

Abstract
Light‐induced interlayer ultrafast charge transfer in 2D heterostructures provides a new platform for optoelectronic and photovoltaic applications. The charge separation process is generally hypothesized to be dependent on the interlayer stackings and interactions, however, the quantitative characteristic and detailed mechanism remain elusive. Here, a systematical study on the interlayer charge transfer in model MoS2/WS2 bilayer system with variable stacking configurations by time‐dependent density functional theory methods is demonstrated. The results show that the slight change of interlayer geometry can significantly modulate the charge transfer time from 100 fs to 1 ps scale. Detailed analysis further reveals that the transfer rate in MoS2/WS2 bilayers is governed by the electronic coupling between specific interlayer states, rather than the interlayer distances, and follows a universal dependence on the state‐coupling strength. The results establish the interlayer stacking as an effective freedom to control ultrafast charge transfer dynamics in 2D heterostructures and facilitate their future applications in optoelectronics and light harvesting.
Keywords: interlayer‐state‐coupling, MoS2/WS2 heterostructures, stacking configurations, TDDFT calculations, ultrafast charge transfer
Since the discovery of graphene and the rise of MoS2 as well as black phosphorus, atomically thin 2D crystals have grown into a huge family of materials ranging from semimetal, semiconductors to insulators.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 Monolayer transition‐metal dichalcogenides denoted as MX2 (e.g., M = Mo, W, and X = S, Se, Te), have been prepared by physical exfoliation and chemical vapor deposition, providing more choices for 2D materials. The MX2 materials share similar crystalline structures and symmetries, but possess distinct electronic properties in bandgaps, photoabsorption, and spin–orbit coupling strength.7, 8, 9 The heterostructures vertically reassembled from different 2D materials form even richer material systems, and thus provide a new platform for investigating new physics12, 13, 14, 15, 16 and exploring new applications.17, 18, 19, 20, 21, 22, 23, 24 The heterostructures of two MX2 are of particular interests because many of them form type II heterojunctions,25, 26, 27 which facilitate the efficient separation of photoexcited electrons and holes28, 29 and therefore exhibit great potentials in the applications of photodetectors,30, 31 photovoltaic cells,32, 33 and light emitters.34
The interlayer charge transfer in MX2 heterostructures is of central importance in their photoresponse, which determines both the speed and efficiency of the charge separation.35, 36 Since the charge transfer is mainly through the overlapping between interlayer electronic states, the charge transfer process is believed to be highly dependent on the interlayer stackings (twisting, translation, and spacing) and interactions. Femtosecond pump–probe spectroscopy experiments reveals that the excited hole in MoS2/WS2 bilayer takes place in an ultrafast time scale.28 Wang et al. first report that collective motion of excitons at the interface leads to plasma oscillations associated with optical excitation in the ultrafast charge transfer in such van der Waals heterostructures, which provides a good insight in this new pheomenon.37 Although both experimental28, 29 and theoretical efforts37, 38 have been made to understand the ultrafast charge transfer process in MX2 heterostructures, the quantitative characteristic and detailed mechanism for the interlayer stacking/interaction dependence still remain elusive.
Here we utilize the state‐of‐the‐art time‐dependent density functional theory method (TDDFT) and demonstrate a systematical study on the interlayer charge transfer in model MoS2/WS2 bilayer system with variable stacking configurations. Our results demonstrate that the interlayer twisting, translation, or spacing can significantly modulate interlayer charge transfer time from 100 to 1000 fs scale, a huge modulation that has not been realized before. Further analysis reveals that the transfer rate in MoS2/WS2 bilayers is governed by the coupling between specific interlayer states (rather than the total coupling strength) and follows a universal exponential dependence on their dipole coupling matrix values. This work establishes a firm correlation between charge transfer dynamics and interlayer stacking/interaction in 2D heterostructures on the atomic level, and thus facilitate their future applications in, for example, high speed optoelectronics and new generations of light harvesting.
The choice of MoS2/WS2 as a model system to study the interlayer charge transfer process in 2D heterostructures is mainly initiated by the ultrafast experiment from Wang and co‐workers at UC Berkeley.28 MoS2/WS2 bilayers have a type II band alignment: the conduction band minimum and valence band maximum at K point reside at MoS2 and WS2 layers, respectively [Figure 1 ; also see Figure S1 in the Supporting Information]. Since MoS2 has an energy bandgap smaller than WS2, we can selectively excite the MoS2 by purposely choosing the excitation light with photon energy in between the bandgap of MoS2 and WS2. After photoexcitation, holes in MoS2 will have the chance to transfer into the WS2 because its valence band maximum is lower than that of WS2; while the electrons will stay in MoS2 since its conduction band minimum is also lower than WS2 (Figure 1b).
Figure 1.
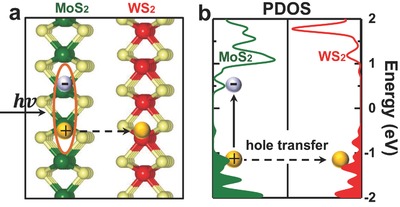
Atomic and electronic structure of the MoS2/WS2 bilayer. a) Side view of MoS2/WS2 bilayers. Green, red, and light yellow spheres represent Mo, W, and S atoms, respectively. b) Projected density of states (PDOS) on MoS2 and WS2 layer. Photoexcited holes will transfer from MoS2 valence bands to WS2.
MoS2/WS2 bilayers have many possible interlayer stacking configurations, i.e., AB1‐2H and AB2‐2H, AA1‐3R and AA2‐3R, and twisted ones.39, 40 Here, we choose the most stable AB1‐2H as an example to show the time evolution of the interlayer charge transfer. In our calculations, we use Ehrenfest dynamics based on TDDFT approach to study excited‐state dynamics, or ab initio TDDFT‐molecular dynamics (MD) methods,41, 42 which is widely proven reliable in describing quantum systems, such as optical absorption spectrum in dye‐sensitized TiO2 nanowire43 and electron injection and electron–hole recombination in dye solar cells.44 Optical transition in MoS2/WS2 bilayer is profoundly determined by the direct excitation of electron–hole pair in the individual layer since the interlayer interactions will only disturb the optical transition slightly.28 As shown in the band structure of MoS2/WS2 bilayer (Figure 2 a), at K point in the first Brillouin zone the |−2> and |1> states are mainly distributed on MoS2 while |−1> state is on WS2. Since the |−2> and |−1> states at K point are located at two different layers (MoS2 and WS2, respectively), they are referred to as interlayer state. It should be noted that |−2> and |−1> states at K point are not sensitive to interlayer coupling, and they do not change much with the number of layers, while the states at Γ point is quite allergic to the interlayer interactions. We note that direct interlayer excitation has a negligible strength as compared to intralayer excitation due to the minimal wavefunction overlap. Once this occurs, the interlayer exciton is formed right away upon excitation, and no charge transfer takes place in a short time <1 ps. By choosing the photon energy to be that of MoS2 bandgap, it will mainly excite the electron from |−2> to |1> state and leaves the holes in MoS2 layer. The holes will transfer into WS2 layers afterward.
Figure 2.
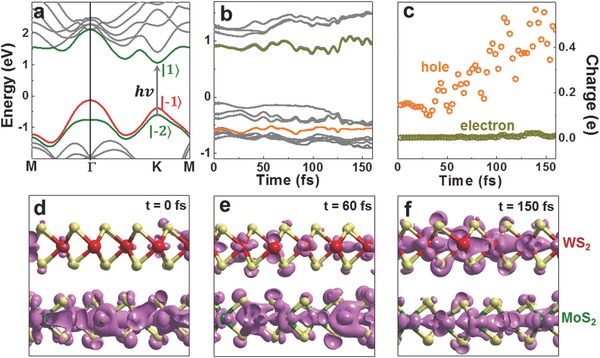
Charge transfer dynamics in AB1‐2H stacked MoS2/WS2 bilayers. a) Energy band dispersion of MoS2/WS2 bilayer in AB1‐2H stacking order. |1>, |−1>, and |−2> indicate these corresponding states at K point in the Brillouin zone. b) Evolution of electronic energy levels after photoexcitation. The orange (dark yellow) curve indicates the energy levels of photoexcited hole (electron) states. c) The fractions of photoexcited holes (electrons) that transferred from MoS2 to WS2 layer. d–f) Snapshots of the spatial distribution of hole density at 0, 60, and 150 fs after photoexcitation at a contour level of 0.02 e Å−3. The upper (lower) layer is WS2 (MoS2).
In the following, we look into the detailed hole transfer dynamics. We simulate the time evolution of interlayer charge transfer by simultaneously solving the time‐dependent Kohn–Sham equation and the Newtonian motion of ions (ionic forces along the classical trajectory evaluated through the Ehrenfest theorem,41, 42 see the Experimental Section for details). In Figure 2b, we show the evolution of Kohn–Sham energy levels of MoS2/WS2 bilayer. At the beginning, the energy of hole states (orange curve) is 0.48 eV below the valence band maximum. As the energy levels evolve in real time accompanied by the ionic motion, the hole states are getting closer to the valence band maximum. At t = 150 fs the difference becomes as small as 0.13 eV and the excited hole states in MoS2 and original occupied states in WS2 get largely mixed, or the hole transfer takes place. In Figure 2d–f, we give out the spatial distribution evolution of the hole states at different time. Immediately after photoexcitation, the holes are mostly distributed in MoS2 (<10% of the states are in WS2, attributing to the weak interlayer state hybridization). At t = 60 fs, ≈25% of the holes have transferred into WS2, and clearly at t = 150 fs, about 50% of the holes are localized in WS2. It means that the hole transfer between MoS2 and WS2 is taking place in an ultrafast way.
To gain quantitative information on charge transfer dynamics, we integrate the hole (electron) density (χ) on the WS2 orbitals at different time after excitation (Figure 2c). Within 150 fs, about half of the holes density has transferred to WS2. While as expected the electrons still stay in MoS2 and would not transfer to WS2 during the whole simulation time. We note that an oscillation of about 30 fs is observed, possibly due to the ultrafast oscillation of electronic states (oscillation of ionic motion is of period much larger than 30 fs and thus can be excluded).37 The A 1g mode was found to be critical to the photoexcited hole dynamics. Here by doing Fourier transformation of dipole moment along the vertical direction of the heterojunction, we find that the A 1g phonon mode indeed plays an important role in the process of ultrafast hole transfer (see the Supporting Information for details). The formation of intralayer exciton prior to interlayer charge transfer and the collective motion of excitons is secondary for the dynamics of photoexcited charge, because the main driving force in such an ultrafast process is the specific state coupling as detailed below.
Now we turn to evaluate the dependence of charge transfer dynamics on interlayer stackings and interactions. There are five typical stacking configurations of AB1‐2H, AB2‐2H, AB3‐2H, AA1‐3R, and AA2‐3R in MoS2/WS2 bilayers. For AB ones, the M‐X bond directions in the two layers are opposite; while for AA ones, the directions are the same. So between AA and AB ones, the bilayers rotate by an angle of π; and between AA1‐3R (AB1‐2H) and AA2‐3R (AB2‐2H), the two layers translate in‐plane by 1.1 Å. From our calculations (Table 1 ) and also previous results,22 AB1‐2H, AB2‐2H, and AA1‐3R are the stable configurations with a smaller interlayer spacing of 6.3 Å; while AA2‐3R is metastable with a larger interlayer spacing of 6.8 Å. The formation energies of AB1‐2H, AB2‐2H, and AA1‐3R is of similar strength, but much lower than in AA2‐3R.21 Both AA2‐3R and AB3‐2H share the similar orientation and interlayer distance (6.8 Å) and hole dynamics. Thus, the hole dynamics of AB3‐2H stacking mode is presented for comparison in Figure S6 (Supporting Information). Since the charge transfer is related to the interlayer electronic coupling, naively one would expect that the shorter the interlayer distance is, the faster the interlayer charge transfer will be, or τAB1 ≈ τAB2 ≈ τAA1 ≪ τAA2.
Table 1.
Calculated parameters for MoS2/WS2 bilayers
| Stacking | d a) [Å] | E b) [meV per atom] | M c) [e Å] | τd) [fs] | |
|---|---|---|---|---|---|
| AB1‐2H | 6.3 | −30.6 | 0.42 | 150 | |
| AB1‐2H | Artificial | 6.0 | −27.5 | 0.62 | 120 |
| AB1‐2H | Artificial | 7.0 | −20.0 | 0.16 | 320 |
| AB1‐2H | Artificial | 7.3 | −16.2 | 0.005 | 1600 |
| AB2‐2H | 6.3 | −28.3 | 0.06 | 1100 | |
| AA1‐3R | 6.3 | −28.7 | 0.02 | 1500 | |
| AA2‐3R | 6.8 | −19.7 | 0.18 | 180 |
d: interlayer spacing
E: formation energy
M: dipole transition matrix element; and
τ: hole transfer lifetime.
In our results, we fit the time evolution data with an exponential equation χ = a + b*exp (−t/τ), where τ is the charge transfer lifetime. As marked in Figure 3 a–d, τAB1 or τAA2 is around 100 fs timescale and τAB2 or τAA1 is around 1000 fs scale (or τAB1 ≈ τAA2 ≪ τAB2 ≈ τAA1). It seems that the charger transfer time has no obvious correlation to interlayer coupling strength, which is quite counter‐intuitive. The timescale is obtained by fitting to the simulated trends of ultrafast charge transfer upon photoexcitation. Direct simulation with trajectories longer than picoseconds is not only computationally unaffordable, but also blocked by the divergence of orbital propagation and break‐down of mean field approach. We found that simulation in a relatively short timescale already gives a very reasonable fitting result of the charge transfer timescale (Figure S5, Supporting Information).
Figure 3.
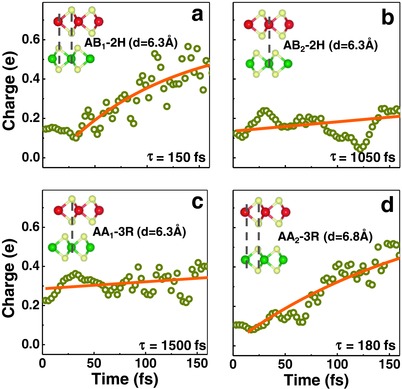
a–d) Hole transfer evolution for MoS2/WS2 in different stacking configurations. The transfer lifetime is fitted by an exponential relation. The insets give out the schematic atomic structure, interlayer spacing, and lifetime. From the consideration of total interlayer distances, one would expect τAB1 ≈ τAB2 ≈ τAA1 ≪ τAA2, but the simulation results show τAB1 ≈ τAA2 ≪ τAB2 ≈ τAA1. It reveals that the charger transfer time has no obvious correlation to the total interlayer coupling strength.
The above observed discrepancy between computation data and expectation drives us to rethink about the interlay couplings. The usual mechanical or electronic interlayer coupling we talked about is actually the total one, which includes all the electronic states. However, the interlayer charge transfer takes place only between some specific interlayer states, and the charger transfer may be only related to the coupling between these specific ones. We therefore evaluate all the coupling elements between different specific interlayer states and eventually figure out the one that is responsible for the charge transfer dynamics, namely, the one between the |−2> and |−1> states at K point in the Brillouin zone. We use the dipole transition matrix element (M) to evaluate the coupling strength between the two states as
| (1) |
where is the position operator along the vertical direction normal to the MX2 plane. Quite interestingly, we found that M AB1 > M AA2 ≫ M AB2 > M AA1, which is positively correlated to 1/τ. Detailed analysis further revealed that 1/τ is exponentially dependent on M, or 1/τ ∝ eM (dashed line in Figure 4 a).
Figure 4.
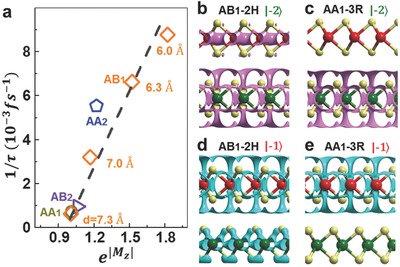
Interlayer‐state‐coupling dependent charge transfer rate in MoS2/WS2 bilayers. a) Dependence of the charge transfer rates (1/τ) on the dipole transition coupling strength (M) between |−2> and |−1> states. AA1‐3R, AA2‐3R, AB1‐2H, and AB2‐2H data are, respectively, shown as circle, pentagon, square, and triangles. The interlayer spacing value of AB1‐2H (including those artificial ones) are labeled beside the data points. All data seat around the same curve, revealing a universal dependence of charge transfer rate on interlayer‐state‐coupling strength. b–e) Spatial distribution of |−2> and |−1> states in AB1‐2H and AA1‐3R stackings. The overlapping between |−2> and |−1> states along vertical direction on both the upper WS2 and lower MoS2 layers in AB1‐2H stacking is finite, while it is nearly zero in AA1‐3R stacking. Thus M AB1 ≫ M AA1 is understandable. The isosurface value is 1 × 10−3 e Å−3 in (b–e).
In ref. 37, using both TDDFT and model Hamiltonian analysis Wang et al. studied dipole transition couplings between different layers in the heterojunctions and showed charge transfer time is dependent on stacking parameters. In the present work, the first principle simulations were further extended to different systems and longer times, where the charge transfer time is determined for every single stacking mode and thus the exponential dependence of charge transfer time on the interlayer‐state‐coupling strength can be plotted.
To further check whether this exponential dependence is universal in determining the interlayer charge transfer dynamics, we artificially tune the interlayer spacing from 6.0 to 7.3 Å in AB1‐2H stacking and obtained the τ and M (see Figure S2 in the Supporting Information for details). Again, all the data seat around the same 1/τ versus eM curve. So, the interlayer coupling between the |−2> and |−1> states is the universal factor that determines the charge transfer dynamics in MoS2/WS2 bilayers. To clarify why the specific coupling strength between |−2> and |−1> states at K point is crucial, we have checked all relative states (occupied or unoccupied states) and found only , is allergic to the photoexcited hole dynamics (with a typical value of 0.5 e Å for AB1‐2H stacking). The couplings between other pairs of states are much smaller (with a typical value of 10−3 e⋅ Å). The dependence of charge transfer rate on the interlayer‐state‐coupling M justifies the importance of M as a measure for interface ultrafast dynamics.
Now we try to give out a physical picture why slight change of interlayer stacking configuration will give out dramatically different M. We use AB1‐2H and AA1‐3R bilayers as an example for illustration, which are both stable with the interlayer spacing of 6.3 Å. We directly draw the spatial distribution of |−2> (Figure 4b) and |−1> states (Figure 4 d) in both WS2 and MoS2 layers. For AB1‐2H stacking, in the upper WS2 layer, |−2> has finite overlapping with |−1> states along vertical direction and so as in the lower MoS2 layer. In contrast, for AA1‐3R stacking, the overlapping in both layers is nearly zero. Since the is an operation that projects electronic states to vertical directions, therefore M AB1 ≫ M AA1 is understandable (see Figure S3 in the Supporting Information for the other two stackings).
In the original experiment by Hong et al., the authors used film transfer method to prepare MoS2/WS2 bilayers, where the interlayer stacking configurations are not well controlled.28 Recently, MX2 bilayers with defined interlayer geometry can be directly grown by chemical vapor deposition methods in several groups.15, 20, 21 And to utilize the two‐color pump–probe optical technique, the charge transfer lifetime can be obtained by evaluating the rising up part of the pump–probe curve.28, 29 Therefore, it is now the right time to experimentally study the charge transfer dynamics in MoS2/WS2 bilayers with different stackings predicted here.
In summary, we employ ab initio TDDFT‐MD methods to investigate the ultrafast interlayer charge transfer dynamics in MoS2/WS2 bilayers. Our study reveals that slight interlayer geometry modulation of twisting, translation, or spacing can tune charge transfer dynamics very effectively, resulting in a change of transfer lifetime spanning from 100 to 1000 fs timescale. A universal exponential relationship between the charge transfer rate and the coupling strength between specific interlayer electronic states is established, based on detailed analysis of stacking‐mode and layer‐spacing modulation on the charge transfer dynamic processes. Based on these findings, one could utilize physical or chemical methods to control the interlayer geometry and therefore to control the charge transfer quantum dynamics, thus facilitating future applications of 2D heterostructures in novel optoelectronic and light harvesting devices.
Experimental Section
First principle calculations of MX2 vertical heterostructures were performed using density functional theory implemented in the Vienna ab initio simulation package45 with Perdew, Burke, and Ernzerhof (PBE) generalized gradient approximation for the exchange–correlation functional.46, 47 Because of the absence of strong chemical bonding between layers, van der Waals density functional in the opt88 form48 was employed for structural optimization. Both lattice constants and atomic positions were relaxed until all residual forces remain less than 10−2 eV Å−1 and the total energy variation is less than 10−4 eV. The Brillouin zone was sampled by a set of 25 × 25 × 1 k‐mesh with an energy cutoff of 400 eV for plane waves. The thickness of vacuum layer was set to be larger than 15 Å, so that interactions between repeated images are avoided. Formation energy (E) of the MoS2/WS2 heterostructure is defined as , where , , are, respectively, the total energies of MoS2/WS2 bilayers, MoS2 and WS2 layers, and N is the number of atoms in the supercells.
The excited‐state real‐time TDDFT simulations were carried out with the time‐dependent ab initio package TDAP42 based on SIESTA.49 Pseudopotentials of the Troullier–Martins type, the PBE exchange–correlation functional, and a local basis set of double‐ζ polarized orbitals were used. It was noted that the band offset between the valence band maximum of WS2 and MoS2 in experiment (0.7 eV) were correctly reproduced by the PBE functional (0.5 eV).50 Although PBE functional usually underestimates the bandgaps, it is accurate enough to describe the spatial distribution of electronic states and the state couplings, which are crucial in the dynamic simulations.51 In addition, very similar band structures based on PBE functional and HSE06 functional are shown in Figure S7 (see the Supporting Information for details). Supercells in rectangle shape containing 108 atoms in the supercells were used to model bilayers with a single k‐point for integration in the Brillouin zone and the states relative to the photoexcited states especially at K point in the first Brillouin zone were approximately folded to the supercells. The time step of all simulations was set to be 0.024 fs. Electron–hole interaction and electron–phonon effects were naturally included in the methods. The initial velocities of ions were assigned according to the equilibrium Boltzmann–Maxwell distribution at a given temperature of 350 K. It should be noted that some energy levels in the supercells were used may be degenerate, so states with similar energy and spatial distributions have been checked in the following dynamic simulations to ensure the reliability of the results.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supplementary
Acknowledgements
J.Z., H.H., and C.L. contributed equally to this work. The authors are grateful to H. Wang, J. Bang, and S. B. Zhang of Department of Physics, Applied Physics, and Astronomy, Rensselaer Polytechnic Institute in sharing the method to calculate dipole transition matrix element. This work was supported by the National Key Research & Development Program of China (Grant Nos. 2016YFA0300902, 2016YFA0300903, and 2015CB921001) and the National Natural Science Foundation of China (Grant Nos. 11222431, 11474006, and 51522201).
Zhang J., Hong H., Lian C., Ma W., Xu X., Zhou X., Fu H., Liu K., Meng S., Adv. Sci. 2017, 4, 1700086 https://doi.org/10.1002/advs.333
Contributor Information
Kaihui Liu, Email: khliu@pku.edu.cn.
Sheng Meng, Email: smeng@iphy.ac.cn.
References
- 1. Geim A. K., Novoselov K. S., Nat. Mater. 2007, 6, 183. [DOI] [PubMed] [Google Scholar]
- 2. Frenzel A., Lui C., Shin Y., Kong J., Gedik N., Phys. Rev. Lett. 2014, 113, 056602. [DOI] [PubMed] [Google Scholar]
- 3. Splendiani A., Sun L., Zhang Y., Li T., Kim J., Chim C.‐Y., Galli G., Wang F., Nano Lett. 2010, 10, 1271. [DOI] [PubMed] [Google Scholar]
- 4. Mak K. F., Lee C., Hone J., Shan J., Heinz T. F., Phys. Rev. Lett. 2010, 105, 136805. [DOI] [PubMed] [Google Scholar]
- 5. Mak K. F., McGill K. L., Park J., McEuen P. L., Science 2014, 344, 1489. [DOI] [PubMed] [Google Scholar]
- 6. Ye Z., Cao T., O'Brien K., Zhu H., Yin X., Wang Y., Louie S. G., Zhang X., Nature 2014, 513, 214. [DOI] [PubMed] [Google Scholar]
- 7. Wang Q. H., Kalantar‐Zadeh K., Kis A., Coleman J. N., Strano M. S., Nat. Nanotechnol. 2012, 7, 699. [DOI] [PubMed] [Google Scholar]
- 8. Xia F., Wang H., Xiao D., Dubey M., Ramasubramaniam A., Nat. Photonics 2014, 8, 899. [Google Scholar]
- 9. Baugher B. W., Churchill H. O., Yang Y., Jarillo‐Herrero P., Nat. Nanotechnol. 2014, 9, 262. [DOI] [PubMed] [Google Scholar]
- 10. Wang H., Zhang C., Rana F., Nano Lett. 2015, 15, 339. [DOI] [PubMed] [Google Scholar]
- 11. Li L., Yu Y., Ye G. J., Ge Q., Ou X., Wu H., Feng D., Chen X. H., Zhang Y., Nat. Nanotechnol. 2014, 9, 372. [DOI] [PubMed] [Google Scholar]
- 12. Geim A. K., Grigorieva I. V., Nature 2013, 499, 419. [DOI] [PubMed] [Google Scholar]
- 13. Hunt B., Sanchez‐Yamagishi J. D., Young A. F., Yankowitz M., LeRoy B. J., Watanabe K., Taniguchi T., Moon P., Koshino M., Jarillo‐Herrero P., Science 2013, 340, 1427. [DOI] [PubMed] [Google Scholar]
- 14. Lee C. H., Lee G. H., Van Der Zande A. M., Chen W., Li Y., Han M., Cui M. X., Arefe G., Nuckolls C., Heinz T. F., Nat. Nanotechnol. 2014, 9, 676. [DOI] [PubMed] [Google Scholar]
- 15. Gong Y., Lin J., Wang X., Shi G., Lei S., Lin Z., Zou X., Ye G., Vajtai R., Yakobson B. I., Nat. Mater. 2014, 13, 1135. [DOI] [PubMed] [Google Scholar]
- 16. Chiu M.‐H., Zhang C., Shiu H.‐W., Chuu C.‐P., Chen C.‐H., Chang C.‐Y. S., Chen C.‐H., Chou M.‐Y., Shih C.‐K., Li L.‐J., Nat. Commun. 2015, 6, 7666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heo H., Sung J. H., Cha S., Jang B.‐G., Kim J.‐Y., Jin G., Lee D., Ahn J.‐H., Lee M.‐J., Shim J. H., Nat. Commun. 2015, 6, 7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rigosi A. F., Hill H. M., Li Y., Chernikov A., Heinz T. F., Nano Lett. 2015, 15, 5033. [DOI] [PubMed] [Google Scholar]
- 19. Rivera P., Schaibley J. R., Jones A. M., Ross J. S., Wu S., Aivazian G., Klement P., Seyler K., Clark G., Ghimire N. J., Nat. Commun. 2015, 6, 6242. [DOI] [PubMed] [Google Scholar]
- 20. Zhang Q., Xiao X., Zhao R., Lv D., Xu G., Lu Z., Sun L., Lin S., Gao X., Zhou J., Angew. Chem., Int. Ed. 2015, 54, 8957. [DOI] [PubMed] [Google Scholar]
- 21. Zhang J., Wang J., Chen P., Sun Y., Wu S., Jia Z., Lu X., Yu H., Chen W., Zhu J., Adv. Mater. 2016, 28, 1950. [DOI] [PubMed] [Google Scholar]
- 22. Han S., Kwon H., Kim S. K., Ryu S., Yun W. S., Kim D., Hwang J., Kang J.‐S., Baik J., Shin H., Phys. Rev. B 2011, 84, 045409. [Google Scholar]
- 23. Zhang L., Zunger A., Nano Lett. 2015, 15, 949. [DOI] [PubMed] [Google Scholar]
- 24. Kang J., Zhang L., Wei S. H., J. Phys. Chem. Lett. 2016, 7, 597. [DOI] [PubMed] [Google Scholar]
- 25. Meier D., Kuschel T., Shen L., Gupta A., Kikkawa T., Uchida K.‐I., Saitoh E., Schmalhorst J.‐M., Reiss G., Phys. Rev. B 2013, 87, 054421. [Google Scholar]
- 26. Kang J., Tongay S., Zhou J., Li J., Wu J., Appl. Phys. Lett. 2013, 102, 012111. [Google Scholar]
- 27. Gong C., Zhang H., Wang W., Colombo L., Wallace R. M., Cho K., Appl. Phys. Lett. 2013, 103, 053513. [Google Scholar]
- 28. Hong X., Kim J., Shi S.‐F., Zhang Y., Jin C., Sun Y., Tongay S., Wu J., Zhang Y., Wang F., Nat. Nanotechnol. 2014, 9, 682. [DOI] [PubMed] [Google Scholar]
- 29. Ceballos F., Bellus M. Z., Chiu H.‐Y., Zhao H., ACS Nano 2014, 8, 12717. [DOI] [PubMed] [Google Scholar]
- 30. Britnell L., Ribeiro R., Eckmann A., Jalil R., Belle B., Mishchenko A., Kim Y.‐J., Gorbachev R., Georgiou T., Morozov S., Science 2013, 340, 1311. [DOI] [PubMed] [Google Scholar]
- 31. Yu W. J., Liu Y., Zhou H., Yin A., Li Z., Huang Y., Duan X., Nat. Nanotechnol. 2013, 8, 952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bernardi M., Palummo M., Grossman J. C., Nano Lett. 2013, 13, 3664. [DOI] [PubMed] [Google Scholar]
- 33. Yu Y., Hu S., Su L., Huang L., Liu Y., Jin Z., Purezky A. A., Geohegan D. B., Kim K. W., Zhang Y., Nano Lett. 2014, 15, 486. [DOI] [PubMed] [Google Scholar]
- 34. Ross J. S., Klement P., Jones A. M., Ghimire N. J., Yan J., Mandrus D., Taniguchi T., Watanabe K., Kitamura K., Yao W., Nat. Nanotechnol. 2014, 9, 268. [DOI] [PubMed] [Google Scholar]
- 35. Falke S. M., Rozzi C. A., Brida D., Maiuri M., Amato M., Sommer E., De Sio A., Rubio A., Cerullo G., Molinari E., Science 2014, 344, 1001. [DOI] [PubMed] [Google Scholar]
- 36. Rozzi C. A., Falke S. M., Spallanzani N., Rubio A., Molinari E., Brida D., Maiuri M., Cerullo G., Schramm H., Christoffers J., Nat. Commun. 2013, 4, 1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang H., Bang J., Sun Y., Liang L., West D., Meunier V., Zhang S., Nat. Commun. 2016, 7, 11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Long R., Prezhdo O. V., Nano Lett. 2016, 16, 1996. [DOI] [PubMed] [Google Scholar]
- 39. van Der Zande A. M., Kunstmann J., Chernikov A., Chenet D. A., You Y., Zhang X., Huang P. Y., Berkelbach T. C., Wang L., Zhang F., Nano Lett. 2014, 14, 3869. [DOI] [PubMed] [Google Scholar]
- 40. Liu K., Zhang L., Cao T., Jin C., Qiu D., Zhou Q., Zettl A., Yang P., Louie S. G., Wang F., Nat. Commun. 2014, 5, 4966. [DOI] [PubMed] [Google Scholar]
- 41. Runge E., Gross E. K., Phys. Rev. Lett. 1984, 52, 997. [Google Scholar]
- 42. Meng S., Kaxiras E., J. Chem. Phys. 2008, 129, 054110. [DOI] [PubMed] [Google Scholar]
- 43. Meng S., Ren J., Kaxiras E., Nano Lett. 2008, 8, 3266. [DOI] [PubMed] [Google Scholar]
- 44. Meng S., Kaxiras E., Nano Lett. 2010, 10, 1238. [DOI] [PubMed] [Google Scholar]
- 45. Kresse G., Furthmüller J., Comput. Mater. Sci. 1996, 6, 15. [Google Scholar]
- 46. Blöchl P. E., Phys. Rev. B 1994, 50, 17953. [DOI] [PubMed] [Google Scholar]
- 47. Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865. [DOI] [PubMed] [Google Scholar]
- 48. Klimeš J., Bowler D. R., Michaelides A., Phys. Rev. B 2011, 83, 195131. [Google Scholar]
- 49. Soler J. M., Artacho E., Gale J. D., García A., Junquera J., Ordejón P., Sánchez‐Portal D. J., J. Phys.: Condens. Matter. 2002, 14, 2745. [DOI] [PubMed] [Google Scholar]
- 50. Hill H. M., Rigosi A. F., Rim K. T., Flynn G. W., Heinz T. F., Nano Lett. 2016, 16, 4831. [DOI] [PubMed] [Google Scholar]
- 51. Ma W., Zhang J., Yan L., Jiao Y., Gao Y., Meng S., Comput. Mater. Sci. 2016, 112, 478. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary