86 |
| W3R | WWRLRRIW | 12.5 | 25 | >200 | 10.77 |
| L4R | WWWRRRIW | 12.5 | 25 | >200 | 10.78 |
| I7R | WWWLRRRW | 3.1 | 6.2 | 150 | 11.07 |
| W8R | WWWLRRIR | 12.5 | 25 | >200 | 10.87 |
C-terminal amidation.
The abbreviations used are SA, S. aureus USA300; EC, E. coli ATCC 25922; WT, wild type; W1R, residue W1 is replaced by an arginine, and so on; tR, the retention time of a peptide measured on a Waters HPLC system (for details, see the Supporting material).
To further appreciate these activity differences, we also compared the growth inhibition as well as membrane permeation power of these peptide variants in Table 1. I7R is almost equally effective to the WT in inhibiting the growth of S. aureus USA300 at 3.1 μM (Figure 2A). The L4R variant is also potent followed by the W2R peptide. The substitution of W1, W2, or W8, however, made the peptide less inhibitory in this experiment. A similar trend was also observed with membrane permeation by propidium iodide (PI) (Figure 2B). The WT is most potent followed by the I7R and W2R variants. These results reinforce the significance of aromatic amino acids W1, W3, and W8 in anchoring the peptide to the membranes of S. aureus USA300.
Figure 2.
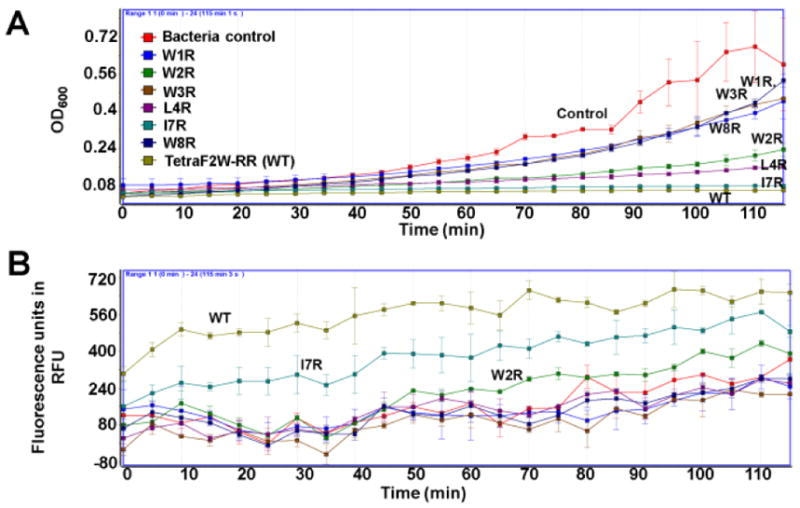
Growth inhibition (A) and propidium iodide-based membrane permeation (B) of S. aureus USA300 in TSB media by 3.1 μM of TetraF2W-RR and its single arginine variants (Table 1) at 37°C with shaking at 100 rpm.5
In nature, most bacteria live in the biofilm form, and the formation of biofilms makes it more resistant to traditional antibiotics. To further understand the activity of these peptides in Table 1, we also compared their ability in disrupting the 24-h biofilms of S. aureus USA300. After peptide treatment (3.1 to 25 μM), biofilms were stained with either crystal violet (Figure 3, left panels) or the XTT-based cell proliferation kit (right panels). Crystal violet staining measures the amount of biofilms. When the C-terminal hydrophobic residues were replaced, the peptide variants showed only slightly reduced antibiofilm capability (Figure 3, E–G). However, substitution of any of the N-terminal Trp residues (panels B–D) had a detrimental effect on antibiofilm ability, although W2R became more effective at 12.5–25 μM (panel C). These plots underscore the importance of the N-terminal Trp triplet in disrupting the S. aureus biofilms.
Figure 3.
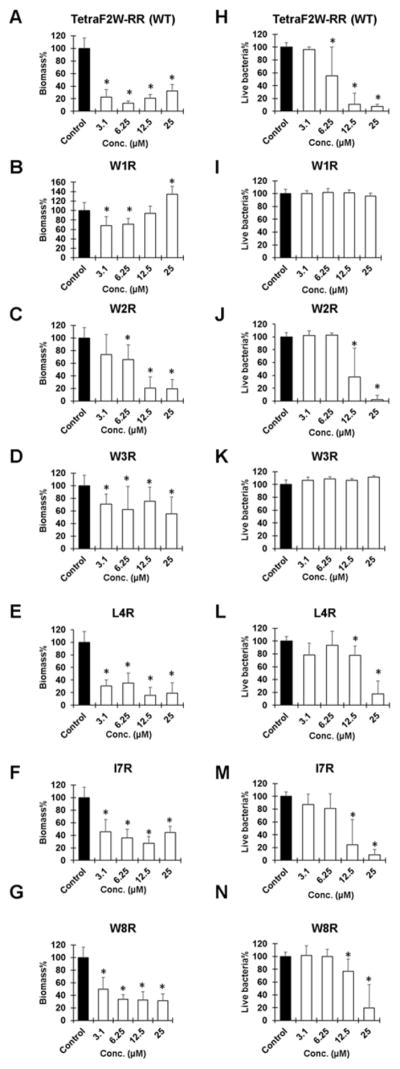
Antibiofilm ability of the Trp-rich peptides (Table 1) against 24-h preformed biofilms of S. aureus USA300. Panels A–G were stained with crystal violet, while panels H–N were stained with XTT (see the text for further details). Thus, there are two panels for each peptide: peptide TetraF2W-RR (WT), panels A & H; W1R, B & I; W2R, C & J; W3R, D & K; L4R, E & L; I7R, F & M; W8R, G & N. P values were calculated based on paired Student’s t-test with two tailed distribution. P values <0.05 were considered significant (*).
Whether S. aureus is killed in biofilms remains to be tested. We estimated dead bacteria by using the XTT assay that reports the amount of live cells. At a low concentration of 3.1 μM, none of the peptides were effective. At 6.2 μM, only the WT was active. When the peptide reached 12.5 μM, nearly all S. aureus USA300 was eliminated by the WT (Figure 3H). In addition, the W2R, L4R, I7R, and W8R variants also displayed some effects, with W2R and I7R (80% killing in Figure 3J&M) being much more effective than L4R and W8R (20% killing in Figure 3, L&N). At 25 μM, these four peptide variants killed nearly all S. aureus in the biofilms based on the XTT staining. The stronger effect of the WT may result from its higher hydrophobicity (longest HPLC retention time in Table 1). Notably, neither W1R nor W3R was able to kill the S. aureus USA300 strain in the biofilms even at 25 μM, indicating that W1 and W3 are critical in both biofilm disruption and bacterial killing based on a combined use of two biofilm staining dyes (Figure 3).
We also compared the cytotoxicity of these peptides using human red blood cells. A concentration dependent hemolysis enabled us to estimate HL50, the concentration that causes 50% hemolysis. The results are included in Table 1. Based on HL50 and MIC values, we also calculated cell selectivity index, which is defined as the ratio of HL50 and MIC. While the cell selectivity index for the WT peptide is 35/3.1=11, it is 150/3.1=48 for I7R. Thus, the hemolytic ability of the I7R variant was reduced (Table 1) by four fold compared to the WT. Therefore, through this structure-activity relationship studies, we have obtained a more selective peptide, the I7R variant, that retains antibacterial activity (the same MIC), making it a more suitable candidate for development of novel antimicrobials to combat MRSA.
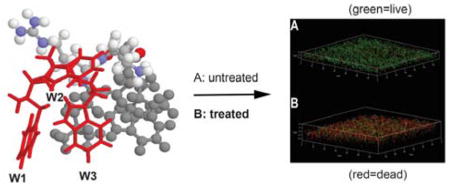
To further verify that the I7R variant remains membrane targeting, we also conducted a live cell experiment by following the entry of a non-membrane permeable dye FITC into S. aureus USA300. Only after peptide treatment, we saw green fluorescent bacteria in 28 sec (Figure S5), indicating membrane permeation by the peptide. To view the dead bacteria directly, we also treated the 24 h preformed biofilms of S. aureus USA300 with the I7R peptide followed by confocal microscopy. Without treatment with the I7R variant, we observed green cells (live). After treatment, the majority of cells are red, indicating that this peptide killed MRSA in biofilms as well (red cells in Figure S6). Similar results were obtained using the WT peptide, indicating I7R, like the WT, killed S. aureus USA300 in the biofilms.
We then attempted to correlate the peptide activity (Table 1) with the 3D structure. W2, being located in the interface of the amphipathic helix, is less significant for membrane binding, consistent with only a two-fold loss in activity (Table 1). Likewise, I7 is close to the interface and its change to an arginine probably has compensated the loss, leading to the same MIC in Table 1 for both bacteria. These two peptide variants also retained antibiofilm capability similar to the WT (Figure 3 right). In contrast, residues W1, W3, L4, and W8 are all located on the hydrophobic surface, critical for anchoring the peptide into bacterial membranes (Figure 1D). It is not surprising that a change of any of these residues into an arginine caused a substantial drop in antibacterial activity (Table 1). The same is true of the biofilm cases, especially when XTT was utilized (Figure 3 right). It is worthwhile to point out that W1, W3, L4 and W8 are also important for hemolysis since their arginine variants became much less toxic (Table 1).
In disrupting the MRSA biofilms, however, W1 and W3 of the peptide play an even more critical role than L4 or W8. When stained with crystal violet, there are more biofilms left for the cases of W1R and W3R variants (Figure 3, B & D) than those of L4R and W8R (Figure 3, E & G). Remarkably, the peptide variants of W1 and W3 completely lost their killing ability when stained with XTT (Figure 3, I & K), indicating that biofilm disruption based on crystal violet did not entirely reflect bacterial killing based on XTT. Interestingly, the three Trp residues at the N-terminus of TetraF2W-RK of the structure assemble into a π configuration (Figure 1F, red), where W2 is the horizontal bar, while W1 and W3 constitute the two legs critical for interdigitation into the membranes. These aromatic rings of the WWW motif stack with each other. For instance, a cross peak between W2 and W3 can be seen in Figure 1B, while a cross peak between W1 and W3 can be seen in Figure S3. Also, W1 and W3 are perpendicular to each other, leading to the ring current shifts of multiple protons (Table S1). It seems that aromatic-aromatic interactions play a role in stabilizing the π-configuration of the WWW structural motif.
In conclusion, we determined the 3D structure for a newly designed Trp-rich peptide (50% Trp). This structure nicely correlates with antibacterial and antibiofilm activities. It also revealed a novel π-configuration for the Trp triplet stabilized by both aromatic-aromatic and aromatic-aliphatic interactions. This π-configuration of the WWW motif, especially the two legs (W1 and W3), is critical for biofilm disruption and bacterial killing in the biofilms. Such a critical structural motif may be utilized to guide our design of novel peptides. To obtain better peptides, the WWW motif at the N-terminus may be retained by varying the C-terminal residues. Our identification of the I7R variant, which retains antimicrobial and antibiofilm activities but with reduced toxicity to human cells, provides an excellent example for this. Consequently, I7R constitutes a better candidate for further development than the WT peptide. In addition, the WWW motif can be applied to peptide ends to enhance antimicrobial activity as demonstrated by Schmidtchen and colleagues.9 Collectively, our study has not only obtained a more promising peptide candidate, but also uncovered a novel π-configuration for a tryptophan triplet via studying the structure-activity relationship of a new Trp-rich peptide that yields novel insight into membrane targeting, MRSA killing, and biofilm disruption. This unique WWW motif is of general interest for engineering potent antibiofilm peptides to combat drug-resistant pathogens.
Supplementary Material
Acknowledgments
We are grateful to the NIH funding support R03 AI128230 and R01 AI105147 to GW. DZ gratefully acknowledges UGC, Govt. of India for the Raman Postdoctoral Fellowship (F.No. 5-133/2016 (IC)). FW is supported by a Chinese Visiting Scholarship.
Footnotes
Notes
The authors declare no competing financial interests.
Experimental details, additional two tables and six figures are provided as Supporting Information, which is available online free of charge.
References
- 1.(a) Zasloff M. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]; (b) Hancock RE, Sahl HG. Nat Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]; (c) Epand RM, Vogel HJ. Biochim Biophys Acta. 1999;1462:11–28. doi: 10.1016/s0005-2736(99)00198-4. [DOI] [PubMed] [Google Scholar]
- 2.(a) Wang G, Li X, Wang Z. Nucleic Acids Res. 2016;42:D1087–D1093. doi: 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang G. Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies. Cambridge: CABI; 2010. [Google Scholar]
- 3.(a) Chan DI, Prenner EJ, Vogel HJ. Biochim Biophys Acta. 2006;1758:1184–1202. doi: 10.1016/j.bbamem.2006.04.006. [DOI] [PubMed] [Google Scholar]; (b) Rozek A, Friedrich CL, Hancock RE. Biochemistry. 2000;39:15765–15774. [PubMed] [Google Scholar]; (c) Schibli DJ, Hwang PM, Vogel HJ. Biochemistry. 1999;38:16749–16755. doi: 10.1021/bi990701c. [DOI] [PubMed] [Google Scholar]
- 4.(a) Wüthrich K. NMR of Proteins and Nucleic Acids. Wiley; New York: 1986. [Google Scholar]; (b) Kay LE, Keifer PA, Saarinen T. J Am Chem Soc. 1992;114:10663–10665. [Google Scholar]; (c) Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]; (d) Garrett DS, Powers R, Gronenborn AM, Clore GM. J Magn Reson. 1991;95:214–220. doi: 10.1016/j.jmr.2011.09.007. [DOI] [PubMed] [Google Scholar]; (e) Cornilescu G, Delaglio F, Bax A. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]; (f) Schwieters CD, Kuszewski J, Tjandra N, Clore GM. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]; (g) Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 5.Fojan P, Gurevich L. Methods Mol Biol. 2017;1548:201–215. doi: 10.1007/978-1-4939-6737-7_14. [DOI] [PubMed] [Google Scholar]
- 6.Mishra B, Lushnikova T, Golla RM, Wang X, Wang G. Acta Biomater. 2017;49:316–328. doi: 10.1016/j.actbio.2016.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Wang G, Li Y, Li X. J Biol Chem. 2005;280:5803–5811. doi: 10.1074/jbc.M410116200. [DOI] [PubMed] [Google Scholar]; (b) Conibear AC, Rosengren KJ, Harvery PJ, Craik DJ. Biochemistry. 2012;51:9718–9726. doi: 10.1021/bi301363a. [DOI] [PubMed] [Google Scholar]; (c) Wang G. Pharmaceuticals. 2013;6:728–758. doi: 10.3390/ph6060728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Wang G. Biochim Biophys Acta. 2007;1768:3271–3281. doi: 10.1016/j.bbamem.2007.08.005. [DOI] [PubMed] [Google Scholar]; (b) Li X, Li Y, Han H, Miller DW, Wang G. J Am Chem Soc. 2006;128:5776–5785. doi: 10.1021/ja0584875. [DOI] [PubMed] [Google Scholar]; (c) Wang G. J Biol Chem. 2008;283:32637–32643. doi: 10.1074/jbc.M805533200. [DOI] [PubMed] [Google Scholar]
- 9.Schmidtchen A, Pasupuleti M, Mörgelin M, Davoudi M, Alenfall J, Chalupka A, Malmsten M. J Biol Chem. 2009;284:17584–17594. doi: 10.1074/jbc.M109.011650. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.