

 This study reports the first use of an iridium(iii) compound and its enantiomer to inhibit the H-Ras/Raf-I PPI in vitro and repress renal cancer xenografts in vivo.
This study reports the first use of an iridium(iii) compound and its enantiomer to inhibit the H-Ras/Raf-I PPI in vitro and repress renal cancer xenografts in vivo.
Abstract
Targeting protein–protein interactions (PPIs) offers tantalizing opportunities for therapeutic intervention for the treatment of human diseases. Modulating PPI interfaces with organic small molecules has been found to be exceptionally challenging, and few candidates have been successfully developed into clinical drugs. Meanwhile, the striking array of distinctive properties exhibited by metal compounds renders them attractive scaffolds for the development of bioactive leads. Here, we report the identification of iridium(iii) compounds as inhibitors of the H-Ras/Raf-1 PPI. The lead iridium(iii) compound 1 exhibited potent inhibitory activity against the H-Ras/Raf-1 interaction and its signaling pathway in vitro and in vivo, and also directly engaged both H-Ras and Raf-1-RBD in cell lysates. Moreover, 1 repressed tumor growth in a mouse renal xenograft tumor model. Intriguingly, the Δ-enantiomer of 1 showed superior potency in the biological assays compared to Λ-1 or racemic 1. These compounds could potentially be used as starting scaffolds for the development of more potent Ras/Raf PPI inhibitors for the treatment of kidney cancer or other proliferative diseases.
Introduction
Protein–protein interactions (PPIs) are indispensable for numerous biological processes such as cell signaling, gene expression, cell division, immunity, and metabolism.1 Up to a staggering 650 000 PPIs are estimated to exist in living systems,2,3 with 367 527 unique PPIs already documented on the BioGRID Database.4 Crucially, however, less than 0.01% of the PPIs constituting the interactome have been targeted with an inhibitor.2,5 Dysfunction of PPIs is implicated in oncogenesis, for example, as PPIs mediate the homo-/heterodimerization of receptor tyrosine kinases to initiate a relay of oncogenic signals to enable cancer progression.6 PPIs also offer a viable target to engage pathogenicities such as Alzheimer’s disease or viral infections that lack conventional targets such as enzymes and G-protein coupled receptors.2,7 As a consequence, targeting PPIs offers tantalizing potential for therapeutic intervention for the treatment of human disease. However, PPIs often consist of extended shallow and non-polar interfaces, thwarting the identification of small-molecule inhibitors. In this context, metal-based compounds have emerged as feasible alternatives to small molecules for the development of PPI modulators.8–16 The distinctive but tunable characteristics of metal compounds, including their molecular geometries, chemical reactivities, and electronic properties, can allow them to adopt unique three-dimensional architectures that can engage hitherto unexplored regions of the chemical space of proteins that are inaccessible to purely organic molecules.17,18
The Ras/Raf/mitogen-activated protein kinase (MEK)/extracellular-signal-regulated kinase (ERK) (Ras/Raf/MEK/ERK) signaling pathway transmits information from membrane receptors to transcription factors,19–23 thereby regulating apoptosis and cell cycle progression.24 Abnormal activation of the Ras/Raf/MEK/ERK pathway enhances tumor initiation, progression, and metastasis and is thus a frequent event in proliferative disorders such as renal cell carcinoma (RCC).25 In particular, constitutive activation of MAP kinases has been implicated in renal cell carcinogenesis,26 while activated H-Ras activity has been detected in human renal cell carcinomas.27 These studies suggest that inhibition of the Ras/Raf/MEK/ERK signaling pathway could be a potential therapeutic strategy to treat RCC. For example, sorafenib is a small molecule inhibitor of several kinases, including the Raf family kinases, which has been approved for the clinical treatment of RCC, hepatocellular carcinoma, and radioactive iodine-resistant advanced thyroid carcinoma.28–30 Recently, rigosertib was identified as the first small molecule Ras/Raf PPI inhibitor that acts as a Ras-mimetic by binding to the Ras-binding domains (RBDs) of Raf kinases, resulting in the inhibition of Raf activation and perturbation of the Ras/Raf/MEK/ERK pathway.31
In continuation of our previous efforts in developing metal compounds to modulate different PPIs,32–36 we were inspired to explore the application of group 9 metal compounds to target the PPI between Ras and Raf as a potential therapeutic strategy to treat renal cancer. In this work, we describe the identification of an iridium(iii)-based compound, 1, that blocks the H-Ras and Raf-1 PPI and inhibits Ras/Raf/MEK/ERK signaling pathway activation both in vitro and in vivo. Isolation and testing of both enantiomers of 1 critically revealed that Δ-1 is the more potent of the two enantiomers at inhibiting the H-Ras/Raf-1 PPI via engaging H-Ras and Raf-1-RBD, and thus it contributes the majority of the biological activity of racemic 1. Moreover, the ability of both racemic 1 and Δ-1 to suppress tumor growth in a murine xenograft model of renal cancer without causing overt toxicity to mice was demonstrated. To our knowledge, compound 1 is the first substitutionally-inert organometallic compound that has been reported to inhibit the Ras/Raf PPI.
Results and discussion
Discovery of an iridium-based compound as an inhibitor of the H-Ras/Raf-1 interaction
An in-house library of 15 structurally diverse, potentially biologically-privileged iridium-based and rhodium-based compounds 1–15 (Fig. 1) were first investigated for their ability to inhibit the H-Ras/Raf-1 PPI using the biomolecular fluorescence complementation (BiFC) assay. In this assay, HEK293T cells were transiently co-transfected with the BiFC plasmid pair (pEGFP-VC-H-RasV12 and pEGFP-VN-Raf-1-RBD) and exposed to 5 μM of the compounds for 6 h. In the absence of the compounds, H-Ras could bind to the Ras-binding domain present in Raf-1 in cells, generating a strong yellow fluorescence that could be detected using confocal imaging. Compounds that disrupt the H-Ras/Raf-1 PPI would be expected to reduce the fluorescence of the HEK293T cells, as was observed with the positive control compound sulindac sulphide.37 Among the library of 15 metal compounds, the iridium(iii) compound 1 showed the greatest effect at disrupting the H-Ras/Raf-1 interaction in cellulo (Fig. S1a and b†). Moreover, we determined that 1 was stable at room temperature for over 48 h as there were no changes of absorbance of 1 in 80% acetonitrile/20% Tris–HCl buffer or cell culture medium (Fig. S2†) after subtraction of the corresponding background absorbance in UV spectroscopy, and no changes of 1H NMR spectra of 1 in 90% DMSO-d6/10% D2O (Fig. S3†).
Fig. 1. Chemical structures of the cyclometallated iridium(iii) and rhodium(iii) compounds 1–34 that were synthesized and evaluated in this study.

The iridium(iii) compound 1 bears the 4,4′-dinonyl-2,2′-bipyridine (dnbpy) N⁁N ligand and two 2-(p-tolyl)pyridine (tpy) C⁁N ligands. To further explore the potential structure–activity relationships (SAR) of these metal compounds against the H-Ras/Raf-1 PPI, a lead-like diverse collection of compounds (16–34, Fig. 1) were designed and synthesized. Iridium(iii) compounds 16–19 contained the dnbpy N⁁N ligand of 1, but varied in the nature of their C⁁N ligands, while conversely iridium(III) compounds 20–24 retained the tpy C⁁N ligands of 1 but possessed different N⁁N ligands. Compounds 25–29 were rhodium(iii) congeners of iridium(III) compounds 1, 16–19, while compounds 30–34 were the rhodium(iii) congeners of 19–24. These compounds were subsequently tested at 5 μM for their ability to disrupt the H-Ras/Raf-1 interaction using the BiFC assay (Fig. S1c and d†). Encouragingly, compound 1 still emerged as the most potent compound, inhibiting the H-Ras/Raf-1 interaction by 80.2% inhibition at 5 μM.
Based on the BiFC results, a preliminary SAR analysis could be performed. Compound 1 was more potent than compounds 16–19, indicating that the tpy C⁁N ligand was superior to the other C⁁N ligands tested. Moreover, based on the fact that compounds 20–24 were among the least potent compounds of the library, the dnbpy N⁁N ligand of 1 was also deemed to be crucial for H-Ras/Raf-1 inhibitory activity. The iridium(iii) center was also important for the activity of 1, as its rhodium(iii) congener 25 showed significantly reduced activity. However, the other rhodium(iii) congeners showed comparable or even slightly higher potency than the iridium(iii) series. Of particular interest is the rhodium(iii) compound 29, bearing the dnbpy N⁁N ligand and 1-phenylisoquinoline (piq) C⁁N ligand, which showed 60.9% inhibition of the H-Ras/Raf-1 PPI, and was the second most active compound of the library. Potentially, the rhodium(iii) compound 29 could also be developed further as an alternative scaffold class for H-Ras/Raf-1 PPI inhibitors. Taken together, the results indicate that the nature of both the C⁁N and N⁁N ligands as well as the character of the metal center are important for the Ras/Raf inhibitory properties of 1. We also checked whether 1 affected the expression of H-Ras and Raf-1 in the transfected HEK293T cells. The results showed that 1 had no significant effect on H-Ras and Raf-1 protein expression in transfected HEK293T cells after incubation for 6 h (Fig. S4†). These findings indicate that 1 disrupts the H-Ras/Raf-1 interaction without affecting their expression levels.
Enantiomer Δ-1 is more potent than Λ-1 at disrupting the H-Ras/Raf-1 interaction
To further elucidate the mechanism of disruption of the H-Ras/Raf-1 interaction by 1, the two enantiomers of 1 (Λ-1 and Δ-1) were synthesized and tested side-by-side using the BiFC assay. Intriguingly, enantiomer Δ-1 (5 μM) effectively suppressed the interaction between H-Ras and Raf-1, whereas Δ-1 showed no obvious effect at the same concentration (Fig. 2). This result demonstrates that the inhibition of the H-Ras/Raf-1 interaction by racemic 1 could be attributed mainly to the action of the Δ-1 enantiomer. In order to examine the accumulation of racemic 1 and its enantiomers in cells, an inductively-coupled plasma mass spectrometry (ICP-MS) assay was performed (Fig. S5†). The compounds tended to localize in the cytoplasm, where H-Ras interacts with Raf-1, and also in the mitochondria. However, no significant differences in cellular distribution were observed between the different compounds.
Fig. 2. Racemic 1, Λ-1 and Δ-1 inhibit the H-Ras/Raf-1 PPI as determined by the BiFC assay. Racemic 1, Λ-1 and Δ-1 (5 μM) were incubated with A498 cells for 6 h and the fluorescence was monitored using confocal imaging.

To further confirm the mechanism of H-Ras/Raf-1 interaction inhibition, a pull-down assay was performed. After overexpression of the Raf-1-Flag and H-Ras-Myc fusion proteins in A498 human kidney carcinoma cells, racemic 1 or enantiomers Δ-1 and Λ-1 (5 μM) were added and cells were incubated for 6 h. In the presence of enantiomer Δ-1 and, to a lesser extent racemic 1, the amount of H-Ras-Myc detected in Flag immunoprecipitates was significantly decreased, suggesting that Δ-1 or racemic 1 could disrupt the H-Ras/Raf-1 interaction in cellulo (Fig. 3a). However, Λ-1 did not inhibit the interaction between H-Ras and Raf-1 in the pull-down assay. In the previous experiments, the G12V mutant of H-Ras was used. We were therefore also interested to investigate whether 1 could also potentially inhibit the interaction between wt-H-Ras and Raf-1. As shown in Fig. S6,† Δ-1 or racemic 1 inhibited the interaction between wt-H-Ras and Raf-1, but with less potency than against the H-RasV12/Raf-1 interaction. We also examined the ability of 1 to inhibit the interaction between H-Ras and other H-Ras effectors, such as RalGDS and PI3K. Slight inhibitions of the H-Ras/RalGDS and H-Ras/PI3K interactions were observed after treatment with 5 μM of Δ-1, but not after treatment with racemic 1 or Λ-1 in A498 cells (Fig. S7†).
Fig. 3. (a) Racemic 1, Λ-1 and Δ-1 (5 μM) disrupt the interaction of H-Ras-Myc and Raf-1-Flag in A498 cells as revealed by a pull-down assay. Protein complexes were immunoprecipitated by the Flag antibody and analyzed with the Myc antibody. (b) Effects of racemic 1, Λ-1 and Δ-1 (5 μM) on inhibiting the H-Ras-Flag and Raf-1-RBD-his PPI in A498 cells as determined by a pull-down assay. (c) Effects of racemic 1, Λ-1 and Δ-1 on inhibiting the H-Ras-Flag or Raf-1-CRD-HA PPIs in A498 cells as determined by the pull-down assay. Significantly different from control at *p < 0.05, **p < 0.005, and ***p < 0.001.

As a critical Ras effector target, Raf-1 contains two Ras-binding sites for activation, the Ras-binding domain (RBD) and the cysteine-rich domain (CRD).38 In order to elucidate the possible mode of action of compound 1, we repeated the pull-down assay with the two separate Ras-binding domain constructs, namely Raf-1-RBD-his and Raf-1-CRD-HA. The results showed that the amount of Raf-1-RBD-his bound to H-Ras-Flag was obviously reduced after incubation with Δ-1 or racemic 1 (5 μM) Flag immunoprecipitates, with Δ-1 again being more potent than racemic 1, while Λ-1 only had a slight effect (Fig. 3b). However, neither racemic 1 nor Λ-1 (5 μM) showed any inhibitory impact on the interaction of H-Ras-Flag/Raf-1-CRD-HA in the treated cells, while Δ-1 only exhibited moderate inhibition at the same concentration (Fig. 3c). Taken together, these findings indicate that Δ-1 and racemic 1 disrupt the interaction of H-Ras and Raf-1 through selective targeting of the Ras-binding domain of Raf-1, rather than the cysteine-rich domain. Moreover, neither of the isolated ligands (tpy and dnbpy) showed an observable effect on the H-Ras/Raf-1 interaction in cells in the BiFC assay (Fig. S8†), suggesting that the coordination of the ligands to the iridium(iii) center to form an intact compound was required for the inhibition of the H-Ras/Raf-1 interaction. Taken together, these findings demonstrate that the H-Ras/Raf-1 inhibitory potency of racemic 1 is largely due to the activity of enantiomer Δ-1 rather than Λ-1. Additionally, these results provide indirect evidence that the interaction between 1 and H-Ras/Raf-1 is shape-driven rather than being mediated through non-specific effects.
Drug target engagement of enantiomer Δ-1
Target engagement, which refers the ability of a small molecule to interact with its intended protein target in a living system,39 is an important parameter of drug efficacy. Therefore, we evaluated the H-Ras and Raf-1 binding ability of Δ-1 using the cellular thermal shift assay (CETSA). After incubation of A498 cell lysates with Δ-1 (5 μM) for 30 min, aliquots were heated at different temperatures ranging from 47 °C to 68 °C for 5 min and then immunoblotted to quantitate the level of H-Ras or Raf-1 remaining in the soluble fraction. A significant increase of the melting temperature of H-Ras from 50 °C to 68 °C and of Raf-1 from 59 °C to 68 °C was observed upon addition of Δ-1 to cell lysates, indicating that Δ-1 could bind and stabilize H-Ras and Raf-1 in the presence of complex cellular debris (Fig. S9a and b†). On the other hand, Δ-1 had no effect on the thermal stability of GAPDH (Fig. S9c†). Furthermore, to evaluate the potential binding target of Δ-1 in the Raf-1 protein, two domains of Raf-1, Raf-1-RBD and Raf-1-CRD, were transfected into the cells (Fig. S9d and e†). As revealed by the result, Δ-1 showed more effectiveness for the stabilization of Raf-1-RBD by targeting Raf-1-RBD than Raf-1-CRD under the same experimental conditions. Moreover, an in vitro thermal shift assay was performed to further evaluate the binding affinity of Δ-1 towards Raf-1 RBD and H-Ras. As shown in Fig. S10,† 10 μM of Δ-1 shifted the melt curves of both Raf-1 RBD (from 53.0 °C to 58.1 °C) and H-Ras (from 49.0 °C to 53.3 °C), suggesting that the compound could stabilize both proteins. A dose experiment with 0, 1, 3, 5, or 10 μM of Δ-1 allowed the determination of K d values for Raf-1-RBD and H-Ras of 4.40 and 2.88 μM, respectively. BSA was used as a negative control to evaluate the non-specific binding of Δ-1 to hydrophobic surfaces, by detecting the changes of UV absorption of Δ-1 (5 μM) at different concentrations of BSA. BSA, having hydrophobic regions on its surface, is widely used as an additive in biological experiments to reduce the non-specific binding of other substances.40 As shown in Fig. S11,† only a slight increase of the absorbance of Δ-1 was observed in different concentrations of BSA ranging from 0.67–6.67 μg mL–1 in PBS buffer. Therefore, Δ-1 is presumed to exhibit only weak non-specific binding to hydrophobic surfaces. Taking these results together, we hypothesize that the inhibition of the H-Ras/Raf-1 PPI observed in the BiFC and pull-down assays could be attributed to the ability of Δ-1 to directly engage H-Ras and Raf-1-RBD in cells.
Iridium(iii) compound 1 and enantiomer Δ-1 block the Ras-dependent signal transduction
The Ras/Raf/MEK/ERK pathway is initiated when ligands bind to cytokine receptors, activating Ras. Activated Ras then recruits Raf-1 by binding the Ras-binding domain present on Raf-1. After a multi-step phosphorylation and dephosphorylation process, activated Raf-1 phosphorylates and activates MEK, which in turn phosphorylates and activates ERK, finally culminating in the activation of transcriptional factor AP-1 which translocates into the nucleus to regulate gene expression.41 We also investigated the ability of 1 and its enantiomers to modulate the various stages of the Ras/Raf/MEK/ERK signaling pathway in cells. Immunoblotting analysis revealed that the phosphorylation of MEK and ERK was significantly reduced after 6 h treatment with racemic 1, Λ-1, Δ-1, or sulindac sulfide (5 μM) in A498 cells (Fig. 4a). Notably, Δ-1 exhibited higher potency at inhibiting phosphorylated MEK and ERK compared to Λ-1, racemic 1, and sulindac sulfide. Moreover, all of the compounds had no impact on the expression of total MEK and ERK. We further investigated the effect of Δ-1 on the transcriptional activity of AP-1 using a luciferase reporter assay. Enantiomer Δ-1 antagonized AP-1 luciferase activity in a dose-dependent manner with an IC50 value of ca. 1.6 μM, and was more potent than racemic 1, which had an IC50 value of ca. 3.4 μM (Fig. 4b). Taken together, these results suggest that racemic 1 and Δ-1 could down-regulate the transcriptional activity of AP-1, presumably via their inhibition of the H-Ras/Raf-1 interaction.
Fig. 4. The effect of compound 1 and its enantiomers (Δ-1 and Λ-1) on the Ras/Raf/MEK/ERK signaling pathway. (a) Compound 1 and its enantiomers (Λ-1 and Δ-1) (5 μM) down-regulate the phosphorylation of MEK and ERK, but have no significant effect on total MEK and ERK content in A498 cells. (b) Racemic 1 and Δ-1 (5 μM) attenuate the transcriptional activity of AP-1 in a dose-dependent manner. Significantly different from control at *p < 0.05, **p < 0.005, ***p < 0.001, and ****p < 0.0001.

Compound 1 and Δ-1 inhibit the proliferation of kidney cancer cells
We next tested the anti-proliferative activity of compound 1 towards a panel of cancer cell lines including human kidney cancer cells (A498 and HEK293), human breast cancer cells (MDA-MB-231, MCF7, T47D, and MCF10A), human lung cancer cells (A549 and H1299), human melanoma cancer cells (A375), human prostatic cancer cells (DU145), human ovarian cancer cells (A2780), and human erythroleukemic cancer cells (K562) using the MTT assay (Table S1†). We found that our lead compound was most toxic towards A498 cells (IC50 of Δ-1: 9.2 ± 1.1 μM) harboring mutant H-Ras and K-Ras oncogenes and showed a similar toxicity towards H1299 cells (IC50 of Δ-1: 10.3 ± 1.2 μM) harboring a mutant K-Ras oncogene. Additionally, the lead compound exhibited moderate cytotoxicity (IC50 of Δ-1: >18 μM) towards A431 cells harboring the activated H-Ras oncogene, DU145 cells harboring the wild-type Ras, and A2780 cells harboring K-Ras oncogenic activation. However, the compound exhibited similarly modest cytotoxicity towards T47D cells and A375 cells that do not have activated H-Ras oncogenes. We reason that the anti-proliferative activity of Δ-1 might be due, at least in part, to the disruption of the H-Ras/Raf-1 PPI, however, we do not preclude the possibility that the compound could also act via other modes of action.
Iridium(iii) compound 1 and enantiomer Δ-1 suppress tumor growth and inhibit oncogenic Ras-induced signaling in a mouse renal xenograft tumor model
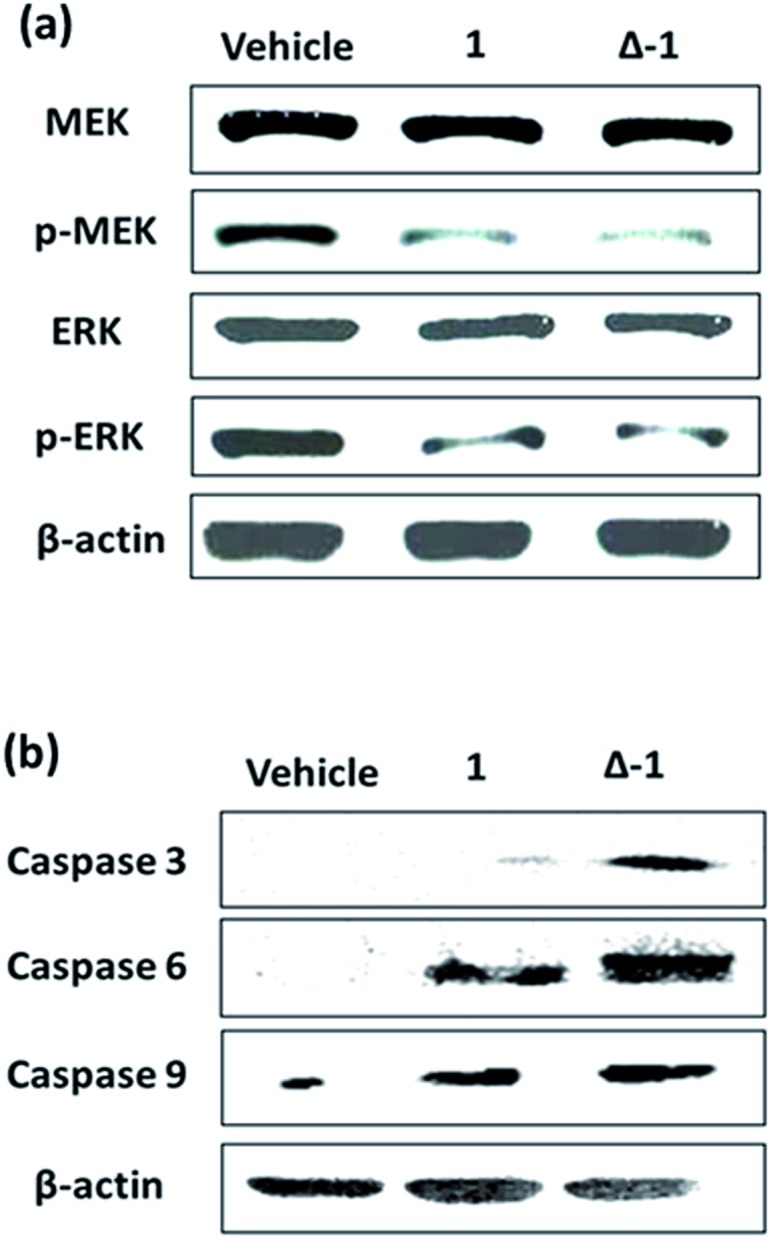
Given the promising anti-proliferative activity exhibited by racemic 1 and Δ-1 in vitro, we investigated the biological efficacy of those compounds in a mouse renal xenograft tumor model. In a preliminary animal toxicity study, high concentrations of compound 1 (140 or 280 mg kg–1) caused significant toxicity to mice in terms of decreases in body weight and changes in the liver and spleen indices (Fig. S12†). Female BALB/cAnN.Cg-Foxn1 nu/CrlNarl were injected subcutaneously with A498 (human kidney cancer) cells and were treated four times a week with an intraperitoneal (i.p.) injection of racemic 1 or Δ-1 (14 mg kg–1) or with vehicle until sacrifice at day 30. Encouragingly, treatment of mice with racemic 1 or Δ-1 resulted in a significant decrease in the estimated tumor volume from day 10 onwards compared to the control group, with Δ-1 being more potent than racemic 1 (Fig. 5a and b). There were no signs of gross toxicity or statistically significant weight loss of the treated mice over the course of the experiment (Fig. 5c). Furthermore, both racemic 1 and Δ-1 inhibited the phosphorylation of MEK and ERK in vivo, without affecting their total protein expression (Fig. 6a). Moreover, racemic 1 and Δ-1 induced the expression of the apoptotic markers caspase 3, 6, and 9 in vivo, suggesting that they could induce apoptosis of tumor tissues (Fig. 6b).
Fig. 5. The effects of racemic 1 and Δ-1 in a kidney cancer xenograft model. Mice harboring A498 (human kidney cancer) tumors were injected with vehicle or with 1 or Δ-1 (14 mg kg–1) four times a week. (a) Photographs of control and treatment mice after 30 days. (b) Average tumor volumes of control group and treatment group over the measurement period. (c) Changes of body weight over the measurement period. Significantly different at *p < 0.05, **p < 0.005, ***p < 0.001, and ****p < 0.0001.

Fig. 6. The effect of racemic 1 and Δ-1 on the phosphorylation of MEK and ERK and expression of caspase 3, 6, and 9 in a kidney cancer xenograft model. Mice harboring A498 (human kidney cancer) tumors were injected with vehicle or with 1 or Δ-1 (14 mg kg–1) four times a week. (a) Racemic 1 and its enantiomer Δ-1 inhibit the phosphorylation of MEK and ERK. (b) Racemic 1 and its enantiomer Δ-1 induce caspase 3, 6, and 9 expression.
Conclusions
This study has identified the iridium(iii) compound 1 as the first kinetically-inert organometallic inhibitor of the H-Ras/Raf-1 PPI. Compound 1 was identified using a preliminary BiFC screening campaign of an in-house library of 15 iridium(iii) and rhodium(iii) compounds (1–15). A further 19 compounds (16–34) were synthesized and tested to explore the SAR surrounding compound 1, and this revealed that the combination of the dnbpy N⁁N ligand and tpy C⁁N ligands together with the iridium(iii) center were important for achieving effective inhibition of the H-Ras/Raf-1 PPI. Moreover, the two enantiomers (Λ and Δ) of compound 1 were isolated for individual evaluation. In addition to using the BiFC assay, we also performed a pull-down experiment to demonstrate that racemic 1 and Δ-1 could target the H-Ras/Raf-1 PPI in cells. Furthermore, the domain pull-down assay and domain CETSA assay indicated that racemic 1 and Δ-1 showed selective binding to Raf-1-RBD over Raf-1-CRD. As the H-Ras/Raf-1 PPI is a crucial signaling hub of the Ras/Raf/MEK/ERK signaling pathway, disrupting this PPI can have knock-on effects on downstream mediators of the pathway. In this study, we demonstrated that compound 1 inhibited the phosphorylation of MEK and ERK and also attenuated the transcriptional activity of AP-1, which we presume is due to its inhibition of the H-Ras/Raf-1 PPI. Furthermore, the specificity of compound Δ-1 was highlighted by CETSA, which revealed that compound Δ-1 directly engaged H-Ras and Raf-1-RBD even in the complicated biological environment of cell lysates, whereas it had no effect on GAPDH. Finally, we demonstrated the ability of racemic 1 and Δ-1 to suppress tumor growth in a mouse xenograft model of human kidney cancer without causing weight loss or overt signs of toxicity to mice over 30 days. Compound 1 also upregulated pro-apoptotic caspase activity in vivo, thereby providing a probable mechanism for the anti-cancer activity of compound 1.
Overall, these findings highlight the exciting potential of iridium(iii) compounds to be developed as effective Ras/Raf PPI inhibitors for the treatment of kidney cancer. Future work will explore the structural basis of Ras/Raf inhibition by 1 using techniques such as X-ray crystallography or molecular modelling, and subsequent structure-based optimization could drive the potency of the compounds from the micromolar down to the nanomolar range. Previous research groups have shown that the two enantiomers of a metal complex can show markedly different biological activities.42–44 However, this work is the first to demonstrate that two enantiomers of iridium(iii)-based metal compounds can show different biological activities both in vitro and in vivo, which is an important consideration for progressing organometallic compounds towards drug-like status. We envisage that iridium(iii) compound 1, as the first metal-based Ras/Raf inhibitor reported in the literature, could potentially be used as a starting scaffold to develop more potent metal-based Ras/Raf PPI inhibitors, including targeting the Ras-binding domain of Raf-1, for the treatment of kidney cancer or other proliferative diseases.
Experimental
Biomolecular fluorescence complementation (BiFC) assay
The BiFC assay was performed in order to investigate the inhibition of the H-Ras/Raf-1 interaction by the compounds. HEK293T and A498 cells co-transfected with pEGFP-VC-H-RasV12 and pEGFP-VN-Raf-1-RBD by TurboFect transfection reagent were seeded in a 6-well plate with a serum-free DMEM medium and incubated for 6 h. The cells were then plated in a complete medium for additional 24 h. After formulating the concentration of each compound to 5 μM, the cells were placed in 8-well chamber slides and treated with the compounds for 6 h. Later, the cells were fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature, and were washed with phosphate-buffered saline (PBS) buffer three times. After 5 min incubation with Hoechst 33342 (10 mg mL–1, 1 : 2000 dilution) at room temperature, the cells were imaged using a Leica TCS SP8 confocal microscope. Fluorescence intensity was determined using ImageJ software.
Cellular thermal shift assay (CETSA)
2 × 106 A489 cells were lysed and collected. 240 μg of cell lysates were diluted and allocated in same aliquots (160 μL) with PBS. Each lysate was treated with 5 μM of compounds for 30 min at room temperature and was then divided into 8 aliquots of 20 μL and placed in separate PCR tubes. The PCR tubes were heated individually at different temperatures ranging from 47 °C to 68 °C (Applied Biosystems 7500, Life Technologies). The heated lysates were then centrifuged for 5 min at 13 000g and the supernatants were subjected to SDS-PAGE followed by immunoblotting with anti-Raf and anti-Ras antibodies (Abcam, 1 : 1000 dilution).
Animal Materials
In this study, the use of animals complied with the Guiding Principles in the Care and Use of Animals of the American Physiology Society and was approved by the Animal Care and Use Committee at the National Kaohsiung Medical University. Female BALB/cAnN.Cg-Foxn1 nu/CrlNarl (4–5 weeks) were purchased from the BioLASCO Experimental Animal Center (Taiwan Co., Ltd). The mice were housed in Plexiglas cages in a temperature-controlled room (22 ± 1°C), on a 12 h/12 h light/dark schedule, and with free access to food and water. After one week, the mice were randomly divided into control and treatment groups.
Xenograft tumor assay
Female BALB/cAnN.Cg-Foxn1 nu/CrlNarl were housed and tested at the animal center (Kaohsiung Medical University, Kaohsiung, Taiwan). Mice were implanted subcutaneously with 1 × 107 A498 cells in 0.1 mL PBS. After the establishment of palpable tumors (the mean tumor volume was around 150–200 mm3), mice were treated four times a week with a intraperitoneal (i.p.) injection of compounds (14 mg kg–1) or vehicle (13% DMSO) in 0.05 mL PBS until sacrifice at 30 day. The diameters of xenograft tumors were measured at 3 day intervals with vernier calipers, and the tumor volume (in mm3) was calculated using the formula: volume = length × width2/2. The treatment and control groups each contained 6 mice.
Additional information on materials, synthesis of compounds, plasmid construction, pull-down assay, luciferase reporter assay, immunoblot analysis, and MTT assay is provided in the ESI.†
Acknowledgments
This work is supported by the Hong Kong Baptist University (FRG2/15-16/002), the Health and Medical Research Fund (HMRF/14130522), the Research Grants Council (HKBU/12301115, HKBU/204612, and HKBU/201913), the National Natural Science Foundation of China (21575121), the Guangdong Province Natural Science Foundation (2015A030313816), the Hong Kong Baptist University Century Club Sponsorship Scheme 2016, the Interdisciplinary Research Matching Scheme (RC-IRMS/15-16/03), the Science and Technology Development Fund, Macao SAR (098/2014/A2), the University of Macau (MYRG2015-00137-ICMS-QRCM, MYRG2016-00151-ICMS-QRCM and MRG044/LCH/2015/ICMS), the National Natural Science Foundation of China (21628502), and Ministry of Science and Technology (MOST 105-2622-E-005-006-CC2; MOST 104-2221-E-005-096-MY2; and MOST 104-2628-E-005-004-MY3). We thank Prof. Kazumasa Ohashi of the Department of Biomolecular Sciences, Graduate School of Life Sciences for the gifts of the BiFC plasmids.
Footnotes
References
- Scott D. E., Bayly A. R., Abell C., Skidmore J. Nat. Rev. Drug Discovery. 2016;15:533–550. doi: 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- Skwarczynska M., Ottmann C. Future Med. Chem. 2015;7:2195–2219. doi: 10.4155/fmc.15.138. [DOI] [PubMed] [Google Scholar]
- Zhang Q. C., Petrey D., Deng L., Qiang L., Shi Y., Thu C. A., Bisikirska B., Lefebvre C., Accili D., Hunter T., Maniatis T., Califano A., Honig B. Nature. 2012;490:556–560. doi: 10.1038/nature11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetta L. Nature. 2010;468:851–854. doi: 10.1038/468851a. [DOI] [PubMed] [Google Scholar]
- Modell A. E., Blosser S. L., Arora P. S. Trends Pharmacol. Sci. 2016;37:702–713. doi: 10.1016/j.tips.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A. A., Khuri F. R., Fu H. A. Trends Pharmacol. Sci. 2013;34:393–400. doi: 10.1016/j.tips.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L. Y., Wang W. R., Fang G. W. Annu. Rev. Pharmacol. Toxicol. 2014;54:435–456. doi: 10.1146/annurev-pharmtox-011613-140028. [DOI] [PubMed] [Google Scholar]
- Gothe Y., Marzo T., Messori L., Metzler-Nolte N. Chem.–Eur. J. 2016;22:12487–12494. doi: 10.1002/chem.201601542. [DOI] [PubMed] [Google Scholar]
- Gothe Y., Marzo T., Messori L., Metzler-Nolte N. Chem. Commun. 2015;51:3151–3157. doi: 10.1039/c4cc10014j. [DOI] [PubMed] [Google Scholar]
- Metzler-Nolte N., Guo Z. J. Dalton Trans. 2016;45:12965. doi: 10.1039/c6dt90135b. [DOI] [PubMed] [Google Scholar]
- Hess J., Keiser J., Gasser G. Future Med. Chem. 2015;7:821–830. doi: 10.4155/fmc.15.22. [DOI] [PubMed] [Google Scholar]
- Ma D. L., Liu L. J., Leung K. H., Chen Y. T., Zhong H. J., Chan D. S. H., Wangand H. M. D., Leung C. H. Angew. Chem., Int. Ed. 2014;53:9178–9182. doi: 10.1002/anie.201404686. [DOI] [PubMed] [Google Scholar]
- Zhong H. J., Lu L. H., Leung K. H., Wong C. C. L., Peng C., Yan S. C., Ma D. L., Cai Z. W., Wang H. M. D., Leung C. H. Chem. Sci. 2015;6:5400–5408. doi: 10.1039/c5sc02321a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu K. Q., Liu Y. K., Huang H. Y., Liu C. F., Zhu H. Y., Chen Y., Ji L. N., Chao H. Dalton Trans. 2016;45:16144–16147. doi: 10.1039/c6dt03328h. [DOI] [PubMed] [Google Scholar]
- Meng X. J., Leyva M. L., Jenny M., Gross I., Benosman S., Fricker B., Harlepp S., Hebraud P., Boos A., Wlosik P., Bischoff P., Sirlin C., Pfeffer M., Loeffler J. P., Gaiddon C. Cancer Res. 2009;69:5458–5466. doi: 10.1158/0008-5472.CAN-08-4408. [DOI] [PubMed] [Google Scholar]
- Bratsos I., Mitri E., Ravalico F., Zangrando E., Gianferrara T., Bergamo A., Alessio E. Dalton Trans. 2012;41:7358–7371. doi: 10.1039/c2dt30654a. [DOI] [PubMed] [Google Scholar]
- Frezza M., Hindo S., Chen D., Davenport A., Schmitt S., Tomco D., Dou Q. P. Curr. Pharm. Des. 2010;16:1813–1825. doi: 10.2174/138161210791209009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D. L., Chan D. S. H., Leung C. H. Acc. Chem. Res. 2014;47:3614–3631. doi: 10.1021/ar500310z. [DOI] [PubMed] [Google Scholar]
- Dhillon A. S., Hagan S., Rath O., Kolch W. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- Niault T. S., Baccarini M. Carcinogenesis. 2010;31:1165–1174. doi: 10.1093/carcin/bgp337. [DOI] [PubMed] [Google Scholar]
- Rusconi P., Caiola E., Broggini M. Curr. Med. Chem. 2012;19:1164–1176. doi: 10.2174/092986712799320510. [DOI] [PubMed] [Google Scholar]
- Cseh B., Doma E., Baccarini M. FEBS Lett. 2014;588:2398–2406. doi: 10.1016/j.febslet.2014.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann H., Karaguni I. M., Carpintero M., Gourzoulidou E., Herrmann C., Brockmann C., Oschkinat H., Muller O. Angew. Chem., Int. Ed. 2004;43:454–458. doi: 10.1002/anie.200353089. [DOI] [PubMed] [Google Scholar]
- Roberts P. J., Der C. J. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- Leicht D. T., Balan V., Kaplun A., Singh-Gupta V., Kaplun L., Dobson M., Tzivion G. Biochim. Biophys. Acta. 2007;1773:1196–1212. doi: 10.1016/j.bbamcr.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka H., Chatani Y., Hoshino R., Ogawa O., Kakehi Y., Terachi T., Okada Y., Kawaichi M., Kohno M., Yoshida O. Cancer Res. 1995;55:4182–4187. [PubMed] [Google Scholar]
- Fujita J., Kraus M. H., Onoue H., Srivastava S. K., Ebi Y., Kitamura Y., Rhim J. S. Cancer Res. 1988;48:5251–5255. [PubMed] [Google Scholar]
- Escudier B., Eisen T., Stadler W. M., Szczylik C., Oudard S., Siebels M., Negrier S., Chevreau C., Solska E., Desai A. A., Rolland F., Demkow T., Hutson T. E., Gore M., Freeman S., Schwartz B., Shan M. H., Simantov R., Bukowski R. M. N. Engl. J. Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- Boudou-Rouquette P., Thomas-Schoemann A., Bellesoeur A., Goldwasser F. Lancet. 2015;385:227–228. doi: 10.1016/S0140-6736(15)60054-X. [DOI] [PubMed] [Google Scholar]
- Llovet J. M., Ricci S., Mazzaferro V., Hilgard P., Gane E., Blanc J. F., de Oliveira A. C., Santoro A., Raoul J. L., Forner A., Schwartz M., Porta C., Zeuzem S., Bolondi L., Greten T. F., Galle P. R., Seitz J. F., Borbath I., Haussinger D., Giannaris T., Shan M., Moscovici M., Voliotis D., Bruix J. N. Engl. J. Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- Athuluri-Divakar S. K., Vasquez-Del Carpio R., Dutta K., Baker S. J., Cosenza S. C., Basu I., Gupta Y. K., Reddy M. V. R., Ueno L., Hart J. R., Vogt P. K., Mulholland D., Guha C., Aggarwal A. K., Reddy E. P. Cell. 2016;165:643–655. doi: 10.1016/j.cell.2016.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C., Wang W. H., Li G. D., Zhong H. J., Dong Z. Z., Wong C. Y., Kwong D. W. J., Ma D. L., Leung C. H. Sci. Rep. 2017;7:42860. doi: 10.1038/srep42860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. J., Wang W. H., Kang T. S., Liang J. X., Liu C. F., Kwong D. W. J., Wong V. K. W., Ma D. L., Leung C. H. Sci. Rep. 2016;6:36044. doi: 10.1038/srep36044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. J., Wang W. H., Zhong Z. F., Lin S., Lu L. H., Wang Y. T., Ma D. L., Leung C. H. Chem. Commun. 2016;52:12278–12281. doi: 10.1039/c6cc06155a. [DOI] [PubMed] [Google Scholar]
- Liu L. J., He B. Y., Miles J. A., Wang W. H., Mao Z. F., Che W. I., Lu J. J., Chen X. P., Wilson A. J., Ma D. L., Leung C. H. Oncotarget. 2016;7:13965–13975. doi: 10.18632/oncotarget.7369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H. J., Leung K. H., Liu L. J., Lu L. H., Chan D. S. H., Leung C. H., Ma D. L. ChemPlusChem. 2014;79:508–511. doi: 10.1002/cplu.201400014. [DOI] [PubMed] [Google Scholar]
- Zeng J., Nheu T., Zorzet A., Catimel B., Nice E., Maruta H., Burgess A. W., Treutlein H. R. Protein Eng. 2001;14:39–45. doi: 10.1093/protein/14.1.39. [DOI] [PubMed] [Google Scholar]
- Drugan J. K., KhosraviFar R., White M. A., Der C. J., Sung Y. J., Hwang Y. W., Campbell S. L. J. Biol. Chem. 1996;271:233–237. doi: 10.1074/jbc.271.1.233. [DOI] [PubMed] [Google Scholar]
- Simon G. M., Niphakis M. J., Cravatt B. F. Nat. Chem. Biol. 2013;9:200–205. doi: 10.1038/nchembio.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Park I. S., Kim H., Woo J. S., Choi B. S., Min D. H. Biosens. Bioelectron. 2015;69:167–173. doi: 10.1016/j.bios.2015.02.030. [DOI] [PubMed] [Google Scholar]
- Chang F., Steelman L. S., Lee J. T., Shelton J. G., Navolanic P. M., Blalock W. L., Franklin R. A., McCubrey J. A. Leukemia. 2003;17:1263–1293. doi: 10.1038/sj.leu.2402945. [DOI] [PubMed] [Google Scholar]
- Song H., Kaiser J. T., Barton J. K. Nat. Chem. 2012;4:615–620. doi: 10.1038/nchem.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaratnam R., Martin E. K., Dorr M., Harms K., Casini A., Meggers E. Inorg. Chem. 2015;54:8111–8120. doi: 10.1021/acs.inorgchem.5b01349. [DOI] [PubMed] [Google Scholar]
- Kang T. S., Mao Z. F., Ng C. T., Wang M. D., Wang W. H., Wang C. M., Lee S. M. Y., Wang Y. T., Leung C. H., Ma D. L. J. Med. Chem. 2016;59:4026–4031. doi: 10.1021/acs.jmedchem.6b00112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.