

Abstract
Carbon-11 labelled carbon dioxide is the cyclotron-generated feedstock reagent for most positron emission tomography (PET) tracers using this radionuclide. Most carbon-11 labels, however, are installed using derivative reagents generated from [11C]CO2. In recent years, [11C]CO2 has seen a revival in applications for the direct incorporation of carbon-11 into functional groups such as ureas, carbamates, oxazolidinones, carboxylic acids, esters, and amides. This review summarizes classical [11C]CO2 fixation strategies using organometallic reagents and then focuses on newly developed methods that employ strong organic bases to reversibly capture [11C]CO2 into solution, thereby enabling highly functionalized labelled compounds to be prepared. Reactions and radiopharmaceuticals that have been translated to the clinic are highlighted.
1. Introduction
Carbon nuclei make up all organic entities and are the most versatile sites for labelling biological molecules of interest without altering their chemical and/or biological profiles. Carbon-11, a nearly pure positron emitter (t½ = 20.38 min, Eavg(β+) = 0.39 MeV) is used extensively for developing radiotracers for positron emission tomography (PET), a non-invasive molecular imaging technique. The development of PET radiopharmaceuticals may provide an ideal methodology to enable diagnosis, monitor disease progression, and evaluate drug therapies in vivo without eliciting a pharmacological response.1 In addition to serving as a clinical tool for disease diagnosis, PET is increasingly relevant for drug development as it can provide quantitative pharmacokinetic, biodistribution, and receptor occupancy data for a drug candidate. While the longer half-life of fluorine-18 (t½ = 109.77 min, Eavg(β+) = 0.25 MeV) offers advantages for synthesis, longer imaging times and mulit-centre trials, carbon-11 can more frequently be substituted into biological molecules without causing chemical alterations that could influence the outcomes of imaging studies. Furthermore, repeat imaging studies in the same subject over a short duration are possible with carbon-11 due to its shorter half-life.
Dozens of reagents have been employed for incorporation of no-carrier-added carbon-11 ([11C]) nuclei in radiotracers.2 Still, the most common strategy used is methylation using [11C]methyl iodide or [11C]methyl triflate.3,4 Carbon-11 labelled carbon dioxide is the product of proton bombardment of nitrogen-14 in the presence of small amounts of oxygen by the 14N(p,α)11C nuclear reaction. Most often, [11C]CO2 is transformed to more reactive species, such as the aforementioned methylating reagents, to facilitate radiolabelling. However, [11C]CO2 itself is an attractive starting material for radiochemists as it is produced directly from the cyclotron in high specific activity. Its use also promises access to high oxidation state functional groups such as carboxylic acids, amides, ureas, carbamates, oxazolidinones and their derivatives without resorting to redox manipulations during radiotracer synthesis. Alternatively, [11C]phosgene (produced from [11C]CO25,6 or [11C]methane7-10) has been used to prepare such functional groups, though it has seen limited adoption due to technical challenges required for its routine production and use.11 As described in a recent review, the high reactivity of [11C]COCl2 has been exploited to generate 11C-labelled intermediates such as isocyanates and carbamoyl chlorides from amines, and chloroformates from alcohols, en route to [11C]ureas, [11C]carbamates and alkyl [11C]carbonates.12 Carbon-11 labelled carbon monoxide, produced by reduction of [11C]CO2,13 has been more widely employed for preparation of amides, esters, ureas, carbamates and acids using transition metal or selenium-mediated reactions or photoinitiated radical methods.11 Research has focused on overcoming the low solubility of [11C]CO by employing micro-autoclaves,14 sequestration reagents such as borane,14 microfluidics,16-18 or soluble Xe(g) carrier.19 This topic has been reviewed elsewhere.2,11,20
The low chemical reactivity of carbon dioxide poses a challenge for direct incorporation into organic molecules. Carbon dioxide generally requires highly reactive nucleophiles or catalysts to effect covalent bond formation and typically large excesses of CO2 are used in industrial scale processes with this feedstock.21 In contrast, [11C]CO2 is the limiting reagent (10 – 100 fold excess of precursor) when used in radiochemical transformations. The low amounts of [11C]CO2 in nitrogen carrier gas obtained from the cyclotron target (typically ∼100 nmol) necessitate an efficient CO2-trapping solution. Carbon-11 CO2 reactions are further complicated by the presence of oxygen (1%) and byproduct nitric oxides in the target gas mixture that often require gas purification steps, particularly if transition metals or sensitive catalysts are to be used. Two predominant strategies exist for purification of [11C]CO2 from target gas. Cryogenic purification consists of depositing the mixture in a vessel cooled by liquid nitrogen. Non-condensable gases are thereby removed, while condensable impurities (such as NOx species) can be removed using chemical traps.22 A second approach is to immobilize the carbon dioxide on a solid material (e.g. molecular sieves such as CarboSpheres23). After stripping off undesired impurities, the CO2 is thermally released from the trap into the reactor. In contrast to [11C]COCl2 and [11C]CO, no additional chemical steps involving elaborate apparatus are required for preparation of [11C]CO2. However, as with any gaseous reagents with short half-lives (e.g., [11C]CO2, [11C]CO, [11C]COCl2, [11C]HCN, [11C]CH3I, [11C]CH3OTf, and [18F]F2) apparatus must be well designed and constructed to allow efficient handling, and be leak-proof. Flow rates must also be appropriate for rapid gas trapping in small volume solutions. Any traps, additives or fixation bases should avoid contamination of the isotope with impurities that could jeopardize the incorporation reaction. Many CO2-fixations proceed rapidly at room temperature and ambient pressure. The reaction set-ups are generally very simple, which contributes to good reproducibility of syntheses and high radiochemical yields relative to starting [11C]CO2 activity are achievable with high specific activity (3 – 6 Ci·μmol-1). CO2 is amenable to transformations into commonly found functional groups, and compounds have been recently advanced for first-in-human trials using [11C]CO2 fixation (vide infra). The purpose of this review is to highlight prominent examples of [11C]CO2 fixation that have been used to expand the chemical scope of labelled compounds and radiopharmaceuticals.
2. [11C]CO2 fixation by basic organometallic reagents
2.1 Grignard reagents
Carbon-11 labelled CO2 has been in use for preparation of labelled carboxylic acids since at least the 1940s (Scheme 1A).24 Radiolabelled amides have also been synthesized by [11C]CO2 fixation with Grignard reagents. Heating the magnesium carboxylate intermediates in the presence of primary or secondary amines has been reported to produce the corresponding [11C]carboxyamides.25 The product amides have also been subsequently reduced in the presence of sodium borohydride, giving access to [11C]tertiary amines.26 More indirectly, carboxylation can be followed by activation using reagents such as thionyl chloride or phthaloyl dichloride to prepare active acylation intermediates such as [11C]acetyl and [11C]propionyl chloride.27-30 While Grignard reagents have proven useful in the synthesis of simple [11C]carboxylic acids and a selection of their derivatives, their high reactivity inherently limits the potential scope of their applications and enforces requirements for careful handling procedures. Since Grignard reagents readily absorb CO2 from the atmosphere and are moisture-sensitive they are ideally prepared fresh and manipulated under an inert atmosphere using anhydrous solvents to obtain high specific activities and reproducibility. The propensity for magnesium salts to precipitate from solution poses challenges for automated synthesis and can necessitate delicate and time consuming filtration steps.
Scheme 1.

11CO2-fixations using strongly basic organometallic reagents such as Grignard reagents (A); organolithium reagents (B and C); and silanamines (D). TMS: trimethylsilyl; asterisk denotes 11C.
2.2 Organolithiums
In the presence of excess methyllithium [11C]CO2 is readily transformed to the dilithium salt of acetone acetal. Hydrolysis leads to [2-11C]acetone, which is a useful intermediate for radiochemistry (Scheme 1B).31-33 Reports using other organolithiums to trap [11C]CO2 have been relatively scarce. [11C]Pyruvic acid was prepared by tandem methylation-carboxylation of an isocyanide in the presence of methyllithium and [11C]CO2, followed by hydrolysis.34 Also of note are the syntheses of radiolabelled glycine and two derivative dipeptides using the lithium salt of methylisocyanide. After [11C]CO2-fixation the isocyanide is hydrolyzed through the formamide to the amine, yielding [1-11C]glycine (Scheme 1C).35 This species could be coupled to an intramolecularly activated amino acid equivalent under basic conditions.
2.3 Silanamines
Silylated amines were developed in the 1980s as fixating agents for [11C]CO2. This work was performed by Ram and co-workers and was applied to various targets including imipramine,36 chlorpromazine,37 SCH-23390,38 and tamoxifen.39 Silanamines were prepared up to one week in advance of radiosynthesis by refluxing a secondary amine with hexamethyldisilazane (HMDS), often in the presence of catalytic quantities of ammonium sulfate. When exposed to [11C]CO2 at elevated temperatures, O-silyl carbamates were produced, which were subsequently reduced in situ by LiAlH4 to produce labelled tertiary methylamines (Scheme 1D).40 Since silyl groups are electron-donating, the silanamine is rendered more nucleophilic, despite the added steric bulk and weak N-Si bond strength.
The above methods all require the use of an unstable organometallic/organosilicon species for [11C]CO2 fixation. These reagents are often quite reactive and pose obstacles to reproducibility and automation in a radiochemical setting. Naturally, they also present major chemoselectivity issues during the preparation of PET radiotracers with complex functionalities, often limiting their utility to synthesis of simple prosthetic groups or very small molecules with restricted functionality. These approaches have been previously reviewed41 and stand in contrast to the recent developments described below.
3. Recently developed methods of [11C]CO2 fixation
Over the past 20 years, and under the impetus of “green” chemistry, approaches to CO2 fixation have expanded tremendously.42 Among the most commonly used fixation agents are guanidines,43 amidines,44,45 and alkali carbonates,46 the last of which are not suitable for [11C]CO2 fixation. Though each of these bases facilitates transamidation of an added amine, the nature of products of CO2 trapped by guanidines or amidines have been the subject of some debate. Initial spectroscopic and crystallographic studies suggested that bicarbonate salts are formed in the presence of adventitious water, perhaps by hydrolysis of carbamic intermediates.43,47-49 More recently, the carbamate anion has been confirmed crystallographically in the context of a bicyclic guanidine.50 Practically, Hooker et al. demonstrated that an amidine, diazabicyclo[5.4.0]undec-7-ene (DBU), was highly efficient at trapping cyclotron-produced [11C]CO2 at ambient temperature and pressure and practical flow rates.51 Wilson et al. later showed that 2-tert-butylimino-2-diethylamino-1,3-dimethyl-perhydro-1,3,2-diazaphosphorine (BEMP) was even more effective in this regard.52 This strategy succeeds by decoupling the [11C]CO2 capture in solution from subsequent covalent bond formation with substrate.
Transition metals have been extensively used for both CO2 trapping in solution and as fixation catalysts for carbon-carbon bond formation.53,54 The mode and efficiency of trapping are likely to be dependent on the nature of the metal centre, as well as the ligand and solvent systems. These topics have been the subjects of reviews in recent years.55,56
4. Functional groups prepared by [11C]CO2 fixation
4.1 Urea
4.1.1 Symmetrical ureas
While early examples of [11C]CO2 fixation centred around formation of carboxylic acids and amides, higher oxidation state functional groups such as ureas and carbamates have become research targets in the past 20 years. Chakraborty et al. reported the first synthesis of [11C]urea by bubbling [11C]CO2 through a THF solution of LHMDS, followed by hydrolysis using aqueous ammonium chloride (Scheme 2A).57 [11C]Urea could then be condensed with diethyl malate in sulfuric acid to yield [11C]uracil.
Scheme 2.

Synthesis of [11C]ureas by [11C]CO2-fixation. (A) Synthesis of parent [11C]urea using LHMDS and aqueous ammonium chloride. (B) Synthesis of symmetrical and unsymmetrical substituted [11C]ureas by activation of an ammonium carbamate intermediate with POCl3. The utility of this reaction was greatly expanded by judicious control of reaction conditions to favour unsymmetrical products. TEA: triethylamine; TMS: trimethylsilyl; asterisk denotes 11C.
The synthesis of both ureas and carbamates from carbon dioxide often requires CO2 to react twice as an electrophile. While we have seen above that in the presence of strong nucleophiles carbon dioxide will react as an electrophile, the resulting carboxylate or carbamate is not electrophilic. It is for this reason that [11C]phosgene has previously been used to prepare labelled ureas.12 An alternative approach is to utilize activating reagents such as phosphoryl chloride or thionyl chloride to generate an acid chloride or isocyanate, which are both highly electrophilic. In its earliest iterations, this strategy was applied to prepare [11C-carbonyl]phenyl isocyanate, which promptly reacted further to form both [11C]diphenyl urea and [11C]diphenyl carbodiimide (Scheme 2B).58 While observation of these products is clear evidence of the desired reactivity, they also illustrate the practical challenges in harnessing it for preparation of unsymmetrical ureas in high radiochemical yield.
4.1.2 Unsymmetrical ureas
By using dilute and carefully balanced solutions of trapping amine, POCl3, and nucleophile, unsymmetrical [11C]ureas can be produced with high selectivity (Scheme 2B).59 Simply using a large excess of POCl3 was found to suppress attack of remaining trapping amine on the 11C-isocyanate generated in situ, but also required even larger excess of the nucleophile, which is also consumed. This results in a high concentration reaction mixture and consequent challenging purification. Fortunately, it was found that the concentration of trapping amine could be reduced without negatively impacting the yield or prolonging the reaction time beyond two minutes. Aliphatic primary amines are ideal substrates for isocyanate formation, while aniline reacts sluggishly.59 Cyclic secondary amines are also well tolerated, presumably through an [11C]carbamoyl chloride intermediate, though more hindered secondary amines must be used in higher concentrations to achieve useful conversions. The scope of amine nucleophiles for attack on [11C]isocyanates or [11C]carbamoyl chlorides was limited primarily to dimethylamine for the discovery investigations, but high yields were also obtained with 4-(2-methoxyphenyl)piperazine.59
Inhibitors of glycogen synthase kinase 3β(GSK-3β) are currently under exploration for diverse cancers and neurological illnesses, with the “GSK-3 hypothesis of Alzheimer's disease” sparking further medicinal chemistry efforts in this area. Vasdev et al. prepared the first PET imaging agent for this target, [11C-methoxy]AR-A014418.60 The room temperature synthesis of an isotopologue of this unsymmetrical urea, [11C-carbonyl]AR-A014418, demonstrates an [11C]CO2 fixation approach to this tracer.61 In this case the terminal nucleophile was the aromatic 2-amino-5-nitrothiazole. This is the first successful example of aromatic amines being employed in this role. The total time for synthesis and formulation was 28 minutes, and we were able to prepare 70 ± 33 mCi of the tracer in 8.3 ± 3.9% uncorrected radiochemical yield (RCY) relative to starting [11C]CO2 with high (4.0 ± 1.1 Ci·μmol-1) specific activity at the end of synthesis (Figure 1). Though we found AR-A014418 to be less potent than initially reported, this methodology affords a general approach to synthesize arrays of 11C-labelled urea-based GSK-3β inhibitors, which would not be possible using standard 11C-methylation strategies.
Fig. 1.
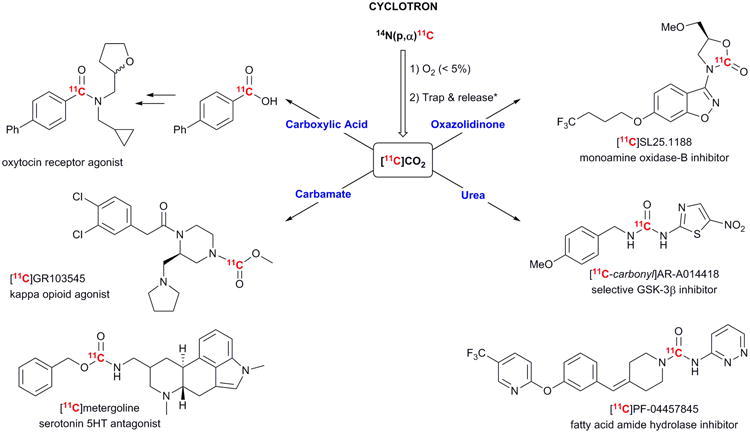
Cyclotron produced [11C]CO2has been recently applied to radiolabelling of structurally complex carboxylic acids and amides, carbamates, oxazolidinones, and ureas. *Trapping is typically done with molecular sieves or liquid nitrogen and release Is achieved by heating.
Pfizer's potent and irreversible fatty acid amide hydrolase (FAAH) inhibitor, PF-04457845, has advanced to clinical trials and has generated recent interest as a scaffold for PET radiotracer development.62 Whereas a [18F]fluoroethyl derivative, [18F]PF-9811, was recently reported,63 we have prepared an isotopologue by [11C]CO2 fixation, namely, [11C-carbonyl]PF-04457845 (Figure 1). The precursors for [11C]CO2 fixation are a non-nucleophilic primary aromatic amine and a cyclic secondary aliphatic amine. To compensate for the higher reactivity of the latter compound, the aromatic amine was employed in 20-fold excess relative to the secondary amine. [11C]CO2 was bubbled into the vial containing the precursors and BEMP in anhydrous CH3CN. POCl3 was later added, followed by an aqueous quench and purification to provide [11C-carbonyl]PF-04457845 in 4.5 ± 1.3% uncorrected radiochemical yield (Figure 1).64 The total synthesis time was 25 ± 2 min and the product had a specific activity of 75.5 ± 8.2 GBq·μmol-1. All operations were performed at room temperature. In contrast to the preparation of [11C]AR-A014418, both amines were present in the reaction mixture during [11C]CO2 fixation. Presumably, the reactive intermediate is a [11C]carbamoyl chloride or a mixed phosphate [11C]anhydride. Our promising preclinical results, coupled with the known pharmacology and toxicology of PF-04457845, should facilitate clinical translation of this radiotracer.
An alternative approach to [11C]isocyanates is by the condensation of phosphinimines with [11C]CO2. Phosphinimines can be prepared from azides or primary amines, and have varying degrees of stability. Commercially available phenyltriphenylphosphinimine was used for [11C]urea preparation with aliphatic and aromatic amines.65 The radionuclide was trapped in a solution of THF at -60 °C in the presence of the phosphinimine and amine. The reaction was heated to 60 °C for 6 minutes to complete the synthesis (Scheme 3). By radio-HPLC, aliphatic amines gave decay corrected unpurified radiochemical conversion yields ranging from 45 – 49%, while aniline unsurprisingly gave a lower yield of 8 ± 1%.
Scheme 3.

Synthesis of [11C]phenylisocyanate and [11C]ureas by fixation with N-phenyl(triphenylphosphin)imine. Asterisk denotes 11C.
4.2 Carbamate
4.2.1 Syntheses using alkyl electrophiles
Similar to ureas, carbamates are an attractive functional group for radiolabelling due to their stability in vivo and role as a versatile linker for ligand fragments. This functional group has previously been labelled using [11C]phosgene and [11C]carbon monoxide,11 so a synthetic strategy based around [11C]CO2 would have extensive applications for the development of PET radiopharmaceuticals. In contrast to ureas, syntheses of O-alkyl carbamates do not necessarily require activation of the carbamate salt intermediate if reactive electrophiles are used. This strategy was exemplified by employing benzyl chloride in a solution of DBU, benzylamine, DMF and bubbled [11C]CO2 (Scheme 4).51 The total reaction time was 10 minutes at 75 °C and achieved a radiochemical yield of 85%. Similar to some of the urea results discussed above, reducing the concentration of benzylamine in this reaction was better tolerated than reducing the concentrations of other reagents. Secondary and sterically hindered amines gave good yields, while anilines were much less reactive. The best electrophiles were benzyl and allyl chlorides since analogous bromides displayed undesired reactivity with amines and DBU. n-Pentyl bromide showed desired reactivity, while more hindered aliphatic electrophiles were troublesome. This technology was used to prepare [11C]metergoline in 32% decay corrected radiochemical yield (RCY) in high specific activity (up to 5 Ci·μmol-1) in a single step. [11C]MS-275, an isotopologue of the histone deacetylase inhibitor entinostat, was also prepared in this way and showed poor blood-brain barrier penetration in nonhuman primates.66 The tracer was synthesized in 25% decay corrected RCY with specific activities of 2.7 – 6.2 Ci· μmol-1. Synthesis and purification required 50 min and afforded > 20 mCi per synthesis.
Scheme 4.

[11C-carbonyl]Carbamates can be synthesized by nucleophilic alkylation of the trapped [11C]CO2 followed by amine substitution (left), or through activation of an intermediate carbamate salt using POCl3 (right). DBU: 1,8-Diazabicyclo[5.4.0]undec-7-ene; Q: fixation base; R: H, phenyl, vinyl, alkyl; X: Cl, Br, I, OTs, sulfate; asterisk denotes 11C.
Methyl [11C-carbonyl]carbamates have also been prepared following a similar strategy with methylating agents such as dimethylsulfate (DMS), methyl iodide (CH3I), or methyl tosylate (CH3OTs) (Scheme 4).52 In this case, BEMP in DMF was found to be the best trapping solution for [11C]CO2. The reactions proceeded with primary or secondary amines. Electron rich 4-methoxyaniline was also tolerated, while acceptable yields of electron deficient 4-nitroaniline could be achieved with increased amine concentration. The reactions also proceeded very rapidly, with maximum yields being reached using one minute of mixing prior to addition of the methylating agent and only 10 seconds afterwards. Increasing the concentration of methylating agent had a negative effect on radiochemical yield. Reversing the order of addition (i.e., first DMS, followed by amine) reportedly produced very low RCYs of product, suggesting the intermediacy of the carbamate salt.
The utility of this process was demonstrated by labelling GR103545, a selective high affinity agonist for the κ-opioid receptor, by [11C]CO2 fixation. The methyl carbamate has previously been labelled using [11C]methylchloroformate,67 phosgene with [11C]CH3OH,68 and both [11C]CH3I68 and [11C]CH3OTf70 with cold CO2 fixation. In this iteration, the “loop” method was employed by lining the inside of a narrow-bore steel tube with a solution of the precursor and BEMP in DMF. Without requiring heating, 2.6 – 3.8 GBq of 108 – 162 GBq·μmol-1 [11C-carbonyl]GR103545 (Figure 1) was produced using 0.1 mg of precursor, an uncorrected 13% RCY at end-of-synthesis (EOS).52 The total synthesis time was 23 minutes.
4.2.2 Syntheses via isocyanates
[11C-carbonyl]Carbamates have also been produced through [11C]isocyanate intermediates.59 Again, appropriate stoichiometry must be selected to prevent symmetrical [11C]urea formation. The isocyanate is quenched with an alcohol to affix the desired O-substituent. While methanol and phenols were originally deployed, in our most recent work we have also successfully used ethanol, isopropanol, and even tert-butanol and hexafluoroisopropanol. It is possible that the reaction is assisted by the strongly basic conditions established by the presence of BEMP.
The synthesis and function of [11C]CURB (Scheme 5A) is illustrative of the importance of this labelling strategy. FAAH is a serine hydrolase that regulates levels of anandamide, an endocannabinoid, in the central nervous system. FAAH is found heterogeneously throughout the brain, and is an attractive target in studies of addiction, obesity, pain, anxiety, and eating disorders.71-73 The O-aryl carbamate scaffold has shown to be an effective one for design of irreversible inhibitors of FAAH, as the serine-241 residue in the active site has been shown to attack the carbonyl of the carbamate, releasing a phenolic fragment (Scheme 5B). With the potential for structure-activity relationship (SAR) studies in mind, it can be appreciated that the conserved carbamate carbonyl is an ideal position for placing a radiolabel.
Scheme 5.

(A) Radiosynthesis of [11C]CURB; (B) Mechanism of FAAH radiolabelling with [11C]CURB; the radiolabel must be placed on the carbonyl or N-alkyl fragment of the tracer to effectively measure levels of FAAH in the brain. Asterisk denotes 11C.
[11C]CURB was prepared by [11C]CO2 fixation using cyclohexylamine and BEMP, followed by [11C]isocyanate formation with POCl3.59 2-Phenyldihydroquinone was used to quench the isocyanate, forming a mixture of regioisomers which are separated by HPLC. The radiotracer was isolated in 8% uncorrected RCY with a specific activity of 2.5 Ci·μmol-1 in 27 minutes from end-of-bombardment (EOB). [11C]CURB shows high brain penetration in rats, and selectivity was ascertained by blocking studies with a known FAAH inhibitor.74 The radiotracer has been translated to human use.75 To optimize radiotracer pharmacokinetics, an SAR study was performed using a small library of O-aryl carbamates. Eight [11C]carbamates were prepared in a manner analogous to [11C]CURB.76 Each of the radiotracers demonstrated brain uptake and specificity for FAAH in conscious rodents. Kinetic analysis in rats showed that [11C]dihydrooxazole carbamates had greater brain uptake, lower non-specific binding, and faster binding to FAAH than the [11C]biphenyl carbamates, such as [11C]CURB. The results of the SAR study and the human kinetic studies using [11C]CURB will allow for design and selection of the optimal FAAH radiotracer.
A camptothecin derivative that displays potent antitumor activity, irinotecan, was radiolabelled with carbon-11.77 Kawamura et al. prepared [11C]irinotecan both by direct [11C]CO2 fixation and using [11C]COCl2 derived from [11C]CO2 by way of [11C]CH4 and [11C]CCl4. Using [11C]CO2 directly, the decay corrected RCY was 16.9 ± 2.9% with specific activity 35 min from EOB of 78 – 136 GBq·μmol-1. Using [11C]COCl2, the decay corrected RCY was 8.8 ± 2.0% with specific activity 35 min from EOB of 77 – 197 GBq·μmol-1. The tracer was used to perform metabolite analysis of the drug in mice.
4.3 Oxazolidinone
[11C]SL25.1188 (Figure 1) was developed as a reversibly binding radiotracer for monoamine oxidase-B (MAO-B). The radiolabel is placed as an [11C-carbonyl]oxazolidinone. The original synthesis employed [11C]phosgene, and suffered from a low 3.5 – 7% decay-corrected RCY with 50 – 70 GBq·μmol-1 specific activity after a 30 – 32 minutes.78 We recently reported the application of the amino-alcohol precursor for [11C]CO2 fixation followed by dehydration and intramolecular cyclization to improve the radiosynthesis.79 After optimization of the fixation base, dehydrating agent, and component concentrations, conditions were developed to perform the labelling at ambient temperature using BEMP and POCl3. The uncorrected RCY was 11.5 ± 0.9% to prepare 98 ± 8 mCi of the radiotracer with 1.0 ± 0.05 Ci·μmol-1 specific activity after a 30 min synthesis. This tracer has since been successfully translated for human use and specific activities of 3.7 ± 0.8 Ci·μmol-1 have been achieved by us (unpublished work).
4.4 Carboxylic Acid
Transition metal-mediated carboxylation of organic compounds has seen significant development in part due to the reputation of CO2 as an environmentally benign feedstock. In particular, catalytic and even metal-free conditions using prefunctionalized organoboron and organozinc reagents have been the focus of much attention.80-82 The advantages these reagents hold over Grignard and organolithium compounds are their functional group tolerance and relative stability to storage under ambient conditions. Boronic esters have recently been applied for [11C]CO2 fixation using a copper catalyst.83 Due to low concentration of [11C]CO2 available in the reaction mixture, which is in sharp contrast to isotopically unenriched CO2-fixation conditions, the reaction parameters required significant optimization efforts. Since alkoxide bases bound [11C]CO2 tightly, TMEDA was employed as both fixation base and presumably a ligand for the copper catalyst. A soluble fluoride additive was also found to dramatically improve RCY. With optimized conditions at 90 – 100 °C, various arylboronic esters were efficiently converted to [11C]carboxylic acids (Scheme 6). Halide, formyl, cyano, and nitro substituents were well tolerated, while protic substituents such as hydroxyl and amino groups, and electron poor heterocycles were challenging substrates. Examples of alkyl, vinyl and alkynyl substrates undergoing [11C]carboxylation were also reported.
Scheme 6.

Cu(I) catalyzes11C-carboxylation of boronic acid esters. The products can be elaborated to 11C-esters and 11C-amides. TMEDA: N,N,N′,N′-tetramethylethylenediamine; asterisk denotes 11C.
The [11C]benzoic acids were also elaborated to a methyl ester (using iodomethane), an amide (via an acid chloride), and a succinic ester (via carbodiimide activation).83 An [11C]amide-labelled oxytocin receptor ligand84 was also reported in 20% decay-corrected RCY with 1.5 Ci·μmol-1 specific activity over 43 minutes synthesis time. Access to various [11C]carboxylic acid derivatives through regiospecific metal catalyzed carboxylation is sure to be widely exploited.
5. Conclusion
The last few years have seen considerable enthusiasm for radiosynthesis using [11C]CO2. Whereas classical [11C]CO2 incorporation has relied on highly basic organometallic reagents, modern strategies employ more functional group tolerant reagents and catalysts for activation. This has enabled the synthesis of more structurally complex radiopharmaceuticals and precise placement of the carbon-11 label within functional groups such as ureas, carbamates, and oxazolidinones under very mild conditions, as well as carboxylic acids, esters and amides. Carbamates and ureas are among the most common classes of compounds generated by combinatorial chemistry and we anticipate that even greater numbers of drug hits of this chemical class will emerge. Most recently, the carbamate radiotracer [11C]CURB and the oxazolidinone, [11C]SL25.1188 have advanced for human neuroimaging studies of FAAH and MAO-B, respectively. We anticipate that future developments in this field will establish mild and robust strategies for labelling a greater number of functional positions, including amides. Enabling technologies such as microfluidics and automated apparatus should soon also be employed for rapid method optimization and widespread adoption of [11C]CO2 fixation for clinical translation.
Acknowledgments
We thank Hogger & Co. Photography for graphical abstract design.
Biographies

Benjamin Rotstein studied chemistry (BScH 2007) at Dalhousie University and University of King's College (Halifax, Nova Scotia). He moved to University of Toronto to continue his education under the supervision of Prof. Andrei Yudin, earning a PhD in 2012. During this time, his research focused on multicomponent reactions and peptide chemistry, and included medicinal chemistry research at GlaxoSmithKline (Durham, North Carolina). In 2012, he began a postdoctoral position with Dr. Neil Vasdev at Harvard Medical School and Massachusetts General Hospital, where he is studying carbon-11 and fluorine-18 radiochemistry and PET imaging.

Neil Vasdev obtained a PhD in Radiochemistry from McMaster University (Canada) with Prof. Raman Chirakal and Prof. Gary Schrobilgen in 2003, followed by a postdoctoral fellowship with Prof. Henry VanBrocklin at the E.O. Lawrence Berkeley National Laboratories (USA). In 2004, he started his independent research career at the University of Toronto and CAMH (Canada). In 2011, he was recruited as an Associate Professor at Harvard Medical School and is the Director of Radiochemistry at Massachusetts General Hospital (USA). He has developed a radiopharmaceutical chemistry research programme aimed at synthesizing new 11C and 18F-labelled probes and translating these for human imaging studies.
References
- 1.Ametamey SM, Horner M, Schubiger PA. Chem Rev. 2008;108:1501. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- 2.Ferrieri RA. In: Handbook of Radiopharmaceuticals. Welch MJ, Redvanly CS, editors. John Wiley & Sons, Ltd.; Chichester: 2003. pp. 229–282. [Google Scholar]
- 3.Bolton R. J Labelled Compd Radiopharm. 2001;44:701. [Google Scholar]
- 4.Allard M, Fouquet E, James D, Szlosek-Pinaud M. Curr Med Chem. 2008;15:235. doi: 10.2174/092986708783497292. [DOI] [PubMed] [Google Scholar]
- 5.Roeda D, Crouzel C, van Zanten B. Radiochem Radioanal Lett. 1978;33:175. [Google Scholar]
- 6.Crouzel C, Roeda D. Int J Appl Radiat Isot. 1983;34:1558. doi: 10.1016/0020-708x(83)90296-x. [DOI] [PubMed] [Google Scholar]
- 7.Landais P, Crouzel C. Appl Radiat Isot. 1987;38:297. [Google Scholar]
- 8.Nishijima K, Kuge Y, Seki K, Ohkura K, Motoki N, Nagatsu K, Tanaka A, Tsukamoto E, Tamaki N. Nucl Med Biol. 2002;29:345. doi: 10.1016/s0969-8051(01)00310-9. [DOI] [PubMed] [Google Scholar]
- 9.Ogawa M, Takada Y, Suzuki H, Nemoto K, Fukumura T. Nucl Med Biol. 2010;37:73. doi: 10.1016/j.nucmedbio.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 10.Branmoullé Y, Roeda D, Dollé F. Tetrahedron Lett. 2010;51:313. [Google Scholar]
- 11.Miller PW, Long NJ, Vilar R, Gee AD. Angew Chem Int Ed. 2008;47:8998. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- 12.Roeda D, Dollé F. Curr Top Med Chem. 2010;10:1680. doi: 10.2174/156802610793176710. [DOI] [PubMed] [Google Scholar]
- 13.Zeisler SK, Nader M, Theobald A, Oberdorfer F. Appl Radiat Isot. 1997;48:1091. [Google Scholar]
- 14.Eriksson J, Åberg O, Långström B. Eur J Org Chem. 2007:455. [Google Scholar]
- 15.Audrain H, Martarello L, Gee A, Bender D. Chem Commun. 2004;558 doi: 10.1039/b314982j. [DOI] [PubMed] [Google Scholar]
- 16.Miller PW, Long NJ, de Mello AJ, Vilar R, Audrain H, Bender D, Passchier J, Gee A. Angew Chem Int Ed. 2007;46:2875. doi: 10.1002/anie.200604541. [DOI] [PubMed] [Google Scholar]
- 17.Miller PW, Audrain H, Bender D, deMello AJ, Gee AD, Long NJ, Vilar R. Chem Eur J. 2011;17:460. doi: 10.1002/chem.201002644. [DOI] [PubMed] [Google Scholar]
- 18.Kealey S, Plisson C, Collier TL, Long NJ, Husbands SM, Martarello L, Gee AD. Org Biomol Chem. 2011;9:3313. doi: 10.1039/c0ob00631a. [DOI] [PubMed] [Google Scholar]
- 19.Eriksson J, van den Hoek J, Windhorst AD. J Labelled Compd Radiopharm. 2012;55:223. [Google Scholar]
- 20.Långström B, Isenko O, Rahman O. J Labelled Compd Radiopharm. 2007;50:794. [Google Scholar]
- 21.Omae I. Coord Chem Rev. 2012;256:1384. [Google Scholar]
- 22.Tewson TJ, Banks W, Franceschini M, Hoffpauir J. Appl Radiat Isot. 1989;40:765. [Google Scholar]
- 23.Mock BH, Vavrek MT, Mulholland GK. Nucl Med Biol. 1995;22:667. doi: 10.1016/0969-8051(94)00150-i. [DOI] [PubMed] [Google Scholar]
- 24.Buchanan JM, Hastins AB, Nesbett FB. J Biol Chem. 1943;150:413. [Google Scholar]
- 25.Aubert C, Huard-Perrio C, Lasne MC. J Chem Soc Perkin Trans 1. 1997:2837. [Google Scholar]
- 26.Perrio-Huard C, Aubert C, Lasne MC. J Chem Soc Perkin Trans 1. 2000;311 [Google Scholar]
- 27.Luthra SK, Pike VW, Brady F. Chem Commun. 1985;1423 [Google Scholar]
- 28.McCarron J, Turton DR, Pike VW, Poole KG. J Labelled Compd Radiopharm. 1996;38:941. [Google Scholar]
- 29.Hwang DR, Simpson NR, Montoya J, Mann JJ, Laurelle M. Nucl Med Biol. 1999;26:815. doi: 10.1016/s0969-8051(99)00056-6. [DOI] [PubMed] [Google Scholar]
- 30.Mäding P, Zessin J, Pleiß U, Füchtner F, Wüst F. J Labelled Compd Radiopharm. 2006;49:357. [Google Scholar]
- 31.Prenant C, Sastre J, Crouzel C, Syrota A. J Labelled Compd Radiopharm. 1987;24:227. [Google Scholar]
- 32.Studenov AR, Berridge MS, Soloviev DV, Matarrese M, Todde S. Nucl Med Biol. 1999;26:431. doi: 10.1016/s0969-8051(99)00003-7. [DOI] [PubMed] [Google Scholar]
- 33.van der Meij M, Carruthers NI, Herscheid JDM, Jablownowski JA, Leysen JE, Windhorst AD. J Labelled Compd Radiopharm. 2003;46:1075. [Google Scholar]
- 34.Kilbourn MR, Welch MJ. Int J Appl Radiat Isot. 1982;33:359. doi: 10.1016/0020-708x(82)90149-1. [DOI] [PubMed] [Google Scholar]
- 35.Bolster JM, Vaalburg W, Elsinga PhH, Woldring MG, Wynberg H. Appl Radiat Isot. 1986;37:985. doi: 10.1016/0883-2889(86)90251-0. [DOI] [PubMed] [Google Scholar]
- 36.Ram S, Ehrenkaufer RE, Jewett DM. Appl Radiat Isot. 1986;37:391. doi: 10.1016/0883-2889(86)90094-8. [DOI] [PubMed] [Google Scholar]
- 37.Ram S, Spicer LD. Appl Radiat Isot. 1989;40:413. doi: 10.1016/0883-2889(89)90207-4. [DOI] [PubMed] [Google Scholar]
- 38.Ram S, Ehrenkaufer RE, Spicer LD. Appl Radiat Isot. 1989;40:425. doi: 10.1016/0883-2889(89)90210-4. [DOI] [PubMed] [Google Scholar]
- 39.Ram S, Spicer LD. J Labelled Compd Radiopharm. 1989;27:662. [Google Scholar]
- 40.Ram S, Ehrenkaufer RE. Tetrahedron Lett. 1985;26:5367. [Google Scholar]
- 41.Ram S, Ehrenkaufer RL. Nucl Med Biol. 1988;15:345. doi: 10.1016/0883-2897(88)90002-5. [DOI] [PubMed] [Google Scholar]
- 42.Sakakura T, Choi JC, Yasuda H. Chem Rev. 2007;107:2365. doi: 10.1021/cr068357u. [DOI] [PubMed] [Google Scholar]
- 43.Stuani Pereira F, Ribeiro deAzevedo E, da Silva EF, Bonagamba TJ, da Silva Agostíni DL, Magalhães A, Job AE, Pérez González ER. Tetrahedron. 2008;64:10097. [Google Scholar]
- 44.Yoshida M, Komatsuzaki Y, Ihara M. Org Lett. 2008;10:2083. doi: 10.1021/ol800663v. [DOI] [PubMed] [Google Scholar]
- 45.Pérez ER, da Silva MO, Costa VC, Rodrigues-Filho UP, Franco DW. Tetrahedron Lett. 2002;43:4091. [Google Scholar]
- 46.Salvatore RN, Shin SI, Nagle AS, Jung KW. J Org Chem. 2001;66:1035. doi: 10.1021/jo001140u. [DOI] [PubMed] [Google Scholar]
- 47.Stuani Pereira F, da Silva Agostíni DL, do Espírito Santo RD, Ribeiro deAzevedo E, Bonagamba TJ, Job AE, Pérez González ER. Green Chem. 2011;13:2146. [Google Scholar]
- 48.Pérez ER, Santos RHA, Gambardella MTP, de Macedo LGM, Rodriguez-Filho UP, Launay JC, Franco DW. J Org Chem. 2004;69:8005. doi: 10.1021/jo049243q. [DOI] [PubMed] [Google Scholar]
- 49.Heldebrant DJ, Jessop PG, Thomas CA, Eckert CA, Liotta CL. J Org Chem. 2005;70:5335. doi: 10.1021/jo0503759. [DOI] [PubMed] [Google Scholar]
- 50.Villiers C, Dognon JP, Pollet R, Thuéry P, Ephritikhine M. Angew Chem Int Ed. 2010;49:3465. doi: 10.1002/anie.201001035. [DOI] [PubMed] [Google Scholar]
- 51.Hooker JM, Reibel AT, Hill SM, Schueller MJ, Fowler JS. Angew Chem Int Ed. 2009;48:3482. doi: 10.1002/anie.200900112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson AA, Garcia A, Houle S, Vasdev N. Org Biomol Chem. 2010;8:428. doi: 10.1039/b916419g. [DOI] [PubMed] [Google Scholar]
- 53.Tsuji Y, Fujihara T. Chem Commun. 2012;48:9956. doi: 10.1039/c2cc33848c. [DOI] [PubMed] [Google Scholar]
- 54.Riduan SN, Zhang Y. Dalton Trans. 2010;39:3347. doi: 10.1039/b920163g. [DOI] [PubMed] [Google Scholar]
- 55.Huang K, Sun CL, Shi ZJ. Chem Soc Rev. 2011;40:2435. doi: 10.1039/c0cs00129e. [DOI] [PubMed] [Google Scholar]
- 56.Fan T, Chen X, Lin Z. Chem Commun. 2012;48:10808. doi: 10.1039/c2cc34542k. [DOI] [PubMed] [Google Scholar]
- 57.Chakraborty PK, Mangner TJ, Chugani HT. Appl Radiat Isot. 1997;48:619. doi: 10.1016/j.apradiso.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 58.Schirbel A, Holschbach MH, Cohen HH. J Labelled Compd Radiopharm. 1999;42:537. [Google Scholar]
- 59.Wilson AA, Garcia A, Houle S, Sadovski O, Vasdev N. Chem Eur J. 2011;17:259. doi: 10.1002/chem.201002345. [DOI] [PubMed] [Google Scholar]
- 60.Vasdev N, Garcia A, Stableford WT, Young AB, Meyer JH, Houle S, Wilson AA. Bioorg Med Chem Lett. 2005;15:5270. doi: 10.1016/j.bmcl.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 61.Hicks JW, Wilson AA, Rubie EA, Woodgett JR, Houle S, Vasdev N. Bioorg Med Chem Lett. 2012;22:2099. doi: 10.1016/j.bmcl.2011.12.139. [DOI] [PubMed] [Google Scholar]
- 62.Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, Beidler D, Liimatta MB, Smith SE, Dudley DT, Sadagopan N, Bhattachar SN, Kesten SJ, Nomanbhoy TK, Cravatt BF, Ahn K. ACS Med Chem Lett. 2011;2:91. doi: 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Skaddan MB, Zhang L, Johnson DS, Zhu A, Zasadny KR, Coelho RV, Kuszpit K, Currier G, Fan KH, Beck EM, Chen L, Drozda SE, Balan G, Niphakis M, Cravatt BF, Ahn K, Bocan T, Villalobos A. Nucl Med Biol. 2012;39:1058. doi: 10.1016/j.nucmedbio.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hicks JW, Parkes J, Sadovski O, Tong J, Houle S, Vasdev N, Wilson AA. Nucl Med Biol. doi: 10.1016/j.nucmedbio.2013.04.008. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Tilburg EW, Windhorst AD, van der Mey M, Herscheid JDM. J Labelled Compd Radiopharm. 2006;49:321. [Google Scholar]
- 66.Hooker JM, Kim SW, Alexoff D, Xu Y, Shea C, Reid A, Volkow N, Fowler JS. ACS Chem Neurosci. 2010;1:65. doi: 10.1021/cn9000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ravert HT, Scheffel U, Mathews WB, Musachio JL, Dannals RF. Nucl Med Biol. 2002;29:47. doi: 10.1016/s0969-8051(01)00285-2. [DOI] [PubMed] [Google Scholar]
- 68.Talbot PS, Narendran R, Butelman ER, Huang Y, Ngo K, Slifstein M, Martinez D, Laruelle M, Hwang DR. J Nucl Med. 2005;46:484. [PubMed] [Google Scholar]
- 69.Shoultz BW, Årstad E, Marton J, Willoch F, Drzezga A, Wester HJ, Henriksen G. Open Med Chem J. 2008;2:72. doi: 10.2174/1874104500802010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nabulsi NB, Zheng MQ, Ropchan J, Labaree D, Ding YS, Blumberg L, Huang Y. Nucl Med Biol. 2011;38:215. doi: 10.1016/j.nucmedbio.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 71.McKinney MK, Cravatt BF. Annu Rev Biochem. 2005;74:411. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 72.Fowler CJ. Fundam Clin Pharmacol. 2006;20:549. doi: 10.1111/j.1472-8206.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- 73.Schlosburg JE, Kinsey SG, Lichtman AH. AAPS J. 2009;11:39. doi: 10.1208/s12248-008-9075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilson AA, Garcia A, Parkes J, Houle S, Tong J, Vasdev N. Nucl Med Biol. 2011;38:247. doi: 10.1016/j.nucmedbio.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 75.Rusjan P, Wilson AA, Mizrahi R, Boileau I, Chavez SE, Lobaugh NJ, Kish SJ, Houle S, Tong J. J Cereb Blood Flow Metab. 2012:1. doi: 10.1038/jcbfm.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilson AA, Hicks JW, Sadovski O, Parkes J, Tong J, Houle S, Fowler CJ, Vasdev N. J Med Chem. 2013;56:201. doi: 10.1021/jm301492y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kawamura K, Hashimoto H, Ogawa M, Yui J, Wakizaka H, Yamasaki T, Hatori A, Xie L, Kumata K, Fujinaga M, Zhang MR. Nucl Med Biol. 2013 doi: 10.1016/j.nucmedbio.2013.03.004. in press. [DOI] [PubMed] [Google Scholar]
- 78.Bramoullé Y, Puech F, Saba W, Valette H, Bottlaender M, George P, Dollé F. J Labelled Compd Radiopharm. 2008;51:153. [Google Scholar]
- 79.Vasdev N, Sadovski O, Garcia A, Dollé F, Meyer JH, Houle S, Wilson AA. J Labelled Compd Radiopharm. 2011;54:678. [Google Scholar]
- 80.Takaya J, Tadami S, Ukai K, Iwasawa N. Org Lett. 2008;10:2697. doi: 10.1021/ol800829q. [DOI] [PubMed] [Google Scholar]
- 81.Kobayashi K, Kondo Y. Org Lett. 2009;11:2035. doi: 10.1021/ol900528h. [DOI] [PubMed] [Google Scholar]
- 82.Zhang L, Cheng J, Ohishi T, Hou Z. Angew Chem Int Ed. 2010;49:8670. doi: 10.1002/anie.201003995. [DOI] [PubMed] [Google Scholar]
- 83.Riss PJ, Lu S, Telu S, Aigbirhio FI, Pike VW. Angew Chem Int Ed. 2012;51:2698. doi: 10.1002/anie.201107263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bellenie BR, Barton NP, Emmons AJ, Heer JP, Salvagno C. Bioorg Med Chem Lett. 2009;19:990. doi: 10.1016/j.bmcl.2008.11.064. [DOI] [PubMed] [Google Scholar]