


Abstract
Chromosome territories (CTs) in higher eukaryotes occupy tissue-specific non-random three-dimensional positions in the interphase nucleus. To understand the mechanisms underlying CT organization, we mapped CT position and transcriptional changes in undifferentiated embryonic stem (ES) cells, during early onset of mouse ES cell differentiation and in terminally differentiated NIH3T3 cells. We found chromosome intermingling volume to be a reliable CT surface property, which can be used to define CT organization. Our results show a correlation between the transcriptional activity of chromosomes and heterologous chromosome intermingling volumes during differentiation. Furthermore, these regions were enriched in active RNA polymerase and other histone modifications in the differentiated states. These findings suggest a correlation between the evolution of transcription program in modifying CT architecture in undifferentiated stem cells. This leads to the formation of functional CT surfaces, which then interact to define the three-dimensional CT organization during differentiation.
INTRODUCTION
Genetic material is hierarchically packaged into the nuclei of higher eukaryotes as chromatin. This is further condensed into chromosomes and these are organized, during interphase, into distinct regions termed chromosome territories (CTs) (1). In humans, gene-rich CTs were found in the nuclear centre and gene-poor at the nuclear periphery (2,3). But such radial organization of CTs is correlated with CT size (4,5). Simultaneous labelling of multiple CTs in different cell types has revealed that CT organization is also cell type-specific (6). This is reflected in higher chromosomal translocation patterns for the adjacent chromosomes (7,8) and is also present in human cancer cells derived from specific tissues. For instance, Burkitt's lymphoma, a B-cell malignancy, is characterized by translocation between chromosome 8 and chromosome 14, whereas acute T cell leukaemia are associated with translocations between chromosome 7 and chromosome 10 or chromosome 10 and chromosome 14 (9). Such chromosomal interactions were also divulged by extensive 3C data (10,11). However, the principles underlying specific relative chromosome organization are not yet clear.
Although chromosome length and gene density may guide the radial organization of CTs, these factors remain constant across multiple cell types in an organism, and hence, are insufficient to explain the cell type-specific organization of CTs. 3C data have uncovered intra-chromosome interactions that result from function driven folding of DNA sequences. These data also predict that intra-chromosome interactions are mediated by certain transcription factors and are required for transcription activity (12). The specific folding of the DNA sequences is active in transcription and mRNA splicing and is hypothesized to induce chromosome intermingling (13–15), which has been probed by imaging and Hi-C techniques (16–18). Using a single gene fluorescence in situ hybridization (FISH) technique to visualize inter-chromosome three-dimensional (3D) interactions between candidate genes, co-clustering of genes within the nucleus at sites of active transcription was revealed (12,19,20). The 3D organization of chromosomes is thus important in the regulation of gene expression and hence, we hypothesized that sites of active transcription can be the organizing centres for CT positioning. Such an idea may also extend to the relative positioning of non-homologous chromosomes, which we have previously shown to be dependent on their transcriptional activity in specific cells (21).
Interestingly, the above-mentioned cell type-specific CT organization evolves from pluripotent stem cells. Stem cells are known to comprise a highly active transcriptome, and exhibit plasticity in the stiffness of their nuclei (22,23) and chromatin dynamics (24). Differentiation results in drastic changes to these properties (25–27) that are accomplished only within a few cell divisions. Since chromosomes can only move in a constrained fashion during interphase (28), the cell type-specific CT organization should accumulate progressively during stem cell differentiation. Therefore quantitative comparisons of the spatio-temporal organization of chromosomes during stem cell differentiation and its correlation to gene expression programs will be important to understand the underlying principles of CT organization.
In this work, we correlated whole genome transcriptome patterns with the spatial organization of chromosomes in undifferentiated ES cells and at the early onset of differentiation. This was compared to that in terminally differentiated NIH3T3 cells. Quantitative confocal imaging of individual chromosomes revealed the chromosome intermingling volume fraction as an important parameter for understanding relative CT organization. The intermingled regions between two heterologous chromosomes were enriched in transcriptionally active gene, phosphorylated RNA Pol II (RNAPII) and regulatory histone modifications. We also found that the radial chromosome positioning also correlates with the chromosome intermingling volume and size. Our results provide evidence to support the differential rearrangement of smaller chromosomal domains on individual chromosomes, which together can lead to large-scale transcription-dependent chromosome positioning and its intermingling during cellular differentiation.
MATERIALS AND METHODS
Cell culture and perturbations
NIH3T3 cells were cultured in DMEM (Gibco, Life Technologies, USA) supplemented with 10% FBS (Gibco, Life Technologies, USA) and 1X pen/strep (100 units of penicillin, 100 μg of streptomycin; Gibco, Life Technologies, USA). E14.1 Mouse embryonic cells were maintained by culturing them on gelatin (0.1%) coated dishes with reconstituted media. Reconstituted media was made from ES cell Knockout DMEM supplemented with 15% Knockout Fetal Bovine Serum, 1 mM sodium pyruvate (Sigma Aldrich, USA), 0.1 mM nonessential amino acids, 2 mM L-Glutamine, 0.1 mM β-mercaptoethanol (Sigma Aldrich, USA) and 500 U/ml leukaemia inhibitory factor (Merck Millipore) and penicillin–streptomycin. All cell culture reagents, unless otherwise indicated, are from Life Technologies. All cells were maintained at 37°C in 5% CO2 incubator. Transcription was inhibited using a transcription initiation inhibitor, α-amanitin (Sigma Aldrich, USA) at 40 μg/ml for either 3 or 12 h. Cells were treated with 100 nM of Trichostatin A (TSA, Sigma Aldrich, USA), a histone deacetylase inhibitor that leads to hyper-acetylation of chromatin during the 60 h incubation period.
Br-UTP incorporation and detection
NIH3T3 cells were cultured on fibronectin-coated slides for 3 h. Cells were briefly washed with hypotonic buffer (10 mM HEPES-pH7.4 and 30 mM KCl). This was followed by incubation with 10 mM Br-UTP (Sigma, B7166) in hypotonic buffer at room temperature. After 5 min, the hypotonic media was replaced with 10% FBS containing DMEM medium and incubated at 37°C and 5% CO2 for 20 min before fixing it with freshly thawed 4% PFA (in PBS) for 10 min. The PFA was neutralized with a brief rinse in 0.1 M Tris HCl followed by gentle washing of the cells with 1X PBS followed by 1 XPBS containing 0.5% Triton X-100 for permeabilization. After blocking the nuclei with 5% BSA (in 2X SSC) for an hour at room temperature, they were subjected to primary and the secondary antibody diluted in 5% BSA solution made in 2X SSC.
Gene FISH probe preparation
Rolling cycle amplification method was used to label Bacterial Artificial Chromosome (BAC) clone containing zyxin sequence (RP23–358011, from SourceBioScience, UK) with biotin or Dig labelled UTP. For the rolling circle amplification, 100 ng of the BAC clone was denatured at 95°C for 5 min in 10 μl of annealing buffer (80 mM TRIS-HCl, pH 8.0 and 20 mM MgCl2) before moving to ice. The denatured DNA was subjected to amplification by six units of phi 29 DNA polymerase (New England Biolabs, catalogue no. M0269S) with 0.2 mM d(AGC)TP, 0.15 mM dTTP, and 0.1 mM labelled UTP and 12.5 μM Exo-Resistant Random Primer (Thermo scientific, catalogue no. S0181). The amplification was carried out at 30°C for 8 h, 65°C for 10 min and kept at 4°C till CviKI-1 digestion to generate DNA fragments of 100–400 bp in size. The digested DNA fragments was precipitated using 3M Sodium Acetate and twice the volume of Ethanol for 1 h in −80°C followed by centrifugation at 13 000 rpm for 20 min on a desktop centrifuge. The pellet of the gene FISH probe was washed with 70% ethanol and re-suspended in 10 mM Tris-HCL pH8.0 and used for FISH.
Chromosome FISH and immunofluorescence staining
For chromosome painting experiments, NIH3T3 cells were cultured on fibronectin-coated slides for 3 h. ES cells were cultured on fibronectin for 4, 12 and 24 h. ES cells cultured on Poly-D-Lysine (PDL) were used as the undifferentiated control. Cultured cells were then washed with 1X PBS to remove cell culture medium followed by treatment with CSK buffer (100 mM NaCl, 300 mM Sucrose, 3 mM MgCl2, 10 mM PIPES with pH6.8) for 5 min. Cells are then fixed with 4% PFA (Paraformaldehyde) for 10 min. PFA was neutralized with 0.1M Tris-HCl and then cells were washed and permeabilized with 0.5% Triton X-100 for 8 min. This was followed by incubation in 20% glycerol overnight and then five or six freeze-thaw cycles in liquid nitrogen. After three rinses in 1X PBST solution (0.1% Triton containing 1X PBS), cells were equilibrated with 0.1 N HCl (made in 1X PBS) for 5 min at room temperature. They were then subjected to 0.002% porcine pepsin (Sigma Aldrich, USA) digestion in 0.01N HCl (diluted in PBS) at 37°C for 4 min and quickly fixed with a 1% PFA solution for 5 min. The cells were briefly rinsed in 1X PBS before being treated with RNAse (from Promega, USA, 200 μg/ml made in 2X SSC-0.3M sodium chloride and 30 mM trisodium citrate) at 37°C for 30 min. Cells were then equilibrated in 50% Formamide (FA) made in 2X SSC (pH 7.4) at room temperature for 2–3 h. This was followed by incubation at 4°C or until further use for hybridization. Hybridization was set up the following day. Chromosome paints with or without DNA FISH probes for zyxin gene, tagged with different fluorophores (Chrombios, Germany) were thawed to room temperature and mixed with hybridization buffer provided by the supplier.
Cells were denatured in 70% FA/2X SSC at 85°C for 2 min and then incubated with the fluorescently labelled mouse whole chromosome FISH probe mix (Cambio, Cambridge, UK) and the slides were then sealed with a Sigmacote (Sigma) coated hydrophobic coverslip and rubber cement for 1–2 days in a moist chamber at 37°C with shaking. At the end of the incubation period, slides were washed thrice, each in 50% FA made in 2X SSC at 45°C and in 0.1X SSC at 60°C. Cells were counterstained with Hoechst 33342 (Sigma, USA) and then mounted with Prolong Gold antifade mounting medium (Life Technologies, USA), sealed with a coverslip and imaged on a Zeiss 510-Meta confocal microscope.
In case, where the transcription markers were detected together with the chromosome positions by FISH, the nuclei were fixed with 4% PFA after the RNAse treatment for overnight at 45°C. The fixed nuclei were then equilibrated in 50% FA in 2X SSC (pH 7.4) at room temperature for 2–3 h and followed by hybridization and post hybridization washes as mentioned before. After the last stringent wash of the 50% FA made in 0.1X SSC at 45°C the nuclei were blocked in 5% BSA solution made in 2X SSC and then subjected to primary and the secondary antibody diluted in 5% BSA solution made in 2X SSC. The primary antibody used here are anti-RNA Polymerase II CTD repeat YSPTSPS phospho S5 (Abcam—ab5131), anti-Macro H2A.2 (Abcam—ab102126), anti-Histone H3 (acetyl K9) (Abcam—ab10812), anti-H3 (trimethyl K9) (Abcam—ab8898), anti-Histone H4 (trimethyl K4) (Abcam—ab1012), anti-BrdU (Sigma, B2531) and anti-SRF (Santa Cruz biotechnology, sc-335). Finally, the cells were counterstained with Hoechst 33342 (Sigma Aldrich, USA) and then mounted with Prolong Gold antifade mounting medium (Life Technologies, USA) and sealed with a coverslip.
Confocal laser scanning microscope imaging
Chromosome FISH slides were imaged using a 100 × 1.4 NA objective mounted on Nikon A1 Confocal microscope (Nikon, USA). Nyquist sampling criteria were followed and pixel sizes of 0.13 and 0.15 μm were used in XY and axial directions, respectively. The pinhole size of one airy unit was selected and the z-section images acquired using a 639 nm excitation wavelength. Imaging was performed in sequential acquisition mode to minimize crosstalk between different fluorophores. 2X line averaging was used to minimize the noise involved.
Image analysis
Analysis of 3D FISH images was performed by a semi-automated algorithm written in MATLAB (Mathworks, USA). Nuclei and chromosomes were manually selected in the maximum projected images and their respective masks were generated. The entire z-stack of the nucleus and chromosome was multiplied using masks to select only the region of interest. The mean and standard deviation of intensity was computed for the masked nucleus and chromosome z-stacks and (mean + standard deviation) was used as threshold for each section. This resulted in faithful selection of the nuclear chromosomal regions.
Estimation of radial chromosome position
Thresholded images of chromosomes and nuclei were used to estimate the chromosome and nuclei centroids. The nucleus was fitted with an ellipse to estimate its orientation and used to reorient the nucleus and the chromosomes along the x-axis. Radial position of the chromosome was measured as the distance between the nuclear centroid and the centroid of the chromosome. This distance is normalized by the radius of the nucleus at the location of the chromosome centroid. The most probable distance for a given chromosome was estimated as the distance with highest frequency in the distribution of its radial distances.
Estimation of intermingling volume
In order to estimate the volume of intermingling, a combined 3D image was generated from the binary images of individual homologues chromosome pairs. For each pair of chromosome analysed, two images were generated. When both of these images were multiplied, only the regions shared between the two chromosomes retain a value of 1 and the other regions are rendered a value of 0. This selected region was identified as the intermingled region and the number of voxels in this region corresponds to the intermingling volume. This intermingling volume is divided with the volume of the nucleus to obtain the normalized intermingling volume (Ivol). Pairs with non-zero intermingling volume are considered positive for intermingling and all such pairs are enumerated for a particular pair of chromosomes.
Estimation of intermingling frequency
The frequency of intermingling, Ifreq, was defined as
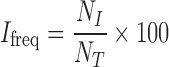 |
where NI is the number of intermingling heterologous chromosomes pairs in a nucleus and NT is the total number of possible heterologous chromosome pairs in a nucleus for a given pair of chromosomes.
Estimation of RNAPII enrichment
The distribution of RNAPII on the chromosome was estimated by dividing the chromosome into a 3-pixel (450 nm) thick shell and interior excluding the shell. The chromosome surface was identified from the binary image of the chromosome and a 3-pixel shell that was generated by differentiation in all the three directions. The shell in the x direction is generated by
 |
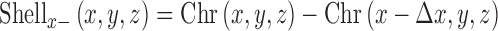 |
Where Chr(x,y,z) represents the binary chromosome image with the centroid at (x,y,z) of the chromosome and Δx is the thickness of shell. Shellx+ and Shellx- denote the differentiation in the positive and negative direction. The complete shell in the x direction is obtained by
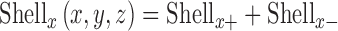 |
Similar shells were generated in y and z directions and denoted as Shelly and Shellz, respectively. The final shell is given by
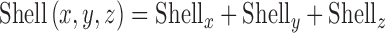 |
This can lead to overcounting of pixels. Hence a binary image was generated by replacing all non-zero pixels by 1. The region interior of the chromosome is generated by
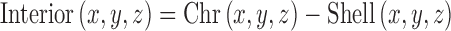 |
The mean and standard deviation of RNAPII intensity was estimated in the shell and the interior of the chromosome. The distribution of RNAPII was estimated by the coefficient of variation (COV) defined as
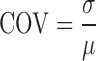 |
In order to estimate the enrichment of RNAPII in the intermingled region, a RNAPII enrichment factor was defined as
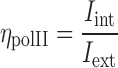 |
where Iint is the intensity of RNAPII in the intermingled region and Iext is the intensity of RNAPII in the chromosome excluding the intermingling region. 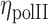 could be affected by the statistics over the small number of pixels in intermingling region as compared to the rest of the chromosome. Hence to eliminate such effects, an image was generated with random intensity with mean and standard deviation similar to the experimental RNAPII image.
could be affected by the statistics over the small number of pixels in intermingling region as compared to the rest of the chromosome. Hence to eliminate such effects, an image was generated with random intensity with mean and standard deviation similar to the experimental RNAPII image. 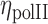 estimated from this was considered as a negative control.
estimated from this was considered as a negative control.
Statistical analysis
A Student's T-test was used as a test of significance wherever the standard deviation in the distributions overlapped. In the case of gene expression profiles, which have a non-Gaussian distribution, a Wilcoxon's signed rank test was performed to test the significance in differences in the activity of chromosomes. Significance of the correlations obtained was tested by false discovery rate (FDR) analysis. Towards this one of the quantities correlated was generated randomly for 10 000 iterations and correlated with the other quantity. The FDR was defined as
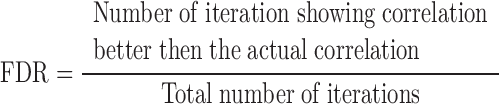 |
Microarray sample preparation and analysis
To perform genome-wide transcriptome analysis, 65 000 cells were seeded on fibronectin coated (FN) and uncoated (CT-control) tissue culture dishes (Nunc, Denmark) and cultured for 3 h. E14.1 cells were either cultured on PDL to maintain them in a differentiated state (CT-control) or were cultured on fibronectin for different periods of time to inducing an early onset of differentiation. The cells were harvested and RNA was isolated using RNAeasy kit (Qiagen). RNA concentration and purity was determined using a Nanodrop® ND-1000 spectrophotometer (Wilmington, DE, USA) and the integrity of RNA was verified. Microarray was performed using the Agilent Gene Whole genome 40 000 microarray chip (AMADID -014868) by Genomax technologies, Singapore. To reduce the noise in gene expression, microarrays were performed in duplicate for NIH3T3 cells and triplicate for ES cells. Typically, each gene on a microarray chip is represented by multiple probes. Hence the expression level of a particular gene was obtained by computing the mean expression of all the probes representing the gene. Similarly expressing genes within replicates were selected using the following condition:
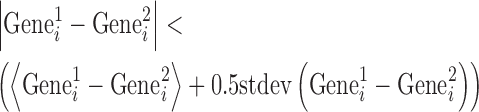 |
where  and
and  represent the activity of ith gene in the first and second replicate, respectively. ‘< >’ and ‘
represent the activity of ith gene in the first and second replicate, respectively. ‘< >’ and ‘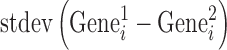 ’ represent averaging and standard deviation over all the gene. In case of ES cells where microarray was performed in triplicates, a T-test was performed and the genes with a P-value < 0.05 were selected. The selected genes indicate that the gene expression is faithfully represented between the biological replicates.
’ represent averaging and standard deviation over all the gene. In case of ES cells where microarray was performed in triplicates, a T-test was performed and the genes with a P-value < 0.05 were selected. The selected genes indicate that the gene expression is faithfully represented between the biological replicates.
Gene expression profiles obtained from microarray experiments have a long-tailed distribution. To estimate the difference between the gene expression profiles of individual chromosomes, the histogram of the individual gene expression profiles was compared. Highly expressed genes were selected by setting a cutoff of 500 in the intensity. The histograms of each chromosome were generated with 10 bins and normalized with the total number of selected genes in the chromosomes. Inter-chromosome activity distance (IADSKL) was generated by selecting a pair of chromosomes and estimating the symmetrized Kullback Liebler divergence given by
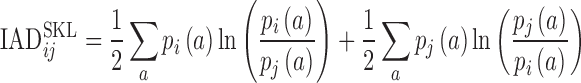 |
RESULTS
Chromosome territories in terminally differentiated cells have higher intermingling than undifferentiated cells
In order to identify changes in chromosome organization that occur in differentiated and undifferentiated cells, we measured the chromosome positions in ES cells and terminally differentiated NIH3T3 cells. ES cells were cultured on PDL-coated glass slides to maintain the undifferentiated stem cell state. Morphologically, the ES cells remained spherical on PDL, whereas NIH3T3 cells were flatter with ellipsoid nucleus. The flattening was quantified as the aspect ratio of the nucleus (ratio of the long axis and short axis of the fitted ellipse). ES cells had more spherical nuclei with aspect ratio near one and in NIH3T3 the aspect ratio was much higher (Supplementary Figure S1). The cells were then subjected to 3D-Fluorescence In situ Hybridization (3D-FISH) (Figure 1A).
Figure 1.

Chromosome intermingling in undifferentiated ES and differentiated NIH3T3 cells. (A) Representative images showing intermingling volume in different chromosome pairs in ES and NIH3T3 cells. (B) Schematic for Ivol, intermingling volume fraction. (C) Quantification of the Ivol for ES cells on PDL and NIH3T3. Scale bar, 5 μm. ** denotes FDR < 0.05.
Several recent studies have quantified chromosome organization by visualizing absolute and relative chromosome positioning. Absolute positioning is measured by radial position of chromosomes based on gene density or size, assuming each chromosome to be an independent entity. Relative chromosome positioning measured as proximities of chromosomes forms an additional layer of organization, where neighbourhood of chromosomes are the organizing entities. This results in a multidimensional interaction map, which is unique for each tissue type. In addition to course-grained chromosome FISH data, 3C techniques have mapped sequence level proximity and revealed a long-range interaction between regulatory elements and genes, which could be present on different chromosomes. Similar interactions can also be visualized as overlap between two CTs in both light and electron microscopy and has been termed chromosome intermingling. Chromosome intermingling leads to frequent chromosome translocations between certain pair of chromosomes in cancer cells (13). Due to the suggested functional importance and implicated role in transcriptional regulation, we analysed chromosome intermingling as a measurement of chromosome organization.
We observed multiple interactions between heterologous chromosomes (Figure 1A) by measuring chromosome intermingling volume, which is the region where two heterologous chromosomes overlap spatially. To quantify these interactions, the intermingling volume fraction for heterologous chromosome pair was defined as (Figure 1B)
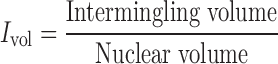 |
The intermingling volume is normalized to nuclear volume, as this does not underestimate the volume of intermingling, if the concerned chromosome is larger in size. As larger chromosomes also contain the largest number of genes, they could also potentially form larger chromosome intermingling volumes according to our hypothesis. Intermingling of selected pairs of heterologous chromosomes in ES cells cultured for 4 h on PDL was compared to those of NIH3T3 cells. When the mean value of Ivol for all the fluorescently labelled heterologous chromosome pairs was pooled, Ivol in NIH3T3 was significantly larger than ES cells (Figure 1C). This showed that the fraction of the individual CTs, which overlapped or interacted with other heterologous chromosomes, was higher in the differentiated cells. The chromatin in the ES cells is known to be more fluidic where the histones are more dynamic (24) and the chromatin shows higher spatial fluctuations (23). Hence we hypothesized that the chromosome intermingling in ES cells can be due to transient interactions, which will lead to smaller intermingling volume as compared to the differentiated state where the chromosome intermingling can be a result of much more stable chromatin interactions.
An extension of this hypothesis was that the interaction between the different heterologous chromosomes in the plastic ES cell state would be similar. To test if the measured intermingling volume fraction for specific pair of chromosomes were significantly different than others, a T-test probability matrix was computed (Supplementary Figure S2A and E). Pairs of chromosomes, which showed significant difference in intermingling volume, are coloured in brown, which has P-values <0.05 in the heat map. The T-test matrix showed that in addition to lower intermingling volume in ES cells a majority of chromosome pairs were similar (Supplementary Figure S2F). This reflected the plastic nature of the chromosome organization in the ES cells as shown earlier (23).
Increase in chromosome intermingling fraction correlates with emergence of transcriptional programs
What could be the underlying reason of the higher intermingling volume observed in the differentiated cells? Transcription co-regulation where genes and regulatory sequence come together to form cis- or trans-interactions can be one of the reasons. Spatial mapping of the co-expressed genes by gene FISH reveals (12) that co-expressed genes present on either homologous or heterologous chromosomes can come together and co-localize with common transcription factors required for their transcription. To test this hypothesis we first measured the transcriptional output of the undifferentiated ES cells and the differentiated NIH3T3 state. ES cells grown on PDL for 4 h and NIH3T3 cells on fibronectin for a similar duration were subjected to RNA extraction and microarray for quantifying its transcriptional landscape (‘Materials and Methods’). A clear difference in the transcriptional activity of chromosomes of NIH3T3 was observed when compared to ES cells by performing microarray for these conditions (Supplementary Figures S3A–B and S4). To further assess the transcriptional state of the cells, we generated an IADSKL matrix by measuring the symmetrized Kullback Liebler divergence between pairs of chromosomes (27), which measures the difference between the distribution of gene expression activity (Supplementary Figure S3C) determined by microarray for a pair of chromosomes (‘Materials and Methods’). The IADSKL is defined as
 |
Where 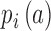 and
and 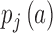 are the probability of gene activity ‘a’ in chromosome i and j, respectively. The summation is performed over all such activities for a pair of chromosomes. High IADSKL values, which are denoted by warmer colours, represent chromosomes with different transcriptional activities, while low IADSKL values, which are displayed by cooler colours in the IADSKL matrices, signify similar transcriptional activities (Figure 2A). To compare the transcriptional state of ES cells with the differentiated NIH3T3 cells, we measured the Pearson Correlation Coefficient (PCC) between the IADSKL matrices of these two states. The PCC value between the ES cell IADSKL matrix and NIH3T3 was very low (Supplementary Figure S5) showing that the transcriptional landscape between the undifferentiated ES cells is very different from that of the differentiated NIH3T3 cells.
are the probability of gene activity ‘a’ in chromosome i and j, respectively. The summation is performed over all such activities for a pair of chromosomes. High IADSKL values, which are denoted by warmer colours, represent chromosomes with different transcriptional activities, while low IADSKL values, which are displayed by cooler colours in the IADSKL matrices, signify similar transcriptional activities (Figure 2A). To compare the transcriptional state of ES cells with the differentiated NIH3T3 cells, we measured the Pearson Correlation Coefficient (PCC) between the IADSKL matrices of these two states. The PCC value between the ES cell IADSKL matrix and NIH3T3 was very low (Supplementary Figure S5) showing that the transcriptional landscape between the undifferentiated ES cells is very different from that of the differentiated NIH3T3 cells.
Figure 2.

Correlation between transcription activity and chromosome intermingling. (A) IADSKL matrix for ES cells and NIH3T3. (B) Scatter plot between the intermingling volume fraction (Ivol) and IADSKL for ES (top) and NIH3T3 (bottom) cells. (C) PCC for correlation between IADSKL of ES and NIH3T3, respectively.
Next, we correlated the intermingling volumes measured in ES cells or NIH3T3 cells with their respective IADSKL values. As shown in the scatter plots of Figure 2B, the IADSKL was weakly correlated with the Ivol in ES cells when compared to the correlation in case of the NIH3T3 cells, where the IADSKL decreased exponentially with increasing Ivol. A very weak correlation of −0.009 between the Ivol and IADSKL in undifferentiated ES cells was found (Figure 2B and C). Whereas in the terminally differentiated NIH3T3 cells, the Ivol was strongly correlated to IADSKL, with a statistically significant PCC value of −0.65 (Figure 2B and C). This correlation between intermingling volume and IADSKL is independent of chromosome size, as correlation between intermingling volume normalized to the sum of participating chromosomes’ volume (as opposed to normalizing with nuclear volume as before) still showed a similar trend with IADSKL (Supplementary Figure S6A).
Intermingling frequencies (Ifreq), which quantify the probability of chromosome interactions (‘Materials and Methods’), were measured for NIH3T3 cells and also showed significant negative correlation with the IADSKL (Supplementary Figure S6B–D). This negative correlation suggested that chromosomes with higher transcriptional activity (low IADSKL values) overlapped more often and to a larger extent in differentiated NIH3T3 cells, whereas such interactions were still undeveloped in the undifferentiated ES cell.
Transcriptional regulators are enriched at the chromosome intermingling regions
The increase in the chromosome intermingling fraction in differentiated cells together with high correlation with the transcriptional activity suggests the functional nature of chromosome intermingling. But the correlation itself does not provide any insight into the molecular mechanism. The undifferentiated ES cell chromatin is known to harbour bivalent histone modifications (29) with both repressive and activating histone modifications which might repress lineage control genes during pluripotency but at the same time keep them poised for activation during differentiation. During differentiation these bivalent histone modifications are lost. The lineage-specific genes get enriched in activating histone modifications and active RNA polymerase where as the other genes are turned off by accumulation of the repressive chromatin marks on the promoters (30). Hence, to further understand the functional nature of the intermingling regions we probed for distribution of transcriptional regulators with respect to the CT organization, which are known to change during differentiation, for transcriptional activation—phosphorylated S2 of Carboxyl Terminal Domain of RNA polymerase II (RNAPII), H3K9 acetylation (H3K9Ac), H3K4 trimethylation (H3K4Me3) (31) and transcription factor serum responsive factor (SRF); and for repression—H3K9 tri methylation (H3K9Me3) (32) and macroH2A.2 (33).
Towards this, we subjected the cells to long fixation protocol to preserve the transcriptional markers before performing the rigorous hybridization of the chromosome probes (‘Materials and Methods’). After the FISH, the transcriptional markers were immuno-localized and imaged in confocal microscope (Figure 3A). First, we measured the distribution of the transcriptional regulators in the differentiated state of NIH3T3 cells. The enrichment of RNAPII at the intermingling regions of the chromosomes was defined as the RNAPII enrichment factor, ηPolII:
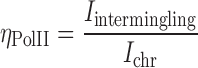 |
where Iintermingling and Ichr are the mean intensities of RNAPII in the intermingled region and the chromosome excluding intermingled region, respectively. As shown in Figure 3B, the mean RNAPII intensities shifted to higher values in regions of chromosome intermingling (Iintermingling) compared to the rest of the chromosome (Ichr). This leads to a much higher ηPolII value than would be expected if RNAPII were not enriched in regions of chromosome intermingling as was the case in the simulated ‘random’ RNAPII intensity of the nucleus (‘Materials and Methods’ and Figure 3E). Interestingly, a significantly lower ηPolII value was obtained from undifferentiated ES cells (inset of Figure 3B and Supplementary Figure S7C). This suggests that the enrichment of RNAPII within intermingled regions was well established only in differentiated cells. The sites marked by RNAPII were indeed regions of the nucleus that are engaged in transcription, as RNAPII foci coincided with the Br-UTP incorporated in live cells ('Methods and Materials' and Supplementary Figure S7A and B). A similar enrichment was observed for markers of activation and repression of transcription, indicating enrichment can be due to segregation of both positively and negatively co-expressed regions of the chromosome (Figure 3E). In our experiments we looked at a pair of the heterologous chromosome intermingling at a given time. Although there are many more unvisualized chromosome intermingling regions are present. This inability of separating all the possible intermingled and non-intermingled regions of the chromosome could contribute to noise in the quantification of the 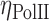 . In spite of this additional noise, the
. In spite of this additional noise, the 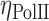 is significantly higher in the intermingling region, suggesting a greater enrichment of the RNAPII.
is significantly higher in the intermingling region, suggesting a greater enrichment of the RNAPII.
Figure 3.

Transcriptional markers are enriched in intermingled regions. (A) A composite image showing Chr 4 (green), Chr 6 (red) and RNAPII (blue). The yellow Region Of Interest shows one of the intermingled regions. Heat map images show the intensity of RNAPII in the intermingled region and regions of Chr 4 and Chr 6, which do not intermingle. (B) Histogram showing the distribution of RNAPII in the intermingled regions and rest of the chromosome. Inset shows ηPolII in ES and NIH3T3 cells (* denotes P < 0.05). (C) Representative colour map showing the distribution of RNAPII in the interior and on a 400 nm thick shell on surface of the chromosome. Colour bar indicates the intensity of RNAPII. (D) A cumulative histogram of RNAPII in the shell (black) and in the interior (red) of the chromosome (distributions are significantly different with P < 10−28). Inset shows the comparison of RNAPII intensity in the regions of the shell occupied by intermingling regions (ShellIntermingled) with the entire shell (Shell). **** denotes P < 10−40. (E) Enrichment factor for random simulated image (random), RNAPII, activation markers—H3K9Ac and H3K4Me3, and repressive markers—macroH2A and H3K9Me3. ** denotes P < 10−4, *** denotes P < 10−10, **** denotes P < 10−25. Scatter plot between ηPolII and IADSKL for NIH3T3 cells. Scale bar, 5 μm. (F) Single confocal Z plane showing of Chr10 (white), Chr4 (cyan), noise-removed RNAPII (green), SRF (red) and nucleus outline (yellow). Scale bar, 5 μm. Inset: zoomed in image of the cropped intermingled regions as marked by the yellow box. Bar graph shows the scatter plot, mean (red line), standard deviation (SD) and standard error of mean (SEM) for the PCC of co-localization between RNAPII and SRF.
To qualitatively estimate the effect of labelled and unlabelled chromosome intermingling regions on the enrichment of RNAPII, we took advantage of the fact that these inter chromosome interactions usually occur at the surface of the chromosomes, which harbour active looping regions of the chromatin (34). We divided the chromosomes into two regions; a 450-nm thick ‘shell’ region and an internal region, to quantify the chromosomal distribution of active RNAPII. The mean intensity of RNAPII was similar in both the interior and shell regions of the chromosome but the shell showed higher heterogeneity in the intensity (Figure 3C). We then estimated the heterogeneity in the distribution of RNAPII by measuring the COV of the marker in the shell and interior regions. After plotting a cumulative histogram of COV values computed over multiple chromosomes in different nuclei, we observed a significant difference between the COV in the shell region and that in the interior region (Figure 3D). This heterogeneity could stem from an enrichment of RNAPII in the intermingled regions (overlapping region between two CTs) of the shell (ShellIntermingling) as compared to non-intermingled regions of the chromosomes (Shell). The intensity of RNAPII in the intermingled regions of the shell was therefore measured, and a significantly higher intensity was observed as compared to other regions of the shells (Figure 3D inset).
We find statistically significant enrichment of activated RNAPII in the intermingling regions, suggesting that these regions are transcriptionally active. We visualized the co-localization of RNAPII, and a transcription factor SRF in the intermingling regions of Chr10–Chr4 in NIH3T3 cells. Chr4 and Chr10 were chosen, as the gene targets SRF are enriched on these two chromosomes. SRF is a known mechano-sensitive protein, which controls cell rounding and differentiation. Figure 3F clearly shows the direct co-localization of the transcription factor with RNAPII in the intermingling regions and a higher PCC of co-localization, further validating the enrichment of the transcriptional regulators in the intermingling region.
Enrichment of transcriptional markers at intermingling regions correlates with transcription
The high correlation between the IADSKL and intermingling volume in the differentiated state corroborated well with the finding that the intermingling regions in the differentiated NIH3T3 cells had higher concentration of the transcriptional markers. Hence, we decided to investigate if the enrichment of active RNAPII is dependent on the intermingling volume and/or transcriptional activity of the chromosomes themselves. We determined the correlation between ηPolII and both Ivol and IADSKL in NIH3T3 cells, where the cell lineage specific transcriptome and the intermingling volumes are the most defined.
We observed a negative correlation between ηPolII and IADSKL with a PCC of −0.5 (Figure 4A) indicating a decrease in the density of RNAPII with increasing IADSKL. On the other hand, we observed a positive correlation between the ηPolII and the Ivol suggesting that the intermingling regions are in fact the sites of transcription (Figure 4B). We also checked if the observed ηPolII was correlated with the size of the heterologous chromosome pair involved. For this we selected a pair of smaller chromosomes (Chr11–Chr18) and a pair of larger chromosomes (Chr1 and Chr2) and computed the ηPolII for the two pairs, as shown in Figure 4C. Interestingly, Chr1 and Chr2 also contain more number of genes as compared to Chr11–Chr18. This result suggests that the larger chromosomes, which have a larger number of transcribing genes, not only may form large intermingling regions but also have a higher density of co-expressed genes.
Figure 4.

Correlation of transcriptional markers enrichment with transcriptional activity and perturbation in differentiated cells. (A) Scatter plot to compute correlation between ηPolII and IADSKL. (B) Scatter plot to compute correlation between ηPolII and Ivol. Red lines indicate linear fit. (C) ηPolII for chromosome intermingling regions of Chr11–Chr18 and Chr1–Chr2. (D) Single confocal Z plane of Chr4 (red), Chr6 (green) and zyxin (white), with nuclear outline (blue). Scale bar, 5 μm. Inset: zoomed-in image of intermingling region as marked by the yellow box. Bar graph shows the quantification of frequency of zyxin in intermingling region. (E) Quantification of ηPolII in chromosome intermingling regions for transcription perturbations (* denotes P < 0.05 and ** denotes P < 0.001). (F) Heterogeneity of RNAPII distribution, quantified as COV on the chromosome surface and interior on untreated and TSA treatment.
To establish position of an active gene with respect to intermingling region, we carried out DNA FISH analysis of a candidate gene zyxin (‘Materials and Methods’), which is regulated by the transcription factor SRF. We found that zyxin is upregulated in spread NIH3T3 cells as opposed to the rounded cells (Supplementary Figure S12B). Hence, this was a good condition where we could test correlation between the gene expression level of zyxin and its position with respect to the chromosome intermingling. The chromosome intermingling was measured for Chr6 and Chr4, as zyxin is present on Chr6, and Chr4 is enriched for other gene targets of SRF. We found that the higher number of cells showed at least one zyxin foci within the intermingling in spread cells as compared with the rounded cells (Figure 4D) which correlated with the higher expression of zyxin in spread cells. This strengthened our hypothesis that the genes in intermingling regions are transcriptionally active.
Transient transcriptional perturbations reveal the structural nature of chromosome intermingling
We asked if the enrichment of active RNAPII is due to more stable structural DNA interactions formed during longer time scale of multiple cell cycles or due to functional modifications of the chromatin required for immediate transcriptional regulation at the chromosome intermingling. To answer this, the cells were treated with global transcriptional perturbants- α-Amanitin, TSA or serum depletion. Despite each perturbant causing a disregulation in transcription, the intermingling region still showed a higher concentration RNAPII than the randomly selected regions in the nucleus (Figure 4E). This suggests that the intermingling regions are formed through a mechanism that is not affected by temporary changes in transcription.
To assess if this variation was due to the heterogeneous organization of the chromatin in the nucleus as euchromatin or heterochromatin regions, we treated the cells with the histone deacetylase inhibitor TSA. This essentially decondenses heterochromatin thereby making the chromatin homogenous and also increases transcription (35,36). The ratio of shell COV (which is a measure of heterogeneity of RNAPII intensity) to interior COV was significantly larger in untreated cells compared to TSA-treated cells, suggesting the structure of chromatin also contributes to the observed heterogeneity of RNAPII distribution within the shell (Figure 4F). But still this was insufficient to decrease the enrichment of the RNAPII in the intermingling region as low as the random region negative control as shown in the graph of Figure 4E. These results suggest that the intermingling regions are structural regions, which are not perturbed by transient transcriptional perturbations and still retain higher concentration of RNAPII.
Radial chromosome organization also correlates with chromosome intermingling volume and size
Till now our results showed that chromosome intermingling regions are the structural intermediates, which connects the transcriptional activity required for specifying tissue specific transcriptome and relative chromosome organization. Next we wanted to determine the role of chromosome intermingling, chromosome size and gene density in the radial positioning of the chromosomes, where the latter two factors have been implicated as factors responsible for radial positioning of chromosomes for many different cell types. In the radial positioning, each chromosome is regarded as an independent entity, hence we first measured the radial positions of individual chromosome, defined as the distance from the nuclear centroid to the chromosome centroid (Figure 5A). These measurements were normalized with respect to the local radius of each nucleus and then plotted as blue points in Figure 5B. The most probable distance (‘Materials and Methods’) of each chromosome from the centre of the nucleus was determined from the distribution measurements and plotted as the red dotted line. In NIH3T3 cells, the larger chromosome 1 was predominantly localized to the nuclear periphery, while the smaller chromosome 18 was primarily located in interior region of the nucleus (Figure 5B and C). But similar distinction for positioning of the larger chromosome 1 and smaller chromosome 19 was not observed for the ES cells. Moreover, when we compared the radial positions of chromosomes by computing a T-test matrix (Supplementary Figure S8), we observed that chromosomes in ES cells are less heterogeneous in their organization as compared to differentiated NIH3T3 cells suggesting the plastic nature of ES cells.
Figure 5.

Radial organization of chromosomes with respect to chromosome size, density and Ivol. (A) Images showing representative chromosomes of ES 4 h and NIH3T3 cell in increasing order of their base pair lengths. (B) Polar plots showing the radial positions of chromosomes 1 and 19 in ES cells, and chromosomes 1 and 18 in NIH3T3 cells, normalized to the nuclear radius at that angular position. Blue dots indicate individual chromosomes and red line indicates the maximum probable position of the distribution. (C) Scatter plot between maximum probable radial position and chromosome length for ES 4 h and NIH3T3 cells. Red line indicates the exponential fit. (D) Intermingling volume for two large chromosomes Chr1–Chr2 and two smaller chromosomes Chr1–Chr2. (E) Scatter plot to quantify the correlation between the mean maximum probable radial distance and the intermingling volume measured for a pair of heterologous chromosomes.
Furthermore, the radial position of each chromosome was strongly correlated with chromosome length in base pairs (Figure 5C: PCC ∼ 0.56, FDR < 0.012) for NIH3T3 cells. This is evident in the size-based colour-coded radial position plots (Supplementary Figure S10E), with the segregation of radial position resulting from the localization of larger chromosomes towards the nuclear periphery. This observation is in agreement with a previously observed correlation in human male fibroblasts, where the radial distribution of chromosomes was suggested to be a general mechanism of chromosome positioning that was conserved across multiple organisms and cell types (4). In contrast, the differences in radial positioning of chromosomes in ES cells (Figure 5B and Supplementary Figure S9), as well as the segregation of the colour-coded probable radial position plots (Supplementary Figure S10A), were significantly less compared to terminally differentiated NIH3T3 cells. This resulted in a lower PCC value for correlation between radial chromosome position and chromosome size in ES cells. Hence, the size-based radial positioning of chromosomes was not significant in the undifferentiated ES cells, but is present in the differentiated NIH3T3 cells.
Previously, the radial positioning of chromosomes was attributed to their gene density—gene-rich chromosomes were associated with the transcriptionally active nuclear centre, while chromosomes with fewer genes were located towards the nuclear periphery (3). To establish whether gene density was a contributing factor in our observations, we correlated the most probable normalized radial position of each chromosome in NIH3T3 cells with their gene density, which was defined by the total number of coding and non-coding genes per base pair from http://www.ensembl.org/Mus_musculus/Location/Genome. In both cases the PCC obtained was lower than the PCC of radial position based on chromosome size (Supplementary Figure S8F). Hence these results suggest that the size-based positioning of the chromosomes is a more general mechanism.
To test if chromosome intermingling has any role to play in radial positioning, we tested if mean most probable radial position of the two heterologous chromosomes for which the Ivol was computed correlated with the Ivol in differentiated NIH3T3 where the radial organization of the chromosomes is well established. The Spearman's rho for the correlation between the most probable radial position and Ivol was 0.687, which was significant as the FDR for this was only 0.007. Again from these observations we conclude that radial position does correlate with the intermingling volume that is well developed in the differentiated NIH3T3 cells. Although with our observations we were able to show that radial positions correlate with both chromosome size and intermingling volume, deconvolving contributions of these two factors in radial chromosome organization requires further inspection.
Both relative and radial chromosome positioning develop together with appearance of chromosome intermingling at early onset of differentiation
The previous sections showed clear differences between the radial and relative positions of chromosomes in the undifferentiated ES cells (on PDL) and differentiated NIH3T3 cells. Both parameters of the chromosome organizations were well established in the differentiated cells of NIH3T3 cells as compared to ES cells. At the same time we observed that the relative position of chromosome correlates well with intermingling volume, enrichment of the RNAPII and transcriptional activity observed in NIH3T3 cells. We next asked the question if these features of the chromosome organizations appear during the early onset of differentiation of the ES cells, as cells move away from the pluripotent state of ES cells.
To investigate the early onset of differentiation, we plated ES cells on fibronectin for 4, 12 or 24 h, which caused visible changes in the morphology of cells and plasticity of chromatin (23). The cells became flatter, the nucleus became more ellipsoid like (similar to NIH3T3 cells) (Figure 6A and B) and the transcriptional landscape also changes during these early onset of differentiation (Supplementary Figures S3 and 4). During the early onset of differentiation the correlation between the radial positions of the chromosome and chromosome size increases as ES cells spend more time on the fibronectin (Figure 6C).
Figure 6.

Radial chromosome organization and chromosome intermingling during early onset of differentiation. (A) DNA marked in blue shows the nuclear morphology. Orthogonal section of each nucleus shown in the inset indicates nuclear height, scale bar, 5 μm. (B) Quantification of the aspect ratio of the nuclei. (C) Spearman's Rho to quantify the correlation between the radial chromosomes and its size in Mbp. (D) Bar graph for intermingling volume, Ivol between heterologous chromosomes. (E) Graph for PCC quantifying the correlation between transcriptional activity, IADSKL and intermingling volume, Ivol for pairs of heterologous chromosomes in different conditions (* denotes P < 10−5).
Similarly, the intermingling volume and its correlation with transcription in the terms of IADSKL shows a general trend of increase with time during this early onset of differentiation (Figure 6D–E and Supplementary Figure S11). But during this time enrichment of RNA Pol II remained at significantly lower as compared to NIH3T3 cells (Supplementary Figure S10), further suggesting that these intermingling regions emerge as structural regions and the enrichment is perhaps a process, which happens during later stages of differentiation.
As we had observed nuclei flattening in differentiating ES cells and terminally differentiated NIH3T3 cells, we asked whether this could be responsible for the size-based radial positioning of chromosomes. To investigate this we labelled chromosomes in a non-adherent T-cell, which, despite being differentiated, retains a spherical nucleus (18,26). The radial position of the six homologous chromosomes showed PCC of 0.7 and suggested the presence of an unidentified mechanism that facilitates the size-based radial positioning of chromosomes (Supplementary Figure S13) and rules out nuclear flattening as one of the possible mechanisms.
DISCUSSION
The 3D organization of CTs within the nucleus has been suggested to be probabilistic, with our current inability to predict the CT positions precisely. Still, CT organization is not completely random, as the radial positions of CT are believed to be determined according to the chromosomes gene density or size. Additionally, in the mammalian cell nucleus, specific heterologous chromosomes are brought together via the adjacent positioning of the Nucleolar Organizing Regions to form the nucleolus and also chromosome neighbourhood changes during differentiation (37). This preferential relative chromosome positioning has also been implicated in an increased frequency of translocation between certain pairs of chromosomes (13). Together, these observations suggest that radial and relative chromosome positioning occurs in an organized and structured manner, albeit by unknown mechanisms. In this work, we measured the positions of CTs during the early stages of differentiation in mouse embryonic stem cells and compared the findings to those from terminally differentiated NIH3T3 cells. This allowed us to record the evolution of CT organization during differentiation and in doing so, gain some insight into the mechanisms that determine CT organization.
Chromosome intermingling regions, which were mapped by 3C and FISH, had been implicated as the functional regions of the chromatin where the co-expressed genes can interact. Hence, we specifically looked at the relationship of the chromosome intermingling and the 3D chromosome organization. To probe cell type-specific CT organization, we measured the intermingling volume and frequencies between heterologous chromosomes. Regions of chromosome intermingling and chromosome surfaces contain a higher density of active genes, which can undergo transcription and splicing (13–15). We found chromosome intermingling volumes to be a more sensitive and statistically significant measure of the relative CT organization then the widely used inter-centroid chromosome distance, which is influenced by the chromosome volume (21). In an earlier study, we found that heterologous chromosomes with similar transcriptional levels are spatially closer when compared to chromosomes with very different transcription levels (21). In support of this, we found that inter-chromosomal interactions, which were quantified through the intermingling volume and frequencies, increased during the early onset of ES cell differentiation and were highest in terminally differentiated NIH3T3 cells. Interestingly, the total intermingling volume was found to be smaller in ES cells as compared to NIH3T3 cells, which again points towards the plastic nature of ES cell nuclear architecture. The intermingling volumes correlated better than the inter-centroid chromosome distances, suggesting that the intermingling volume and frequency measurements are better reflections of the degree of transcription and probabilistic nature of chromosome interactions (21).
Inter-chromosome contacts between heterologous chromosomes from both mouse and human genomes have been extensively mapped using 3C techniques (10) and implicated in chromosome pair translocation and the regulation of transcription. Indeed, we found that intermingling regions in NIH3T3 cells, which can be described as inter-chromosome contacts, were enriched with markers of transcription activation and repression. The intermingling regions, which were enriched in activated RNAPII, also contained Br-UTP incorporation and SRF, a transcription factor relevant for differentiation of ES cells. We also found zyxin, a target gene of SRF, to be present in the intermingling regions more frequently when transcriptionally more active. These evidence support our hypothesis of intermingling region as important functional regions in nucleus.
Undifferentiated ES cells, as well as ES cells in the early stages of differentiation, possessed intermingling regions that showed an enrichment of transcriptionally active phosphorylated RNAPII. The absolute value of RNAPII enrichment factor was however smaller than that observed in NIH3T3 cells for similar heterologous chromosome pairs. RNAPII enrichment was higher between chromosomes with a similar transcriptional activity (or low IADSKL). This negative correlation suggests that the intermingling regions are the basis of such transcriptional activity. This idea was strengthened by the positive correlation between the RNAPII enrichment factor and the intermingling volume. Furthermore, transient perturbation of transcription by administration of histone deacetylase (a transcription upregulator), α-amanitin (a transcriptional repressor) or withdrawal of serum (global transcription repressor) could not completely abolish the enrichment of transcriptional markers at the intermingling regions. This suggests that these are structurally stable regions formed during differentiation that remain unchanged during the cell cycle.
We also investigated the potential role of the chromosome intermingling in the radial organization of the chromosomes. Previous studies have indicated that CT positioning is dependent on both gene density (2,3) and chromosome size (4,5). Our findings corroborate the studies of Bolzer et al. and Sun et al., with CT organization in the terminally differentiated NIH3T3 cells being strongly correlated to chromosome size. However, the radial position of each chromosome was found to be less dependent on the gene density than on the chromosome size. This correlation between chromosome size and position was absent in stably undifferentiated ES cells as well as in ES cells in the early stages of differentiation (4, 12 or 24 h on fibronectin), although considerable changes in the level of transcription were observed during these early stages of differentiation. Consistent with previous studies, nuclei flattened out as differentiation progressed in ES cell (23). By measuring the radial position of CTs in free-floating naïve T cells from mice, which retain a spherical nucleus despite being differentiated, we showed a strong correlation between chromosome size and the radial position of CTs. Our observation that CT positioning is determined independently of nuclear morphology corroborates with a previous study which reported changes in the radial position of chromosome 12 during differentiation of flat pre-adipocyte cells to the more rounded adipocyte cells (37). Interestingly, we also observed that heterologous chromosomes in the differentiated NIH3T3 cells, which share similar radial positions, also intermingle more. Together, these results suggest that the plasticity of nuclear organization in ES cells (23) might result in the radial organization of CTs being non-specific until differentiation commences, at which point a specific radial and relative chromosome organization is induced by developing the chromosome intermingling regions.
In conclusion, we have shown that radial and relative chromosome positioning evolves during the process of differentiation (Figure 7). In the differentiated state, CTs are radially arranged according to the chromosome size, though a weak correlation with chromosome gene density was also observed. The relative position of the chromosomes may result from transcription regulation dependent interactions, which leads to co-clustering of DNA sequences and intermingling of adjacent chromosomes. These intermingled regions are enriched with markers of both transcription activation and repression. Whether these different markers of transcription are segregated in the intermingled region requires further investigation. Since these CT interactions build up, step by step, during differentiation, it remains to be seen how the information of specific interactions between CTs is transmitted and accumulated through cell divisions to develop the cell type-specific nuclear architecture. Identification of RNA- or protein-based molecular ties which brings structural stability to intermingling regions will be the key to understand the mechanisms of chromosome organization.
Figure 7.

Schematic showing the evolution of chromosome intermingling during differentiation. (A) Organization of chromatin and CTs in undifferentiated ES cells, differentiating ES cells and terminally differentiated NIH3T3 cells. (B) Evolution of chromosome intermingling through the process of differentiation. (C) Evolution of radial chromosome organization through differentiation.
Supplementary Material
Acknowledgments
We would like to thank Tsvi Tlusty for helpful discussions. We also thank Caroline Uhler for providing useful inputs on the manuscript. We would like to thank Steven Wolf, for critical reading of the manuscript and Larisa Bulavina, for help in creating the schematic of the experimental results from Mechanobiology Institute (MBI), Singapore.
Author contributions: S.M., K.V.I., N.J., Y.J.W. and G.V.S. designed the experiments. S.M., K.V.I., N.J., Y.J.W. and M.N. performed the experiments. S.M., K.V.I., N.J., M.N., Y.J.W. and G.V.S. analysed data and wrote the paper.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Mechanobiology Institute, NUS-Singapore and Singapore Ministry of Education Academic Research Fund Tier3 [MOE2012-T3–1–001]; all the open lab core facilities of Mechanobiology Institute. Funding for open access charge: Mechanobiology Institute, NUS-Singapore.
Conflict of interest statement. None declared.
REFERENCES
- 1.Cremer T., Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2001;2:292–301. doi: 10.1038/35066075. [DOI] [PubMed] [Google Scholar]
- 2.Boyle S., Gilchrist S., Bridger J.M., Mahy N.L., Ellis J.A., Bickmore W.A. The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum. Mol. Genet. 2001;10:211–219. doi: 10.1093/hmg/10.3.211. [DOI] [PubMed] [Google Scholar]
- 3.Tanabe H., Muller S., Neusser M., von Hase J., Calcagno E., Cremer M., Solovei I., Cremer C., Cremer T. Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. Proc. Natl Acad. Sci. U.S.A. 2002;99:4424–4429. doi: 10.1073/pnas.072618599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolzer A., Kreth G., Solovei I., Koehler D., Saracoglu K., Fauth C., Muller S., Eils R., Cremer C., Speicher M.R., et al. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 2005;3:e157. doi: 10.1371/journal.pbio.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun H.B., Shen J., Yokota H. Size-dependent positioning of human chromosomes in interphase nuclei. Biophys. J. 2000;79:184–190. doi: 10.1016/S0006-3495(00)76282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parada L.A., McQueen P.G., Misteli T. Tissue-specific spatial organization of genomes. Genome Biol. 2004;5:R44. doi: 10.1186/gb-2004-5-7-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brianna Caddle L., Grant J.L., Szatkiewicz J., van Hase J., Shirley B.J., Bewersdorf J., Cremer C., Arneodo A., Khalil A., Mills K.D. Chromosome neighborhood composition determines translocation outcomes after exposure to high-dose radiation in primary cells. Chromosome Res. 2007;15:1061–1073. doi: 10.1007/s10577-007-1181-7. [DOI] [PubMed] [Google Scholar]
- 8.Meaburn K.J., Misteli T., Soutoglou E. Spatial genome organization in the formation of chromosomal translocations. Semin. Cancer Biol. 2007;17:80–90. doi: 10.1016/j.semcancer.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rabbitts P.H. Genetic changes in the development of lung cancer. Br. Med. Bull. 1994;50:688–697. doi: 10.1093/oxfordjournals.bmb.a072917. [DOI] [PubMed] [Google Scholar]
- 10.Miele A., Dekker J. Long-range chromosomal interactions and gene regulation. Mol. BioSyst. 2008;4:1046–1057. doi: 10.1039/b803580f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y., McCord R.P., Ho Y.J., Lajoie B.R., Hildebrand D.G., Simon A.C., Becker M.S., Alt F.W., Dekker J. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schoenfelder S., Sexton T., Chakalova L., Cope N.F., Horton A., Andrews S., Kurukuti S., Mitchell J.A., Umlauf D., Dimitrova D.S., et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 2009;42:53–61. doi: 10.1038/ng.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Branco M.R., Pombo A. Intermingling of chromosome territories in interphase suggests role in translocations and transcription-dependent associations. PLoS Biol. 2006;4:e138. doi: 10.1371/journal.pbio.0040138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cmarko D., Verschure P.J., Martin T.E., Dahmus M.E., Krause S., Fu X.D., van Driel R., Fakan S. Ultrastructural analysis of transcription and splicing in the cell nucleus after bromo-UTP microinjection. Mol. Biol. Cell. 1999;10:211–223. doi: 10.1091/mbc.10.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fanucchi S., Shibayama Y., Burd S., Weinberg M.S., Mhlanga M.M. Chromosomal contact permits transcription between coregulated genes. Cell. 2013;155:606–620. doi: 10.1016/j.cell.2013.09.051. [DOI] [PubMed] [Google Scholar]
- 16.Boyle S., Rodesch M.J., Halvensleben H.A., Jeddeloh J.A., Bickmore W.A. Fluorescence in situ hybridization with high-complexity repeat-free oligonucleotide probes generated by massively parallel synthesis. Chromosome Res. 2011;19:901–909. doi: 10.1007/s10577-011-9245-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagano T., Lubling Y., Stevens T.J., Schoenfelder S., Yaffe E., Dean W., Laue E.D., Tanay A., Fraser P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ragoczy T., Telling A., Sawado T., Groudine M., Kosak S.T. A genetic analysis of chromosome territory looping: diverse roles for distal regulatory elements. Chromosome Res. 2003;11:513–525. doi: 10.1023/a:1024939130361. [DOI] [PubMed] [Google Scholar]
- 19.Chambeyron S., Da Silva N.R., Lawson K.A., Bickmore W.A. Nuclear re-organisation of the Hoxb complex during mouse embryonic development. Development. 2005;132:2215–2223. doi: 10.1242/dev.01813. [DOI] [PubMed] [Google Scholar]
- 20.Spilianakis C.G., Lalioti M.D., Town T., Lee G.R., Flavell R.A. Interchromosomal associations between alternatively expressed loci. Nature. 2005;435:637–645. doi: 10.1038/nature03574. [DOI] [PubMed] [Google Scholar]
- 21.Iyer K.V., Maharana S., Gupta S., Libchaber A., Tlusty T., Shivashankar G.V. Modeling and experimental methods to probe the link between global transcription and spatial organization of chromosomes. PLoS One. 2012;7:e46628. doi: 10.1371/journal.pone.0046628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Talwar S., Jain N., Shivashankar G.V. The regulation of gene expression during onset of differentiation by nuclear mechanical heterogeneity. Biomaterials. 2014;35:2411–2419. doi: 10.1016/j.biomaterials.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 23.Talwar S., Kumar A., Rao M., Menon G.I., Shivashankar G.V. Correlated spatio-temporal fluctuations in chromatin compaction states characterize stem cells. Biophys. J. 2013;104:553–564. doi: 10.1016/j.bpj.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhattacharya D., Talwar S., Mazumder A., Shivashankar G.V. Spatio-temporal plasticity in chromatin organization in mouse cell differentiation and during Drosophila embryogenesis. Biophys. J. 2009;96:3832–3839. doi: 10.1016/j.bpj.2008.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Laat W., Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature. 2013;502:499–506. doi: 10.1038/nature12753. [DOI] [PubMed] [Google Scholar]
- 26.Peric-Hupkes D., Meuleman W., Pagie L., Bruggeman S.W., Solovei I., Brugman W., Graf S., Flicek P., Kerkhoven R.M., van Lohuizen M., et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajapakse I., Perlman M.D., Scalzo D., Kooperberg C., Groudine M., Kosak S.T. The emergence of lineage-specific chromosomal topologies from coordinate gene regulation. Proc. Natl Acad. Sci. U.S.A. 2009;106:6679–6684. doi: 10.1073/pnas.0900986106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strickfaden H., Zunhammer A., van Koningsbruggen S., Kohler D., Cremer T. 4D chromatin dynamics in cycling cells: Theodor Boveri's hypotheses revisited. Nucleus. 2010;1:284–297. doi: 10.4161/nucl.1.3.11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vastenhouw N.L., Schier A.F. Bivalent histone modifications in early embryogenesis. Curr. Opin. Cell Biol. 2012;24:374–386. doi: 10.1016/j.ceb.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aranda P., Agirre X., Ballestar E., Andreu E.J., Roman-Gomez J., Prieto I., Martin-Subero J.I., Cigudosa J.C., Siebert R., Esteller M. Epigenetic signatures associated with different levels of differentiation potential in human stem cells. PLoS One. 2009;4:e7809. doi: 10.1371/journal.pone.0007809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koch C.M., Andrews R.M., Flicek P., Dillon S.C., Karaoz U., Clelland G.K., Wilcox S., Beare D.M., Fowler J.C., Couttet P., et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 2007;17:691–707. doi: 10.1101/gr.5704207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peters A.H., Kubicek S., Mechtler K., O'Sullivan R.J., Derijck A.A., Perez-Burgos L., Kohlmaier A., Opravil S., Tachibana M., Shinkai Y., et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell. 2003;12:1577–1589. doi: 10.1016/s1097-2765(03)00477-5. [DOI] [PubMed] [Google Scholar]
- 33.Doyen C.M., An W., Angelov D., Bondarenko V., Mietton F., Studitsky V.M., Hamiche A., Roeder R.G., Bouvet P., Dimitrov S. Mechanism of polymerase II transcription repression by the histone variant macroH2A. Mol. Cell. Biol. 2006;26:1156–1164. doi: 10.1128/MCB.26.3.1156-1164.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheuermann M.O., Tajbakhsh J., Kurz A., Saracoglu K., Eils R., Lichter P. Topology of genes and nontranscribed sequences in human interphase nuclei. Exp. Cell Res. 2004;301:266–279. doi: 10.1016/j.yexcr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 35.Banerjee B., Bhattacharya D., Shivashankar G.V. Chromatin structure exhibits spatio-temporal heterogeneity within the cell nucleus. Biophys. J. 2006;91:2297–2303. doi: 10.1529/biophysj.105.079525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toth K.F., Knoch T.A., Wachsmuth M., Frank-Stohr M., Stohr M., Bacher C.P., Muller G., Rippe K. Trichostatin A-induced histone acetylation causes decondensation of interphase chromatin. J. Cell Sci. 2004;117:4277–4287. doi: 10.1242/jcs.01293. [DOI] [PubMed] [Google Scholar]
- 37.Kuroda M., Tanabe H., Yoshida K., Oikawa K., Saito A., Kiyuna T., Mizusawa H., Mukai K. Alteration of chromosome positioning during adipocyte differentiation. J. Cell Sci. 2004;117:5897–5903. doi: 10.1242/jcs.01508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.