

Abstract
Expression of the retained C-terminal extracellular portion of the ovarian cancer glycoprotein MUC16 induces transformation and tumor growth. However, the mechanisms of MUC16 oncogenesis related to glycosylation are not clearly defined. We establish that MUC16 oncogenic effects are mediated through MGAT5-dependent N-glycosylation of two specific asparagine sites within its 58 amino acid ectodomain. Oncogenic signaling from the C-terminal portion of MUC16 requires the presence of Galectin-3 and growth factor receptors co-localized on lipid rafts. These effects are blocked upon loss of either Galectin-3 expression or activity MGAT5. Using synthetic MUC16 glycopeptides, we developed novel N-glycosylation site directed–monoclonal antibodies that block Galectin-3–mediated MUC16 interactions with cell surface signaling molecules. These antibodies inhibit invasion of ovarian cancer cells, directly blocking the in vivo growth of MUC16-bearing ovarian cancer xenografts, elucidating new therapeutic modalities.
INTRODUCTION
The CA125 antigen, recognized by the OC125 antibody, is a heavily glycosylated epitope encoded by MUC16.1,2 MUC16 is a complex tethered mucin consisting of an O-glycosylated amino –terminal domain, a large, glycosylated tandem repeat domain, a 58 amino acid (AA) ectodomain between the cell membrane and the putative cleavage site, a hydrophobic transmembrane region, and a short intracellular tail.3,4 We demonstrated that the MUC16 ectodomain, recognized by specific antibodies (e.g., 4H11, 4A5), transforms immortalized 3T3 cells, while the cytoplasmic domain has limited effect.5,6 The 58 AA MUC16 ectodomain includes three potential N-glycosylation sites, which may represent regulatory sites for MUC16 interaction with other cell surface molecules. However, the mechanisms by which MUC16 C-terminal N-glycosylation might promote oncogenic behavior are not understood.7 MUC1, MUC3, MUC4, and MUC16 are all heavily glycosylated tethered glycoproteins characterized by the presence of transmembrane domains and diverse signaling mechanisms, and each is overexpressed in different spectra of epithelial cancers.8
At the cell surface, lectins perform important biological functions through binding glycans, and are highly dependent on specific glycan species. Galectin-3, which regulates ligand residency, signaling intensity and cellular behavior, interacts with appropriately N-glycosylated cell surface receptors, such as epidermal growth factor receptor (EGFR), and platelet-derived growth factor receptor (PDGFR).9,10 Effects of N-glycosylation depend on both the number of N-glycosylation sites on growth-enhancing receptors and on the relative abundance of specific N-glycosylated species that bind Galectin-3.11 However, relationships of cancer-associated mucins with classic receptor tyrosine kinases are not well delineated. To unravel the specific cancer biology of MUC16/CA125, we have investigated the role of glycosylation-based mechanisms in the ectodomain with potential partners involved in MUC16 oncogenicity. We show that site-specific N-glycosylation of the MUC16 ectodomain plays a key role in mucin-induced transformation, mediating complex cell surface interactions. Employing synthetic glycopeptides, we have developed new blocking monoclonal antibodies (MAbs) directed to the ectodomain N-glycosylation sites inhibiting the glycosylation-dependent effects of MUC16 on invasion and in vivo tumor growth. These MAbs interfere with MUC16–Galectin-3 cell surface interactions destabilizing signal transduction, blocking MUC16-driven oncogenic behaviors.
RESULTS AND DISCUSSION
MUC16 oncogenic effects are dependent on MUC16 ectodomain glycosylation
In earlier studies, forced expression of the terminal 114 AA sequence of the C-terminal MUC16 ectodomain (MUC16c114) produced tunicamycin-sensitive in vitro/in vivo oncogenic behaviors of 3T3 mouse fibroblasts, including a significant increase in MUC16-driven matrigel invasion, oncogene activation, and rapid tumor growth in vivo.6 This sequence includes the ectodomain, the transmembrane domain and the cytoplasmic tail. To identify the responsible MUC16 sequence elements, we employed human MUC16 expression vectors to transfect two MUC16-negative human ovarian cell lines (SKOV3 and A2780) for forced MUC16 expression.6 Like our prior 3T3 studies, stable expression of MUC16c114 in both A2780 and SKOV3 cell lines led to high levels of cell surface MUC16 expression but no growth change in vivo (Supplemental Figure 1A). MUC16 expression resulted in a greater than two-fold increase in matrigel invasion, which was inhibited by Swainsonine and Kifunensine (Figure 1A). To interrogate the glycosylation-dependent mechanisms of MUC16 oncogenic transformation, several strategies were devised to interfere with potential N-glycosylation–dependent effects. First, we created a lectin-blocking fusion protein by combining the sugar-binding domain of Galectin-3 with a truncated pFUSE-human IgG1–Fc2 sequence (117–244LGALS3-pFUSE).12 This mimics Galectin-3 sugar binding but lacks the ability to form Galectin-3 pentamers, preventing formation of the stabilizing Galectin-3 gel/matrix. A second sugar-binding fusion protein (11-119LGALS1-pFUSE) was created as a negative control with the sugar-binding domain of Galectin-1. We also prepared a soluble MUC16 ectodomain by linking the same pFUSE vector to the 58 AA MUC16 ectodomain sequence. This created a “dummy receptor” fusion protein to bind potential MUC16-interacting molecules from the extracellular space (MUC16c57-114-pFUSE).6 As shown in Figure 1A, exogenous exposure to either the MUC16c57-114-pFUSE dummy receptor protein or the 117-244LGALS3-pFUSE protein blocked MUC16-induced increase in matrigel invasion. However, the 11-119LGALS1-pFUSE protein had no inhibitory effect on invasiveness. We also examined MUC16 effects on matrigel invasion in wild-type human ovarian cells with established MUC16/CA125 protein expression (Figure 1B). These cell lines expressed full-length MUC16 with multiple tandem repeats, and released CA125 antigen into the cell culture supernatant. Swainosine, Kifunensine, the MUC16c57-114-pFUSE protein, and the 117-244LGALS3-pFUSE protein all substantially decreased matrigel invasion in cell lines bearing native MUC16. As with the MUC16 transfection models in Figure 1A, the 11-119LGALS1-pFUSE and the empty pFUSE proteins had no effect on matrigel invasion in any of the MUC16-positive cell lines. To confirm N-glycosylation dependence, we hypothesized that MUC16 invasiveness would be inhibited by short hairpin RNA (shRNA)-mediated loss of the glycosylation enzyme MGAT5 or inhibiting Galectin-3.13 MGAT5 catalyzes the formation of tetra-antennary N-glycans binding Galectin-3 with the highest affinity.14 Tumor growth is inhibited in mice deficient in either MGAT5 or Galectin-3. Stable expression of targeted shRNA reduced MGAT5 or Galectin-3 (LGALS3) protein levels, markedly decreasing invasion of both SKOV3 and A2780 cells bearing the retained MUC16 ectodomain sequences (Supplemental Figure 1B). In contrast, shRNA targeting Galectin-1 was ineffective in blocking MUC16-induced invasion.
Figure 1. Effect of MUC16 Expression on SKOV3 and A2780 Ovarian Cancer Cells.
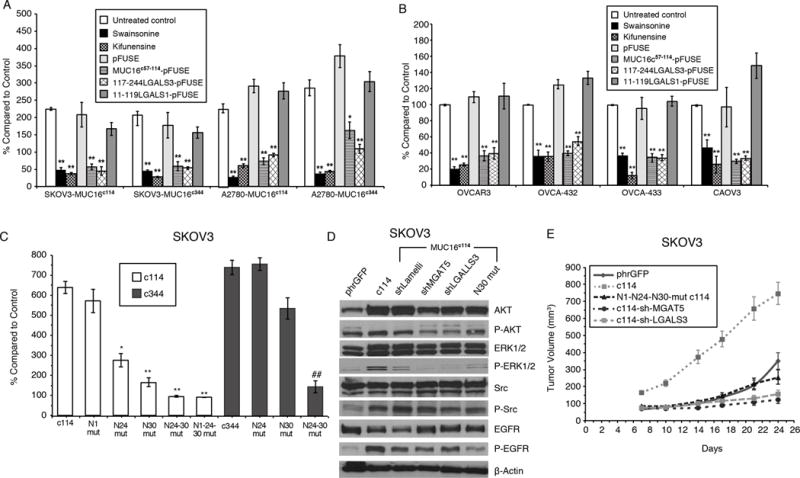
A) MUC16 enhancement of matrigel invasion assay SKOV3 and A2780 cells depends on N-glycosylation. Cells were exposed to Swainsonine(1 ug/ml), Kifunensine (1ug/ml), control pFUSE protein, MUC16c57-114-pFUSE fusion protein, 117-244LGALS3-pFUSE, or 11-119LGALS1-pFUSE fusion protein (all 5 ug/ml) and compared to vector controls. Results (n=3) are expressed as percentages compared by paired t test to parent SKOV3 orA2780 phrGFP control after 48 h (Mean ± SE). (*=p,0.01; ** p< 0.0001) In both SKOV3 and A2780 cell lines, MUC16c114 or MUC16c344 cells were more invasive than the control phrGFP cells (** = p<0.0001), and these invasive properties were unaffected by exposure to the pFUSE vector-only protein. Each study was replicated ≥ 3 times.
B) Matrigel invasion assay for wild-type ovarian cancer cell lines depends on Galectin-3. OVCAR3, OVCA-432, OVCA-433, and CAOV3 cells were exposed to Swainsosine (1 ug/ml), Kifunensin (1ug/m), control pFUSE protein, MUC16c57-114-pFUSE fusion protein, 117-244LGALS3-pFUSE, or 11-119LGALS1-pFUSE fusion protein (all 5 ug/ml). Invasion was measured in triplicate (n=3) and normalized against untreated control cells. Results (n=3) are expressed as percentages compared by paired t test to parental control cells after 48 h (Mean ± SE). (*=p,0.01; ** p< 0.0001) Two or more replicates were performed for each condition.
C) Loss of proximal N-glycosylation sites impair matrigel invasion. SKOV3-MUC16c114 and SKOV3-MUC16c344 transfected cell lines were tested for MUC16-based increased invasion following N→A mutations of N-glycosylation sites at each of the N1, N24, and N30 positions. Invasion was measured in triplicate (n=3), with 3 or more independent replicates. Results are expressed as percentages compared to SKOV3phr cells without MUC16 expression. Results (n=3) are expressed as percentages compared by paired t test to parental control cells after 48 h (Mean ± SE). (*=p,0.01; ** p< 0.0001)
D) MUC16-induced oncogene activation on AKT, MAPK, and SRC signaling pathways indicating MUC16 increased phosphorylation of AKT (S473), ERK1/2 (pT202/Y204), SRC(y416), and EGFR(pY1068) in SKOV3-MUC16c114 cells. MGAT5 (shMGAT5), Galectin-3 (shLGALS3) knockdowns, and N30A mutation all reduced MUC16c114-induced oncogene activation.
E) MUC16 N-glycosylation–dependent tumor growth in vivo. In vivo growth of SKOV3-MUC16c114 expressing cell line was much more aggressive (p<0.0001 by paired t test) compared to the SKOV3-phrGFP control. However, SKOV3-MUC16(N1-N24-N30)mut-c114 glycosylation -impaired transfectants did not show any growth enhancement compared to SKOV3-phrGFP vector control tumors. SKOV-3-MUC16c114 cells with shRNA of LGALS3 or MGAT5 were also similar to control cells
Having implicated N-glycosylation driving MUC16 oncogenic behavior, we next examined asparagine site specificity by systematic deletion of the three MUC16 ectodomain N-glycosylation positions. Deleting all three MUC16 ectodomain N-glycosylation sites (asparagine residues at N1, N24, and N30 of the MUC16c114 sequence) eliminated all observed MUC16 glycosylation-dependent behaviors in 3T3 cells.6 Each mutation was then examined individually in MUC16 transfection models of SKOV3 cells. An asparagine to alanine (N→A) mutation (N1) of the farthest disposed asparagine, adjacent to the cleavage site, had no effect on SKOV3 invasion (Figure 1C). In contrast, N→A mutations of either of the more membrane proximal N24 and N30 asparagines decreased invasion. Preservation of the asparagine closest to the membrane surface (N30) was the most critical for invasion enhancement, and effects only modestly increased by additional N→A mutation of the N24 residue. We examined larger MUC16 constructs with 344 AAs from the MUC16 C-terminal (MUC16c344) in the same models to determine the contribution of seven other N-glycosylation sites. When longer expression vectors bearing MUC16 mutations at N24c344, N30c344, or N24-N30c344 were transfected into SKOV3 lines, matrigel invasion was significantly reduced compared with the original vector (Figure 1C). Although these larger constructs have seven additional N-glycosylation sites distal to the cleavage site, mutating the crucial N24 and N30 sites still markedly decreased matrigel invasion. Similar effects were observed in A2780 cells bearing MUC16 sequences (Supplemental Figure 1C).
Expression of MUC16 in SKOV3 cells also increased phosphorylation of several oncogenes, including pERK1/2, pSRC, and EGFR (Figure 1D). However, shRNA knockdowns of MGAT5 (shMGAT5), Galectin-3 (shLGALS3) and N→A mutation of N30 all impair MUC16c114-induced oncogene activation. Similar effects occured in A2780-MUC16 cells (Supplemental Figure 1D). Finally, we modeled the effects of knocking down MGAT5, LGALS3, and mutation of the MUC16c114 ectodomain N-glycosylation sites in xenograft tumor growth in nude mice using the SKOV3-MUC16c114 cell line (Figure 1E), demonstrating complete abrogation of SKOV3-MUC16c114–induced tumor growth after shMGAT5 introduction, shLGALS3 introduction, or loss of N-glycosylation sites.
Interactions between EGFR and MUC16
It is postulated that stabilization of growth-enhancing receptors such as EGFR depends on protein N-glycosylation and galectins via formation of stabilizing N-glycan-galectin lattices. We hypothesized that MUC16 expression enhanced invasion through EGFR stabilization.15,16 EGFR is primarily expressed on cell surface lipid rafts, and a MUC16-EGFR interaction implies that MUC16 is also present in the lipid rafts.17 This was confirmed in MUC16-positive cell lines, with MUC16 localized to cholera exotoxin-marked cell surface lipid raft regions (Figure 2A). The effects of SKOV3-MUC16c114 on EGFR expression and stability were examined. EGFR presence on the surface of SKOV3 cells increased when transfected with SKOV3-MUC16c114 compared with vector-only controls measured by fluorescence-activated cell sorting (FACS) analysis (Figure 2B). Moreover, MUC16c114 expression nearly doubled cell surface EGFR amounts when new EGFR protein synthesis was inhibited by cycloheximide (CHX) treatment, suggesting a MUC16-directed reduction in EGFR turnover. In contrast, stability of the MUC16c114 ectodomain (measured by 4H11 positivity) was unchanged by CHX. We also compared total EGFR by Western Blot analysis in SKOV3-MUC16c114 and SKOV3-phrGFP cells treated with CHX for 24 h normalized against β-Actin. Densitometry curves (Figure 2C) indicated that the presence of MUC16c114 stabilizes total cellular EGFR amounts compared with the phrGFP-vector control. Confirming these results, we employed a tetracycline-inducible MUC16c114 system. Tetracycline exposure-induced MUC16c114-dependent matrigel invasion was similar to that of the stable transfectants (Figure 2D). We also examined the requirement role of EGFR by stable expression of two EGFR-targeted shRNA constructs (shEGFR) introduced into tetracycline-inducible SKOV3-MUC16c114 cells markedly reducing EGFR expression. In both clonal lines, the shEGFR-transfected cells showed markedly decreased matrigel invasion compared to parental controls. Tetracycline-induced expression of MUC16c114 in shEGFR cells had distinctly reduced matrigel invasion compared with SKOV3-MUC16c114 cells despite robust MUC16 expression (Figure 2D insert), supporting the dependence of SKOV3-MUC16c114-induced matrigel invasion on increased EGFR expression (Figure 2D).
Figure 2. MUC16 expression increases EGFR expression and stability.
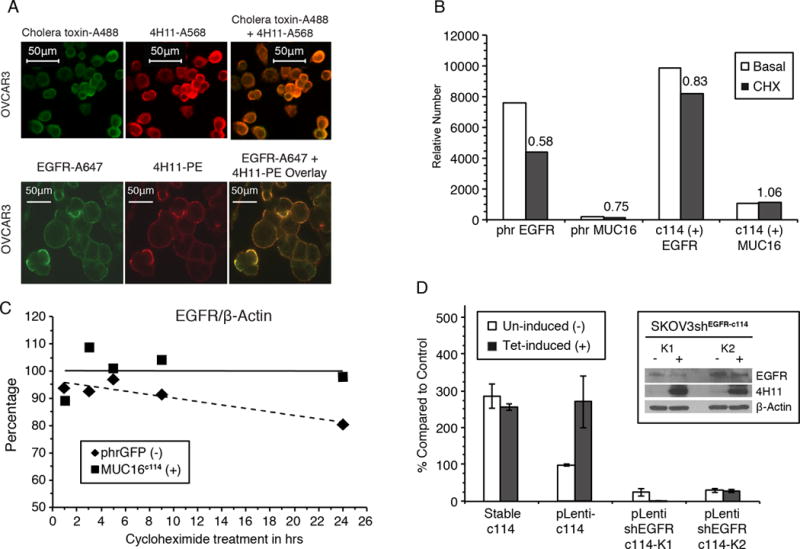
A) Cell surface MUC16 is localized in lipid rafts and co-localizes with EGFR. Cholera toxin localizes to lipid rafts on the cell surface (green label) (Alexa488) and co-localizes with the red-labeled anti-MUC16 (Alexa568) on the cell surface of OVCAR 3 cell line. In the same cell line, EGFR (Alexa647 green label) also co-localizes with MUC16 (Phycoerythrin).
B) MUC16 increased EGFR expression. Cells were labeled wth anti-EGFR-A647 and relative number estimated by FACS geometric mean fluorescence. Relative cell surface EGFR expression was reduced to 58% of untreated levels upon 24 h of CHX exposure in SKOV3-phrGFP. In contrast, in the SKOV3-MUC16c114 cells, there was an increase in EGFR geometric mean fluorescence, which decreased to 83% of that of the control after CHX exposure. MUC16c114 mean fluorescence is not reduced by CHX.
C) Expression of MUC16c114 stabilizes total EGFR after cycloheximide. SKOV3-cells with and without MUC16 expression were exposed to cycloheximide and expression of EGFR species were compared at various times. Densitometry of the EGFR/β-actin ratio illustrates that there is a steady loss of EGFR over time in SKOV3-phrGFP cells during CHX exposure. In contrast, the total level of EGFR in SKOV3-MUC16c114 cells is maintained, showing EGFR stabilization compared to the MUC16-negative control cell line.
D) MUC16c114 enhancement ofmatrigel invasion is dependent on EGFR. Tetracycline induction of SKOV3-MUC16c114(tet) cells resulted in an invasive phenotype similar to the stable SKOV3-MUC16c114 (SKOV3c114). When a short hairpin RNA knockdown of EGFR (shEGFR) was introduced into SKOV3-MUC16c114(tet) cells, tetracycline induced expression of MUC16 (4H11 positive protein in insert) but did not increase matrigel invasion (n=3).
Galectin-dependent co-localization of MUC16 and other cell surface proteins
Our results in whole cells illustrate that MUC16 stabilization of EGFR is linked to ovarian cancer cell invasion, with dependence on the presence of an appropriately N-glycosylated MUC16 protein ectodomain interacting in concert with both Galectin-3 and EGFR. We confirmed this critical interaction with purified proteins in co-precipitation studies to illuminate the necessary tripartite protein interaction. For this, we used purified MUC16c57-114-pFUSE as the glycosylated MUC16 part of the interaction, Fc for precipitation, mixed with purified EGFR and Galectin-3 (LGALS3) proteins. All three proteins were produced in human cell lines for appropriate glycosylation. Each of the three proteins detected are shown in the direct immunoblot in the left three lanes in Figure 3A. MUC16c57-114-pFUSE protein bound to the Protein A/G PLUS-conjugated beads and was present in the eluate when separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel. Galectin-3 also bound to the beads and was present in the column eluate with trace amounts of co-precipitated EGFR. Galectin-3 also co-precipitated when MUC16 c57-114-pFUSE was present. However, significant amounts of EGFR were only detected in the combined eluate when both MUC16c57-114-pFUSE and Galectin-3 were present.
Figure 3. Protein-protein interactions of MUC16c114 with EGFR and Integrin β1 require Galectin-3.
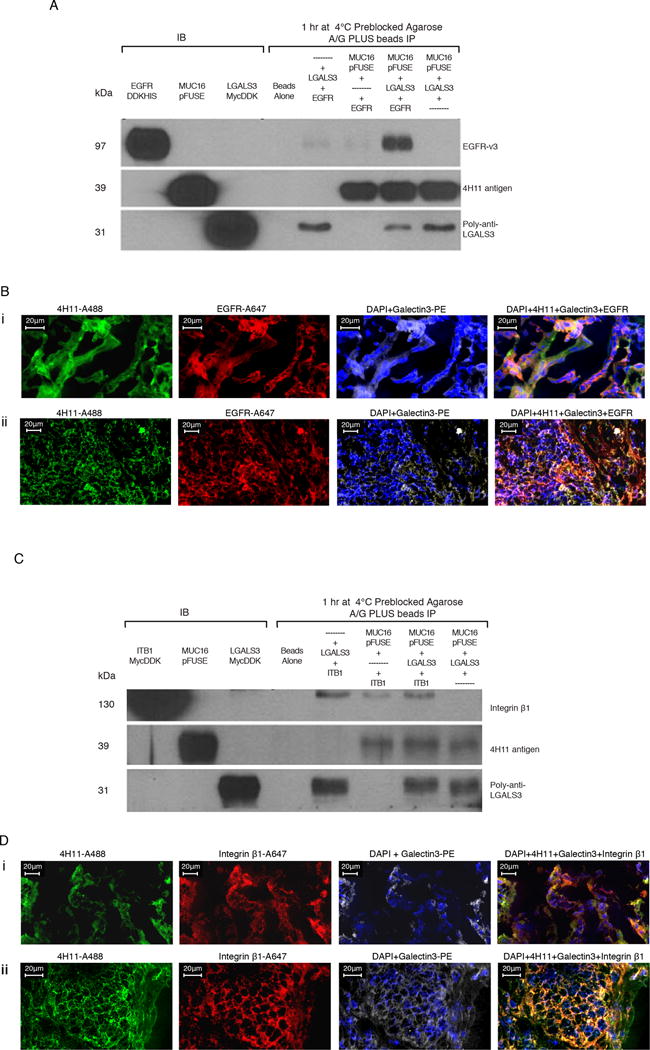
A) Immunoprecipitation (IP) of EGFR, MUC16c57-114-pFUSE, and Galectin-3. Triple immunoprecipitation studies were performed as described in the methods. In control lanes (1-3), each single protein is present by immunoblot. In the immunopreciptation lanes(5-8), the anti-MUC16 4H11antibody binds to theMUC16c57-114-pFUSE and is precipitated bound to the A/G beads. LGALS3 is present in the lane positive for MUC16c57-114-pFUSE, but not EGFR alone. EGFR is detected only when both LGALS3 and MUC16c57-114-pFUSE are present.
B) MUC16, Galectin-3, and EGFR protein co-localization in ovarian cancer explants and human ovarian cancer sections. Immunofluorescence co-localizationof a section from an ovarian cancer explant with EGFR-A647 (red), Galectin-3-PE (white) and 4H11-PE (green) for MUC16, or a combination of all three with DAPI (blue) in the last panel is performed as described in the methods. The lower panels show similar co-localization studies in a snap frozen tumor section from a patient with ovarian cancer.
C) Interaction between MUC16 and Integrin β1 requires Galectin-3. Triple immunoprecipitation studies were perfomred as in 3A. In the combination lanes (5–8) anti MUC16 antibody 4H11staining shows that MUC16c57-114-pFUSE is consistently bound to the A/G beads. LGALS3 binds in the lane positive for MUC16c57-114-pFUSE, but Integrin β1 is detected only when both LGALS3 and MUC16c57-114-pFUSE are present.
D) MUC16, Galectin-3, and Integrin β1 protein co-localization in ovarian cancer explants and human ovarian cancer sections. Immunofluorescence staining of an ovarian cancer cell explant with Integrin β (red), Galectin-3 (white) and 4H11-PE (green) for MUC16, or a combination of all three with DAPI (blue) in the last panel. The lower panels show similar co-localization studies in a snap frozen tumor section from a patient with ovarian cancer.
Because many growth-enhancing receptors are heavily glycosylated, we reasoned that lectin-dependent MUC16 cell surface effects might include other N-glycosylated lipid raft proteins beyond EGFR. Based on the observation that MUC16 ectodomain activated the SRC oncogene, we also examined Integrin β1, frequently associated with SRC activation and cancer progression. Often altered in cancer, Integrins participating in “outside-in” signaling initiated by stromal-epithelial interactions, triggering proto-oncogene tyrosine kinase (SRC) phosphorylation and downstream effects. Analogously with EGFR, purified MUC16c57-114-pFUSE bound to Integrin β1 in a Galectin-3–dependent manner, requiring all three proteins for this heterotrimeric interaction (Figure 3C).
To ascertain how these effects manifest in more complex human tumor tissue, we investigated these interactions in six different human ovarian tumor explants and six snap frozen tumors from de-identified ovarian cancer patients undergoing debulking surgery. EGFR, MUC16, and Galectin were clearly co-localized in both an example ovarian cancer explant (Figure 3Bi) and a surgical excision specimen (Figure 3Bii). Additional explants and surgical samples are shown in Supplemental Figures 2 and 3. We also confirmed co-localization of MUC16 and Integrin β1 by immunofluorescence in both ovarian cancer explants and sections taken from patients undergoing primary cancer debulking surgery (Figures 3Di and Dii). Additional co-localization studies for explants (N=5) and primary surgery sections (N=5) are shown in Supplemental Figures 2 and 3. Figure 3Di also confirms co-localization for MUC16, Galectin-3, and Integrin β1 in the same human ovarian cancer explant. We also examined primary sections of ovarian cancers from de-identified patients who had undergone primary ovarian cancer debulking surgery. When the link between MUC16, Galectin-3, and Integrin β1 was examined, co-localization is observed (Figure 3Dii and Supplemental Figure 3).
Homogeneous synthetic N-glycopeptides as epitope mimics for mAb development
Having identified N-glycosylation at the N24 and N30 sites of MUC16c114 as a central requirement for invasiveness, we analyzed the glycan profile of a purified SKOV3-MUC16c114 with N1 and N24 N→A mutations, leaving a single asparagine residue (N30) available for N-glycosylation. Glycome analysis shows heterogeneous N-glycoforms. However, common to all glycoforms is the chitobiose (GlcNAc2) disaccharide as the minimal unit directly linked to asparagine, which can be fucosylated, or function as the core for a larger mannosylated glycan (Man3GlcNAc2) (Supplemental Figure 4). Building on previous results, we hypothesized that antibodies targeted against a MUC16 ectodomain epitope encompassing the crucial N24-N30 glycosylation sites might inhibit MUC16 interaction with the galectin lattice, abrogating adverse effects of MUC16 expression, including matrigel invasion and in vivo tumor growth. Exploring this hypothesis, we designed synthetic peptide antigens of various lengths (55, 18, and 15 AAs) sequentially within the MUC16 ectodomain, which were glycosylated with chitobiose at the N24 and N30 sites (Figure 4A). Chitobiose was employed as a simplified, model N-glycan, and thought to promote better steric exposure of the underlying peptide eliciting glycan-directed antibodies retaining peptide specificity.18,19 Details are described in the Supplemental Information.
Figure 4. Glycan-MUC16 ectodomain antibody characterization.
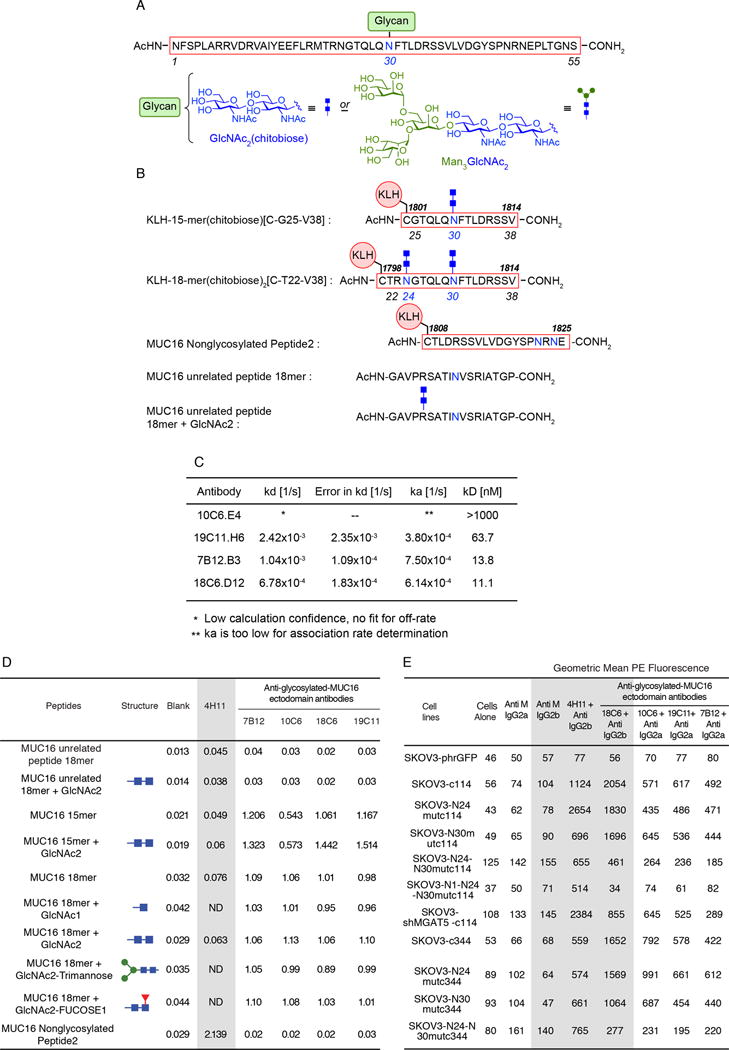
A) Immunogen Structures. Schematic structure of 55-mer MUC16 N-glycopeptide antigen with one chitobiose (GlcNAc2) at the N30 position initially used in mouse immunization for raising antibodies.
B) Schematic structures of shorter MUC16 N-glycopeptide antigens conjugated to KLH. KLH-conjugated 15-mer peptide bearing one chitobiose at the N30 position, and KLH-conjugated 18-mer peptide bis-glycosylated with two chitobioses at the N24 and N30 sites, respectively. These N-glycopeptide–KLH constructs were used to immunize mice to raise monoclonal antibodies against the GlcNAc2–peptide epitope within the MUC16 ectodomain. Sequences of the non-glycosylated MUC16 ectodomain peptide epitope of the 4H11 monoclonal antibody, as well as irrelevant MUC16-unrelated (glyco)peptides used in testing are also shown.
C) Binding affinities of GlcNAc2 mouse monoclonal as determined by using ForteBio Octet QK. Five μg/mL of biotinylated glycopeptide was loaded onto the Streptavidin biosensor. After washing off excess antigen, mouse antibodies were tested at 10 μg/mL for association and dissociation steps, respectively. Binding parameters were calculated using 1:1 binding site model, partial fit.
D) ELISA table comparing peptide reactivity of five anti-MUC16 antibodies. Reactivity of 4H11 and four lead GlcNAc2-MUC16-ectodomain monoclonal antibodies to various MUC16 and GlcNAc2-glycosylated peptides were examined by sandwich ELISA as described. No glycan-MUC16 ectodomain cross reactivity was seen with the non-glycosylated MUC16 peptide 2, or either of the unrelated peptides. Similarly, 4H11 had essentially no affinity for the GlcNAc2-MUC16 15-mer or (GlcNAc2)2-18-mer N-glycopeptides.
E) Affinity of 4H11 and 4 GlcNAc2-MUC16 ectodomain monoclonal antibodies for SKOV3-MUC16 transfections with N-glycosylation site modifications as measured by geometric mean PE fluorescence. All of the cell lines (except the SKOV3-phrGFP line, which is MUC16-negative) retained 4H11 binding, regardless of glycosylation modification, thus confirming MUC16 protein on the cell surface. When both the N24 and N30 sites of glycosylation were lost, there was a reduction of glycan-MUC16 antibody binding for both the MUC16c114 and the MUC16c344 transfectants while 4H11 reactivity persisted.
To focus an immune response against a smaller-sized epitope encompassing the relevant glycosylation site, we also synthesized shorter, 15- and 18-mer glycopeptides bearing one (N30) and two chitobiose glycans (N24 and N30), respectively (Figure 4A), the latter by analogy to the cluster presentation of the Tn antigen (GalNAc-α-O-Ser/Thr), shown to be required for binding to some mAbs.20 These glycopeptides were conjugated to a maleimide-derivatized Keyhole Limpet Hemocyanin (KLH) carrier protein through an N-terminal cysteine generating immunogens for mouse vaccination.
Mouse vaccination with synthetic glycopeptides/glycoconjugates and mAb development
Mouse vaccination and sera collection were performed using our methods, and protocols are described in the Supplemental Information.5 Supernatants were selected and screened for reactivity by enzyme-linked immunosorbent assay (ELISA) against the individual glycopeptides. Although multiple supernatants were reactive with the GlcNAc2-15-mer and (GlcNAc2)2-18-mer glycopeptides, none of the ELISA screened hybridoma supernatants demonstrated high selectivity for glycosylated over non-glycosylated peptides. Importantly, MUC16 specificity was maintained, and none of the positive supernatants were reactive with glycosylated MUC16-irrelevant negative control peptides. No overlap was seen with the peptide sequence recognized by the 4H11 mAb which did not bind with the 15/18-mer MUC16 glycopeptides. Serial subcloning afforded multiple glycosylation site primary MAbs that were reactive with MUC16-glycosylated epitopes and the homologous non-glycosylated sequences, but not with glycosylated negative control peptides. Of this pool, four high-affinity antibodies were selected and further purified for additional binding characterization (affinities are noted in the antibody affinity table [Figure 4C]).
Characterization of glycosylation site–directed anti-MUC16 MAbs
Confirmatory ELISA studies for four representative antibodies were compared to 4H11 antibody binding, which recognizes the adjacent MUC16 ectodomain peptide backbone. Unrelated chitobiose glycopeptides exhibited no significant binding by any of the anti-glycan-MUC16 antibodies selected. Binding to MUC16-derived 15-mers, the aglycosylated peptide, and the corresponding chitobiose glycopeptide, revealed that all candidate antibodies showed similar binding affinities for glycosylated and unglycosylated MUC16 peptides. Notably, additional synthetic glycopeptides including a single GlcNAc, Man3GlcNAc2, and a fucosylated chitobiose (GlcNAc2Fuc), maintained the antibody reactivity found with antibodies raised to chitobiose-linked MUC16 peptides (Figure 4D, ELISA table).
Next, we tested each antibody for affinity specificity on an extended panel of cell lines expressing differentially glycosylated MUC16 peptides (Figure 4E, FACS Table). Here, we compared SKOV3-phrGFP transfectants with the SKOV3-MUC16c114 (intact N-glycosylation), SKOV3-MUC16(N24)c114 (no N24 glycosylation site), SKOV3-MUC16(N30)c114 (no N30 glycosylation site), and SKOV3-MUC16(N1-N24-N30)c114 (no MUC16 N-glycosylation sites at all) mutants. Like the ELISA data, the results indicated that MUC16-specific targeting was present. However, loss of both N24 and N30 glycosylation sites in the MUC16 ectodomain markedly reduced glycosylation site targeted antibody binding in whole cells, but reactivity to the 4H11 antibody was retained. Cells bearing a MUC16 C-terminal chain extended to 344 AAs (e.g., SKOV3-MUC16c344) also demonstrated a similar requirement for N24 or N30 glycosylation.
Functional characterization of anti-glycosylation site antibodies
We examined immunohistochemical staining of the glycosylation-targeted anti-MUC16c114 antibodies in ovarian cancer tissue microarrays, which bound to serous ovarian cancer cells in paraffin-fixed tissue with limited interaction with other stromal tissue, similar to 4H11 behavior (Supplemental Figure 6). We characterized the MUC16 ectodomain antibodies for their effect on matrigel invasion in ovarian cancer cell lines. All of the glycosylation site–directed mAbs showed broad inhibition of matrigel invasion, and were inhibitory in ovarian cancer cells expressing SKOV3-MUC16c114 or the native, full-length MUC16 present in OVCA-433 and CAOV3 (Figure 5A). Importantly, this suggests that antibodies targeting the N24/N30 glycosylation sites inhibit the enhanced invasiveness of full-length MUC16 expression in ovarian cancer cell lines. This demonstrates antibody recognition of the key N-glycosylation sites of native MUC16 and the resulting inhibition is unaffected by other, more distal MUC16 N-glycosylation sites. Antibody inhibition of the N24/30 binding of Galectin-3 to MUC16 impairs EGFR cell surface stabilization (Supplemental Figure 5).
Figure 5. Glycan-MUC16 ectodomain antibody function.
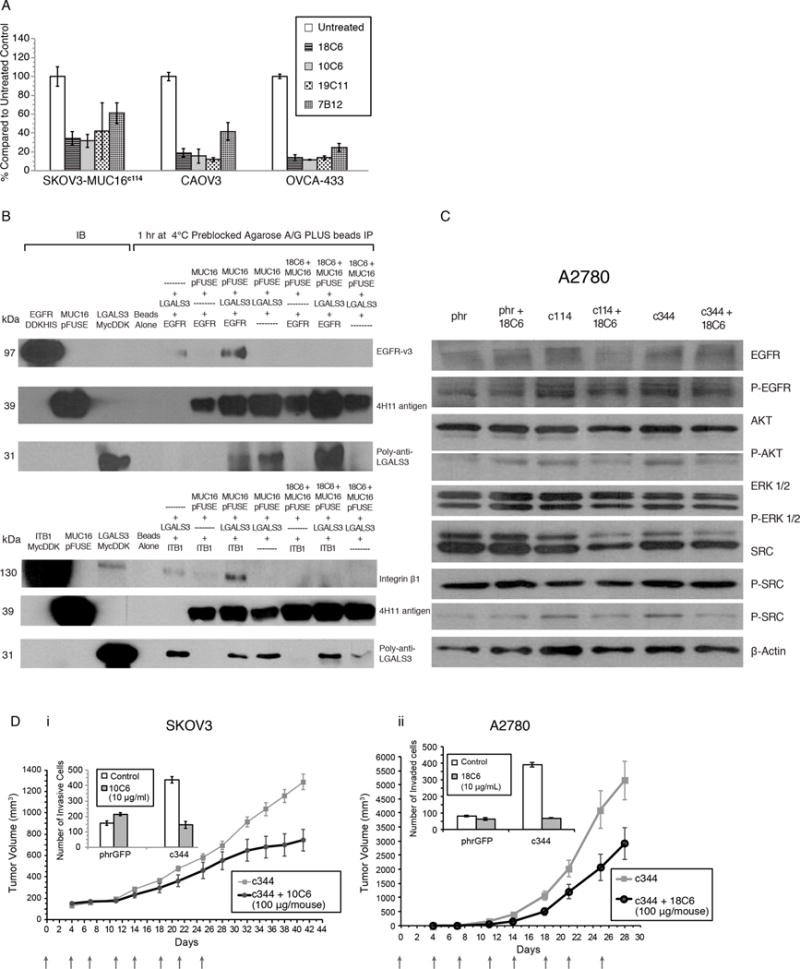
A) Anti-gylcosylation site antibodies block MUC16-enhanced matrigel invasion. Each of the four anti–glycan-MUC16-ectodomain antibodies inhibit the invasion of three different MUC16-positive ovarian cancer cell lines. Results are expressed as percentages compared to the untreated control. All of the inhibitory effects were significant compared to untreated control (p<0.001).
B) Immunoprecipitation (IP) of EGFR, MUC16c57-114-pFUSE, and Galectin-3 is blocked by anti glycosylation site antibodies. The presence of an anti-MUC16 N-glycosylation antibody (18C6) (lanes 9-11) completely eliminates interactions between MUC16–Galectin-3 and EGFR (lanes 5-8). The 18C6 antibody (lanes 9-11) also blocks interaction between MUC16, Galectin-3 and Integrin β1 proteins (lanes 5–8).
C) Anti-glycosylation site antibody 18C6 blocks MUC16-induced oncogene expression. As in Figure 1D, the pAKT, pERK1/2, pSRC, and pEGF receptor (pEGFR) signaling pathways were examined, indicating increased phosphorylation of AKT (S473), ERK1/2 (pT202/Y204), pSRC(y416), and pEGFR(y1068) in A2780-MUC16c114 and A2780-MUC16c344 cells compared to the phr control cells. Eighteen-hour exposure of 18C6 antibody to A2780-MUC16c114 and A2780-MUC16c344 cells blocks MUC16c114 and MUC16c344 oncogene activation in A2780 cells.
D) Anti-glycosylation site antibody blocks SKOV3-MUC16c344 and A2780-MUC16c344 tumor growth in athymic female nude mice. SKOV3-MUC16c344 or A2780-MUC16c344 tumor cells were each introduced into the flank of 20 nu/nu mice. Ten mice were treated intravenously from day 0 with purified 10C6 (Panel 5Di) or 18C6 (Panel 5Dii) GlcNAc2–MUC16 monoclonal antibody at 100 μg/mouse twice per week for 4 weeks. Tumors were measured by calipers twice/week. The differences in mean tumor volume were significantly decreased (p=0.0004) with 10C6 monoclonal antibody-treated mice bearing SKOV3-MUC16c344. The mice bearing A2780-MUC16c344 tumors were treated twice per week with the 18C6 antibody. The mean tumor volume was significantly decreased (p=0.02) with 18C6 monoclonal antibody-treated mice bearing A2780-MUC16c344 tumors compared to untreated A2780-MUC16c344 tumors. The inserts in the figure show the matrigel invasion assay with the same cell lines performed in the presence and absence of purified 10C6 or 18C6 monoclonal antibody.
We then confirmed MUC16 glycosylation targeting antibodies block MUC16–Galectin-3–growth factor interactions. Co-precipitation studies using purified MUC16c114, Galectin-3, and EGFR protein were performed in the presence and absence of anti-MUC16 glycosylation antibodies. As before, MUC16 c114–EGFR and MUC16 c114–Integrin β1 co-precipitation required Galectin-3. However, when the anti-MUC16 glycosylation site antibody 18C6 was added, neither EGFR nor Integrin β1was co-precipitated with MUC16 c114-pFUSE, even in the presence of Galectin-3 (Figure 5B). The glycosylation site–directed antibodies also inhibited MUC16-dependent oncogene activation in A2780 ovarian cancer cells, demonstrated by Western Blot analysis (Figure 5C). Finally, we tested the effects of glycosylation site-targeted anti-MUC16 antibodies on the growth of MUC16-positive xenografts in immunocompromised mice. We chose the SKOV3-MUC16c344 and the A2780-MUC16c344 cells because multiple N-and O-glycosylation sites are present in both extended constructs. The 10C6 antibody decreased matrigel invasion by MUC16c344 cells (Figure 5Di insert) significantly reducing the growth of SKOV3-MUC16c344 tumor cells in the mouse flank. Confirming, mice with MUC16-bearing tumors from A2780-MUC16c344–expressing cells were treated with a different MUC16 glycosylation site antibody, 18C6. As with SKOV3-MUC16c344, the anti-MUC16 glycosylation site antibody inhibited matrigel invasion, and reduced A2780-MUC16c344 tumor growth compared to untreated controls (Figure 5Dii).
The role of mucins in cancer transformation is not completely understood. It is well described that N-glycosylation patterns play important roles in cancer growth and progression through interactions with the tumor microenvironment.11 Many cytokine receptors and solute transporters are N-glycosylated, interacting with lectins regulating receptor function.21 For example, interactions between Galectin-1 and N-glycosylation sites on vascular endothelial growth factor receptor-2 (VEGFR2) sustain angiogenesis in tumor cells even during anti-angiogenic therapy. Glycoprotein diversity is influenced by environmentally dependent hexosamine flux through Golgi-based glycosylation and glycosylation enzyme expression.22 Growth factor receptors such as EGFR are preferentially glycosylated in a nutrient-dependent manner, with heavily glycosylated receptors preferentially delivered to cell surface lipid rafts helping drive cellular growth.23 Contrastingly, inhibitory receptors such as transforming growth factor-β (TGF-β) have fewer potential N-glycosylation sites and are presented on the cell surface later, with consequent inhibition of growth stimuli.10 In ovarian cancer, invasive behavior is linked to EGFR expression, and EGFR signaling is dependent both on receptor glycosylation state and the affinity of the N-glycosylated species for lectins, but this relationship to MUC16 has not be previously described15,24 Although the concentration of tri – antennary and tetra-antennary forms was below the detection limit in our assays (supplement figure 4), the much higher affinity of Galectin-3 for polylactosamine binding to these complex N-glycans forms is probably consistent with the dependence we see in our MGAT5 knockdown cell experiments. The role of Galectin-3 in cancer has been previously recognized, with development of anti–Galectin-3 targeting also proposed.25, 26, 27
Our results reveal novel insights into the role of MUC16/CA125 overexpression in ovarian cancer, including the following: (1) the extracellular glycosylation state of MUC16 helps drive invasive ovarian cancer behavior; (2) MUC16-driven invasion and tumor xenograft growth depend on specific MUC16 ectodomain N-glycosylation sites; and (3) MUC16 heteromolecular interaction with Galectin-3 and EGFR is required for oncogenic effects. Through the formation of lectin-mediated, low-affinity multi-molecular complexes, MUC16 is able to enhance the “outside-in” signal transduction in a glycosylation-dependent manner. We demonstrate that MUC16 is present in ovarian cancer cell lipid rafts promoting growth through Galectin-3–dependent interactions with growth factor receptors such as EGFR and Integrin β1. Interventions compromising these specific N-glycosylation sites through blocking with inhibitory Galectin-3 constructs, or interfering with complex N-glycosylation by suppression of MGAT5, all impair the tumor-promoting effects of MUC16 expression (Figure 6). Further, MUC16 was consistently co-localized with EGFR and Integrin β1 in ovarian cancer cell lines, human ovarian tumor explants, and tumor samples from clinical ovarian cancer resections. A similar MUC1, Galectin-3, and EGFR interaction in breast cancer cells has been demonstrated in human tumor breast tissue.28
Figure 6. Targeting glycosylation-dependent MUC16 function.
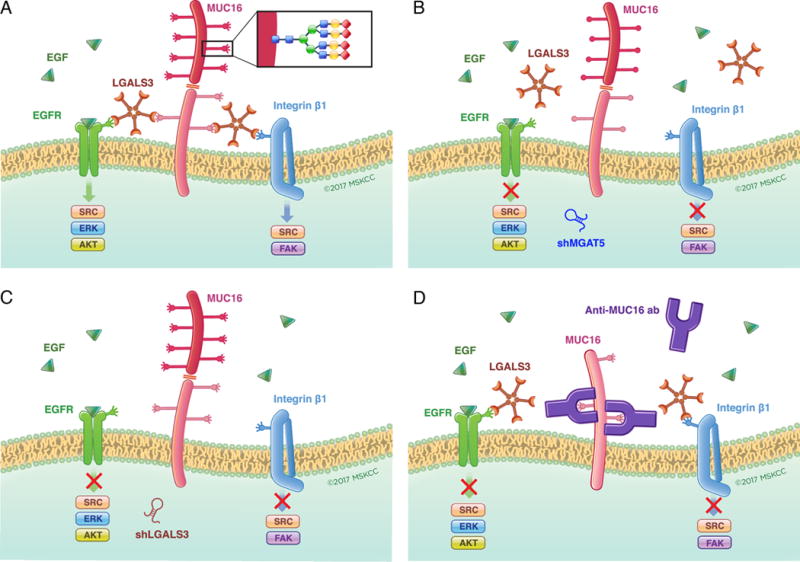
In Panel A, the dependence of MUC16 action on the presence of high complexity N-glycosylation at specific asparagine residues, Galectin-3 pentamers and growth factor receptors is graphically summarized. Panels B and C illustrate how loss of tumor-derived Galectin-3 or the tumor cell glycosylation enzyme MGAT5 may each block this interaction through depletion of a key component of the glycosylation-galectin-growth factor receptor interaction. Panel D illustrates how exogenous anti-MUC16 glycosylation site antibody interferes with galectin binding to MUC16 N-glycosylation sites to block-dependent oncogenic behaviors.
These mechanistic insights into MUC16-transforming mechanisms were applied to develop new MAbs. We successfully utilized simplified MUC16 N-glycan constructs to prepare antibodies against the chitobiose-bearing (at the N24/N30 sites) MUC16c114 glycopeptide epitope of the human MUC16 ectodomain. These new antibodies blocked MUC16-enhanced invasion, oncogene activation, and in vivo tumor growth. Importantly, they also inhibited MUC16 oncogenic properties in several ovarian cancer cells with expressed full-length MUC16, and in models with truncated C-terminal MUC16 expression. The antibody specificity for defined MUC16 sequences and the limited tissue distribution of MUC16 ectodomain antigen provides potentially tissue-specific therapeutic application. Our prior antibodies (4H11) directed to adjacent sequences within the MUC16 ectodomain did not inhibit in vivo tumor growth.5 The two key N-glycosylation sites are conserved in the mouse, rat, hamster, and bovine sequences of MUC16, suggesting common normal biologic function, and the same N-glycosylation sites were preserved in all of the high-grade serous ovarian cancer tumors from The Cancer Genome Atlas (TCGA).29 These results provide novel extracellular targets for MUC16-based therapeutic strategies in ovarian cancer. High-affinity, glycosylation site-specific, humanized antibodies for clinical application are currently in development.
Materials and Methods
Synthesis of MUC16c114, MUC16c344, and Glycosylated Fusion Protein DNA Constructs
The construction of MUC16 vectors and the chimeric proteins MUC16c57-114-pFUSE and 117-244LGALS3-pFUSE were produced as previously described.6
Cell culture, Transfection, and Cell Line Characterization
The SKOV3, A2780, CAOV3, OVCAR3, OV432, and OV433 cell lines were obtained and maintained according to our prior work.5,6
Synthesis of Glycopeptides
MUC16 glycopeptide synthesis is detailed in the Supplemental Chemistry Information section.
Matrigel invasion
Basement membrane invasion was determined in matrigel invasion chambers (BD Biosciences, Bedford, MA).6
Tumor growth in athymic nude mice
Cell lines were introduced into the flank of 10 athymic female nude mice per group, and routine animal care was provided by the Memorial Sloan Kettering Cancer Center (MSK) Antitumor Assessment Core Facility. Tumor measurements were taken twice per week, and tumor growth was recorded as per MSK RARC guidelines. All of the mouse experiments were performed under animal protocols #09-03-003 and 15-04-005 and the NIH Guide of the Care and Use of Laboratory Animals.
Mouse immunization protocol
The immunization protocol started with chitobiose-containing 55-mer MUC16 glycopeptide (GlcNAc2-55-mer), which was administered to five BALB/c mice every 3 weeks in the presence of complete adjuvant. The fourth immunization was carried out with a mixture of KLH-conjugated, mono-glycosylated 15-mer (GlcNAc2-15-mer-KLH) and bis-glycosylated 18-mer ([GlcNAc2]2-18-mer-KLH) MUC16 constructs, and sera were analyzed for reactivity against the GlcNAc2-55-mer and the un-conjugated, chitobiose-bearing 15/18-mer glycopeptides. Mice were further immunized with the shorter KLH-conjugates two more times every 3 weeks, and the serum responses were analyzed by ELISA.5
Western Blot analysis
Detailed methods for Western Blot analysis are reported in our prior publications.5,6
Galectin-Mediated MUC16 Protein–Protein Interactions
MUC16c57-114-pFUSE glycosylated protein was combined with LSGALS3 protein (0.13 μg) and EGFR (0.13 μg) or Integrin β1 (0.13 ug), and then pre-blocked Protein A/G PLUS Agarose beads were added. IP pellets were washed extensively, boiled in loading buffer, and separated by 10% SDS-PAGE gel electrophoresis, then transferred onto nitrocellulose membrane. The membranes were probed either with anti-EGFR-v3, anti-Integrin β1, anti-4H11-HRP, or polyclonal anti-LGALS3 antibodies.
Immunofluorescence staining
The procedures for the immunofluorescence studies are included the supplemental material.
Statistical Analysis
Statistical Analyses for matrigel invasion studies and xenograft growth studies were performed as previously described.5,6
Supplementary Material
Acknowledgments
The authors acknowledge the help and support of MSK core facilities, especially the Monoclonal Antibody Facility, Flow Cytometry, Molecular Cytology, Anti-Tumor Animal Assessment Facility, Organic Synthesis Facility, Research Animal Resource Center (RARC), and Medical Graphics. The authors also acknowledge Dr. Parastoo Azadi, (Complex Carbohydrate Research Center, University of Georgia) for help in N-glycosylation profiling, which was supported by the NIH-funded grant entitled ‘Research Resource for Biomedical Glycomics’ (P41GM10349010) from the Complex Carbohydrate Research Center. We thank Dr. Barney Yoo for technical assistance with the KLH conjugation of the synthetic constructs. We also thank Dr. Baptiste Aussedat for providing Man3GlcNAc2. We also thank George Monemvasitis for his expert editorial assistance. This research was supported by NIH grant 1P01CA190174-01A1 and the Ovarian Cancer Research Foundation Award to Dr. David R. Spriggs. Dr. Alberto Fernández-Tejada thanks the European Commission (Marie Curie International Outgoing Fellowship) for postdoctoral funding. This work used core facilities funded by the NIH Core Grant P30CA008748.
Footnotes
Conflict of Interest Statement
Memorial Sloan Kettering Cancer Center filed a patent application for MUC16 glycosylated ectodomain monoclonal antibodies on March 17, 2015, and the outcome is pending.
Author Contributions
Conceptualization, T.D.R., D.R.S., S.J.D., and J.S.L.; Methodology, T.D.R., A.F.T., A.J.A., D.R.S., S.J.D., D.A.L. and J.S.L.; Investigation, T.D.R., A.F.T., N.R., X.Y., S.T., A.W., A.J.A., K.J.P., B.N., N.O. and J.X.; Data Interpretation, T.D.R., D.R.S., S.J.D., and J.S.L.; Writing, T.D.R., A.F.T. and D.R.S.; Writing - Review & Editing, T.D.R., A.F.T., N.R., X.Y., S.T., A.J.A., K.J.P., B.N., J.X., J.S.L., S.J.D., and D.R.S.; Visualization, D.R.S., and T.D.R.; Supervision, D.R.S., and T.D.R.; Funding Acquisition, D.R.S., S.J.D., and J.S.L.
References
- 1.Bast RC, Jr, et al. Reactivity of a monoclonal antibody with human ovarian carcinoma. J Clin Invest. 1981;68:1331–7. doi: 10.1172/JCI110380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yin BW, Dnistrian A, Lloyd KO. Ovarian cancer antigen CA125 is encoded by the MUC16 mucin gene. Int J Cancer. 2002;98:737–40. doi: 10.1002/ijc.10250. [DOI] [PubMed] [Google Scholar]
- 3.O’Brien TJ, et al. The CA 125 gene: an extracellular superstructure dominated by repeat sequences. Tumour Biol. 2001;22:348–66. doi: 10.1159/000050638. [DOI] [PubMed] [Google Scholar]
- 4.Yin BW, Lloyd KO. Molecular cloning of the CA125 ovarian cancer antigen: identification as a new mucin, MUC16. J Biol Chem. 2001;276:27371–5. doi: 10.1074/jbc.M103554200. [DOI] [PubMed] [Google Scholar]
- 5.Dharma Rao T, et al. Novel monoclonal antibodies against the proximal (carboxy-terminal) portions of MUC16. Appl Immunohistochem Mol Morphol. 2010;18:462–72. doi: 10.1097/PAI.0b013e3181dbfcd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rao TD, et al. Expression of the Carboxy-Terminal Portion of MUC16/CA125 Induces Transformation and Tumor Invasion. PLoS One. 2015;10:e0126633. doi: 10.1371/journal.pone.0126633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bast RC, Jr, Spriggs DR. More than a biomarker: CA125 may contribute to ovarian cancer pathogenesis. Gynecol Oncol. 2011;121:429–30. doi: 10.1016/j.ygyno.2011.04.032. [DOI] [PubMed] [Google Scholar]
- 8.Kufe DW. Mucin in Cancer: function, prognosis, and therapy. Nat Rev Cancer. 2009;9:874–85. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dennis JW, Lau KS, Demetriou M, Nabi IR. Adaptive regulation at the cell surface by N-glycosylation. Traffic. 2009;10:1569–78. doi: 10.1111/j.1600-0854.2009.00981.x. [DOI] [PubMed] [Google Scholar]
- 10.Lau KS, et al. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. 2007;129:123–34. doi: 10.1016/j.cell.2007.01.049. [DOI] [PubMed] [Google Scholar]
- 11.Lau KS, Dennis JW. N-Glycans in cancer progression. Glycobiology. 2008;18:750–60. doi: 10.1093/glycob/cwn071. [DOI] [PubMed] [Google Scholar]
- 12.Ruvolo PP. Galectin 3 as a guardian of the tumor microenvironment. Biochim Biophys Acta. 2016;1863:427–37. doi: 10.1016/j.bbamcr.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Granovsky M, et al. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat Med. 2000;6:306–12. doi: 10.1038/73163. [DOI] [PubMed] [Google Scholar]
- 14.Hirabayashi J, et al. Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochim Biophys Acta. 2002;1572:232–54. doi: 10.1016/s0304-4165(02)00311-2. [DOI] [PubMed] [Google Scholar]
- 15.Lajoie P, et al. Plasma membrane domain organization regulates EGFR signaling in tumor cells. J Cell Biol. 2007;179:341–56. doi: 10.1083/jcb.200611106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gui TSK. The Epidermal Growth Factor Receptor as a Target in Epithelial Ovarian Cancer. Cancer Investigation. 2012;36:490–6. doi: 10.1016/j.canep.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Lajoie P, Goetz JG, Dennis JW, Nabi IR. Lattices, rafts, and scaffolds: domain regulation of receptor signaling at the plasma membrane. J Cell Biol. 2009;185:381–5. doi: 10.1083/jcb.200811059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernandez-Tejada A, Vadola PA, Danishefsky SJ. Chemical synthesis of the beta-subunit of human luteinizing (hLH) and chorionic gonadotropin (hCG) glycoprotein hormones. J Am Chem Soc. 2014;136:8450–8. doi: 10.1021/ja503545r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Z, et al. A general strategy for the chemoenzymatic synthesis of asymmetrically branched N-glycans. Science. 2013;341:379–83. doi: 10.1126/science.1236231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazal D, et al. Monoclonal antibodies toward different Tn-amino acid backbones display distinct recognition patterns on human cancer cells. Implications for effective immuno-targeting of cancer. Cancer Immunology Immunotherapy. 2013;62:1107–1122. doi: 10.1007/s00262-013-1425-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dennis JW, Brewer CF. Density-dependent lectin-glycan interactions as a paradigm for conditional regulation by posttranslational modifications. Mol Cell Proteomics. 2013;12:913–20. doi: 10.1074/mcp.R112.026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Croci DO, et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell. 2014;156:744–58. doi: 10.1016/j.cell.2014.01.043. [DOI] [PubMed] [Google Scholar]
- 23.Partridge EA, et al. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science. 2004;306:120–4. doi: 10.1126/science.1102109. [DOI] [PubMed] [Google Scholar]
- 24.Alper O, et al. Epidermal growth factor receptor signaling and the invasive phenotype of ovarian carcinoma cells. J Natl Cancer Inst. 2001;93:1375–84. doi: 10.1093/jnci/93.18.1375. [DOI] [PubMed] [Google Scholar]
- 25.Mirandola L, et al. Cancer testis antigens: novel biomarkers and targetable proteins for ovarian cancer. Int Rev Immunol. 2011;30:127–37. doi: 10.3109/08830185.2011.572504. [DOI] [PubMed] [Google Scholar]
- 26.Lu H, et al. Galectin-3 regulates metastatic capabilities and chemotherapy sensitivity in epithelial ovarian carcinoma via NF-kappaB pathway. Tumour Biol. 2016;37:11469–77. doi: 10.1007/s13277-016-5004-3. [DOI] [PubMed] [Google Scholar]
- 27.Gu Kang H, et al. Galectin-3 supports stemness in ovarian cancer stem cells by activation of the Notch1 intracellular domain. Oncotarget. 2016 doi: 10.18632/oncotarget.11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramasamy S, et al. The MUC1 and galectin-3 oncoproteins function in a microRNA-dependent regulatory loop. Mol Cell. 2007;27:992–1004. doi: 10.1016/j.molcel.2007.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.