

Abstract
During embryogenesis, the epicardium undergoes proliferation, migration, and differentiation into several cardiac cell types which contribute to the coronary vessels. This process requires epithelial to mesenchymal transition (EMT) and directed cellular invasion. The Type III Transforming Growth Factor-beta Receptor (TGFβR3) is required for epicardial cell invasion and coronary vessel development. Using primary epicardial cells derived from Tgfbr3+/+ and Tgfbr3−/− mouse embryos, high-molecular weight hyaluronan (HMWHA) stimulated cellular invasion and filamentous (f-actin) polymerization are detected in Tgfbr3+/+ cells, but not in Tgfbr3−/− cells. Furthermore, HMWHA-stimulated cellular invasion and f-actin polymerization in Tgfbr3+/+ epicardial cells are dependent on Src kinase. Src activation in HMWHA-stimulated Tgfbr3−/− epicardial cells is not detected in response to HMWHA. RhoA and Rac1 also fail to activate in response to HMWHA in Tgfbr3−/− cells. These events coincide with defective f-actin formation and deficient cellular invasion. Finally, a T841A activating substitution in TGFβR3 drives ligand-independent Src activation. Collectively, these data define a TGFβR3–Src–RhoA/Rac1 pathway that is essential for hyaluronan-directed cell invasion in epicardial cells.
Keywords: Hyaluronan, Type III TGFβ receptor, Epicardium, EMT, Src kinase
1. Introduction
The coronary vasculature is required for proper development and function of the heart. During embryonic development the formation of the coronary vessels is dependent upon the transfer of cells from the proepicardium to the surface of the heart to form the epicardium [1]. The epicardium is an epithelial sheet that covers the myocardium, and secreted growth factors from the myocardium stimulate synthesis of extracellular matrix molecules including hyaluronan (HA) in the subepicardial space [2,3]. These matrix and growth factors induce Epithelial to Mesenchymal Transition (EMT), in which transformed epicardial cells migrate through the subepicardial space, invade the myocardium and differentiate into vascular smooth muscle cells and cardiac fibroblasts [4]. The EMT process is defined by loss of polarity and cell–cell contacts in epithelial cells, and adopting an elongated fibroblast-like morphology. These transformed cells are then competent to undergo cellular invasion. This EMT process is a requirement for development of several organs including the embryonic heart [5]. As EMT is required for heart development, perturbations in this process can lead to congenital defects leading to adult cardiovascular disease, which is the leading cause of death in the United States [6].
HA is a long-chain glycosaminogly can extracellular matrix molecule synthesized by the hyaluronan synthase family of enzymes (Has1, Has2, Has3) [7] and serves structural functions as well as stimulating biochemical signaling cascades in the developing heart. The Has2−/− knockout phenotype is embryonic lethal at 9.5 days of gestation due to blocked cardiac development as a consequence of deficient cardiac EMT [8]. These knockout embryos lack HA and fail to complete endocardial cushion EMT and maturation with lethality occurring before epicardial development. Since lethality precedes formation of the epicardium in the Has2−/− embryo, we used in vitro techniques to determine the role of HA in epicardial cell invasion. Well-characterized mouse epicardial cell lines [9] were used to decipher the mechanisms of HA-triggered epicardial EMT and invasive cell motility. Prior work in our laboratory has shown that high-molecular weight hyaluronan (HMWHA) can induce epicardial cell invasion and EMT, and is required for TGFβ2-induced epicardial cell invasion and EMT [10,11].
HA can engage cell surface receptors CD44 and RHAMM [12] to stimulate intracellular signaling that can modulate epithelial character and cell invasion [10,13]. Hyaluronan-mediated intracellular signal transduction executed through CD44 can enhance canonical TGFβ Type I receptor signaling [14]. Hyaluronan is a driver of Src-dependent cell motility via activation of Rho GTPase family members and filamentous actin polymerization in several tumor-derived cell lines [15,16]. The Rho family of GTPases modulates f-actin polymerization to form distinct structures required for invasive cell motility: Rac1 induces lamellipodia formation at the leading edge of motile cells, cdc42 forms filopodial structures extending beyond leading edge, and RhoA required for turnover of focal adhesions [17].
The Type III TGFβ receptor (TGFβR3) lacks catalytic activity and functions in TGFβ ligand presentation to Type I and II TGFβ receptors to stimulate receptor activation [18,19]. It has previously been demonstrated that TGFβR3 is required for endocardial [20] as well as epicardial cellular invasion [21]. Tgfbr3−/− mice die at E14.5 as a result of failed coronary vessel development associated with decreased epicardial cell invasion into the myocardium [22]. We have previously shown have shown that Tgfbr3−/− epicardial cells do not invade in response to HMWHA [21], however the molecular mechanism underlying this phenotype is unknown. TGFβR3 is also known to regulate cancer cell migration, by augmenting filamentous actin polymerization via TGFβ1 dependent activation of Rac1 and cdc42 GTPases [23]. Src is a ubiquitously expressed non-receptor tyrosine kinase that has been extensively identified as a driver of cell invasion in many cell systems [24]. Src activation has not previously been reported to be involved inTGFβR3 signal transduction, but has been suggested to be activated via Type I TGFβ receptor-dependent pathway in a TGFβ1-dependent manner [25]. How these effectors function in epicardial cells and whether they are responsive during HA directed cell invasion have not yet been reported.
This study reveals that HA mediated activation of cellular invasion and filamentous actin polymerization are dependent on TGFβR3. Furthermore, Src kinase is required for HA mediated epicardial cell invasion, and filamentous actin polymerization. In the absence of TGFβR3, HA-stimulated activation of Src kinase, Rac1 and RhoA GTPases are deficient concomitant with defective cellular invasion. These data establish a novel role for TGFβR3 and Src as central signaling nodes for HA directed epicardial cell invasion.
2. Materials and methods
2.1. Cell lines and reagents
Conditionally immortal murine epicardial cells were originally provided by Dr. Joey Barnett (Vanderbilt Medical University) as described [9]. Cell culture conditions were used as previously described [11]. Epicardial cells used in this study were isolated from wild-type (Tgfbr3+/+) mouse embryos and embryos lacking TGFβR3 (Tgfbr3−/−). Src kinase inhibitor PP2 was purchased from EMD Millipore (#529573). High-molecular weight hyaluronan (HMWHA) was purchased from R&D Systems (#GLR002).
2.2. Western blotting
Whole cell lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene fluoride membrane (PVDF). After blocking in 5% Bovine Serum Albumin (BSA), membranes were probed with different primary and secondary antibodies in 3% BSA. Antibodies for detection of pY416Src (6943S), Src (2109S), and TGFβR3 (2519S) were purchased from Cell Signaling Technologies (Danvers, MA). Antibodies for the detection of β-Tubulin (sc-9104), Rac1 (sc-217), and cdc42 (sc-087) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). RhoA (ARH03) and Actin (AAN01) antibodies were purchased from Cytoskeleton (Aurora, CO). Densitometry was analyzed by ImageJ, all stimulated conditions were set relative to unstimulated conditions for each cell line and siRNA treatment as internal controls.
2.3. Cellular invasion assays
To assess cellular invasion, epicardial cells were cultured in DMEM containing 10% FBS on 1 mg/mL Rat Tail Type I Collagen gels in 4-well tissue culture dishes (BD Biosciences, Franklin Lakes, New Jersey). Cells were seeded at 40,000 cells/gel for each experimental condition, and allowed to attach overnight. Cells were stimulated with 300 μg/mL HMWHA for 30 min, and allowed to undergo cellular invasion for 24 h. Cells were fixed with 4% paraformaldehyde in phosphate buffered saline (PBS), permeabilized with 0.5% TritonX-100, and subject to AlexaFlour594-conjugated Phalloidin staining to visualize filamentous actin and cell morphology (Life Technologies A12381). Invaded, transformed epicardial cells were manually counted by observers blinded to the conditions in three independent experiments.
2.4. Detection of filamentous actin
2.4.1. Phalloidin staining
Tgfbr3+/+ and Tgfbr3−/− epicardial cells were seeded on glass cover-slips at an equal and sub-confluent density (70% confluency) and allowed to adhere overnight. Cells were subject to overnight serum starvation (DMEM, 0% FBS), and stimulated with 300 μg/mL HMWHA for 60 min. Immunofluorescent visualization of filamentous actin was accomplished using AlexaFlour594 Phalloidin (Life Technologies A12381) and a Leica DMLB microscope; images were documented using a Retiga 200R camera and ImagePro Plus 5.1 software.
2.4.2. Filamentous actin assay
Tgfbr3+/+ and Tgfbr3−/− epicardial cells were seeded at 800,000 cells per 10 cm tissue culture dish (70% confluence) and allowed to adhere overnight. Cells were grown in serum free conditions for 18 h prior to stimulation with 300 μg/mL HMWHA for 60 min. The extent of filamentous actin (F-actin) polymerization was assess by the F-Actin/G-Actin In Vivo Bioassay Biochem Kit (to be referred to as the f/g actin assay) according to the manufacturer's instructions (Cytoskeleton BK037). Briefly, whole cell lysates were subjected to ultracentrifugation (100,000 ×g) to fractionate globular (g) and filamentous (f) actin fractions. These fractions were then subjected to SDS-PAGE and Western blot detection of actin to allow for comparison of filamentous actin in each treatment. Densitometry of g and f actin bands was performed using ImageJ, and the f/g actin ratio was obtained dividing f band density by g band density. f/g ratios under stimulated conditions were plotted relative to unstimulated f/g ratios from Tgfbr3+/+ conditions.
2.5. Detection of GTP-bound RhoA, Rac1, and cdc42
Tgfbr3+/+ and Tgfbr3−/− epicardial cells were seeded at 800,000 cells per 10 cm tissue culture dish (70% confluence). Following 18 h of serum starvation, cells were stimulated with 300 μg/mL HMWHA for indicated time points. 1 mg of whole cell lysate was incubated with PAK-PDB beads (Cytoskeleton #PAK02A) for 1 h to bind activated (GTP-bound) Rac1 and cdc42 for each experimental condition. 1 mg of whole cell lysate was incubated with RBD beads (Cytoskeleton #BK036) for 1 h to bind GTP-bound RhoA for each experimental condition. Beads with precipitated GTP-bound RhoA, Rac1, and cdc42 and 25 μg of total lysate (for loading control) under each experimental condition were subjected to SDS-PAGE and Western blot detection of Rac1, cdc42, and RhoA.
2.6. siRNA knockdown experiments
Tgfbr3+/+ epicardial cells were plated at 50,000 cells per well of a 6-well tissue culture plate (50% confluence) in DMEM containing 10% FBS without antibiotics and were cultured for 48 h. Cells were then transfected with 2 μg Control siRNA or 2 μg siRNA targeting mouse TGFβR3 (Santa Cruz Biotechnologies, sc-40225), and 8 μL of X-treme Gene siRNA transfection reagent (Roche). The media was changedtore-move the reagent 16 h after transfection. 48 h post transfection, cells were stimulated with HMWHA and whole cell lysates were subjected to Western blot analysis. Validation of TGFβR3 knockdown was accomplished via Western blot analysis.
2.7. Expression of DNA Vectors
HEK293 cells were plated at 50,000 cells per well of a 6-well plate (50% confluence) in DMEM containing 10% FBS without antibiotics and cultured for 48 h. Cells were then transfected with 2 μg pAdTrack empty vector or pAdTrack vectors containing TGFβR3-T841A (constitutively active TGFβR3) using 3 μL of X-treme gene HP DNA transfection reagent (Roche). The media was changed to DMEM containing 10% FBS without antibiotics 24 h after transfection. 48 h after transfection, whole cell lysates were subjected to SDS-PAGE and Western blot analysis to detect the indicated molecules.
2.8. Statistical analysis
All graphs represent mean values, with error bars showing the standard deviation of the mean. Experiments were performed in duplicate and repeated a minimum of three times. Statistical significance was assessed using a 2-way Student's t-test, with p values below 0.05 considered significant.
3. Results
3.1. TGFβR3 and Src are required for HMWHA induced cellular invasion
Tgfbr3+/+ and Tgfbr3−/− epicardial cells cultured on collagen gels were stimulated with HMWHA for 30 min and the extent of cellular invasion was assessed after 24 h (Fig. 1). Tgfbr3+/+ cells stimulated with HMWHA adopt an elongated mesenchymal phenotype and undergo cellular invasion into the collagen gel (Fig. 1C). In contrast, Tgfbr3−/− epicardial cells do not lose epithelial character or execute cellular invasion in the presence of HMWHA (Fig. 1D). Invaded epicardial cells were enumerated (Fig. 1E). Tgfbr3+/+ epicardial cells stimulated with HMWHA demonstrate a level of cell invasion that is 3 times the level seen in unstimulated Tgfbr3+/+ cells (Fig. 1E). Thus, consistent with our prior reports, TGFβR3 is required for HMWHA-stimulated epicardial cell invasion.
Fig. 1.
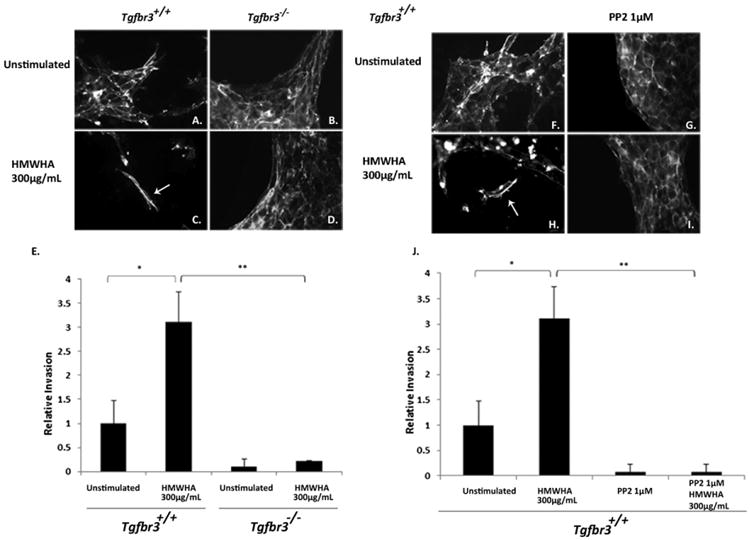
TGFβR3 and Src are required for HMWHA-stimulated cellular invasion. (A–D) Tgfbr3+/+ and Tgfbr3−/− epicardial cells on collagen gels stimulated with 300 μg/mL HMWHA for 24 h. Phalloidin staining used to visualize cell morphology. (E) Enumeration of invaded epicardial cells (*p < 0.05 **p < 0.005). (F–I) Tgfbr3+/+ epicardial cells on collagen gels stimulated with 300 μg/mL HMWHA after 24 h preincubation with the Src kinase inhibitor PP2. (J) Enumeration of invaded epicardial cells (*p < 0.05 **p < 0.005).
In parallel experiments, Tgfbr3+/+ epicardial cells were preincubated with 1 μM PP2, a Src inhibitor, for 1 h prior to HMWHA stimulation. The extent of cellular invasion was visualized by phalloidin-detection of filamentous actin to observe invaded cell morphology and allow for enumeration of invaded cells into the collagen gel. Tgfbr3+/+ epicardial cells executed EMT and invaded the collagen in response to HMWHA (Fig. 1H). In contrast, Tgfbr3+/+ cells stimulated with HMWHA in the presence of PP2 (Fig. 1I) did not invade into the collagen matrix and retained an epithelial cell morphology. Invaded cells were quantified, and Tgfbr3+/+ cells undergo a level of cell invasion that is 3 times higher in response to HMWHA when compared to unstimulated Tgfbr3+/+ cells (Fig. 1J) or cells subjected to preincubation with PP2. Thus, Src kinase activity is required for HMWHA-stimulated epicardial cell invasion.
3.2. TGFβR3 is required for HMWHA-stimulated f-actin polymerization
Induction of filamentous actin stress fibers is essential for cell migration and invasion [27]. Phalloidin staining was used to visualize filamentous actin polymerization (f-actin) in order to determine the role of TGFβR3 in HMWHA-stimulated cell motility. Unstimulated Tgfbr3+/+ cells have low basal levels of organized stress fibers (Fig. 2A). Upon stimulation with HMWHA, Tgfbr3+/+ cells form f-actin into stress fibers compared to unstimulated Tgfbr3+/+ cells (Fig. 2B). In contrast, unstimulated Tgfbr3−/− cells have high basal levels of stress fibers (Fig. 2C). Stimulation with HMWHA results in actin filament disassembly in Tgfbr3−/− cells (Fig. 2D), but is not accompanied by the re-polymerization of actin filaments necessary for cell invasion.
Fig. 2.
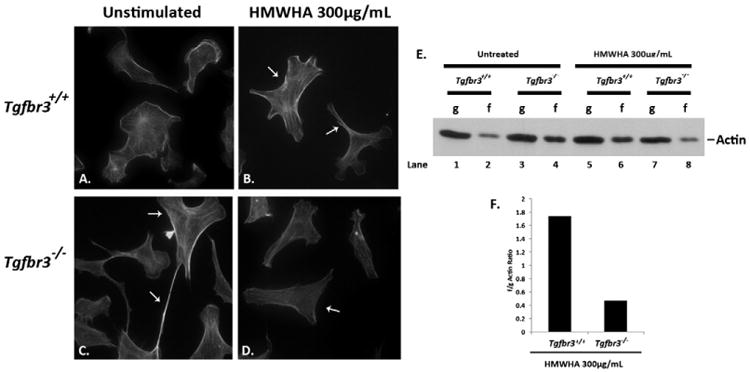
Defective actin dynamics in Tgfbr3−/− epicardial cells in the presence of HMWHA. (A–D) Detection of filamentous actin in Tgfbr3+/+ epicardial cells without (A) and with HMWHA 300 μg/mL stimulation for 60 min (B). Detection of filamentous actin in Tgfbr3−/− epicardial cells without (C) and with HMWHA 300 μg/mL stimulation (D) for 60 min. (E) Globular (g) or filamentous (f) actin detection by Western blot following ultracentrifugation of lysates from vehicle incubated and HMWHA-stimulated epicardial cells. (F) Representative graph of f/g actin ratios for HMWHA 300 μg/mL stimulated Tgfbr3+/+ and Tgfbr3−/− epicardial cells.
The failure of Tgfbr3−/− cells to mobilize filamentous actin stress fibers upon HA stimulation was independently confirmed using an f/g actin assay. A substantial deficiency in the ability of Tgfbr3−/− cells to mobilize actin was detected following stimulation with HMWHA (Fig. 2E). Unstimulated Tgfbr3+/+ cells have low levels of f-actin (Fig. 2E, Lane 2), whereas Tgfbr3−/− cells have high levels of f-actin in the unstimulated conditions (Fig. 2E, Lane 4). There is detection of increased f-actin in Tgfbr3+/+ cells following HMWHA stimulation indicating normal f-actin polymerization (Fig. 2E, Lane 6). In contrast, Tgfbr3−/− cells show a decrease in f-actin following HMWHA stimulation (Fig. 2E, Lane 8). The f/g actin ratio was determined for Tgfbr3+/+ and Tgfbr3−/− cells stimulated with or without HMWHA. Following HA-stimulation, Tgfbr3+/+ cells have an f/g ratio of 1.7 versus 0.47 for Tgfbr3−/− cells demonstrating a requirement for TGFβR3 to mediate HMWHA-stimulated f-actin polymerization.
3.3. HMWHA stimulated f-actin polymerization is dependent on Src
The role of Src in HMWHA-stimulated f-actin polymerization was tested in Tgfbr3+/+ epicardial cells. When stimulated with HMWHA, epicardial cells mobilize polymerization of f-actin as visualized by phalloidin staining (Fig. 3B). In the presence of PP2, HMWHA-stimulated polymerization of f-actin is significantly inhibited (Fig. 3D), a result also reflected in the f/g actin ratio calculated from performing the f/g actin assay (Fig. 3E). Tgfbr3+/+ cells incubated with increasing concentrations of PP2, and subsequently stimulated with HMWHA, exhibit concentration-dependent inhibition of f-actin polymerization (Fig. 3E, Lanes 10, 12 compared to Lane 8). Cells stimulated with HMWHA alone have an f/g actin ratio of 2.1, whereas cells stimulated with HMWHA in the presence of 1 μM PP2 or 10 μM have f/g actin ratios of 1.2 and 0.8, respectively. These data suggest that Src kinase is required for HMWHA-stimulated filamentous actin polymerization and stress fiber formation in epicardial cells.
Fig. 3.
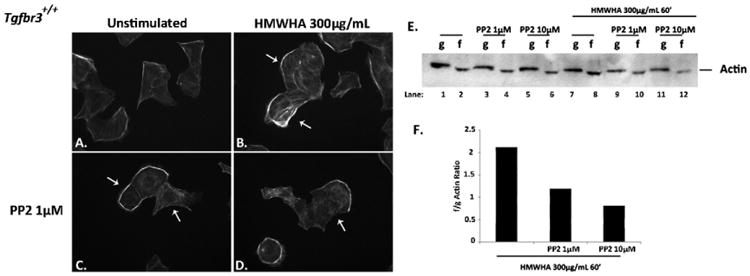
Src is required for HMWHA-induced filamentous actin polymerization. (A–D) Tgfbr3+/+ epicardial cells were dosed without (A, B) or with Src kinase inhibitor (PP2 1 μM) (C, D) for 60 min, followed by 300 μg/mL HMWHA stimulation in the presence (D) or absence (B) of PP2 for 60 min, and subjected to phalloidin staining to visualize filamentous actin. (E) Globular (g) or filamentous (f) actin detection by Western blot following ultracentrifugation of lysates from vehicle incubated and HMWHA-stimulated epicardial cells with or without PP2. (F) Representative graph of the f/g actin ratio for HMWHA stimulated vehicle incubated epicardial cells with or without PP2.
3.4. TGFβR3 is required for Src activation by HA
Although these findings reveal that TGFβR3 and Src are separately required for HMWHA-stimulated f-actin stress fiber formation and cell invasion, the data do not demonstrate that these effectors function in the same pathway. Therefore, experiments were performed to determine whether TGFβR3 is upstream of Src. HMWHA induces the phosphorylation of Src at tyrosine 416 (activating kinase activity) in Tgfbr3+/+ cells 2.25 fold over unstimulated control (Fig. 4A Lane 3), but not in Tgfbr3−/− epicardial cells (Fig. 4A Lane 6). The phosphorylation of Src Y416 does not increase over basal levels in Tgfbr3−/− cells following HMWHA-stimulation (Fig. 4B). To address the requirement of TGFβR3 for HMWHA-stimulated Src activation, additional experiments were carried out with siRNA targeting TGFβR3 in Tgfbr3+/+ epicardial cells to knockdown TGFβR3 expression. Src Y416 phosphorylation was induced 1.9 fold by HMWHA over the levels seen in Tgfbr3+/+ cells transfected with control siRNA (Fig. 4C Lane 3). Tgfbr3+/+ cells transfected with siRNA targeting TGFβR3, and subsequently stimulated with HMWHA, did not induce Src Y416 phosphorylation above the levels seen in unstimulated Tgfbr3+/+ cells transfected with the control siRNA (Fig. 4C, D Lane 4). The knockdown of TGFβR3 by siRNA was confirmed by Western blot analysis (Fig. 4E, F). These data suggest that TGFβR3 is required for HMWHA-dependent activation of Src kinase and establishes a novel role for TGFβR3 in the coordination of hyaluronan specific signal transduction.
Fig. 4.
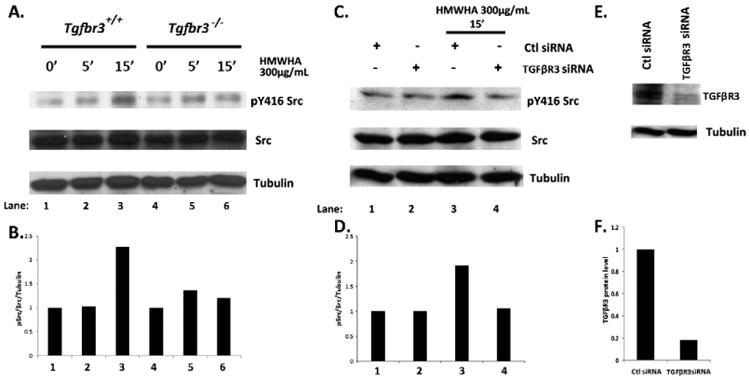
Attenuated Src-activation in the absence of TGFβR3. (A) Tgfbr3+/+ and Tgfbr3−/− epicardial cells were stimulated with 300 μg/mL HMWHA for 5 and 15 min, whole cell lysates were subjected to Western blot detection of pY416 Src. (B) Densitometry of pY46Src. (C) Epicardial cells were transfected with siRNA to TGFβR3, stimulated with HMWHA for 15min, whole cell lysates were subjected to Western blot detection of pY416 Src. (D) Densitometryof pY416Src. (E–F) Confirmation of TGFβR3 knockdown by siRNA in epicardial cells validated by Western blot.
3.5. TGFβR3 regulates Rac1, RhoA, and cdc42
The Rho family of GTPases is a modulator of the actin cytoskeleton and is directly upstream of f-actin polymerization and formation of stress fibers related to distinct cell membrane structures [17]. RhoA has been implicated in both TGFβ- and BMP2-stimulated epicardial EMT in a TGFβR3-dependent fashion [27]. Since Tgfbr3−/− epicardial cells are deficient in forming filamentous actin stress fibers in the presence of HMWHA, the activation of the Rho family of GTPases was tested to determine if these are similarly dysregulated between Tgfbr3+/+ and Tgfbr3−/− cells. Tgfbr3+/+ epicardial cells induce GTP-Rac1 (8 fold over unstimulated) and GTP-RhoA (5.5 fold over unstimulated) in response to HMWHA (Fig. 5A, B Lane 2; Fig. 5C, D Lane 2). In contrast, Tgfbr3−/− epicardial cells fail to activate Rac1 or RhoA (Fig. 5A, B Lane 4; Fig. 5C, D Lanes 4–6). HMWHA stimulation of Tgfbr3+/+ epicardial cells decreases GTP-cdc42 levels (Fig. 5E, F Lane 2), whereas Tgfbr3−/− retains higher constitutive levels of GTP-cdc42 under unstimulated conditions relative to Tgfbr3+/+ controls (Fig. 5E, F Lane 3). Furthermore, Tgfbr3−/− cells fail to decrease GTP-cdc42 following stimulation by HMWHA (Fig. 5E, F Lane 4). This constitutive and unregulated activation of cdc42 GTPase results in increased formation of filopodial structures in Tgfbr3−/− cells relative to Tgfbr3+/+ as visualized by phase contrast microscopy (Fig. 5G) and manual counting of filopodia (Fig. 5H). Taken together these data demonstrate that TGFβR3 is required for HMWHA-stimulated activation of RhoA and Rac1 as well as the modulation of cdc42 activity.
Fig. 5.
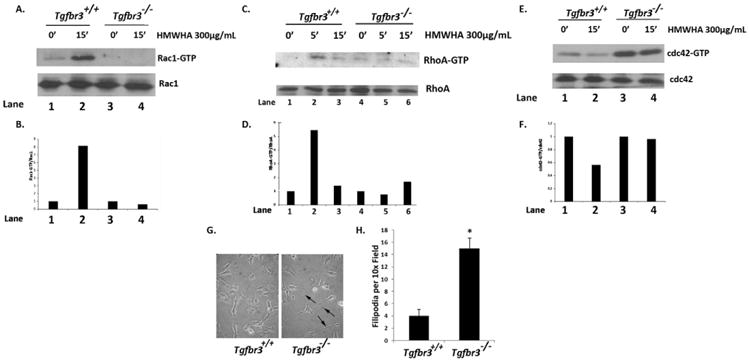
Defective Rac1, RhoA and cdc42 activity in Tgfbr3−/− epicardial cells. (A–B) Detection of GTP-Rac1 in Tgfbr3+/+ and Tgfbr3−/− epicardial cells incubated with 300 μg/mL HMWHA for 15 min. (C–D) Detection of GTP-Rac1 in Tgfbr3+/+ and Tgfbr3−/− epicardial cells incubated with 300 μg/mL HMWHA for 5 and 15 min. (E–F) Detection of GTP-cdc42 in Tgfbr3+/+ and Tgfbr3−/− epicardial cells incubated with 300 μg/mL HMWHA for 15 min. (G) Phase contrast microscopy visualizing filopodia of Tgfbr3+/+ and Tgfbr3−/− epicardial cells. (H) Quantification of filopodia in Tgfbr3+/+ and Tgfbr3−/− epicardial cells (*p = 5.90973E–08).
3.6. T841A-TGFβR3 drives ligand-independent Src activation
Given that TGFβR3 is required for HMWHA-stimulated activation of Src kinase and the formation of filamentous actin stress fibers, we investigated whether TGFβR3 activity alone could drive phosphorylation of Src. TGFβR3 (TGFβR3-T841A) has been shown to prevent β-Arrestin binding to TGFβR3 resulting in inhibited receptor internalization and continued signaling [28,29] HEK293 cells were transfected with pAdTrack vector alone or pAdTrack vector containing the TGFβR3-T841A. TGFβR3-T841A induces stimulus-independent phosphorylation of Y416 of Src to levels 2-fold over controls (Fig. 6A–B, Lane 2). Cells expressing TGFβR3-T841A also became elongated fibroblast-like consistent with the loss of epithelial character relative to control (Fig. 6C). These data suggest that TGFβR3 is a direct upstream regulator of Src kinase activation.
Fig. 6.
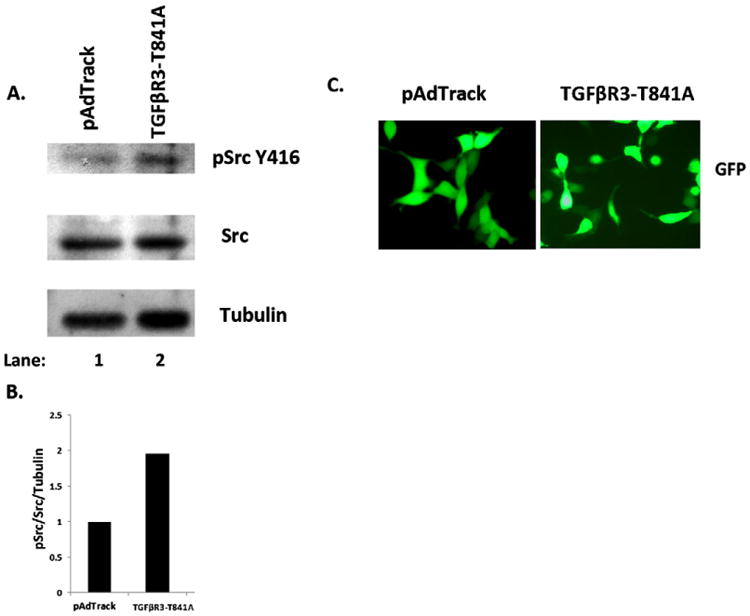
TGFβR3 drives Src phosphorylation. (A–B) Western blot detection of pY416 Src in whole cell lysates obtained from HEK293 cells expressing the constitutively active mutant (TGFβR3-T841A) of TGFβR3. (C) Confirmation of transfection of TGFβR3-T841A and pAdTrack vectors in HEK293 cells by fluorescence microscopy.
4. Discussion
Mobilization and polymerization of filamentous actin structures are the driving forces of cellular migration and invasion [26]. These events can be triggered by HMWHA which is a long chain glycosaminoglycan matrix molecule that serves structural and signaling functions in the developing heart [30]. Previous work in our laboratory has shown that HMWHA can trigger epicardial cell invasion and differentiation [10] and that this occurs in a TGFβR3-dependent manner [18]. However, a molecular explanation for this requirement has not been elucidated. In this study, the activation of Src kinase as a noncanonical effector downstream of TGFβR3 was investigated. Specifically, invasion assays and experiments assessing filamentous actin dynamics were performed in parallel to assess the roles of TGFβR3 and Src in HMWHA-stimulated cell invasion.
The loss of TGFβR3 expression in human ovarian cancer cells leads to enhanced cell migration [23], yet we show that Tgfbr3−/− epicardial cells are deficient in cell invasion when stimulated with HMWHA (Fig. 1). Similarly, TGFβR3 is required for chick endocardial invasion [31] during heart valve development where HMWHA is present and required [30], though a direct requirement for TGFβR3 in HMWHA-stimulated endocardial cell invasion has not been addressed. These observations may highlight a differential role for TGFβR3 in developing embryonic tissues versus terminally differentiated malignantly transformed tumors with respect to invasive cell motility. The HMWHA receptor CD44 has been shown to interact with Type I TGFβ receptors and enhance the canonical (Smad-dependent) TGFβ signaling [32]. This supports the notion that HMWHA promotes receptor complex formation for activation of multiple signaling pathways; however, a role for TGFβR3 in HMWHA-stimulated Src activity had not been demonstrated. Here we report that Tgfbr3−/− cells do not activate Src when stimulated with HMWHA; an observation further confirmed with siRNA knockdown of TGFβR3 in Tgfbr3+/+ cells which also failed to activate Src.Wedonot postulate that TGFβR3 directly binds HMWHA, but that TGFBR3 serves in a receptor complex with HA binding receptors to coordinate signal transduction. It should be noted that we have previously reported that CD44 expression levels were not different between Tgfbr3+/+ and Tgfbr3−/− epicardial cells [21]. Therefore CD44 expression alone is not sufficient to drive HA-stimulated Src, Rac1, or RhoA activation. CD44 may require TGFβR3 to initiate crosstalk with Src-activating receptors such as Type I TGFβ receptors [25] or receptor tyrosine kinases such as FGFR1 [33].
The data from this study establish that TGFβR3 and Src are required for HMWHA-stimulated filamentous actin polymerization in epicardial cells. We postulate that actin filament severing pathways stimulated by HMWHA in Tgfbr3−/− cells remain intact, but due to defective Src, Rac1, and RhoA activation (Figs. 4, 5), these cells fail to repolymerize actin into stress fibers required for cellular invasion. The activation of Rac1 and RhoA GTPases is detected in HMWHA-stimulated Tgfbr3+/+ cells, concomitant with invasive cell motility. However, activation of these GTPases in HMWHA-stimulated Tgfbr3−/− cells is strikingly absent, as is f-actin reorganization under identical conditions. Cdc42 activation has been reported to be activated by HMWHA in cancer cells [34], while our study shows Tgfbr3+/+ cells attenuating Cdc42 during HMWHA stimulation. In contrast, HMWHA-stimulated Tgfbr3−/− cells fail to downregulate Cdc42 activity, an effect not observed in Tgfbr3+/+ cells. This indicates that TGFβR3 modulates Cdc42 activity. Since Tgfbr3−/− cells lost the ability to regulate Rho, Rac1 and Cdc42, HMWHA stimulation causes a shift in the Rho family GTPase activation profile to one indicative of cell motility dependent on TGFβR3. Ligand dependent inhibition of RhoA was shown to be required for epicardial and endocardial EMT and cell invasion [27,35]. In these studies we find low induction of RhoA activity relative to robust Rac1 activation in Tgfbr3+/+ cells and postulate that Rac1 is the major GTPase in driving HMWHA-stimulated f-actin polymerization. It should be noted that Rac1 and cdc42 were not required or sufficient (via expression of dominant negative and constitutively active mutants, respectively) to drive endocardial or epicardial invasion alone [27,35] suggesting that the activation of other signaling cascades upstream of Rho GTPases is required for cell invasion. Overexpression of TGFβR3 in ovarian cancer cells with reduced TGFβR3 leads to constitutive activation of cdc42 through β-Arrestin [21,23] but we observe the opposite effect of cdc42 in Tgfbr3−/− cells relative to Tgfbr3+/+ cells. We suggest that the basis of TGFβR3 arrested motility is dependent on activation of cdc42, in agreement with our findings that Tgfbr3−/− cells fail to decrease cdc42 activation and remain non-invasive in response to HA.
Substitution of Threonine 841 for Alanine in TGFβR3 prevents β-Arrestin binding to TGFβR3, resulting in inhibited receptor internalization, relief of NFκB activity suppression, but no effect on ligand independent activation of the canonical effector Smad2 [28,29]. This observation suggests that noncanonical TGFβR signal transduction may contribute to induction of cell motility which we examined in this study. As T841A-TGFβR3 can drive ligand independent cellular invasion in epicardial cells (Barnett Unpublished), and previous work in our laboratory has shown NFkB activity to be required for HMWHA directed cellular invasion [13], T841A-TGFβR3 was tested to see if it can drive Src activation independent of stimulus. Indeed we detected Src activation and observed that T841A-TGFβR3 drives cell transformation (Fig. 6) suggesting an important and previously unrecognized function of TGFβR3 signaling. These experiments constitute the first to report that TGFβR3 is upstream of HMWHA-stimulated Src tyrosine kinase activation. Collectively, these studies identify a novel TGFβR3–Src–Rac1–RhoA signaling axis that is required for HMWHA regulation of epicardial invasive cell motility.
Acknowledgments
The TGFβR3-T841A construct was generously provided by Dr. Joey Barnett and Dr. Gerard Blobe. We also thank all members of the Camenisch laboratory and Dr. Nicholas Mastrandrea for helpful discussions, and F. Bear for comments on this manuscript.
References
- 1.Mu H, Ohashi R, Lin P, Yao Q, Chen C. Vasc Med. 2005 Feb;10(1):37–44. doi: 10.1191/1358863x05vm584ra. [DOI] [PubMed] [Google Scholar]
- 2.Kalman F, Viragh S, Modis L. Anat Embryol (Berl) 1995 May;191(5):451–464. doi: 10.1007/BF00304430. [DOI] [PubMed] [Google Scholar]
- 3.Munoz-Chapuli R, Macias D, Gonzalez-Iriarte M, Carmona R, Atencia G, Perez-Pomares JM. Rev Esp Cardiol. 2002 Oct;55(10):1070–1082. doi: 10.1016/s0300-8932(02)76758-4. [DOI] [PubMed] [Google Scholar]
- 4.Perez-Pomares JM, de la Pompa JL. Circ Res. 2011 Dec 9;109(12):1429–1442. doi: 10.1161/CIRCRESAHA.111.245589. [DOI] [PubMed] [Google Scholar]
- 5.Lim J, Thiery JP. Development. 2012 Oct;139(19):3471–3486. doi: 10.1242/dev.071209. [DOI] [PubMed] [Google Scholar]
- 6.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Circulation. 2013 Jan 1;127(1):e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weigel PH, DeAngelis PL. J Biol Chem. 2007;282(51):36777. doi: 10.1074/jbc.R700036200. [DOI] [PubMed] [Google Scholar]
- 8.Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A, Jr, et al. J Clin Invest. 2000 Aug;106(3):349–360. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Austin AF, Compton LA, Love JD, Brown CB, Barnett JV. Dev Dyn. 2008 Feb;237(2):366–376. doi: 10.1002/dvdy.21421. [DOI] [PubMed] [Google Scholar]
- 10.Craig EA, Parker P, Austin AF, Barnett JV, Camenisch TD. Cell Signal. 2010 Jun;22(6):968–976. doi: 10.1016/j.cellsig.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Craig EA, Austin AF, Vaillancourt RR, Barnett JV, Camenisch TD. Exp Cell Res. 2010 Dec 10;316(20):3397–3405. doi: 10.1016/j.yexcr.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turley EA, Noble PW, Bourguignon LY. J Biol Chem. 2002 Feb 15;277(7):4589–4592. doi: 10.1074/jbc.R100038200. [DOI] [PubMed] [Google Scholar]
- 13.Craig EA, Parker P, Camenisch TD. Glycobiology. 2009 Aug;19(8):890–898. doi: 10.1093/glycob/cwp064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meran S, Luo DD, Simpson R, Martin J, Wells A, Steadman R, et al. J Biol Chem. 2011 May 20;286(20):17618–17630. doi: 10.1074/jbc.M111.226563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bourguignon LY, Wong G, Earle C, Krueger K, Spevak CC. J Biol Chem. 2010 Nov 19;285(47):36721–36735. doi: 10.1074/jbc.M110.162305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bourguignon LY, Zhu H, Shao L, Chen YW. J Biol Chem. 2001 Mar 9;276(10):7327–7336. doi: 10.1074/jbc.M006498200. [DOI] [PubMed] [Google Scholar]
- 17.Sit ST, Manser E. J Cell Sci. 2011 Mar 1;124(Pt 5):679–683. doi: 10.1242/jcs.064964. [DOI] [PubMed] [Google Scholar]
- 18.Hill CR, Sanchez NS, Love JD, Arrieta JA, Hong CC, Brown CB, et al. Cell Signal. 2012 May;24(5):1012–1022. doi: 10.1016/j.cellsig.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopez-Casillas F, Wrana JL. J Massague, Cell. 1993 Jul 2;73(7):1435–1444. doi: 10.1016/0092-8674(93)90368-z. [DOI] [PubMed] [Google Scholar]
- 20.Townsend TA, Robinson JY, Deig CR, Hill CR, Misfeldt A, Blobe GC, et al. Cells Tissues Organs. 2011;194(1):1–12. doi: 10.1159/000322035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanchez NS, Hill CR, Love JD, Soslow JH, Craig E, Austin AF, et al. Dev Biol. 2011 Oct 15;358(2):331–343. doi: 10.1016/j.ydbio.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Compton LA, Potash DA, Brown CB, Barnett JV. Circ Res. 2007 Oct 12;101(8):784–791. doi: 10.1161/CIRCRESAHA.107.152082. [DOI] [PubMed] [Google Scholar]
- 23.Mythreye K, Blobe GC. Proc Natl Acad Sci U S A. 2009 May 19;106(20):8221–8226. doi: 10.1073/pnas.0812879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bjorge JD, Jakymiw A, Fujita DJ. Oncogene. 2000 Nov 20;19(49):5620–5635. doi: 10.1038/sj.onc.1203923. [DOI] [PubMed] [Google Scholar]
- 25.Hutcheson JD, Ryzhova LM, Setola V, Merryman WD. J Mol Cell Cardiol. 2012 Nov;53(5):707–714. doi: 10.1016/j.yjmcc.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pollard TD, Borisy GG. Cell. 2003 Feb 21;112(4):453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez NS, Barnett JV. Cell Signal. 2012 Feb;24(2):539–548. doi: 10.1016/j.cellsig.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen W, Kirkbride KC, How T, Nelson CD, Mo J, Frederick JP, et al. Science. 2003 Sep 5;301(5638):1394–1397. doi: 10.1126/science.1083195. [DOI] [PubMed] [Google Scholar]
- 29.You HJ, How T, Blobe GC. Carcinogenesis. 2009 Aug;30(8):1281–1287. doi: 10.1093/carcin/bgp071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A, et al. J Clin Invest. 2000;106(3):349–360. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Townsend TA, Robinson JY, How T, DeLaughter DM, Blobe GC, Barnett JV. Cell Signal. 2012 Jan;24(1):247–256. doi: 10.1016/j.cellsig.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bourguignon LY, Singleton PA, Zhu H, Zhou B. J Biol Chem. 2002 Oct 18;277(42):39703–39712. doi: 10.1074/jbc.M204320200. [DOI] [PubMed] [Google Scholar]
- 33.Knelson EH, Gaviglio AL, Tewari AK, Armstrong MB, Mythreye K, Blobe GC. J Clin Invest. 2013 Nov 1;123(11):4786–4798. doi: 10.1172/JCI69657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bourguignon LY, Gilad E, Rothman K, Peyrollier K. J Biol Chem. 2005 Mar 25;280(12):11961–11972. doi: 10.1074/jbc.M411985200. [DOI] [PubMed] [Google Scholar]
- 35.Townsend TA, Wrana JL, Davis GE, Barnett JV. J Biol Chem. 2008 May 16;283(20):13834–13841. doi: 10.1074/jbc.M710607200. [DOI] [PMC free article] [PubMed] [Google Scholar]