

Abstract
Acireductone dioxygenase (ARD) from the methionine salvage pathway (MSP) is a unique enzyme that exhibits dual chemistry determined solely by the identity of the divalent transition metal ion (Fe2+ or Ni2+) in the active site. The Fe2+ containing isozyme catalyzes the on-pathway reaction using substrates 1,2 dihydroxy-3-keto-5-methylthiopent-1-ene (acireductone) and dioxygen to generate formate and the ketoacid precursor of methionine, 2-keto-4-methylthiobutyrate, whereas the Ni2+ containing isozyme catalyzes an off-pathway shunt with the same substrates, generating methylthiopropionate, carbon monoxide and formate. The dual chemistry of ARD was originally discovered in the bacterium Klebsiella oxytoca, but it has recently been shown that mammalian ARD enzymes (mouse and human) are also capable of catalyzing metal-dependent dual chemistry in vitro. This is particularly interesting since carbon monoxide, one of the products of off-pathway reaction, has been identified as an anti-apoptotic molecule in mammals. In addition, several biochemical and genetic studies have indicated an inhibitory role of human ARD in cancer. This comprehensive review describes the biochemical and structural characterization of the ARD family, the proposed experimental and theoretical approaches to establishing mechanisms for the dual chemistry, insights into the mechanism based on comparison with structurally and functionally similar enzymes and the applications of this research to the field of artificial metalloenzymes and synthetic biology.
Graphical Abstract
1. Introduction
Acireductone dioxygenases (ARDs) are a unique family of enzymes that exhibit different chemical and physical properties for the same polypeptide based solely upon the identity of the metal in the active site. If iron (Fe2+) is bound, ARD catalyzes the penultimate step in the methionine salvage pathway (MSP), oxidatively generating the ketoacid precursor of methionine, 2-keto-4-(methylthio)butyric acid. However, if nickel (Ni2+) is bound, an off-pathway product, carbon monoxide, is generated, along with 3-methylthiopropionic acid.1 The off-pathway chemistry is supported by the Co2+ and Mn2+ reconstituted forms, as well. This dual chemistry was first identified for a bacterial (Klebsiella oxytoca originally identified as K. pneumoniae) ARD.2 Recently it was shown that this dual ARD activity is also exhibited by mammalian enzymes (mouse and human ARD isozymes).3–4 In addition to its enzymatic function, several studies have indicated a role of ARD in carcinogenesis and tumor metastasis. ARD is also implicated in hepatitis C infection5, Down’s syndrome (DS)-associated congenital heart defects (DS-CHD)6 and fecundity in Drosophila.7 In plants, ARD expression is associated with development and fruit ripening, and so is of great interest to plant biologists.8 ARD is likely a multi-functional (“moonlighting”) enzyme involved in both regulatory and enzymatic functions. The field of ARD research has seen significant progress over the past 10 years. The goal of this review is to describe this remarkable enzyme family, including the mechanistic and functional aspects of the dual chemistry and the implications of this research on the fields of metalloproteins, disease progression and synthetic biology.
ARDs belong to a structural class known as cupins (from the Latin cupula for “small barrel”), which are formed from several turns of antiparallel β-sheet into a β-helix, a motif also called a “jellyroll”. The cupin motif typically exhibits asymmetry, with one end of the β-helix more “closed” and the other more “open”. The active sites of cupin enzymes are typically found in the open end. Many other non-heme iron-dependent oxygenases, including oxalate decarboxylase and oxalate oxidase, share the cupin motif, as do most ketoglutarate-dependent oxidases (Figure 1).9–12
Figure 1. β-barrel structures in cupins.

a) Oxalate oxidase (PDB ID: 2ET1), b) Clavaminic acid synthase (PDB ID: 1GVG) is an example of a ketoglutarate-dependent oxidase, c) homoprotocatechuate 2,3-dioxygenase (PDB ID: 3OJT) a non-heme iron-dependent dioxygenase, d) Oxalate decarboxylase (PDB ID: 1L3J). The beta sheets are colored red and the helixes are blue.
Biological implications of ARD functions
The observation of off-pathway chemistry with mammalian ARD enzymes is particularly interesting in that carbon monoxide (CO) has both signaling and anti-apoptotic properties in vivo.13 We suspect that ARD gain-of-function as a result of off-pathway chemistry may be a novel source of CO in the cell (currently heme oxygenase is the only confirmed source of biogenetic CO in normal human tissue).14 Also interesting is the observation of ARD gene up- (and down-) regulation in a variety of carcinomas,15 along with the evidence that ARD may regulate matrix metalloproteinase MT1-MMP, which is associated with tumor metastasis.16 A search of the NCBI GEO gene expression profiles for ADI1, the gene that encodes ARD, yields several hundred comparisons showing either up- or down-regulation of this gene in cancer cell lines compared with normal tissue or when treated with a chemotherapy agent. This leads to the intriguing possibility that the off-pathway chemistry catalyzed by the Ni2+, Co2+ or Mn2+-bound enzyme may be relevant in pathology in mammals. The binding of non-ferrous divalent metals in human ARD active site could help protect tumor cells from apoptosis by producing CO. Ni is toxic in mammals, and to date no native Ni-binding metalloenzyme has been identified in mammals.17
Mechanistic considerations
There have been inorganic models18 and computational studies19 of the mechanism of the metal-based functional switching in ARD, but to date there is no conclusive evidence precluding any of the proposed mechanisms. In order to understand how the metal can modulate the function of the enzyme, we have characterized structures of both the bacterial and mammalian ARD enzymes.3, 20–21 Published structures of enzyme-substrate analog and enzyme-product/product analog complexes provided insights into the mechanistic details of the enzyme.3 A comparison of ARD to structurally and functionally similar cupin enzymes provided further insights into potential mechanisms that may help rationalize dioxygen activation that could not be explained by the postulated mechanisms.
Metal sensing in biological systems
To date, only the bacterial ARD isozymes from K. oxytoca have been characterized structurally with Fe and Ni metals bound to the enzyme.20–21 While both structures show the same basic cupin fold, repacking of hydrophobic cores upon metal replacement result in displacement and disruption of helices and a profound change in active site accessibility. Preliminary results on the human ARD enzyme suggest that these same changes are observed in eukaryotic ARDs as well.4 These changes take place despite the fact that the same protein-based ligands are used to bind both Fe2+ and Ni2+ in the bacterial enzyme, and both ions adopt the same pseudo-octahedral geometry in the active site. As such, the ARD isozymes provide a remarkable example of how two metal ions of similar size and identical charge can be distinguished by a folded protein. Understanding how these structural changes are propagated through the protein structure has obvious implications for a vast array of biological machinery that must make such distinctions: these include metallochaperones, metal-dependent response factors and transcription regulators and ion channels, as well as the more obvious examples of metalloenzymes.
Enzyme engineering
In enzymes requiring a metal cofactor for activity, the replacement of the native metal with an abiological metal provides unique opportunities for enzyme engineering. A recent study has reported replacement of iron in Fe-porphyrin proteins with abiological noble metals to create enzymes that catalyze reactions not duplicated in nature.22 Understanding the ARD dual chemistry sets the stage for exploring the role of metals in biocatalysis to create artificial metalloenzymes that perform novel reactions. As we and others have observed, metals can not only modulate the reactivity of a metalloenzyme, but can also expand the scope of reactions catalyzed by native metalloenzymes.
2. The methionine salvage pathway
The methionine salvage pathway (MSP) is ubiquitous in plants, animals and bacteria.23 The MSP recycles the terminal thiomethyl group of methionine, an essential amino acid, and regulates the concentration of several metabolic intermediates in the pathway. In the first step of the MSP, S-adenosylmethionine (SAM) is formed enzymatically from methionine and ATP (Figure 2). SAM is a critical source of electrophilic methyl and ethylene groups in biosynthetic processes, and is involved in the synthesis of polyamines, spermine and spermidine in animals and ethylene in plants. Polyamines are required for cell growth and proliferation24 while ethylene is required for ripening of fruits and vegetables.25 Defects in polyamine regulation are associated with defects in cell cycle, cell growth and DNA replication. Inhibition of polyamine synthesis arrests DNA replication,26 and their synthesis is downregulated in senescent and mature tissue.27 Elevated polyamine levels are associated with suppression of apoptosis and tumor formation.28–29 Neoplastic cells from colorectal cancers are found to have higher levels of polyamines than normal tissue.30 Methylthioadenosine (MTA) is formed as a byproduct from SAM during polyamine synthesis in animals and ethylene synthesis in plants, and is the first committed intermediate in the MSP.24, 31 MTA is an inhibitor of both polyamine synthesis and transmethylation reactions,24, 32 so intracellular levels of MTA are tightly regulated. The MSP is critical in maintaining MTA homeostasis by returning it through a series of reactions to methionine, thereby “salvaging” the thiomethyl group of the original methionine.33 As such, this pathway is sometimes referred to as the MTA cycle. ARD catalyzes the penultimate step in the MSP, the oxidative decomposition of substrate acireductone (1, 2-dihydoxy-3-keto-5-(thiomethyl) pent-1-ene) to formate and 2-keto-4-(thiomethyl) butyrate (KMTB), the keto-acid precursor of methionine. The ARD substrate acireductone is formed by the action of E1 enolase phosphatase, a member of the haloacid dehalogenase superfamily, on 1-phosphonooxy-2, 2-dihydroxy-3-oxo-5-methylthiopentane (E1 substrate).
Figure 2. The Methionine Salvage Pathway in Klebsiella oxytoca.

Reproduced with permission from Ref. 3. Copyright 2016 American Chemical Society.
3. ARD from Klebsiella oxytoca
The first ARD to be identified and structurally characterized, KoARD, was isolated from the bacterium Klebsiella oxytoca. The enzyme (referred to in the early publications as “E2”) was identified by the Abeles group in the course of mapping the MSP in that organism. It was noted that 14C labeling of methionine resulted in the formation of radiolabeled methylthiopropionate (MTP), suggesting that a previously unknown shunt reaction from the MSP was occurring.34–36 That group later identified both the on- and off-pathway activities as the result of a single polypeptide with different physical and catalytic properties depending upon the metal bound to the enzyme, referred to by those workers as “one protein, two enzymes”.1 They found that this enzyme exhibits different activity depending on the identity of the metal ion cofactor. KoARD with Fe2+-bound (“E2’”) was found to catalyze on-pathway MSP chemistry leading to production of formate and the keto-acid precursor to methionine, whereas Ni2+-bound KoARD (E2) catalyzed an off-pathway shunt leading to production of carbon monoxide, formate, and 3-(methylthio) propionic acid (MTP).1–2 Both the Fe2+ and Ni2+-bound forms of the protein were isolated from Klebsiella,35 and later also obtained upon overexpression in E. coli. Both forms of the enzymes are monomers and co-elute during size exclusion chromatography, but can be separated by ion exchange or hydrophobic interaction chromatography, suggesting that the differences between the two isoenzymes were structural rather than due to differences in oligomerization state. KoARD shows tighter binding to Ni2+ compared to Fe2+ since Fe can be removed from the folded enzyme by dialysis with EDTA, while Ni requires denaturation before EDTA dialysis for removal.37 Addition of equimolar amounts of Fe2+ and Ni2+ to the apoenzyme produced > 80% of Ni-containing form within a minute. The activities of the two enzymes could be interconverted by exchanging Fe2+ and Ni2+.37 The enzyme was also found to be promiscuous, with Co2+ and Mn2+ give Ni-like activity and Mg2+ yielding a low level of Fe-like activity (although the possibility of trace Fe2+ contamination of Mg2+ cannot be discounted).37 The Ni2+ and Fe2+-bound KoARDs represented the only known pair of naturally occurring metalloenzymes with distinct chemical and physical properties determined solely by the identity of the metal ion in the active site. The purpose of the off-pathway reaction catalyzed by Ni2+-bound ARD in K. oxytoca is still unknown. The prevalence of Ni2+-bound ARD in native K. oxytoca suggests that it may be physiologically necessary and may be involved in the regulation of the MSP, or that CO production by Klebsiella has a symbiotic role in wild ecosystems. 38–40 CO is a substrate for carbon monoxide dehydrogenase, which catalyzes the reversible oxidation of CO to CO2, which could allow symbiotes (or Klebsiella) to use carbon monoxide as a source of reducing equivalents.41–43 CO is also known to be a signaling molecule for a transcriptional regulator, CooA, in bacteria.44 CO production from methylthioribose has also been reported for Bacillus subtilis, indicating the presence of a stable off-MSP shunt.45
A detailed analysis of enzyme kinetics found no evidence for a stable M+2-O2 complex in either the Fe or Ni isoforms of KoARD.37 The same paper described the use of model substrates to probe mechanism, with a convenient synthetic route described separately (Scheme 1).46 Spectroscopic evidence suggested that the initial enzyme-substrate complex was a chelate between the metal and the dianion 5e. Based upon slow inactivation of the enzyme by suicide substrate 5b it was proposed that the reaction proceeds by a one-electron transfer from substrate to O2, generating the peroxide radical species 6 (Scheme 2).
Scheme 1.

Synthetic route to ARD substrates (Adapted from Ref. 46)
Scheme 2.

Model reactions showing feasibility of peroxy intermediates in Ni KoARD activity (Adapted from Ref. 46).
The feasibility of a peroxo intermediate in Ni-bound KoARD was tested by reaction of hydrogen peroxide with several model compounds. The symmetric β-diketone 6 was shown to form the cyclic peroxide 7, and the bis-hydroxy compound 8 decomposed upon treatment with hydrogen peroxide to form two equivalents of acetic acid along with a molecule of CO (Scheme 3).44
Scheme 3.

Proposed mechanism for inactivation of ARD via a radical intermediate generated by 1-electron transfer from substrate analog 5b (Adapted from Ref. 46).
4. Structures of KoARD isoforms and structural differences arising from differential metal binding
The first ARD structures to be determined were those of the Ni-KoARD (PDB ID: 1ZRR) and Fe-KoARD (PDB ID: 2HJI), both by solution NMR spectroscopy.20–21, 47 ARD belongs to the cupin superfamily, which is characterized by a conserved β-barrel (the “cupin barrel”) consisting of anti-parallel β-sheets that gives rise to a classic β-helix “jelly roll” motif. Due to the paramagnetism of the metal ion, the active site of the Ni2+-bound KoARD could not be characterized directly by NMR, and so was modeled using the structure of another cupin, jack bean canavalin, with the model supported experimentally by X-ray absorption spectroscopy (XAS).21 The structure of the Fe2+-bound KoARD is a model based on the structure of a stable soluble metal-free mutant of ARD, H98S, which is isostructural with Fe-KoARD as determined by a comparison of 1D 1H NMR, NOESY and 2D 1H, 15N heteronuclear single quantum coherence (HSQC) spectra.20
Remarkably, it was observed that the same four protein residues were involved in ligation, regardless of the identity of the metal ion bound. These protein-derived ligands include three histidine (His) residues, His96, His98 and His140 and one glutamate (Glu102), all of which were later found to be strictly conserved in other ARD homologues. This was surprising, in that it might be expected that the very different chromatographic and enzymatic behaviors of the Fe- and Ni-bound forms of ARD, both monomers, could result from different ligands or a change in the number of ligands used for different metals. However, mutation of any of the conserved ligand residues in KoARD resulted in inactive enzymes that did not bind any metal. Furthermore, the XAS data for both the Fe- and Ni-bound KoARD isozymes supported similar distorted octahedral geometries for both metal ligation spheres.48
Given that the Ni2+- and Fe2+-bound forms of bacterial ARD make use of the same ligand set, the reasons for the chromatographic separability and different thermal stabilities of the isozymes must be sought elsewhere. As can be seen from Figure 3, the most readily observed structural difference between the two forms is the C-terminal 310 helix (Q in Figure 3) forming a cap on top of the β-barrel in the Ni-bound enzyme, whereas the same region of the polypeptide is disordered in the Fe-bound form, which is missing secondary structural features O-Q. O is a single turn of helix, and P is a short additional strand edge to the β-barrel. The loss of these features renders the Fe-bound KoARD active site significantly more accessible and open to solvent than in the Ni form. Also, the E,F helices on the opposite face of the barrel pivot by ~20° between the Fe- and Ni-bound ARD isoforms. This results in a significant repacking of a secondary hydrophobic core on that face of the barrel, as well as a change in the order of a loop of polypeptide D at the N-terminal end of the E helix system. In the Ni2+-bound enzyme, this loop is largely disordered, while in the Fe2+-bound form, it becomes more structured. We refer structural entropy switch: In the Ni-bound ARD, the C-terminus is ordered, while the D loop is disordered. In the Fe form, the opposite situation is observed.20 The origins of these conformational shifts are not obvious, but we have hypothesized how they might be driven. While structural entropy switch: In the Ni-bound ARD, the C-terminus is ordered, while the D loop is disordered. In the Fe form, the opposite situation is observed.20 The origins of these conformational shifts are not obvious, but we have hypothesized how they might be driven. While to this phenomenon (increased order in one location with decreasing order at another) as a 0251681792 both Fe2+ and Ni2+ are nearly identical in size (r = ~70 pm), the XAS data indicate that the Ni-N/O bond lengths in ARD are on average ~0.1 Å (10 pm) shorter than the corresponding bonds to Fe. While this is not a large change, the fact that three of the four protein-based ligands (His96 on the edge strand following helix H, Glu102 on strand I and His140 on strand M) originate within the β-barrel, and are thus restrained from changing position as a result of the shorter bond lengths in NiARD, the bond shortening may be amplified by pulling the Ni deeper into the barrel mouth, dragging the fourth ligand (His98) closer to the barrel opening than it is in the Fe2+-bound form, and stabilizing the loop containing His98 as an edge of the β-barrel. This in turn results in the formation of the short antiparallel β-strand P between the C-terminal loop and the new barrel edge. As a result, the active site of the Ni2+-bound form is considerably less accessible than that of the Fe-bound enzyme, as shown in Figure 4. Note that Trp162, which is disordered in the Fe enzyme, is ordered and close to the metal center in the Ni form. Phe92 is also more distant from the metal center in the Fe-KoARD than in the Ni isozyme. This rationalizes the observation that Fe2+ can be removed from the active site relatively easily with chelating agents, while the Ni2+ form must first be denatured to remove the metal, and furthermore explains the significant increase in thermostability observed upon Ni+2 binding in all ARD enzymes characterized to date.3–4 This hypothesis, while not fully tested, also provides some insight into how metal selectivity might be achieved in other metal binding proteins, including transporters and regulatory transcription activators and repressors, with only standard amino acid-based ligands.
Figure 3. Structures of Fe- and Ni-bound KoARD.

Secondary structural features are labeled corresponding to primary sequence: A: Ala2-Phe6, B: Ser14-Ser18, C: Ala21-Lys31, D: Val33-Thr49, E: Ala50-Ala60, F: Ile61-Gly69, G: Ser72-Ile76, H: Pro83-Asn94, I: Glu102-Glu108, J: Leu112-His116, K: Val121-Leu125, L: Leu131-Val134, M: His140-Met144, N: Thr151-Phe156, O: Asn158-Gly161, P: Thr162-Ile163, Q: Ile171-Tyr175.
Figure 4. Active sites of Ni- and Fe-bound KoARD.

Close-up views of the actives sites of Ni-KoARD (1ZRR, top) and Fe-KoARD (2HJI, bottom). Secondary structures O and P in the Ni enzyme are disordered in the Fe enzyme and are not shown in the bottom panel. The Ni view is scaled slightly smaller than the Fe view so that relevant features can be displayed.
5. ARD homologs from plants and Drosophila
ARD has now been characterized from several eukaryotes. The MSP from plants has been studied extensively, as MTA is a byproduct of ethylene biosynthesis. Ethylene is a hormone involved in fruit and vegetable ripening. The OsARD1 gene from rice (Oryza sativa) was identified as a primary ethylene response gene.8 The expression of this gene is strongly up-regulated at low ethylene levels as an early response to ethylene production under hypoxic conditions resulting from submergence. Recombinant Fe2+ bound-OsARD1 protein is a trimer, and performs on-pathway MSP chemistry. The Ni+2-reconstituted OsARD1 exhibits off-pathway chemistry albeit with much lower activity than Fe-OsARD1, and polymerizes beyond the trimeric state. For these reasons, Ni-OsARD1 is deemed unlikely to represent a physiologically relevant form of ARD in rice. A second gene OsARD2 was also identified which was constitutively expressed and unlike OsARD1, its expression was not induced by deep water submergence. OsARD2 has an 85% identity and 93% sequence similarity to OsARD1.
ARD from Arabidopsis thaliana (AtARD1) was identified as an effector of Gβ (AGB1) which is a subunit of the heterotrimeric G protein complex. The activation of G protein complex promotes interactions with other proteins during cell signaling and communication to the cytoplasm. Arabidopsis plants lacking the AGB1 mRNA transcript have several abnormal developmental phenotypes, including aberrant leaf shape, increased root mass, silique morphology, and hypersensitivity to infection.49–51 AtARD1 expression suppresses Gβ-null mutant phenotypes and controls embryonic hypocotyl length by modulating cell division.52 As with other ARD homologues, recombinant Fe2+-bound AtARD1 catalyzes on-pathway MSP chemistry. Enzymatic studies of AtARD1 in the presence and absence of AGB1 showed that ARD enzymatic activity is stimulated by AGB1 in vitro. In addition to AtARD1, the A. thaliana genome encodes AtARD2, AtARD3 and AtARD4. The roles of these other gene products are unknown at this time, nor is there any information regarding the conditions under which they are induced (or even if they are induced at all).52 However, comparison of the results for rice and A. thaliana suggests that many plants have more than one ARD gene.
A role of Salvia miltiorrhiza (red sage) acireductone dioxygenase (SmARD) gene in the defense response under different abiotic stressors like drought, cold and salt has been found.53 Similarly, wheat (Triticum aestivum) ARD (TaARD)54 and potato (Solanum tuberosum) ARD (StARD)55 have been shown to be involved in the defense response against stressors such as wounding. ARD is likely involved in ethylene synthesis and ethylene signaling in response to biotic and abiotic stresses in these plants as well.
The presence of a functional ARD gene product has been shown to be required for normal fecundity in Drosophila.7 In dietary restriction conditions, the egg production of acireductone dioxygenase 1 (ΔADI1) mutant flies was reduced compared to that of control flies. This fecundity defect in mutant flies was rescued by either methionine supply or reintroduction of ADI1 gene. A functional homolog of human ADI1 was also able to rescue the fecundity defect.
6. Mammalian ARD homologs and “moonlighting” functions of ARD: Links to carcinogenesis, congenital defects and viral susceptibility
Mammalian ARDs from mouse (MmARD) and human (HsARD) have now been investigated both biochemically and structurally in order to relate metal ion identity and three-dimensional structure to enzyme function.3–4 Both MmARD and HsARD have been shown to perform the same metal-dependent dual chemistry in vitro as observed with KoARD.1, 3–4 Unlike the bacterial forms, where both isoforms displayed similar turnover rates, the Fe2+-bound forms of the mammalian enzymes showed several-fold higher activity than the Ni2+, Co2+ or Mn2+ forms. However, like KoARD, the Fe2+-bound forms catalyze on-pathway chemistry, while the Ni2+, Co2+ and Mn2+ forms catalyze off-pathway chemistry. Also, as with KoARD, the thermal stability of these mammalian enzymes is a function of the metal ion identity, with Ni2+-bound enzyme being the most stable, followed by Co2+ and Fe2+, and Mn2+-bound enzyme being the least stable.3–4
The human ARD homologue HsARD has been shown to serve regulatory functions in addition to its enzymatic function in the MSP. Seiki et al. showed that HsARD binds to the cytoplasmic tail of membrane-type 1 matrix metalloproteinase (MT1-MMP) and inhibits MT1-MMP-mediated cellular invasiveness.16 A recent study by Pratt et al. has indicated a role of HsARD in regulating the intracellular function of MT1-MMP-mediated autophagy in brain tumors.56 In this study, an inverse correlation was observed between the expression levels of ARD and MT1-MMP in clinically validated grade I to grade IV brain tumor tissues. The functional impact of ARD on MT1-MMP mediated autophagy signaling was further tested in glioblastoma cells. Using FRET microscopy and surface plasmon resonance it was shown that the cytoplasmic domain of MT1-MMP and ARD interacted with a dissociation coefficient of 12 μM.56 Oram et al. have shown that ADI1 encoding ARD (GenBank ID: 55256, ADI1, 5 exons on chromosome 2, NC_000002.12, 3497919.3519579, complement) is down-regulated in rat prostate and human prostate cancer cell lines and its enforced expression induces apoptosis.15 These researchers have also shown that cultured gastric carcinoma cells and fibrosarcoma cells have downregulated ADI1.15, 57 A recent clinical study testing the impact of environmental carcinogens on gene expression differences in humans found ADI1 as a gene having a direct link to cancer development.58 An N-terminal truncated variant of HsARD called SipL was shown to be implicated in the replication of hepatitis C virus in non-permissive cell lines. At the time of publication of that work, the enzymatic function of the protein was unclear, and so it was identified as SipL (submergence-induced protein like) due to sequence homology with what was later identified as OsARD. This truncated version of HsARD has the first 63 amino acids missing.5 Based on structural homology with KoARD, this deletes the N-terminal B helix and the edge strands A, C and D from the narrow end of the cupin barrel (Figure 3), but leaves the metal binding site and the major part of the barrel intact. On the other hand, MmARD and HsARD share an 86% sequence identity, and assuming that the SipL variant folds similar to the full-length MmARD, the entire the β-barrel is present in the SipL variant. It is unknown if SipL is a metal-binding protein or if it exhibits enzymatic activity.
A recent clinical study related the two known functions of HsARD. This study showed that the physical interaction between MT1-MMP and ADI1 led to suppression of HCV infection and this inhibitory effect could be reversed by ADI1 overexpression.59 A recent study to identify the pathways disrupted in Down’s syndrome (DS) and DS-associated congenital heart defects (DS-CHD) has indicated that ADI1 has higher expression in fetuses trisomic for Hsa21 than in normal fetuses. Based on this finding, the authors predicted that the methionine salvage pathway is significantly altered and could function as an indicator of DS-CHD.56
6.1. Mammalian ARD structures
The first crystallographic structure determined for an ARD was that of the mouse homolog MmARD (PDB ID: 1VR3), published by the Joint Center for Structural Genomics (JCSG). However, the identity of the metal ion cofactor in that structure was not determined.60 Recently, low resolution structures of ARD from Bacillus anthracis (2.97 Å) (PDB ID: 4QGM) and HsARD (3.05 Å) (PDB ID: 4QGN) have been deposited (Figure 5). The complete details of these structures have not been published and it is unknown if the proteins used in these structures were enzymatically active, nor have the identities of the metals in the active sites been confirmed.61–62 In the B. anthracis ARD (BaARD) structure, the bound metal is proposed to be Cd2+, while in the HsARD structure, the bound metal ion is proposed to be Fe3+. This is somewhat surprising, in that in our hands, oxidation results in the loss of metal and inactivation of the enzyme. Nevertheless, in all of these structures, the strict conservation of the coordinating ligands originating from the protein was confirmed (Figure 8).
Figure 5. Crystal structures of ARD enzymes from B. anthracis, H. sapiens and M. musculus.

Coordinated metal ions are shown as red spheres. a) BaARD (PDB ID: 4QGM), b) HsARD (PDB ID: 4QGN), c) MmARD (PDB ID: 1VR3)
Figure 8. Sequence alignment of ARD homologs using ClustalOmega.

The metal binding residues His88, His90, Glu94 and His133 are colored blue, the hydrophobic residues Phe84, Phe105, Phe135 and Ala145 found to be interacting with KMTB are colored red. Arg96 and Arg147 are colored green. Phe149 and Trp155 may be involved in conformational gating are shown in magenta. (All residue numbers refer to the MmARD sequence). The gap in the eukaryotic sequences (corresponding to the sequence APTAETVIAA in Klebsiella is due to a shortened flexible loop D between strand C and helix E (Fig. 3).
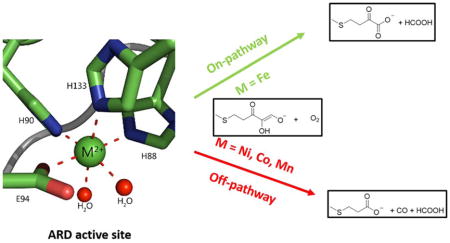
We have recently described the crystallographic structures of Ni2+-bound (PDB ID: 5I91) and Co2+-bound MmARD (PDB ID: 5I8Y) (Figure 6) and found the structures of the two proteins to be similar (RMSD=0.06Å).3 Both Ni and Co exhibit octahedral co-ordination geometry in the ARD active site, using the Nε atoms of imidazole protein ligands His88, His90, His133 and monodentate coordination by the carboxylate of Glu94 (Figure 6). In addition to the protein ligands, there are two distinct water (or hydroxide) ligands bound to the metal ion center, whereas the previously reported structure of MmARD showed undefined electron density in the corresponding regions. This is consistent with the NMR solution structure of Ni2+-bound KoARD in which two water molecules were modeled as ligands to fit EXAFS data in addition to the protein-based ligands.21
Figure 6. X-ray crystal structure of Ni-MmARD (PDB ID: 5I91).

a) The crystallographic structure of Ni-MmARD. The Nickel atom is shown as a green sphere and its protein ligands His88, His90, His133 and Glu94 represented as sticks and two water molecules are shown as red dots. b) Stereo view of nickel binding in the MmARD active site. Glu94 and His88 provide the axial ligands, with His90, His133 and two water/hydroxide ligands occupying equatorial positions, yielding an octahedral coordination geometry. The metal-ligand distances (in Å) are show as red dotted lines. (Reproduced with permission from Ref 3. Copyright 2016 American Chemical Society).
Ni-MmARD was crystallized with on-pathway product 2-keto-4-(methylthio) butyric acid (KMTB) (PDB ID: 5I91) (Figure 7) and off-pathway product analog valeric acid (VA) (PDB ID: 5I8S). Both KMTB and VA are found in the active site. Neither compound ligates the metal, but both are within hydrogen bonding distance of the two water/hydroxide ligands bound to the metal as well as the guanidinium group of Arg96, which is strictly conserved in all known ARD sequences. Both ligands have non-polar contacts with Phe135, Phe105, Phe84 and Ala145 (all of which are also strictly conserved as well as with Val143 and Ile98 (Figure 8). Obviously, a substrate-bound ARD structure would be desirable for mechanistic insights. However, acireductone is highly sensitive to oxidation, and to date this has not been accomplished. We attempted to incorporate substrate into the ARD structure, by generating desthio-acireductone in situ in an anaerobic cuvette, and adding this to a drop containing a Ni-MmARD crystal under anaerobic conditions. While the crystals diffracted and a structure was determined, no ligand was detected in the active site.3
Figure 7. The active site of Ni-MmARD showing substrate-analog and product binding.


The Ni atom is shown as a green sphere, the ligands and active site residues are shown as sticks and waters/hydroxides are shown as small red spheres. Hydrogen bonding distances are shown (in Å) as red dotted lines. Hydrophobic residues in the active site are shown in yellow. a) Stereo view of KMTB bound to Ni-MmARD. KMTB is within hydrogen bonding distance of Arg96 and the two water molecules ligated to Ni. The residues Phe84, Phe105, Phe135, Ala145, Val143 and Ile98 interact with the alkyl group of KMTB. b) Stereo view of D-Lactic acid (D-LA) bound to Ni-MmARD. D-LA replaces both equatorial water ligands, coordinating in a bidentate manner with Ni2+ via the carboxylate and hydroxyl oxygens. D-LA is within hydrogen bonding distance of Arg96. Residues Phe84, Phe105 and Phe135 interact with the alkyl group of D-LA. (Adapted from Ref 3. Copyright 2016 American Chemical Society).
We were able to obtain a structure of substrate analog D-lactic acid (D-LA) bound to Ni-MmARD solved to a resolution of 1.7 Å (PDB ID: 5I8T). D-LA was present in the active site and, unlike the product KMTB and product analog VA, D-LA was seen to coordinate directly to the metal (Figure 7). The two water molecules bound to the metal ion in the KMTB-bound or VA-bound protein structures were replaced by one D-LA carboxylate oxygen and the D-LA hydroxyl oxygen, forming a five-membered ring equatorial chelate complex with the Ni2+. The carboxylate oxygen of D-LA not ligating the metal is within hydrogen bonding distance of Nω atom of Arg96.3 D-LA has a similar arrangement of oxygen atoms as acireductone, but lacks the hydrophobic (methylthio) ethylene moiety of the native substrate. The recently deposited structure of Fe3+-HsARD (4QGN) has L-selenomethionine in the active site and it is coordinated to the metal ion in a manner similar to that of D-LA.62
6.2 Comparison of NMR and crystallographic approaches to ARD structures
The fact that NMR was used to determine the original KoARD structures reflected the difficulties encountered in crystallizing KoARD rather than a preference for any particular methodology. Still, it is fortunate that the Fe2+- and Ni2+-bound KoARD isoforms are readily distinguishable spectroscopically: Their peripheral structures are quite different, particularly in the C-terminal regions, D loops and E, F helices of the two structures (see Figure 4). These differences have allowed us to make some structure-based hypotheses as to the origins of both the physical and functional differences between the two isoforms. On the other hand, the NMR structures suffer from a lack of clarity regarding the precise structures around the (paramagnetic) metals in both forms of KoARD. While we have described NMR methods for making sequential resonance assignments in these regions, 20–21, 47 standard NMR structural restraints (nuclear Overhauser effects, residual dipolar couplings and chemical shifts) are still lacking. For detailed structural investigations of metal binding geometry and arrangements of side chains in the active sites of ARD enzymes, crystallographic structures are essential. Because both of the recently published structures of MmARD with defined metal occupancies are of enzymes that exhibit off-pathway activity (Co2+ and Ni2+), there is still little clarity regarding any structural differences between the Fe2+ and Ni2+ forms of the mammalian enzymes and how these relate to on- versus off-pathway activity in the MSP. Recently we have shown using solution NMR spectra of 15N-labelled Fe2+, Ni2+ and Co2+ -bound HsARD isozymes that, like KoARD, the different metals impart significant structural differences to the overall protein structure.4 Preliminary analysis of multidimensional NMR spectra of Fe-HsARD indicates that, like Fe-KoARD, the C-terminal polypeptide is largely disordered.4
7. Proposed enzymatic mechanisms for ARD
Enzyme kinetic studies of both Fe2+ and Ni2+-bound forms of KoARD indicate a sequential mechanism where both acireductone and oxygen must bind to the enzyme prior to product release.37 Spectroscopic analysis indicates that oxygen does not bind ARD in the absence of substrate. Acireductone first binds to the enzyme as a di-anion followed by oxygen binding to form a ternary complex.37 Since the acireductone slowly reacts with oxygen non-enzymatically to give Fe-KoARD like products, it is possible that the redox nature of metal and metal-oxygen activation may not be important. Neither Ni-KoARD nor Fe-KoARD exhibit an EPR signal under aerobic or anaerobic conditions, nor in the presence or absence of the substrate acireductone. Both Ni2+ and Fe2+ are expected to have integer spins (S=1 and S=2, respectively), and hence EPR signals are difficult to observe due to the forbidden nature of the transitions. Still, the lack of any O2 effect on either EPR or NMR spectra suggest that if redox activity is required at all, it may be transitory.37 Since acireductone binds the metal as a dianion, the metal likely activates the substrate by acting as a Lewis acid. The following sections discuss various approaches to understanding the mechanism(s) of ARD activity.
7.1 Chelate Hypothesis
The chelate hypothesis postulates that different ARD chemistries arise from differences in the nature of the cyclic peroxide formed at the metal center. A five-membered ring involving coordination of C-1 and C-2 oxygen atoms of acireductone would form in the case of Fe-KoARD, while a six-membered ring involving C-1 and C-3 oxygen atoms is formed in the case of Ni-KoARD (Scheme 4). The NMR structures reveal a difference in the active site tertiary structure with Ni-KoARD featuring a closed form and Fe-KoARD showing an open form of the structure.20 Docking studies show that the substrate acireductone could coordinate via a five-membered chelate in Fe-KoARD, but in the more congested Ni-ARD active site, it would bind via a less sterically demanding six-membered chelate ring.20 These binding modes would activate acireductone at the C1 and C2 positions in Fe-KoARD but at the C1 and C3 positions in Ni-KoARD, leading to on-pathway and off-pathway reactions, respectively. The chelate hypothesis is compatible with 18O and 14C incorporation in the products as determined using 18O2 and 14C-labelled acireductone.20–21, 37
Scheme 4. Proposed mechanisms of Ni-ARD and Fe-ARD using the chelate hypothesis.

The results of incorporation of 18O and 14C labeling-studies are indicated by the red O atoms and the asterisk. Reproduced with permission from Ref 3. Copyright 2016 American Chemical Society.
7.2 Small molecule models of ARD active sites
Berreau et al. have synthesized small molecule models of the metal centers of ARD in order to investigate possible mechanisms for ARD dual chemistry. They challenged the chelate hypothesis, showing that both Ni and Fe can form six-membered ring complexes with substrate homologues and give Ni-like products in anhydrous solvents. The on-pathway chemistry is observed with the Fe-center only in the presence of water. They attributed this to the hydration of the tri-ketone reaction intermediate formed in the first step of the reaction.19–20, 63 The working hypothesis is that the hydration of the triketone intermediate is mediated by Fe, but not by Ni. The crystal structures of the solvate complexes are pentacoordinate when M = Fe, with a single solvent molecule bound, but hexacoordinate with two solvent molecules when M = Ni (Schemes 5 and 6). This differing coordination preferences of Fe and Ni may be responsible for the differences in Lewis acid activation of the triketone intermediate. These results are compatible with the fact that the Ni-KoARD active site is less accessible to solvent than the active site in Fe-KoARD.64
7.3 Computational studies
Sparta et al. examined the mechanisms of ARD used mixed quantum-classical molecular dynamics simulations coupled with density functional theory. Their calculations show that, if the conserved Arg is included in the QM region of the simulations, a six-membered chelate of the metal by the substrate has the lowest energy in the cases of both Ni2+ and Fe2+. As a result of these calculations, the differences in the chemistry are attributed to the redox-active nature of Fe2+ relative to Ni2+, allowing Fe-ARD to form an intermediate partially stabilized by charge transfer to Fe2+, whereas Ni simply acts as a Lewis acid (Scheme 7).19 While these calculations do not account for the non-enzymatic and Mg2+-ARD catalyzed oxidative decomposition of acireductone, which give Fe-ARD like-products, there has not been a systematic investigation of either of those reactions: In particular, it is not known whether trace metals (in particular, iron) are responsible for the observed reactivity in either case. As such, the results of the computational work cannot be disregarded.
Scheme 7. Proposed mechanism of Ni-KoARD and Fe-KoARD using computational studies.

(Adapted from Ref. 19)
7.4 Mechanistic implications from crystallographic results
The crystallographic structures of Ni-MmARD and Co-MmARD provide the first detailed structural information regarding the mode of product binding and the possible modes of substrate binding in the active site of Ni2+ or Co2+ -bound MmARD. As noted above, D-lactic acid (D-LA) was found to chelate the metal centers so as to form a five-membered ring, as proposed in the chelate hypothesis, demonstrating that such structures are stable. Of course, the five-membered ring is an intermediate in the on-pathway reaction according to the chelation mechanism, rather than the off-pathway reaction catalyzed by the Ni or Co-bound forms in which this was observed. Still, the fact that the five-membered chelate is observed suggests that, for now, none of the proposed mechanisms can be eliminated from consideration.
Other mechanistic insights can be gleaned from the crystallographic structures. For example, Arg96 is a strongly conserved (Figure 8) active site residue and is within hydrogen bonding distance of both substrate analog and product. (All residue numbers refer to the MmARD sequence). It has been suggested that this conserved Arg serves as a general base for deprotonation of substrate upon binding.3 While the guanidine functionality is usually too basic to serve as a general base in enzymatic reactions (pKa ~ 12.5), the side chain of Arg147, also strongly conserved, is parallel to the side-chain of Arg 96, and so may modulate the of pKa of Arg96 sufficiently so that the residue can act as a general base at physiological pH.17 This parallel arrangement is also seen in the KoARD active site (Arg104 and Arg154, in the KoARD sequence). Other conserved residues in the active sites of ARD orthologs include Phe135, Phe105, Phe84 and Ala145. It is likely that these residues are involved in correct substrate orientation. Previous studies on KoARD have shown that the conserved tryptophan residue Trp162 (KoARD sequence) is ordered and adjacent to the active site entrance in Ni-KoARD, but disordered in the Fe-bound form. It was proposed that the side chain indole of Trp162 drives a preference for the binding mode leading to off-pathway products in Ni-KoARD, and its absence in the Fe form allows for a more sterically demanding mode leading to the on-pathway products.20 A mutant of the analogous Trp residue in Arabidopsis homolog of ARD (AtARD1) (Trp166) was tested for enzymatic activity in the Fe-bound form and it was found that this mutant (W166A) was a more efficient enzyme than the wild-type AtARD1 enzyme, suggesting that the Trp may serve a gating function, reducing on-pathway activity when it occludes the active site.52 In the MmARD structure, this residue (Trp155) also resides in the loop region preceding the long C-terminal helix, and may be involved in conformational changes upon substrate binding (Figure 9). Phe149, also a conserved residue in this loop region may be involved in assisting Trp155 in conformational gating.
Figure 9. Surface representation of MmARD showing residues likely involved in conformational gating.

KMTB, Phe149 and Trp155 are shown in stick representation, Ni is shown as a green sphere and the water molecules shown as red spheres. Trp155 resides in the loop region preceding the long C-terminal helix, and may be involved in conformational changes upon substrate binding. Phe149 may be involved in the assisting Trp155 in conformational gating. a) Overall structure, b) Overall structure showing the C-terminal helix and the loop preceding it in a cartoon representation, c) Active site structure showing Trp155, d) Active site structure showing Trp155 & Phe149.
8. Enzymes structurally and functionally similar to ARD
The ARD enzymes are similar to other cupin superfamily members, having a characteristic β-barrel structure where the active site of the protein is located. The His3-Glu1 metal binding motif found in ARD is also seen in other cupin enzymes, including in oxalate oxidase, oxalate decarboxylase and quercetinase. Scheme 8 shows the reactions catalyzed by these enzymes.
Scheme 8. Reactions catalyzed by oxalate decarboxylase (OxDC), oxalate oxidase (OxOx) and quercetinase (QueD).

As can be seen, of the three enzymes in Scheme 8, only QueD is a dioxygenase, while OxDC catalyzes the disproportionation of oxalic acid under acidic conditions to yield formic acid and CO2, OxOx catalyzes the two-electron reduction of O2 to hydrogen peroxide with concomitant oxidation of oxalate to two equivalents of CO2. Intriguingly, QueD oxidation of the flavinoid quercetin results on the formation of CO, much like the off-pathway chemistry of ARD. However, only the QueD from Streptomyces appears to use Ni2+ as a co-factor.65–66 OxDC and OxOx are both bicupins, containing one Mn2+ per cupin subunit. As with ARD, the metal is present in an octahedral geometry with the two open sites occupied by non-protein ligands.10–11 OxOx from B. subtilis has been studied extensively. The active site of the enzyme contains Mn2+ bound in a His3-Glu1 ligation scheme essentially identical to that observed in ARD, with two water molecules completing the octahedral coordination sphere. A structure of a substrate analog (glycolate) bound to OxOx combined with EPR data were used to provide insights into the mechanism of OxOx catalysis.67 The reaction cycle begins with monodentate binding of oxalate via displacement of one water molecule from the Mn2+ center. The substrate binding allows for O2 binding to the metal by weakening the interaction of the metal ion with the second water ligand. The bound O2 accepts an electron from Mn2+ to form a Mn3+ superoxide metallo-radical. The reaction then proceeds as shown in Scheme 9. In the proposed mechanism, the metal not only binds both substrates (oxalate and O2), organizing them for reaction, but also transiently reduces dioxygen to superoxide. It is important to note that this mode of glycolate binding observed in the protein crystal is distinct from that observed in small molecule inorganic complexes of Mn2+ and glycolate. In the small molecule model studies, glycolate exhibits bidentate coordination.68 The protein complex may exhibit the extended, monodentate coordination due to steric barriers disfavoring bidentate coordination, with specific stabilization of monodentate ligation through steric interactions with active site residues. A similar mode of substrate binding, where oxalate binds in an end-on conformation providing an empty coordination site for dioxygen ligand was observed in oxalate decarboxylase.69
Scheme 9. Proposed catalytic mechanism of oxalate oxidase.

The protein ligands, His88, His90, His135, and Glu95 for Mn2+ are not shown. (Figure adapted from Ref. 67)
8.1. Enzymes related to Ni-ARD
Quercetinase (QueD) is a flavonol 2, 4-dioxygenase involved in the oxidative degradation of quercetin, a common constituent of foodstuffs such as berries, tomatoes and black tea. It is a CO-releasing dioxygenase and is probably the closest enzyme to Ni-ARD in terms of the chemistry it performs (Scheme 8). While fungal quercetinases are exclusively copper dependent enzymes, 70–74 bacterial quercetinases from B. subtilis and Streptomyces were reported to be promiscuous enzymes similar to KoARD. In B. subtilis QueD, Co2+, Cu2+, and Mn2+ can support catalysis but the catalytic efficiency is the highest for Mn2+.75–76 Streptomyces QueD was found to be active with Ni2+, Co2+ and Mn2+ but in contrast to B. subtilis QueD, was most active with Ni2+.77 Fe2+ was shown to be a poor cofactor in both the bacterial species. Although these bacterial QueDs are promiscuous, no changes in the enzymatic chemistry based on the identity metal cofactor as seen in ARD enzymes have been reported.77 Only CO and 2-protocatechuoylphloroglucinol carboxylic acid, were detected for Co2+-bound QueD in the B. subtilis study or for the Co2+ and Ni2+ isoforms from Streptomyces. Characterization of bacterial QueDs used recombinant proteins expressed in E. coli. As the native protein from B. subtilis has not been purified and characterized, the identity of the preferred metal cofactor for this QueD remains unknown. On the other hand, QueD was isolated from the wild-type Streptomyces sp. strain FLA and was found to contain mainly nickel and zinc.77 Hence Ni2+ is most likely the physiologically relevant cofactor of QueD from Streptomyces sp. FLA.
The resting state and substrate/inhibitor bound structures of QueDs from Aspergillus japonicas and B. subtilis have been solved.70, 75, 78–79 However, the catalytic mechanism of these enzymes is still under debate: Whether O2 directly binds the metal ion prior to reaction with substrate or reacts directly with the activated substrate is unknown. There are several reasons to believe that direct binding of dioxygen to the metal ion may not be important: As with acireductone, quercetin can react spontaneously with O2 in the absence of QueD, and bacterial QueDs are promiscuous, which suggests that redox activity of the metal cofactor is not essential for catalysis. Small molecule models of QueD activity do not require an open coordination site at the metal for breakdown of quercetin with dioxygen. Furthermore, cofactor-independent dioxygenases are known to catalyze the cleavage of heteroaromatic rings with dioxygen in the absence of metal cofactors.80–82
Given the structural and functional similarities between Ni-KoARD and Streptomyces QueD, the mechanism of QueD activity is likely applicable to the details of Ni-KoARD function. Structures of Streptomyces Ni-QueD in the resting state and bound to substrates quercetin and O2 have been determined by crystallographic cryotrapping approach, providing direct insight into how quercetin and O2 are activated at the Ni2+ ion (Scheme 10).65 The Ni2+ ion is present in an octahedral geometry coordinated by three His residues (His69, His71 and His115) and a Glu residue (Glu76) in all three states. In the resting state, the two non-protein ligands are water molecules. Upon binding, quercetin replaces one water molecule. This binding rotates the carboxylate group of Glu76 by 90°, which then forms a hydrogen bond with quercetin and stabilizes the enzyme-substrate complex. This conformational change weakens the interaction between Ni2+ and the second water ligand, which is then replaced by side-on (π) O2 binding to Ni2+. The reaction then proceeds with cleavage of the C2–C3 and C3–C4 bonds of quercetin. This is the first direct experimental evidence for direct substrate-O2-metal interaction in this class of enzymes. There has been no experimental evidence to date to indicate the formation of a stable M+2-O2 complex in either Fe- or Ni-ARD.
Scheme 10. Proposed catalytic mechanism of Streptomyces sp. FLA QueD.

The protein ligands, His69, His71 and His115 for Ni2+ are not shown. (Figure adapted from Ref. 65)
8.2 Enzymes related to Fe-ARD
A comprehensive search in the MetalPDB83 database for dioxygenases with Fe2+ with octahedral coordination geometry including Glu and His ligation finds homoprotocatechuate 2,3-dioxygenase (HPCD), 3-hydroxyanthranilate 3,4-dioxygenase (HAD) and homogentisate 1,2-dioxygenase (HGDO) as likely matches to Fe-ARD. These enzymes present an active site with Fe2+ present in an octahedral coordination, but through a His2-Glu1 motif rather than the His3-Glu1 found in ARD. Water molecules occupy the non-protein ligation sites in the octahedron. Crystal structures of the resting HPCD enzyme-substrate and enzyme-substrate-O2 complexes indicate that the ferrous ion is involved in binding both the substrate and O2 prior to reaction. The mechanism appears to require first binding of the substrate, displacing the water ligands to the iron. Substrate binding increases the affinity of the metal center for O2 binding, with the reaction then proceeding subsequent to O2 binding. 84–86 Scheme 11 shows the proposed mechanism for HPCD, an example of an extradiol catechol dioxygenase.
Scheme 11. Proposed catalytic mechanism of homoprotocatechuate 2,3-dioxygenase which is an extradiol dioxygenase.

(R = -CH2COOH) (Figure adapted from Ref. 9)
Crystal structures show that the substrates for HAD and HPCD are both bound in bidentate fashion whereas the substrate for HGDO exhibits monodentate binding to the ferrous ion. Conversely, O2 binds the ferrous ion side-on in HGDO and HPCD and end-on in HAD (Scheme 12).
Scheme 12. Ternary enzyme-substrate-dioxygen complexes for HPCD, HAD and HGDO observed by X-ray crystallography.

9. Summary and Discussion
9.1 Metal-dependent function of ARD and potential roles in carcinogenesis
ARD from Klebsiella oxytoca (KoARD) is currently the only known enzyme that, as isolated from the native organism, performs two different chemistries based on the metal ion cofactor bound to the protein in the active site. 1–2 The function of the off-pathway chemistry in Klebsiella oxytoca is unknown, it can be speculated to be involved in the regulation of the MSP. We have shown that mammalian ARDs (mouse and human) also exhibit this metal ion-dependent on- and off-pathway chemistry in vitro where the Fe2+-bound forms catalyze on-pathway and the Ni2+, Co2+ and Mn2+-bound forms catalyze off-pathway chemistry.3–4 Unlike ARD in Klebsiella oxytoca, where Ni-KoARD has a higher activity than Fe-KoARD37, the mammalian ARDs exhibit the opposite behavior, with the Fe2+-bound form of the enzymes exhibiting several fold higher activity than the Ni2+, Co2+ and Mn2+-bound forms. In all cases tested to date, the Ni2+-bound ARDs are the most thermostable, followed by Co2+, Fe2+ and Mn2+ respectively. A eukaryotic ARD from Oryza sativa L. (OsARD) has also been studied biochemically and the Ni2+-bound OsARD (which exhibits the off-chemistry) polymerizes and has a much reduced activity relative to Fe2+-bound OsARD (exhibits on-pathway chemistry).8
Since Fe2+-bound mammalian enzymes exhibit the maximum on-pathway enzymatic activity and many oxidoreductases use Fe2+ as a metal ion co-factor,87 we suspect that the Fe2+-bound form is the native form of ARD under normal conditions in mammals. Immunoprecipitation experiments to isolate native ARD from mice liver can be used to identify the native metal cofactor. The reduced activity of Ni2+-bound mammalian and eukaryotic ARD enzymes suggests that off-pathway chemistry catalyzed by the Ni2+-bound enzyme may not be relevant in a healthy organism and may be only seen only in pathology. However, given the increased thermal stability of the Ni2+-bound enzyme relative to the Fe2+-bound form, formation of Ni-ARD could represent a pathological kinetic trap in vivo in mammalian systems. Given the very low expected concentrations of nickel in vivo (< 2 μg/L in serum or other biological fluids)88, and the known carcinogenicity of soluble nickel compounds,89 it is possible that the formation of a stable Ni-ARD may be involved in tumor development and/or other pathological conditions in humans and other mammals.
This is an especially intriguing possibility due to the known up- and down-regulation of the ADI1 gene in multiple types of carcinoma.15, 56–58 Multiple independent studies have indicated a role for HsARD in cancer. Seiki, et al. have shown that HsARD binds the cytoplasmic tail of MMP14 and inhibits cell migration, metastasis and tissue invasion.16 Oram et al., have shown that ADI1 is down-regulated in rat and human prostate cancer cell lines and the enforced expression of ADI1 causes apoptosis.15 Besides the role of HsARD in regulating the activity of MMP, the metal-dependent dual-chemistry of mammalian ARD enzymes is particularly interesting in that carbon monoxide (CO) which is a product of the off-pathway chemistry, is known to be an anti-apoptotic molecule and has been proposed to act as a signaling molecule analogous to NO.13, 90–92 This leads to the intriguing possibility that binding of Ni2+, Co2+ or Mn2+ in the ARD active site could lead to inappropriate gain-of-function in carcinogenesis, helping to protect transformed cells from apoptosis by producing CO and/or loss-of-function in regulating MMP14 activity. Experiments with rat liver homogenates indicated presence of only on-pathway ARD activity, with CO formation not detected.2
9.2 Implications for the mechanism of ARD-catalyzed dioxygenation reactions
The Abeles and Pochapsky groups have comprehensively characterized the dual chemistry of KoARD in terms of mechanistic enzymology.1, 37, 48 Kinetic analyses have indicated that both Ni-KoARD and Fe-KoARD follow a sequential mechanism, i.e. both substrates must bind before the products are released. UV-visible spectroscopy indicates that acireductone substrate binds to both Fe-KoARD and Ni-KoARD (ES complex formation) regardless of whether oxygen is present. Oxygen does not bind to either of the enzymes in the absence of substrate, but reaction is rapid enough once added to the ES complex that binding of oxygen by the metal (if it is occurring) cannot be easily measured. Titration studies of the model substrate used in these experiments indicated that its maximum absorption depends on its ionization state. The pKa values of the model substrate are 4.0 and 12.2 and its λmax shifts from 305 nm in singly deprotonated form (pH 7) to 345 nm as a dianion. Although the authors suggested that acireductone binds as a dianion to KoARD (in both forms) based on spectroscopic shifts, they were unable to see the formation of a complex between Ni2+ or Fe2+ and acireductone di-anion in solution spectroscopically at pH 13.0.37 Hence the precise nature of the interaction of the substrate with enzyme can only be determined by high-resolution structural studies. However, NMR and XAS experiments on KoARD showed that acireductone directly ligates the metal center in the ES complex.93
Several hypotheses for the dual chemistry of KoARD have been proposed. The chelate hypothesis postulates that the difference in the chemistry is due to a difference in the coordination modes of substrate to the metal ion center, where Fe2+ forms a five-membered ring and Ni2+ forms a six-membered ring leading to different products.20–21, 37 Small molecule model studies and computational studies challenged the chelate hypothesis, showing that both Ni and Fe can form six-membered ring.18–19, 63, 94 However, the D-LA-bound Ni-MmARD structure demonstrates that five-membered rings can indeed form involving the Ni2+ ion and a substrate analog. Furthermore, although several potential ligands were tested, no six-membered bidentate metal-ligand complexes were observed in our MmARD crystallographic structures. While not conclusive evidence for a five-membered ring intermediate in ARD catalysis, these results suggest that the mechanism is still an open question. What can be concluded from the current state of knowledge regarding the mechanism of ARD catalysis is that binding of the acireductone substrate is the first step in catalysis. However, how (or even if) O2 binding to the metal is required for activation by ARD is still not known. None of the proposed mechanisms require an assumption of metal-oxygen binding, although they all presume a bidentate ES complex. Although reaction of O2 with singlet compounds typically requires activation of the ground-state triplet dioxygen, the acireductone functionality (as typified by ascorbic acid) is a good reducing agent, and some triplet character may be generated on the bound acireductone via interaction with the paramagnetic metal ion with which it is complexed in the ARD active site, making a single electron transfer to O2 to generate a radical cation anion pair less forbidden.20–21, 37 For the most part, mononuclear non-heme Fe2+-containing-dioxygenases (e.g., extradiol catechol dioxygenases) require direct O2 binding to Fe2+ in the for activation. However, Fe3+-containing dioxygenases (intradiol catechol dioxygenases) can activate the substrate, rather than dioxygen, in order to facilitate reaction. 95
Unlike ARD, the structure of Ni2+-bound Streptomyces QueD ES-O2 complex does indicate a direct metal-O2 interaction.65 The structural data suggests that before the reaction proceeds, Ni2+ activates both the substrate and dioxygen through direct binding, although the electronic state of the ternary complex remains unresolved. The authors speculate the bound O2 to be a superoxide species based on the O-O bond length. The authors thus propose that the reaction mechanism of Streptomyces Ni-QueD is similar to that of Fe2+-dependent dioxygenases (extradiol catechol dioxygenases) such as HPCD. The presence of Ni2+ to activate O2 is unexpected since Ni2+ does not typically have accessible redox chemistry under physiological conditions. Similar to the mechanistic proposition for HPCD, it is likely that one or two electrons may pass from the Ni2+-bound substrate to the O2 with either no change in the oxidation state of Ni or a transient change that persists for much less than the freezing time for rapid freeze quench (RFQ) crystallography experiments or EPR experiments.86 Although there are several similarities between Streptomyces Ni-QueD and Ni-MmARD, the structure of the ES complex in Ni-QueD shows monodentate binding to the Ni2+ ion, displacing a solvent ligand. The other solvent ligand is replaced upon dioxygen binding to Ni2+. There is no spectroscopic data to support one mode of substrate binding (monodentate or bidentate) over the other in Ni-MmARD. However, the structure of D-lactic acid bound to Ni-MmARD suggests that bidentate binding is possible. If the substrate binds in a bidentate fashion, there is no open coordination site for dioxygen binding unless a protein ligand dissociates from the metal. Spectroscopic studies on KoARD suggested bidentate binding of the substrate, but the authors state that monodentate binding cannot be ruled out.37 Furthermore, EXAFS data of the resting state and ES complex of KoARD suggests that one His could be displaced by substrate binding,93 leaving room for dioxygen binding, even if the substrate remains bound in bidentate fashion.
A structure of the ternary ES-O2 complex would be extremely useful for obtaining mechanistic details for Ni-MmARD. However, given the sensitivity of acireductone to oxidation, it is unlikely that such a structure will be obtained with current methods. The structure of Ni-MmARD bound to an oxidatively stable competitive inhibitor might provide details on the substrate binding modes, including the possibility of O2 binding. Another important advance would be to crystallize an Fe2+-bound ARD isoform. The Fe2+-bound MmARD is oxidatively unstable and requires anaerobic handling due to oxidation from the Fe+2 to Fe+3 state. The Fe3+ form of the enzyme is unstable and exhibits a tendency to autoproteolysis. Although Fe-MmARD was purified and set for crystallization anaerobically, repeated attempts to crystallize it have failed. However, multidimensional NMR spectra of Fe-HsARD have been recently obtained and analyzed, and sequential resonances assigned. Preliminary results suggest that, as with Fe-KoARD, the C-terminal peptide of Fe-HsARD is disordered, and significant structural differences exist between the Fe- and Ni-bound forms of HsARD (Figure 10).4
Figure 10. Solution NMR data of Mn2+, Fe2+, Co2+ and Ni2+-bound HsARD isozymes.

Left: Upfield region of 800 MHz 1H NMR spectra of Mn-HsARD (pink), Fe-HsARD (red), Co-HsARD (blue), and Ni-HsARD (green) showing methyl resonances of amino acid side chains ring current-shifted by nearby aromatic residues. The different spectral patterns indicate differences in side chain packing in hydrophobic cores. Right: Overlay of the 2D 1H, 15N HSQC spectra of Mn-HsARD (pink), Co-HsARD (blue), Fe-HsARD (red) and Ni-HsARD (green). Different shift patterns indicate differences in local hydrogen bonding and structure. All spectra were obtained at 25 °C, pH 7.0 at 800 MHz 1H observe frequency. Reproduced with permission from Ref. 4. Copyright 2017 Oxford University Press.
The 1D and 2D NMR spectra of metal isoforms of HsARD in Figure 10 shows that, although the signals from Mn2+-bound HsARD (pink in both spectra) are quite broad due to paramagnetic relaxation, they are virtually superimposable on the corresponding signals from Ni-HsARD (in green). This suggests that the structures of the Mn2+ and Ni2+ HsARD isozymes are very similar. On the other hand, the octahedral complexes of Fe2+ and Ni2+ with N/O ligation both have integer electronic spins (S=2 and S=1, respectively) and have similar 1H pseudocontact shift patterns and magnitudes in model complexes.96 As such, the spectral differences between the Fe2+ and Ni2+ HsARD isoforms likely reflect genuine structural differences, highly reminiscent of what was observed in KoARD.20 The Co2+-HsARD (S=3/2) probably exhibits significantly different pseudocontact 1H shifts than the Ni2+-bound enzyme, again based on the results from the model complexes. So, while the similarity between the crystallographic structures of Ni-MmARD and Co-MmARD (RMSD = 0.06 Å) may be to some extent crystallographically artifactual, resolving this issue must await the results of a more complete NMR structural characterization of these HsARD isoforms.
9.3 Moonlighting function(s) of HsARD
ARD has been shown to serve regulatory functions in mammals in addition to its enzymatic function in the MSP. HsARD binds the cytoplasmic tail of membrane-type 1 matrix metalloproteinase (MT1-MMP) and acts as a negative regulator by inhibiting MT1-MMP-mediated cellular invasiveness.16 ADI1 encoding ARD is downregulated in rat prostate and human prostate cancer cell lines, cultured gastric carcinoma cells, fibrosarcoma cells and glioblastoma cells. Enforced ARD expression induces apoptosis.15 ADI1 gene expression has been identified as having a direct link with carcinogenesis in a clinical study testing the impact of environmental carcinogens on gene expression differences in humans.58 Yeh, et al. have identified an N-terminal truncated version of HsARD which is implicated in the replication of hepatitis C virus in non-permissive cell lines.5 The MT1-MMP and ADI1 interaction suppresses HCV infection and this inhibition can be reversed by ADI1 overexpression.59 ADI1 is also required for normal Drosophila fecundity.7 ADI1 expression is impacted in Down’s syndrome (DS) and DS-associated congenital heart defects (DS-CHD) with higher expression seen in fetuses trisomic for Hsa21 than in normal control fetuses. These multiple roles for ARD in human physiology and pathology leads to interesting questions: Do each of these functions have a different mechanism or are they inter-related? Are these functions performed in different subcellular compartments? Are these non-enzymatic functions dependent on the enzymatic activity of ARD? Future work can be addressed towards delineating some of these functions of ARD and identifying regions of the protein that are involved in non-enzymatic functions of ARD.
10. Perspective
10.1 Challenges in working with promiscuous metalloproteins
The biochemical and mechanistic study of metalloproteins can be complicated due to several reasons, including difficulty in obtaining a homogeneous purified sample with only a single metal bound, difficulties in identification of the native metal. This becomes even more challenging in the case of a promiscuous binder such as ARD, in which any metal impurity at any step of expression, isolation or purification can complicate analysis. Having met this challenge in the case of ARD expression, purification and characterization, we will summarize our findings for the edification of interested researchers.
10.2 Isolation of metalloprotein samples with a single metal bound
Biochemical and structural characterization of metalloproteins can be both unsatisfying and ambiguous if the sample is a mixture of several metal bound forms (and, if the metals are redox active, this ambiguity can be further exacerbated by mixed oxidation states!). The incorporation of a single metal ion in a recombinantly expressed metalloprotein can be experimentally challenging. We have used several approaches to obtain a single-metal bound form of ARD enzymes. These include preparation of the apoprotein and reconstitution with the desired metal48 and expression of the protein in minimal media in the presence of excess of a single metal.3–4 In order for the first method to work, the apoprotein must be either stably folded in the absence of metal or refolded in high yield upon addition of metal. In the case of ARD, we have found that while the bacterial enzyme can be refolded and repurified, yields tend to be low. Expression of the protein in media enriched with the desired metal has allowed us to obtain sufficient quantities of ARD highly enriched in a single metal for all of our experimental needs, including crystallography and NMR.3
Another issue that can hamper characterization of metalloproteins is the common use of metal affinity tags for purification. The His tag used routinely for purification of overexpressed proteins is not recommended for metalloproteins, for obvious reasons. The affinity of the tag for metal ions such as Ni2+ and Co2+ can complicate metal analysis and, in the case of the ARD enzyme family, introduce uncertainty regarding the reasons for product distributions. We have found that the Strep tag (WSHPQFEK) tag with affinity for streptavidin provides a high degree affinity purification for ARD enzymes.3 Affinity purification minimizes the number of purification steps, which can be critical if the metal is relatively weakly bound to the protein and might be lost during purification. This is particularly useful for the purification of Fe2+-bound ARD enzymes, as oxidative loss of ferrous iron is a serious issue. In all cases, we test the recombinant metalloprotein for metal content using ICP mass spectroscopy to determine the homogeneity of the sample. Furthermore, anomalous scattering measurements provide unambiguous information regarding metal identity in crystalline enzymes. Since each metal has a distinct anomalous scattering wavelength, X-ray diffraction data collected slightly below and above the absorption edge can be used for unambiguous metal identification. To confirm the identity of the active site metals as Ni or Co in MmARD, X-ray datasets were collected 100 eV below and above the absorption peaks of the K-absorption edges of each of these metals.3 Strong peaks were observed in the expected metal position in the anomalous electron density maps calculated using the above absorption edge X-ray data whereas much weaker peaks were observed in the corresponding position calculated using the below absorption edge X-ray data. This sharp change in anomalous scattering on passing through the absorption edges of Ni or Co confirmed the metal identity for each case of MmARD structure determinations.
10.3 What is the in vivo metal cofactor of my metalloprotein?
Determination of the physiologically relevant metal ion of a metalloprotein can be a challenging task. This is especially true when the metal is loosely bound, or (as is often the case) the protein is expressed heterologously. There are several examples of proteins whose native metal identity is still under debate (examples discussed below). Purification of the native enzyme from the wild-type organism and conducting metal analysis on the purified protein is the ideal method to establish metal identity, but this is not always possible due to low yields. Recombinant heterologous expression is the most common method of obtaining proteins for biochemical studies.
There are approaches that can help in the identification of the in vivo metal cofactor, even in the absence of isolation of the native enzyme. Comparison of biochemical properties such as activity and stability profiles of the native enzyme (in fresh cell lysates) with those of purified recombinant proteins bound to various single metal ions can provide an idea of the likely native metal cofactor. Metal abundance in the native organism and metal coordination preference is also an important factor to consider in such studies. However, this is not necessarily definitive, given that metal sequestration and delivery is often complex and highly evolved. For example, ferrous iron was shown to be the likely native metal of the enzyme peptide deformylase (PDF). PDF catalyzes the removal of N-terminal formyl groups from nascent ribosome-synthesized polypeptides. It was proposed that PDF from E coli is Fe2+-dependent (Fe-EcPDF) since the activity and stability (oxidative instability) of recombinant Fe-EcPDF were similar to those of native EcPDF.97 But the assumption that this was true for all bacterial PDFs was shown to be incorrect by Nguyen et al. who studied PDF in Borrelia burgdorferi (BbPDF) and Lactobacillus plantarum (LpPDF) which grow in Fe-limited conditions.98 In their study, they showed that purified native BbPDF contains approximately one mol Zn2+/mol protein and no detectable levels of iron, and that recombinant BbPDF when expressed in medium containing both zinc and iron, selectively binds zinc, while EcPDF preferentially binds Fe2+ when overexpressed in rich medium and binds Zn2+ only in iron depleted medium. A number of other Fe2+-dependent enzymes such as methionyl aminopeptidase99, S-ribosylhomocysteinase (LuxS)100, UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC)101, and histonedeacetylase-8 (HDAC8)102, γ-carbonic anhydrase103, cytosine deaminase104, and atrazine chlorohydrolase105 were initially misidentified as Zn2+ metalloenzymes because they were purified under aerobic conditions leading to oxidation of Fe2+ to Fe3+ followed by dissociation of Fe3+ and replacement with Zn2+.
The identity of the in vivo metal cofactor is clearly important for developing effective therapeutic inhibitors. Most inhibitors of metalloenzymes bind to the catalytic metal ion and affect not only the catalytic activity but also substrate affinities. Consequently, the identity of the native physiological metal cofactor is essential for the design of effective and specific inhibitors. One such example is histone deacetylase 8 (HDAC8), which has been validated as a cancer target.106 Human HDAC8 retains catalytic activity with a number of divalent metal ions in the order of specific activity: Co2+ > Fe2+ > Zn2+ > Ni2+. It is postulated that Fe2+ may be the native metal in human HDAC due to the oxidative instability exhibited by this enzyme. The affinity of the HDAC8 inhibitor suberoylanilide hydroxamic acid (SAHA) and the enzyme kinetic parameters are influenced by the metal ion identity with 5-fold lower KM value for Fe2+-bound HDAC8 compared to Zn2+ -bound HDAC8.102
We expect that this question will be critical if HsARD is to be targeted for any disease state functions. If Fe2+ is the co-factor under normal physiological conditions (an assumption that seems reasonable but is not yet proven), what metal(s) are present when the enzyme is expressed in tumors or in disease tissue? Is the off-pathway chemistry observed in these tissues, or is the role of HsARD in these states not a function of the metal, but unrelated? These questions are currently the subjects of our investigations.
10.4. Metal-dependent chemistry in metalloproteins
The various structural genomics initiatives have produced structures of many metalloproteins, but the identity of the metal ion in these structures are often ambiguous. In fact, given that heterologous expression systems combined with affinity labels such as His tags are ubiquitously used for protein preparations in high-throughput projects, this is a serious issue. As we and others have seen with ARD, the metal ion not only determines the enzymology but can have strong influence on structure as well. There have been several examples in the literature where authors have studied the impact of different metals on enzyme kinetics of a metalloprotein.98, 100, 102, 107 It is often assumed that a metalloenzyme will perform the same chemistry irrespective of the metal in the active site, and changing the metal will only change the kinetic parameters of the enzyme, but not the product(s). The dual chemistry exhibited by ARD clearly shows that this assumption can be misleading. It is critical that future metalloenzyme research tests the effect of different metals not only on enzyme kinetics but product distributions as well.
10.5. Metal sensing in biological systems
Metal sensing and metal homeostasis is a critical function for all organisms. Metal ions are ubiquitous in the environment, and many are toxic if present in the cytosol in high concentration, or injurious if not present in sufficient amounts. It is no surprise then that extensive metal sensing and transport systems have evolved in order to remove toxic metals or take in and appropriately compartmentalize required metals.108–109 Still, relatively little is known regarding the mechanisms by which one metal ion is distinguished from another, especially in cases where ions have the same charge and similar sizes. Although many metal-containing co-factors (e.g., heme, chlorophyll, cobalamin) are pre-formed when finally bound to their protein partners, this still requires that the co-factors themselves be properly synthesized with the correct metal. Alkali metal and alkaline earth ions are sometimes bound via amide carbonyl oxygens (as in selective ion channels110 or when stabilizing local structural motifs111). In these cases, the protein backbone conformations appear to be pre-arranged to selectively bind cations of the appropriate size and charge. Serine and tyrosine hydroxyl groups are often involved in Ca2+ binding, which triggers major conformational changes in calmodulin.112–113 However, the vast majority of proteins incorporating transition metals use a common set of ligands: histidine (imidazole), aspartate and glutamate (carboxylate) and cysteine or methionine (thiolate and thioether, respectively). Within this group, histidine can ligate through either imidazole nitrogen, but due to steric considerations, Nε ligation is the most common. Carboxylates also have the possibility of mono- or bidentate ligation, with the increased flexibility of the glutamate side chain relative to aspartate providing a variety of steric possibilities for metal-ligand interactions. Depending upon identity and oxidation state, many transition metals have a preferred coordination geometry that pre-disposes certain ligand arrangements within a polypeptide to bind a particular metal selectively. However, this is not the only determinant, as we have found that Zn2+ does not form a stable monomeric complex with KoARD, while the Streptomyces QueD can incorporate zinc.20, 77 Fierke and co-workers found that Zn2+ can replace Fe2+ in a bacterial deacetylase (LpxC) depending upon cellular conditions, and that both are active enzymes, although ferrous ion is bound more tightly than zinc and results in a more efficient enzyme.101, 114
The response of a protein to the binding of a metal can vary from the protein being disordered in the absence of the metal (e.g., zinc finger motifs and many ferredoxins115) to a more general “tightening” of the structure, as seen in some sensor proteins and globins.116–117 This “tightening” is usually reflected in increased denaturation temperatures and decreased amide H/D exchange rates, and/or decreases in hydrodynamic radii.111, 116 Metal binding at an interface between subunits can modulate affinity for second-site metal and interaction partners in activators, promoters and inhibitors of transcription.118–119 In the case of KoARD, the apo-enzyme appears to fold in much the same manner as the Fe2+-bound enzyme, suggesting that the “default” fold of KoARD is pre-disposed to bind iron without significant structural rearrangement. This is inferred from the observation that the metal-free mutant H98S KoARD is stably folded and does not exhibit significantly more dynamic line broadening in NMR spectra than the WT enzyme with metals bound.20
What renders the metal sensitivity of the ARD enzymes unusual is the fact that the off-pathway (Ni2+-bound) isoforms of all of the ARD enzymes characterized to date exhibit significantly higher thermal stability than any other metal-bound form, including the presumed “default” Fe2+-bound isoforms. In the case of KoARD, we have proposed that the fixed arrangement of three of the four protein-based ligands (His96, His140 and Glu102) within the cupin barrel combined with Ni-N/O bonds on average 0.1 Å shorter than the corresponding Fe-N/O bonds pulls the fourth ligand (His98) deeper into the active site. This allows the formation of stable hydrogen bonds not present in the iron isoform, and dramatically increases the ordering of the polypeptide around the active site. In turn, this ordering triggers a substantial re-arrangement of a hydrophobic core region and rearrangement of helices on the distal face of the cupin barrel, as well as the formation of a C-terminal helix on the distal face. Conversely, a region that is relatively well ordered in the Fe2+-bound KoARD (the D loop) is disordered in the Ni2+-bound enzyme. Based upon spectral evidence, this is also likely to be true for HsARD as well, suggesting that the metal-dependent structural switching is evolutionarily conserved.
10.6 Synthetic biology and artificial metalloproteins
The field of artificial metalloenzymes has expanded significantly over the last decade to explore some of the unaddressed challenges in synthetic organic chemistry. Transition metal complexes catalyze a broad range of chemical reactions used to prepare industrially important chemicals.120 In general, transition metal catalysts provide only the first coordination sphere to modulate reactivity. Metalloenzymes, on the other hand, can modulate both the first and second coordination spheres to provide substrate specificity and product selectivity. This makes modulation of native metalloenzyme function to provide useful high-value products an attractive goal. There are several different approaches in creating artificial metalloenzymes, including introduction of a novel function through site specific modification and directed evolution,121–122 introduction of an organometallic catalyst into a scaffold protein, and introduction of a novel function by replacement of the native metal with an abiotic metal.123
Enzymes have evolved to achieve the catalytic efficiency and selectivity required for controlled biochemical processes. It is becoming clear that not just the active site, but the entire enzyme structure is involved in this control.124 As such, re-engineering to improve or modify an enzyme to reach high efficiencies must take this requirement into account. The most straightforward way of mimicking natural selection is to use directed evolution, with iterative rounds of enzyme diversification and functional screening. Cytochrome P450s have been modified and evolved using directed evolution protocols to catalyze a wide range of non-natural reactions.125 Similarly, Kim and coworkers conferred β-lactamase activity to glyoxalase II using directed evolution.126 Seelig and Szostak found that randomization of two loops found in the zinc-finger domain of a retinoid-X-receptor protein induced RNA ligase activity.127 Hayashi and coworkers engineered myoglobin to obtain peroxidase activity by introducing site specific mutations and replacing the porphyrin ring with a “double winged” cofactor (replacing the propionate groups of the heme with nonpolar aromatic moieties).128 The normally non-catalytic myoglobin has also been engineered to perform carbene insertion into N-H bonds,129 alkenes130 and S-H bonds131 and nitrene transfer reactions to effect C-H amination132 by introducing a substrate binding cleft through site specific mutations.
Another approach is to incorporate an organometallic catalyst into a protein scaffold to create artificial metalloenzymes. This, it is hoped, will combine the substrate selectivity of enzymes with the efficiency of homogeneous transition-metal catalysts. Pioneered by Wilson and Whitesides,133 the streptavidin-biotin system has been researched extensively for non-covalent anchoring of biotinylated organometallic moieties within streptavidin to form artificial metalloenzymes. Several reviews have described this system, which has been used for a variety of enantioselective transformations including hydrogenation, trans-olefination, metathesis, allylic alkylation and sulfoxidation.125, 134–138 Replacement of the native organometallic cofactor with an abiotic cofactor has also been used as a strategy to introduce novel function in heme proteins because of the ability of the porphyrin to accommodate metals other than iron, or, alternatively, bind other polycylic metal co-factors into the heme binding pocket. The porphyrin ring in myoglobin has been substituted with a variety of artificial cofactors, including substituted porphyrins such as porphycene132, 139 metal-Schiff base complexes,140–142 metallo-salophen,140, 142 metallo-salen141, 143 and Rh phebox144 complexes. These substituted Mb-hybrid systems have activities exhibited by the synthetic metal catalysts. A variety of other protein hosts have also been used for covalent anchoring of artificial metallocofactors which include albumins,145–148 proteases,149–153 lipases,121, 154–155 anhydrases156–159 and other proteins.160–161 Organometallic cofactors have also been introduced into cage proteins, providing an interior cavity for immobilization of the catalyst. Ferritin has been shown a good candidate for immobilizing a range of metal catalysts and serving as a catalytic reaction space.162–165
A third approach, replacement of the native metal with an abiological metal is relatively unexplored. ARD is the only example of a natural metalloprotein which exhibits completely different chemistries when bound to different metals. Replacement of the native metal with an abiotic metal is attractive in that it is a simple approach and can be used to explore novel chemistries not typically seen in enzymes. There are a few examples in literature describing this approach. The Zn2+-binding carboxypeptidase A (CPA) was substituted by Kaiser and coworkers with Cu2+, converting it into an oxidase. This Cu-CPA oxidase catalyzes the oxidation of ascorbic acid to dehydroascorbic acid, but lacks the peptidase and esterase activities of the native enzyme.166 In another case, Zn2+ ion of human carbonic anhydrase (hCA) was removed and replaced with Mn2+ and Rh2+. hCA replaced with Mn2+ catalyzed an enantioselective artificial epoxidase167 while hCA replaced with Rh2+ catalyzed hydrogenation and regioselective hydro- formylation reactions.168–169 A recent study has reported replacement of iron in Fe-porphyrin proteins with abiological noble metals to create enzymes that catalyze reactions not catalyzed by native Fe-enzymes or other metalloenzymes.22 Artificial metalloenzymes obtained by replacement of the native metal with an abiotic or non-native metal can serve as a unique opportunity for protein engineering to expand the repertoire of reactions catalyzed by native metalloenzymes.
10.7 Conclusions
It has been over twenty years since ARD was first discovered in bacteria2, 35–36, and a little over ten years since it was discerned that humans and other eukaryotes encode a similar enzyme. It is worth noting that the motivation leading to ARD’s discovery was the observation that many human cancer cell lines have a methionine dependence, and identifying the origins of that dependence could lead to potential therapies.170 In retrospect, it is intriguing that the ADI1 gene product was first identified not as an enzyme, but as a regulator of matrix metalloproteinase activity, viral infection and apoptosis. The context-dependent nature of those regulatory functions was (and remains) puzzling, but in light of the recent confirmation that HsARD also exhibits the dual chemistries of the bacterial enzyme, there is some hope that the context dependence of other, non-enzymatic, functions of ARD might be related to the structural and chemical changes brought by binding different metals in the ARD active site.
Scheme 5. Proposed mechanism of Ni-ARD chemistry using small molecule model of Ni-KoARD active site.

(Figure adapted from Ref. 94)
Scheme 6. Proposed mechanism of Fe-ARD chemistry using small molecule model of Fe-KoARD active site.

(Figure adapted from Ref. 94)
Acknowledgments
Funding Sources
This work was supported in part by a grant from National Institutes of Health (GM26788 to GAP and DR).
Biographies
Aditi Deshpande received her Bachelors in Chemical Engineering from Institute of Chemical Technology, India and Masters in Chemical Engineering from University of Delaware. She worked for 5 years in the biotech industry in process development and manufacturing of biologics. In 2010, she joined Dagmar Ringe and Gregory Petsko’s research group at the Brandeis University where she investigated the metal-dependent behavior of mammalian acireductone dioxygenase enzymes. She received her Ph.D. in biochemistry & biophysics in 2016 from Brandeis University and is currently working as a scientist at Allena Pharmaceuticals. Her research interests include enzymology, protein structure, biocatalysis and design of artificial metalloenzymes.
Thomas was born in 1954 in Cleveland, Ohio. He received his B.Sc. in analytical chemistry from the University of Pittsburgh in 1977, and worked for several years in industry before joining William Pirkle’s research group at the University of Illinois. While there, he invented a series of novel chiral stationary phases and provided spectroscopic evidence to support their mechanism of enantioselectivity. He received his Ph.D. in organic chemistry in 1986, and did post-doctoral research with Stephen Sligar at Illinois and Peter Wright at Scripps. He began his career as assistant professor of organic chemistry at Brandeis University in 1989. He is currently Professor of Chemistry and Biochemistry at Brandeis. His research interests are in enzyme structure and function, macromolecular recognition and the molecular basis of Parkinson’s disease.
Dr. Ringe received her B.A. degree in Chemistry from Barnard College in 1963 and Ph.D. degree in Organic Chemistry from Boston University in 1968. She is currently the Harold and Bernice Davis Professor of Aging and Neurodegenerative Diseases at Brandeis University, and Adjunct Professor Neurology at Harvard Medical School. Her research is aimed at the study of the molecular mechanisms of enzymes and the functions of proteins, with a focus on the determination of function for proteins of unknown function and the development of methods for intervention on that function. The tools being used include X-ray crystallography, molecular biology, kinetics, organic synthesis, genetics, and computational approaches.
There are a number of problems to which these methods are being applied, and they include: the structural basis for efficient enzyme catalysis of proton and hydride transfer; the role of the metal ions in bridged bimetallo-enzyme active sites; direct visualization of proteins in action by time-resolved protein crystallography; the structural basis for reaction selectivity in enzymes; the evolution of new enzyme activities from old ones and the development of new pathways. The most important of these problems is the study of neurodegenerative diseases, especially Parkinson’s, Alzheimer’s and Lou Gehrig’s diseases, with a view toward determining the functions of gene products associated with such diseases, finding common pathways that underlie them, and the search for and design of specific inhibitors or activators as potential drugs to treat these diseases. A number of methods are being used, from development of genetic and in vitro screens to structure-based drug design methods.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Dai Y, Wensink PC, Abeles RH. One protein, two enzymes. J Biol Chem. 1999;274(3):1193–1195. doi: 10.1074/jbc.274.3.1193. [DOI] [PubMed] [Google Scholar]
- 2.Wray JW, Abeles RH. The methionine salvage pathway in Klebsiella pneumoniae and rat liver. Identification and characterization of two novel dioxygenases. J Biol Chem. 1995;270(7):3147–3153. doi: 10.1074/jbc.270.7.3147. [DOI] [PubMed] [Google Scholar]
- 3.Deshpande AR, Wagenpfeil K, Pochapsky TC, Petsko GA, Ringe D. Metal-Dependent Function of a Mammalian Acireductone Dioxygenase. Biochemistry. 2016;55(9):1398–1407. doi: 10.1021/acs.biochem.5b01319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deshpande AR, Pochapsky TC, Petsko GA, Ringe D. Dual chemistry catalyzed by human acireductone dioxygenase. Protein Eng Des Sel. 2017;30(3):197–204. doi: 10.1093/protein/gzw078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeh CT, Lai HY, Chen TC, Chu CM, Liaw YF. Identification of a hepatic factor capable of supporting hepatitis C virus replication in a nonpermissive cell line. J Virol. 2001;75(22):11017–11024. doi: 10.1128/JVI.75.22.11017-11024.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen YQ, Li T, Guo WY, Su FJ, Zhang YX. Identification of altered pathways in Down syndrome-associated congenital heart defects using an individualized pathway aberrance score. Genet Mol Res. 2016;15(2) doi: 10.4238/gmr.15027601. [DOI] [PubMed] [Google Scholar]
- 7.Chou HY, Lin YH, Shiu GL, Tang HY, Cheng ML, Shiao MS, Pai LM. ADI1, a methionine salvage pathway enzyme, is required for Drosophila fecundity. J Biomed Sci. 2014;21:64. doi: 10.1186/s12929-014-0064-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sauter M, Lorbiecke R, Ouyang B, Pochapsky TC, Rzewuski G. The immediate-early ethylene response gene OsARD1 encodes an acireductone dioxygenase involved in recycling of the ethylene precursor S-adenosylmethionine. Plant J. 2005;44(5):718–729. doi: 10.1111/j.1365-313X.2005.02564.x. [DOI] [PubMed] [Google Scholar]
- 9.Fetzner S. Ring-cleaving dioxygenases with a cupin fold. Appl Environ Microbiol. 2012;78(8):2505–2514. doi: 10.1128/AEM.07651-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woo EJ, Dunwell JM, Goodenough PW, Marvier AC, Pickersgill RW. Germin is a manganese containing homohexamer with oxalate oxidase and superoxide dismutase activities. Nat Struct Biol. 2000;7(11):1036–1040. doi: 10.1038/80954. [DOI] [PubMed] [Google Scholar]
- 11.Anand R, Dorrestein PC, Kinsland C, Begley TP, Ealick SE. Structure of oxalate decarboxylase from Bacillus subtilis at 1.75 A resolution. Biochemistry. 2002;41(24):7659–7669. doi: 10.1021/bi0200965. [DOI] [PubMed] [Google Scholar]
- 12.Martinez S, Hausinger RP. Catalytic Mechanisms of Fe(II)- and 2-Oxoglutarate-dependent Oxygenases. J Biol Chem. 2015;290(34):20702–20711. doi: 10.1074/jbc.R115.648691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verma A, Hirsch DJ, Glatt CE, Ronnett GV, Snyder SH. Carbon monoxide: a putative neural messenger. Science. 1993;259(5093):381–384. doi: 10.1126/science.7678352. [DOI] [PubMed] [Google Scholar]
- 14.Wu L, Wang R. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev. 2005;57(4):585–630. doi: 10.1124/pr.57.4.3. [DOI] [PubMed] [Google Scholar]
- 15.Oram SW, Ai J, Pagani GM, Hitchens MR, Stern JA, Eggener S, Pins M, Xiao W, Cai X, Haleem R, Jiang F, Pochapsky TC, Hedstrom L, Wang Z. Expression and function of the human androgen-responsive gene ADI1 in prostate cancer. Neoplasia. 2007;9(8):643–651. doi: 10.1593/neo.07415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uekita T, Gotoh I, Kinoshita T, Itoh Y, Sato H, Shiomi T, Okada Y, Seiki M. Membrane-type 1 matrix metalloproteinase cytoplasmic tail-binding protein-1 is a new member of the Cupin superfamily. A possible multifunctional protein acting as an invasion suppressor down-regulated in tumors. J Biol Chem. 2004;279(13):12734–12743. doi: 10.1074/jbc.M309957200. [DOI] [PubMed] [Google Scholar]
- 17.Pochapsky TC, Ju T, Dang M, Beaulieu R, Pagani G, OuYang B. Nickel in Acireductone Dioxygenase. In: Sigel A, Sigel H, Sigel RKO, editors. Metal Ions in Life Sciences: Nickel and Its Surprising Impact in Nature. 1. John Wiley & Sons, Ltd; Chichester, England: 2007. pp. 473–500. [Google Scholar]
- 18.Berreau LM, Borowski T, Grubel K, Allpress CJ, Wikstrom JP, Germain ME, Rybak-Akimova EV, Tierney DL. Mechanistic studies of the O2-dependent aliphatic carbon-carbon bond cleavage reaction of a nickel enolate complex. Inorg Chem. 2011;50(3):1047–1057. doi: 10.1021/ic1017888. [DOI] [PubMed] [Google Scholar]
- 19.Sparta M, Valdez CE, Alexandrova AN. Metal-dependent activity of Fe and Ni acireductone dioxygenases: how two electrons reroute the catalytic pathway. J Mol Biol. 2013;425(16):3007–3018. doi: 10.1016/j.jmb.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Ju T, Goldsmith RB, Chai SC, Maroney MJ, Pochapsky SS, Pochapsky TC. One protein, two enzymes revisited: a structural entropy switch interconverts the two isoforms of acireductone dioxygenase. J Mol Biol. 2006;363(4):823–834. doi: 10.1016/j.jmb.2006.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pochapsky TC, Pochapsky SS, Ju T, Mo H, Al-Mjeni F, Maroney MJ. Modeling and experiment yields the structure of acireductone dioxygenase from Klebsiella pneumoniae. Nat Struct Biol. 2002;9(12):966–972. doi: 10.1038/nsb863. [DOI] [PubMed] [Google Scholar]
- 22.Key HM, Dydio P, Clark DS, Hartwig JF. Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature. 2016;534(7608):534–537. doi: 10.1038/nature17968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abeles R, Frey P, Jencks W. Biochemistry. Jones & Bartlett Publishers; Boston, MA: 1992. pp. 685–689. [Google Scholar]
- 24.William-Ashman HG, Pegg AE. Polyamines in Biology and Medicine. Marcel Dekker; New York: 1981. pp. 43–73. [Google Scholar]
- 25.Taiz L. Plant Physiology. Benjamin/Cummins Publishin Co; Redwood City, CA: 1991. [Google Scholar]
- 26.Oredsson SM. Polyamine dependence of normal cell-cycle progression. Biochem Soc Trans. 2003;31(2):366–370. doi: 10.1042/bst0310366. [DOI] [PubMed] [Google Scholar]
- 27.Gerner EW, Meyskens FL., Jr Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer. 2004;4(10):781–792. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- 28.Marton LJ, Pegg AE. Polyamines as targets for therapeutic intervention. Annu Rev Pharmacol Toxicol. 1995;35:55–91. doi: 10.1146/annurev.pa.35.040195.000415. [DOI] [PubMed] [Google Scholar]
- 29.Pegg AE. Polyamine metabolism and its importance in neoplastic growth and a target for chemotherapy. Cancer Res. 1988;48(4):759–774. [PubMed] [Google Scholar]
- 30.Martinez ME, O’Brien TG, Fultz KE, Babbar N, Yerushalmi H, Qu N, Guo Y, Boorman D, Einspahr J, Alberts DS, Gerner EW. Pronounced reduction in adenoma recurrence associated with aspirin use and a polymorphism in the ornithine decarboxylase gene. Proc Natl Acad Sci U S A. 2003;100(13):7859–7864. doi: 10.1073/pnas.1332465100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams-Ashman HG, Seidenfeld J, Galletti P. Trends in the biochemical pharmacology of 5′-deoxy-5′-methylthioadenosine. Biochem Pharmacol. 1982;31(3):277–288. doi: 10.1016/0006-2952(82)90171-x. [DOI] [PubMed] [Google Scholar]
- 32.Schlenk F. Methylthioadenosine. Adv Enzymol Relat Areas Mol Biol. 1983;54:195–265. doi: 10.1002/9780470122990.ch4. [DOI] [PubMed] [Google Scholar]
- 33.Shapiro SK, Barrett A. 5-Methylthioribose as a precursor of the carbon chain of methionine. Biochem Biophys Res Commun. 1981;102(1):302–307. doi: 10.1016/0006-291x(81)91521-7. [DOI] [PubMed] [Google Scholar]
- 34.Furfine ES, Abeles RH. Intermediates in the conversion of 5′-S-methylthioadenosine to methionine in Klebsiella pneumoniae. J Biol Chem. 1988;263(20):9598–9606. [PubMed] [Google Scholar]
- 35.Wray JW, Abeles RH. A bacterial enzyme that catalyzes formation of carbon monoxide. J Biol Chem. 1993;268(29):21466–21469. [PubMed] [Google Scholar]
- 36.Myers RW, Wray JW, Fish S, Abeles RH. Purification and characterization of an enzyme involved in oxidative carbon-carbon bond cleavage reactions in the methionine salvage pathway of Klebsiella pneumoniae. J Biol Chem. 1993;268(33):24785–24791. [PubMed] [Google Scholar]
- 37.Dai Y, Pochapsky TC, Abeles RH. Mechanistic studies of two dioxygenases in the methionine salvage pathway of Klebsiella pneumoniae. Biochemistry. 2001;40(21):6379–6387. doi: 10.1021/bi010110y. [DOI] [PubMed] [Google Scholar]
- 38.Daniels L, Fuchs G, Thauer RK, Zeikus JG. Carbon monoxide oxidation by methanogenic bacteria. J Bacteriol. 1977;132(1):118–126. doi: 10.1128/jb.132.1.118-126.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Brien JM, Wolkin RH, Moench TT, Morgan JB, Zeikus JG. Association of hydrogen metabolism with unitrophic or mixotrophic growth of Methanosarcina barkeri on carbon monoxide. J Bacteriol. 1984;158(1):373–375. doi: 10.1128/jb.158.1.373-375.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rother M, Metcalf WW. Anaerobic growth of Methanosarcina acetivorans C2A on carbon monoxide: an unusual way of life for a methanogenic archaeon. Proc Natl Acad Sci U S A. 2004;101(48):16929–16934. doi: 10.1073/pnas.0407486101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Volbeda A, Fontecilla-Camps JC. Structural bases for the catalytic mechanism of Ni-containing carbon monoxide dehydrogenases. Dalton Trans. 2005;(21):3443–3450. doi: 10.1039/b508403b. [DOI] [PubMed] [Google Scholar]
- 42.Kung Y, Drennan CL. A role for nickel-iron cofactors in biological carbon monoxide and carbon dioxide utilization. Curr Opin Chem Biol. 2011;15(2):276–283. doi: 10.1016/j.cbpa.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ragsdale SW. Nickel and the carbon cycle. J Inorg Biochem. 2007;101(11–12):1657–1666. doi: 10.1016/j.jinorgbio.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gullotta F, di Masi A, Coletta M, Ascenzi P. CO metabolism, sensing, and signaling. Biofactors. 2012;38(1):1–13. doi: 10.1002/biof.192. [DOI] [PubMed] [Google Scholar]
- 45.Sekowska A, Denervaud V, Ashida H, Michoud K, Haas D, Yokota A, Danchin A. Bacterial variations on the methionine salvage pathway. BMC Microbiol. 2004;4:9. doi: 10.1186/1471-2180-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Heinsen MH, Kostic M, Pagani GM, Riera TV, Perovic I, Hedstrom L, Snider BB, Pochapsky TC. Analogs of 1-phosphonooxy-2,2-dihydroxy-3-oxo-5-(methylthio)pentane, an acyclic intermediate in the methionine salvage pathway: a new preparation and characterization of activity with E1 enolase/phosphatase from Klebsiella oxytoca. Bioorg Med Chem. 2004;12(14):3847–3855. doi: 10.1016/j.bmc.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 47.Pochapsky TC, Pochapsky SS, Ju T, Hoefler C, Liang J. A refined model for the structure of acireductone dioxygenase from Klebsiella ATCC 8724 incorporating residual dipolar couplings. J Biomol NMR. 2006;34(2):117–127. doi: 10.1007/s10858-005-5735-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chai SC, Ju T, Dang M, Goldsmith RB, Maroney MJ, Pochapsky TC. Characterization of metal binding in the active sites of acireductone dioxygenase isoforms from Klebsiella ATCC 8724. Biochemistry. 2008;47(8):2428–2438. doi: 10.1021/bi7004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ullah H, Chen JG, Temple B, Boyes DC, Alonso JM, Davis KR, Ecker JR, Jones AM. The beta-subunit of the Arabidopsis G protein negatively regulates auxin-induced cell division and affects multiple developmental processes. Plant Cell. 2003;15(2):393–409. doi: 10.1105/tpc.006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Llorente F, Alonso-Blanco C, Sanchez-Rodriguez C, Jorda L, Molina A. ERECTA receptor-like kinase and heterotrimeric G protein from Arabidopsis are required for resistance to the necrotrophic fungus Plectosphaerella cucumerina. Plant J. 2005;43(2):165–180. doi: 10.1111/j.1365-313X.2005.02440.x. [DOI] [PubMed] [Google Scholar]
- 51.Trusov Y, Sewelam N, Rookes JE, Kunkel M, Nowak E, Schenk PM, Botella JR. Heterotrimeric G proteins-mediated resistance to necrotrophic pathogens includes mechanisms independent of salicylic acid-, jasmonic acid/ethylene- and abscisic acid-mediated defense signaling. Plant J. 2009;58(1):69–81. doi: 10.1111/j.1365-313X.2008.03755.x. [DOI] [PubMed] [Google Scholar]
- 52.Friedman EJ, Wang HX, Jiang K, Perovic I, Deshpande A, Pochapsky TC, Temple BR, Hicks SN, Harden TK, Jones AM. Acireductone dioxygenase 1 (ARD1) is an effector of the heterotrimeric G protein beta subunit in Arabidopsis. J Biol Chem. 2011;286(34):30107–30118. doi: 10.1074/jbc.M111.227256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hao G, Wang J, Shi R, Zhang X. Cloning, molecular characterization and expression of acireductone dioxygenase (ARD) gene from Salvia miltiorrhiza. Zhongguo Zhong Yao Za Zhi. 2011;36(3):346–350. [PubMed] [Google Scholar]
- 54.Xu L, Jia J, Lv J, Liang X, Han D, Huang L, Kang Z. Characterization of the expression profile of a wheat aci-reductone-dioxygenase-like gene in response to stripe rust pathogen infection and abiotic stresses. Plant Physiol Biochem. 2010;48(6):461–468. doi: 10.1016/j.plaphy.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 55.Kim JH, Kim HS, Lee YH, Kim YS, Oh HW, Joung H, Chae SK, Suh KH, Jeon JH. Polyamine biosynthesis regulated by StARD expression plays an important role in potato wound periderm formation. Plant Cell Physiol. 2008;49(10):1627–1632. doi: 10.1093/pcp/pcn115. [DOI] [PubMed] [Google Scholar]
- 56.Pratt J, Iddir M, Bourgault S, Annabi B. Evidence of MTCBP-1 interaction with the cytoplasmic domain of MT1-MMP: Implications in the autophagy cell index of high-grade glioblastoma. Mol Carcinog. 2016;55(2):148–160. doi: 10.1002/mc.22264. [DOI] [PubMed] [Google Scholar]
- 57.Oram S, Jiang F, Cai X, Haleem R, Dincer Z, Wang Z. Identification and characterization of an androgen-responsive gene encoding an aci-reductone dioxygenase-like protein in the rat prostate. Endocrinology. 2004;145(4):1933–1942. doi: 10.1210/en.2003-0947. [DOI] [PubMed] [Google Scholar]
- 58.Espin-Perez A, de Kok TM, Jennen DG, Hendrickx DM, De Coster S, Schoeters G, Baeyens W, van Larebeke N, Kleinjans JC. Distinct genotype-dependent differences in transcriptome responses in humans exposed to environmental carcinogens. Carcinogenesis. 2015;36(10):1154–1161. doi: 10.1093/carcin/bgv111. [DOI] [PubMed] [Google Scholar]
- 59.Chang ML, Huang YH, Cheng JC, Yeh CT. Interaction between hepatic membrane type 1 matrix metalloproteinase and acireductone dioxygenase 1 regulates hepatitis C virus infection. J Viral Hepat. 2015 doi: 10.1111/jvh.12486. [DOI] [PubMed] [Google Scholar]
- 60.Xu Q, Schwarzenbacher R, Krishna SS, McMullan D, Agarwalla S, Quijano K, Abdubek P, Ambing E, Axelrod H, Biorac T, et al. Crystal structure of acireductone dioxygenase (ARD) from Mus musculus at 2.06 angstrom resolution. Proteins. 2006;64(3):808–813. doi: 10.1002/prot.20947. [DOI] [PubMed] [Google Scholar]
- 61.Milaczewska AM, Chruszcz M, Shabalin IG, Cooper DR, Borowski T, Anderson WF, Minor W. Acireductone dioxygenase from Bacillus anthracis with cadmium ion in active center. http://www.rcsb.org/pdb/explore/explore.do?structureId=4QGM.
- 62.Milaczewska AM, Chruszcz M, Petkowski JJ, Niedzialkowska E, Minor W, Borowski T. Human acireductone dioxygenase with iron ion and L-methionine in active center. http://www.rcsb.org/pdb/explore/explore.do?structureId=4QGN.
- 63.Szajna E, Arif AM, Berreau LM. Aliphatic carbon-carbon bond cleavage reactivity of a mononuclear Ni(II) cis-beta-keto-enolate complex in the presence of base and O2: a model reaction for acireductone dioxygenase (ARD) J Am Chem Soc. 2005;127(49):17186–17187. doi: 10.1021/ja056346x. [DOI] [PubMed] [Google Scholar]
- 64.Maroney MJ, Ciurli S. Nonredox nickel enzymes. Chem Rev. 2014;114(8):4206–4228. doi: 10.1021/cr4004488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jeoung JH, Nianios D, Fetzner S, Dobbek H. Quercetin 2,4-Dioxygenase Activates Dioxygen in a Side-On O2 -Ni Complex. Angew Chem Int Ed Engl. 2016;55(10):3281–3284. doi: 10.1002/anie.201510741. [DOI] [PubMed] [Google Scholar]
- 66.Merkens H, Kappl R, Jakob RP, Schmid FX, Fetzner S. Quercetinase QueD of Streptomyces sp FLA, a Monocupin Dioxygenase with a Preference for Nickel and Cobalt. Biochemistry. 2008;47(46):12185–12196. doi: 10.1021/bi801398x. [DOI] [PubMed] [Google Scholar]
- 67.Opaleye O, Rose RS, Whittaker MM, Woo EJ, Whittaker JW, Pickersgill RW. Structural and spectroscopic studies shed light on the mechanism of oxalate oxidase. J Biol Chem. 2006;281(10):6428–6433. doi: 10.1074/jbc.M510256200. [DOI] [PubMed] [Google Scholar]
- 68.Lis T. Structure of manganese(II) glycolate dihydrate. Acta Crystallogr Sect B Struct Sci. 1980;36:701–703. [Google Scholar]
- 69.Zhu W, Easthon LM, Reinhardt LA, Tu C, Cohen SE, Silverman DN, Allen KN, Richards NG. Substrate Binding Mode and Molecular Basis of a Specificity Switch in Oxalate Decarboxylase. Biochemistry. 2016;55(14):2163–2173. doi: 10.1021/acs.biochem.6b00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fusetti F, Schroter KH, Steiner RA, van Noort PI, Pijning T, Rozeboom HJ, Kalk KH, Egmond MR, Dijkstra BW. Crystal structure of the copper-containing quercetin 2,3-dioxygenase from Aspergillus japonicus. Structure. 2002;10(2):259–268. doi: 10.1016/s0969-2126(02)00704-9. [DOI] [PubMed] [Google Scholar]
- 71.Tranchimand S, Ertel G, Gaydou V, Gaudin C, Tron T, Iacazio G. Biochemical and molecular characterization of a quercetinase from Penicillium olsonii. Biochimie. 2008;90(5):781–789. doi: 10.1016/j.biochi.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 72.Oka T, Simpson FJ. Quercetinase, a dioxygenase containing copper. Biochem Biophys Res Commun. 1971;43(1):1–5. doi: 10.1016/s0006-291x(71)80076-1. [DOI] [PubMed] [Google Scholar]
- 73.Oka T, Simpson FJ. Degradation of rutin by Aspergillus flavus. Quercetinase: isolation of a low molecular-weight form containing less carbohydrate. Can J Microbiol. 1972;18(7):1171–1175. doi: 10.1139/m72-182. [DOI] [PubMed] [Google Scholar]
- 74.Hund HK, Breuer J, Lingens F, Huttermann J, Kappl R, Fetzner S. Flavonol 2,4-dioxygenase from Aspergillus niger DSM 821, a type 2 CuII-containing glycoprotein. Eur J Biochem. 1999;263(3):871–878. doi: 10.1046/j.1432-1327.1999.00574.x. [DOI] [PubMed] [Google Scholar]
- 75.Gopal B, Madan LL, Betz SF, Kossiakoff AA. The crystal structure of a quercetin 2,3-dioxygenase from Bacillus subtilis suggests modulation of enzyme activity by a change in the metal ion at the active site(s) Biochemistry. 2005;44(1):193–201. doi: 10.1021/bi0484421. [DOI] [PubMed] [Google Scholar]
- 76.Schaab MR, Barney BM, Francisco WA. Kinetic and spectroscopic studies on the quercetin 2,3-dioxygenase from Bacillus subtilis. Biochemistry. 2006;45(3):1009–1016. doi: 10.1021/bi051571c. [DOI] [PubMed] [Google Scholar]
- 77.Nianios D, Thierbach S, Steimer L, Lulchev P, Klostermeier D, Fetzner S. Nickel quercetinase, a “promiscuous” metalloenzyme: metal incorporation and metal ligand substitution studies. BMC Biochem. 2015;16:10. doi: 10.1186/s12858-015-0039-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Steiner RA, Kalk KH, Dijkstra BW. Anaerobic enzyme.substrate structures provide insight into the reaction mechanism of the copper-dependent quercetin 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2002;99(26):16625–16630. doi: 10.1073/pnas.262506299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Steiner RA, Kooter IM, Dijkstra BW. Functional analysis of the copper-dependent quercetin 2,3-dioxygenase. 1. Ligand-induced coordination changes probed by X-ray crystallography: inhibition, ordering effect, and mechanistic insights. Biochemistry. 2002;41(25):7955–7962. doi: 10.1021/bi0159736. [DOI] [PubMed] [Google Scholar]
- 80.Steiner RA, Janssen HJ, Roversi P, Oakley AJ, Fetzner S. Structural basis for cofactor-independent dioxygenation of N-heteroaromatic compounds at the alpha/beta-hydrolase fold. Proc Natl Acad Sci U S A. 2010;107(2):657–662. doi: 10.1073/pnas.0909033107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fetzner S. Cofactor-independent oxygenases go it alone. Nat Chem Biol. 2007;3(7):374–375. doi: 10.1038/nchembio0707-374. [DOI] [PubMed] [Google Scholar]
- 82.Thierbach S, Bui N, Zapp J, Chhabra SR, Kappl R, Fetzner S. Substrate-assisted O2 activation in a cofactor-independent dioxygenase. Chem Biol. 2014;21(2):217–225. doi: 10.1016/j.chembiol.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 83.Andreini C, Cavallaro G, Lorenzini S, Rosato A. MetalPDB: a database of metal sites in biological macromolecular structures. Nucleic Acids Res. 2013;41(Database issue):D312–319. doi: 10.1093/nar/gks1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jeoung JH, Bommer M, Lin TY, Dobbek H. Visualizing the substrate-, superoxo-, alkylperoxo-, and product-bound states at the nonheme Fe(II) site of homogentisate dioxygenase. Proc Natl Acad Sci U S A. 2013;110(31):12625–12630. doi: 10.1073/pnas.1302144110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang Y, Colabroy KL, Begley TP, Ealick SE. Structural studies on 3-hydroxyanthranilate-3,4-dioxygenase: the catalytic mechanism of a complex oxidation involved in NAD biosynthesis. Biochemistry. 2005;44(21):7632–7643. doi: 10.1021/bi047353l. [DOI] [PubMed] [Google Scholar]
- 86.Kovaleva EG, Lipscomb JD. Structural basis for the role of tyrosine 257 of homoprotocatechuate 2,3-dioxygenase in substrate and oxygen activation. Biochemistry. 2012;51(44):8755–8763. doi: 10.1021/bi301115c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Waldron KJ, Rutherford JC, Ford D, Robinson NJ. Metalloproteins and metal sensing. Nature. 2009;460(7257):823–830. doi: 10.1038/nature08300. [DOI] [PubMed] [Google Scholar]
- 88.Sunderman FW., Jr Biological monitoring of nickel in humans. Scand J Work Environ Health. 1993;19(Suppl 1):34–38. [PubMed] [Google Scholar]
- 89.Zambelli B, Ciurli S. Nickel and human health. Met Ions Life Sci. 2013;13:321–357. doi: 10.1007/978-94-007-7500-8_10. [DOI] [PubMed] [Google Scholar]
- 90.Gunther L, Berberat PO, Haga M, Brouard S, Smith RN, Soares MP, Bach FH, Tobiasch E. Carbon monoxide protects pancreatic beta-cells from apoptosis and improves islet function/survival after transplantation. Diabetes. 2002;51(4):994–999. doi: 10.2337/diabetes.51.4.994. [DOI] [PubMed] [Google Scholar]
- 91.Liu XM, Chapman GB, Wang H, Durante W. Adenovirus-mediated heme oxygenase-1 gene expression stimulates apoptosis in vascular smooth muscle cells. Circulation. 2002;105(1):79–84. doi: 10.1161/hc0102.101369. [DOI] [PubMed] [Google Scholar]
- 92.Thom SR, Fisher D, Xu YA, Notarfrancesco K, Ischiropoulos H. Adaptive responses and apoptosis in endothelial cells exposed to carbon monoxide. Proc Natl Acad Sci U S A. 2000;97(3):1305–1310. doi: 10.1073/pnas.97.3.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Al-Mjeni F, Ju T, Pochapsky TC, Maroney MJ. XAS investigation of the structure and function of Ni in acireductone dioxygenase. Biochemistry. 2002;41(21):6761–6769. doi: 10.1021/bi012209a. [DOI] [PubMed] [Google Scholar]
- 94.Allpress CJ, Grubel K, Szajna-Fuller E, Arif AM, Berreau LM. Regioselective aliphatic carbon-carbon bond cleavage by a model system of relevance to iron-containing acireductone dioxygenase. J Am Chem Soc. 2013;135(2):659–668. doi: 10.1021/ja3038189. [DOI] [PubMed] [Google Scholar]
- 95.Que L, Jr, Ho RY. Dioxygen Activation by Enzymes with Mononuclear Non-Heme Iron Active Sites. Chem Rev. 1996;96(7):2607–2624. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 96.Crans DC, Yang L, Gaidamauskas E, Kahn R, Jin W, Simonis U. Applications of paramagnetic NMR spectroscopy for monitoring transition metal complex stoichiometry and speciation. In: Telser J, editor. Paramagnetic Resonance of Metallobiomolecules. American Chemical Society; Washington D.C: 2003. ACS Symposium Series 858. [Google Scholar]
- 97.Rajagopalan PT, Datta A, Pei D. Purification, characterization, and inhibition of peptide deformylase from Escherichia coli. Biochemistry. 1997;36(45):13910–13918. doi: 10.1021/bi971155v. [DOI] [PubMed] [Google Scholar]
- 98.Nguyen KT, Wu JC, Boylan JA, Gherardini FC, Pei D. Zinc is the metal cofactor of Borrelia burgdorferi peptide deformylase. Arch Biochem Biophys. 2007;468(2):217–225. doi: 10.1016/j.abb.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.D’Souza VM, Holz RC. The methionyl aminopeptidase from Escherichia coli can function as an iron(II) enzyme. Biochemistry. 1999;38(34):11079–11085. doi: 10.1021/bi990872h. [DOI] [PubMed] [Google Scholar]
- 100.Zhu J, Dizin E, Hu X, Wavreille AS, Park J, Pei D. S-Ribosylhomocysteinase (LuxS) is a mononuclear iron protein. Biochemistry. 2003;42(16):4717–4726. doi: 10.1021/bi034289j. [DOI] [PubMed] [Google Scholar]
- 101.Gattis SG, Hernick M, Fierke CA. Active site metal ion in UDP-3-O-((R)-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC) switches between Fe(II) and Zn(II) depending on cellular conditions. J Biol Chem. 2010;285(44):33788–33796. doi: 10.1074/jbc.M110.147173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gantt SL, Gattis SG, Fierke CA. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry. 2006;45(19):6170–6178. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- 103.Tripp BC, Smith K, Ferry JG. Carbonic anhydrase: new insights for an ancient enzyme. J Biol Chem. 2001;276(52):48615–48618. doi: 10.1074/jbc.R100045200. [DOI] [PubMed] [Google Scholar]
- 104.Porter DJ, Austin EA. Cytosine deaminase. The roles of divalent metal ions in catalysis. J Biol Chem. 1993;268(32):24005–24011. [PubMed] [Google Scholar]
- 105.Seffernick JL, McTavish H, Osborne JP, de Souza ML, Sadowsky MJ, Wackett LP. Atrazine chlorohydrolase from Pseudomonas sp. strain ADP is a metalloenzyme. Biochemistry. 2002;41(48):14430–14437. doi: 10.1021/bi020415s. [DOI] [PubMed] [Google Scholar]
- 106.Van den Wyngaert I, de Vries W, Kremer A, Neefs J, Verhasselt P, Luyten WH, Kass SU. Cloning and characterization of human histone deacetylase 8. FEBS Lett. 2000;478(1–2):77–83. doi: 10.1016/s0014-5793(00)01813-5. [DOI] [PubMed] [Google Scholar]
- 107.Huang X, Kocabas E, Hernick M. The activity and cofactor preferences of N-acetyl-1-D-myo-inosityl-2-amino-2-deoxy-alpha-D-glucopyranoside deacetylase (MshB) change depending on environmental conditions. J Biol Chem. 2011;286(23):20275–20282. doi: 10.1074/jbc.M111.234229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gunther V, Lindert U, Schaffner W. The taste of heavy metals: gene regulation by MTF-1. Biochim Biophys Acta. 2012;1823(9):1416–1425. doi: 10.1016/j.bbamcr.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 109.Helmann JD. Specificity of metal sensing: iron and manganese homeostasis in Bacillus subtilis. J Biol Chem. 2014;289(41):28112–28120. doi: 10.1074/jbc.R114.587071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lockless SW, Zhou M, MacKinnon R. Structural and thermodynamic properties of selective ion binding in a K+ channel. PLoS Biol. 2007;5(5):e121. doi: 10.1371/journal.pbio.0050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.OuYang B, Pochapsky SS, Pagani GM, Pochapsky TC. Specific effects of potassium ion binding on wild-type and L358P cytochrome P450(cam) Biochemistry. 2006;45(48):14379–14388. doi: 10.1021/bi0617355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huang Y, Zhou YB, Castiblanco A, Yang W, Brown EM, Yang JJ. Multiple Ca2+-Binding Sites in the Extracellular Domain of the Ca2+-Sensing Receptor Corresponding to Cooperative Ca2+ Response. Biochemistry. 2009;48(2):388–398. doi: 10.1021/bi8014604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yamniuk AP, Vogel HJ. Calmodulin’s flexibility allows for promiscuity in its interactions with target proteins and peptides. Mol Biotechnol. 2004;27(1):33–57. doi: 10.1385/MB:27:1:33. [DOI] [PubMed] [Google Scholar]
- 114.Hernick M, Gattis SG, Penner-Hahn JE, Fierke CA. Activation of Escherichia coli UDP-3-O-[(R)-3-hydroxymyristoyl]-N-acetylglucosamine deacetylase by Fe2+ yields a more efficient enzyme with altered ligand affinity. Biochemistry. 2010;49(10):2246–2255. doi: 10.1021/bi902066t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kazanis S, Pochapsky TC, Barnhart TM, Pennerhahn JE, Mirza UA, Chait BT. Conversion of a Fe2S2 ferredoxin into a Ga3+ rubredoxin. J Am Chem Soc. 1995;117(24):6625–6626. [Google Scholar]
- 116.Ashraf KU, Josts I, Mosbahi K, Kelly SM, Byron O, Smith BO, Walker D. The Potassium Binding Protein Kbp Is a Cytoplasmic Potassium Sensor. Structure. 2016;24(5):741–749. doi: 10.1016/j.str.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 117.Cocco MJ, Kao YH, Phillips AT, Lecomte JTJ. Structural comparison of apomyoglobin and metaquomyoglobin-pH titration of histidines by NMR spectroscopy. Biochemistry. 1992;31(28):6481–6491. doi: 10.1021/bi00143a018. [DOI] [PubMed] [Google Scholar]
- 118.Foster AW, Pernil R, Patterson CJ, Robinson NJ. Metal specificity of cyanobacterial nickel-responsive repressor InrS: cells maintain zinc and copper below the detection threshold for InrS. Mol Microbiol. 2014;92(4):797–812. doi: 10.1111/mmi.12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Osman D, Piergentili C, Chen J, Sayer LN, Uson I, Huggins TG, Robinson NJ, Pohl E. The Effectors and Sensory Sites of Formaldehyde-responsive Regulator FrmR and Metal-sensing Variant. J Biol Chem. 2016;291(37):19502–19516. doi: 10.1074/jbc.M116.745174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hartwig JF. Organotransition metal chemistry. University Science Books; Mill Valley, CA: 2010. [Google Scholar]
- 121.Basauri-Molina M, Verhoeven DG, van Schaik AJ, Kleijn H, Klein Gebbink RJ. Ring-Closing and Cross-Metathesis with Artificial Metalloenzymes Created by Covalent Active Site-Directed Hybridization of a Lipase. Chemistry. 2015;21(44):15676–15685. doi: 10.1002/chem.201502381. [DOI] [PubMed] [Google Scholar]
- 122.Farwell CC, Zhang RK, McIntosh JA, Hyster TK, Arnold FH. Enantioselective Enzyme-Catalyzed Aziridination Enabled by Active-Site Evolution of a Cytochrome P450. ACS Cent Sci. 2015;1(2):89–93. doi: 10.1021/acscentsci.5b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lewis JC. Artificial Metalloenzymes and Metallopeptide Catalysts for Organic Synthesis. ACS Catal. 2013;3:2954–2975. [Google Scholar]
- 124.Colthart AM, Tietz DR, Ni Y, Friedman JL, Dang M, Pochapsky TC. Detection of substrate-dependent conformational changes in the P450 fold by nuclear magnetic resonance. Sci Rep. 2016;6:22035. doi: 10.1038/srep22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hyster TK, Ward TR. Genetic Optimization of Metalloenzymes: Enhancing Enzymes for Non-Natural Reactions. Angew Chem Int Ed Engl. 2016;55(26):7344–7357. doi: 10.1002/anie.201508816. [DOI] [PubMed] [Google Scholar]
- 126.Park HS, Nam SH, Lee JK, Yoon CN, Mannervik B, Benkovic SJ, Kim HS. Design and evolution of new catalytic activity with an existing protein scaffold. Science. 2006;311(5760):535–538. doi: 10.1126/science.1118953. [DOI] [PubMed] [Google Scholar]
- 127.Seelig B, Szostak JW. Selection and evolution of enzymes from a partially randomized non-catalytic scaffold. Nature. 2007;448(7155):828–831. doi: 10.1038/nature06032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sato H, Hayashi T, Ando T, Hisaeda Y, Ueno T, Watanabe Y. Hybridization of modified-heme reconstitution and distal histidine mutation to functionalize sperm whale myoglobin. J Am Chem Soc. 2004;126(2):436–437. doi: 10.1021/ja038798k. [DOI] [PubMed] [Google Scholar]
- 129.Sreenilayam G, Fasan R. Myoglobin-catalyzed intermolecular carbene N-H insertion with arylamine substrates. Chem Commun (Camb) 2015;51(8):1532–1534. doi: 10.1039/c4cc08753d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bordeaux M, Tyagi V, Fasan R. Highly diastereoselective and enantioselective olefin cyclopropanation using engineered myoglobin-based catalysts. Angew Chem Int Ed Engl. 2015;54(6):1744–1748. doi: 10.1002/anie.201409928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tyagi V, Bonn RB, Fasan R. Intermolecular carbene S-H insertion catalysed by engineered myoglobin-based catalystsdagger. Chem Sci. 2015;6(4):2488–2494. doi: 10.1039/c5sc00080g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bordeaux M, Singh R, Fasan R. Intramolecular C(sp(3))H amination of arylsulfonyl azides with engineered and artificial myoglobin-based catalysts. Bioorg Med Chem. 2014;22(20):5697–5704. doi: 10.1016/j.bmc.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wilson ME, Whitesides GM. Conversion of a protein to a homogeneous asymmetric hydrogenation catalyst by site-specific modification with a diphosphinerhodium(I) moiety. J Am Chem Soc. 1978;100(1):306–307. [Google Scholar]
- 134.Palomo JM, Filice M. New emerging bio-catalysts design in biotransformations. Biotechnol Adv. 2015;33(5):605–613. doi: 10.1016/j.biotechadv.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 135.Sauer DF, Gotzen S, Okuda J. Metatheases: artificial metalloproteins for olefin metathesis. Org Biomol Chem. 2016;14(39):9174–9183. doi: 10.1039/c6ob01475e. [DOI] [PubMed] [Google Scholar]
- 136.Heinisch T, Ward TR. Artificial Metalloenzymes Based on the Biotin-Streptavidin Technology: Challenges and Opportunities. Acc Chem Res. 2016;49(9):1711–1721. doi: 10.1021/acs.accounts.6b00235. [DOI] [PubMed] [Google Scholar]
- 137.Ward TR. Artificial metalloenzymes based on the biotin-avidin technology: enantioselective catalysis and beyond. Acc Chem Res. 2011;44(1):47–57. doi: 10.1021/ar100099u. [DOI] [PubMed] [Google Scholar]
- 138.Ringenberg MR, Ward TR. Merging the best of two worlds: artificial metalloenzymes for enantioselective catalysis. Chem Commun (Camb) 2011;47(30):8470–8476. doi: 10.1039/c1cc11592h. [DOI] [PubMed] [Google Scholar]
- 139.Oohora K, Kihira Y, Mizohata E, Inoue T, Hayashi T. C(sp3)-H bond hydroxylation catalyzed by myoglobin reconstituted with manganese porphycene. J Am Chem Soc. 2013;135(46):17282–17285. doi: 10.1021/ja409404k. [DOI] [PubMed] [Google Scholar]
- 140.Ohashi M, Koshiyama T, Ueno T, Yanase M, Fujii H, Watanabe Y. Preparation of artificial metalloenzymes by insertion of chromium(III) Schiff base complexes into apomyoglobin mutants. Angew Chem Int Ed Engl. 2003;42(9):1005–1008. doi: 10.1002/anie.200390256. [DOI] [PubMed] [Google Scholar]
- 141.Ueno T, Koshiyama T, Ohashi M, Kondo K, Kono M, Suzuki A, Yamane T, Watanabe Y. Coordinated design of cofactor and active site structures in development of new protein catalysts. J Am Chem Soc. 2005;127(18):6556–6562. doi: 10.1021/ja045995q. [DOI] [PubMed] [Google Scholar]
- 142.Ueno T, Ohashi M, Kono M, Kondo K, Suzuki A, Yamane T, Watanabe Y. Crystal structures of artificial metalloproteins: tight binding of FeIII(Schiff-Base) by mutation of Ala71 to Gly in apo-myoglobin. Inorg Chem. 2004;43(9):2852–2858. doi: 10.1021/ic0498539. [DOI] [PubMed] [Google Scholar]
- 143.Carey JR, Ma SK, Pfister TD, Garner DK, Kim HK, Abramite JA, Wang Z, Guo Z, Lu Y. A site-selective dual anchoring strategy for artificial metalloprotein design. J Am Chem Soc. 2004;126(35):10812–10813. doi: 10.1021/ja046908x. [DOI] [PubMed] [Google Scholar]
- 144.Satake Y, Abe S, Okazaki S, Ban N, Hikage T, Ueno T, Nakajima H, Suzuki A, Yamane T, Nishiyama H, Watanabe Y. Incorporation of a Phebox Rhodium Complex into apo-Myoglobin Affords a Stable Organometallic Protein Showing Unprecedented Arrangement of the Complex in the Cavity. Organometallics. 2007;26(20):4904–4908. [Google Scholar]
- 145.Kokubo T, Sugimoto T, Uchida T, Tanimoto S, Okano M. The bovine serum albumin–2-phenylpropane-1,2-diolatodioxo-somium(VI) complex as an enantioselective catalyst for cis-hydroxylation of alkenes. J Chem Soc, Chem Commun. 1983;(14):769–770. [Google Scholar]
- 146.Bertucci C, Botteghi C, Giunta D, Marchetti M, Paganelli S. Aqueous Biphasic Hydroformylation Catalysed by Protein-Rhodium Complexes. Adv Synth Catal. 2002;344(5):556–562. [Google Scholar]
- 147.Mahammed A, Gray HB, Weaver JJ, Sorasaenee K, Gross Z. Amphiphilic corroles bind tightly to human serum albumin. Bioconjug Chem. 2004;15(4):738–746. doi: 10.1021/bc034179p. [DOI] [PubMed] [Google Scholar]
- 148.van de Velde F, Arends IW, Sheldon RA. Vanadium-catalysed enantioselective sulfoxidations: rational design of biocatalytic and biomimetic systems. Top Catal. 2000;13:259–265. [Google Scholar]
- 149.Levine HL, Nakagawa Y, Kaiser ET. Flavopapain: synthesis and properties of semi-synthetic enzymes. Biochem Biophys Res Commun. 1977;76(1):64–70. doi: 10.1016/0006-291x(77)91668-0. [DOI] [PubMed] [Google Scholar]
- 150.Deuss PJ, Popa G, Botting CH, Laan W, Kamer PC. Highly efficient and site-selective phosphane modification of proteins through hydrazone linkage: development of artificial metalloenzymes. Angew Chem Int Ed Engl. 2010;49(31):5315–5317. doi: 10.1002/anie.201002174. [DOI] [PubMed] [Google Scholar]
- 151.Talbi B, Haquette P, Martel A, de Montigny F, Fosse C, Cordier S, Roisnel T, Jaouen G, Salmain M. (Eta6-arene) ruthenium(II) complexes and metallo-papain hybrid as Lewis acid catalysts of Diels-Alder reaction in water. Dalton Trans. 2010;39(24):5605–5607. doi: 10.1039/c001630f. [DOI] [PubMed] [Google Scholar]
- 152.Reetz MT. Directed evolution of selective enzymes and hybrid catalysts. Tetrahedron. 2002;58(32):6595–6602. [Google Scholar]
- 153.Panella L, Broos J, Jin J, Fraaije MW, Janssen DB, Jeronimus-Stratingh M, Feringa BL, Minnaard AJ, de Vries JG. Merging homogeneous catalysis with biocatalysis; papain as hydrogenation catalyst. Chem Commun (Camb) 2005;(45):5656–5658. doi: 10.1039/b512138h. [DOI] [PubMed] [Google Scholar]
- 154.Kruithof CA, Casado MA, Guillena G, Egmond MR, van der Kerk-van Hoof A, Heck AJ, Klein Gebbink RJ, van Koten G. Lipase active-site-directed anchoring of organometallics: metallopincer/protein hybrids. Chemistry. 2005;11(23):6869–6877. doi: 10.1002/chem.200500671. [DOI] [PubMed] [Google Scholar]
- 155.Dijkstra HP, Sprong H, Aerts BN, Kruithof CA, Egmond MR, Klein Gebbink RJ. Selective and diagnostic labelling of serine hydrolases with reactive phosphonate inhibitors. Org Biomol Chem. 2008;6(3):523–531. doi: 10.1039/b717345h. [DOI] [PubMed] [Google Scholar]
- 156.Monnard FW, Heinisch T, Nogueira ES, Schirmer T, Ward TR. Human carbonic anhydrase II as a host for piano-stool complexes bearing a sulfonamide anchor. Chem Commun (Camb) 2011;47(29):8238–8240. doi: 10.1039/c1cc10345h. [DOI] [PubMed] [Google Scholar]
- 157.Monnard FW, Nogueira ES, Heinisch T, Schirmer T, Ward TR. Human carbonic anhydrase II as host protein for the creation of artificial metalloenzymes: the asymmetric transfer hydrogenation of imines. Chem Sci. 2013;4:3269–3274. [Google Scholar]
- 158.Zhao J, Kajetanowicz A, Ward TR. Carbonic anhydrase II as host protein for the creation of a biocompatible artificial metathesase. Org Biomol Chem. 2015;13:5652–5655. doi: 10.1039/c5ob00428d. [DOI] [PubMed] [Google Scholar]
- 159.Key HM, Clark DS, Hartwig JF. Generation, Characterization, and Tunable Reactivity of Organometallic Fragments Bound to a Protein Ligand. J Am Chem Soc. 2015;137(25):8261–8268. doi: 10.1021/jacs.5b04431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Reetz MT, Rentzsch M, Pletsch A, Taglieber A, Hollmann F, Mondiere RJ, Dickmann N, Hocker B, Cerrone S, Haeger MC, Sterner R. A robust protein host for anchoring chelating ligands and organocatalysts. Chembiochem. 2008;9(4):552–564. doi: 10.1002/cbic.200700413. [DOI] [PubMed] [Google Scholar]
- 161.Davies RR, Distefano MD. A Semisynthetic Metalloenzyme Based on a Protein Cavity That Catalyzes the Enantioselective Hydrolysis of Ester and Amide Substrates. J Am Chem Soc. 1997;119(48):11643–11652. [Google Scholar]
- 162.Abe S, Ebihara T, Enomoto S, Furuno K, Gando Y, Ichimura K, Ikeda H, Inoue K, Kibe Y, Kishimoto Y, et al. Precision measurement of neutrino oscillation parameters with KamLAND. Phys Rev Lett. 2008;100(22):221803. doi: 10.1103/PhysRevLett.100.221803. [DOI] [PubMed] [Google Scholar]
- 163.Abe S, Hirata K, Ueno T, Morino K, Shimizu N, Yamamoto M, Takata M, Yashima E, Watanabe Y. Polymerization of phenylacetylene by rhodium complexes within a discrete space of apo-ferritin. J Am Chem Soc. 2009;131(20):6958–6960. doi: 10.1021/ja901234j. [DOI] [PubMed] [Google Scholar]
- 164.Abe S, Hikage T, Watanabe Y, Kitagawa S, Ueno T. Mechanism of accumulation and incorporation of organometallic Pd complexes into the protein nanocage of apo-ferritin. Inorg Chem. 2010;49(15):6967–6973. doi: 10.1021/ic1003758. [DOI] [PubMed] [Google Scholar]
- 165.Ueno T, Abe M, Hirata K, Abe S, Suzuki M, Shimizu N, Yamamoto M, Takata M, Watanabe Y. Process of accumulation of metal ions on the interior surface of apo-ferritin: crystal structures of a series of apo-ferritins containing variable quantities of Pd(II) ions. J Am Chem Soc. 2009;131(14):5094–5100. doi: 10.1021/ja806688s. [DOI] [PubMed] [Google Scholar]
- 166.Yamamura KaK ET. Studies on the oxidase activity of copper(II) carboxypeptidase A. J Chem Soc Chem Commun. 1976;(20):830–831. [Google Scholar]
- 167.Okrasa K, Kazlauskas RJ. Manganese-substituted carbonic anhydrase as a new peroxidase. Chemistry. 2006;12(6):1587–1596. doi: 10.1002/chem.200501413. [DOI] [PubMed] [Google Scholar]
- 168.Jing Q, Okrasa K, Kazlauskas RJ. Stereoselective hydrogenation of olefins using rhodium-substituted carbonic anhydrase--a new reductase. Chemistry. 2009;15(6):1370–1376. doi: 10.1002/chem.200801673. [DOI] [PubMed] [Google Scholar]
- 169.Fernandez-Gacio A, Codina A, Fastrez J, Riant O, Soumillion P. Transforming carbonic anhydrase into epoxide synthase by metal exchange. Chembiochem. 2006;7(7):1013–1016. doi: 10.1002/cbic.200600127. [DOI] [PubMed] [Google Scholar]
- 170.Hoffman RM. Development of recombinant methioninase to target the general cancer-specific metabolic defect of methionine dependence: a 40-year odyssey. Expert Opin Biol Ther. 2015;15(1):21–31. doi: 10.1517/14712598.2015.963050. [DOI] [PubMed] [Google Scholar]