

Abstract
Novel ligands that target Toll-like receptors and other innate recognition pathways represent a potent strategy for modulating innate immunity to generate anti-tumor immunity. While many of the current clinically successful immunotherapies target adaptive T-cell responses, both pre-clinical and clinical studies suggest that adjuvants have the potential to enhance the scope and efficacy of cancer immunotherapy. Radiation may be a particularly good partner to combine with innate immune therapies, since it is a highly efficient means to kill cancer cells, but may fail to send the appropriate inflammatory signals needed to act as an efficient endogenous vaccine. This may explain why although radiation therapy is a highly used cancer treatment, true abscopal effects – regression of disease outside the field without additional systemic therapy – are extremely rare. This review focuses on efforts to combine innate immune stimuli as adjuvants with radiation, creating a distinct and complementary approach from T cell targeted therapies to enhance anti-tumor immunity.
Keywords: radiation, adjuvant, innate, immunotherapy, TLR, STING, IFN, TNF
Introduction
Ionizing radiation has the ability to induce various types of cell death, including apoptosis, necrosis, necroptosis, and autophagy, which have been shown to have both immunosuppressive or immunogenic effects1. Radiation directed to one tumor site can induce the regression of tumor(s) at other distant site(s), a phenomenon known as the abscopal effect. However, despite approximately 500,000 radiation treatments per year in the USA, Abuodeh et al. recently described that there have been only 21 cases of abscopal tumor regression of solid tumors reported in the literature over the past 45 years, excluding recent reports of radiation therapy combined with systemic immunotherapy2 This suggests that radiation therapy alone does not generate clinically significant systemic immunity. Since radiation therapy remains an effective means to induce cell death and provide antigen to the immune system, we have to consider why radiation does not generate systemic immunity as a single agent.
Most of the recent studies that have validated radiation therapy as an effective partner for immunotherapy in preclinical and clinical settings have utilized immunotherapies that block T cell checkpoint regulatory molecules, such as antagonistic anti-CTLA4 and anti-PD1 antibodies that block inhibitory signals on T cells to unleash full T cell effector function3–8, or agonistic antibodies that target 4-1BB and OX40, costimulatory molecules that are present for a short period after antigen stimulation. Ligation of these co-stimulatory molecules results in expansion of antigen-stimulated T cells9–14 including tumor-specific T cells and drive their differentiation into effector and memory T cells with anti-tumor potential15–17. One of the classic tenets in immunology states that T cells require three signals in order to generate effective immunity. Signal 1 comes from antigen bound to MHC class I or MHC class II signaling through the T cell receptor. Signal 2 is a co-stimulatory signal (B7.1 and B7.2 on antigen presenting cells (APC) binding to CD28 on T cells), and Signal 3 is a cytokine that helps shape the subsequent immune response. Thus, antigen without adjuvant fails to generate effective adaptive immunity because it provides signal 1 without signals 2 or 3 resulting in tolerance or anergy. While radiation provides dying cells as a source of antigen (Signal 1), we propose that radiation does not provide sufficient co-stimulation (signal 2) or cytokine release (signal 3) to efficiently activate the adaptive immune system. While cytokines are induced in tumors following radiation therapy, the poor efficacy of radiation alone as an endogenous vaccine is most clear when compared to strong exogenous vaccines: in preclinical experiments, our work has shown that the T cell response to antigens from dying cancer cells can be an order of magnitude lower than the response to antigens expressed in bacteria or viruses.
Dying cells can provide adjuvant in the form of DAMPs (danger associated molecular patterns) by expressing heat shock proteins18–20, releasing HMGB121,22 or translocating calreticulin23, and lysis of tumor cells has been associated with the adjuvant activity of IL-3324 and uric acid25,26. However, M2 polarized macrophages, the dominant myeloid cell in most tumor environments, respond to adjuvant by secreting cytokines such as VEGF, IL-10 and TGFβ27–29, which are viewed as tumor supporting or immunosuppressive molecules. Moreover, irradiated cancer cells have been shown to drive undifferentiated macrophages towards M2 polarization27,30–32 despite the adjuvant content of irradiated cells. These macrophages limit the efficacy of radiation therapy in a range of mouse models33–37, and preventing M2 polarization enhances radiation tumor control by radiation therapy27,38. Taken together, these data suggest that the endogenous adjuvant activity of irradiated cancer cells is often insufficient to overcome the preexisting suppressive environment of the tumor and may even enhance the suppressive M2 macrophage environment. Since any immune response generated following tumor radiation very rarely influences tumors outside the treatment field2, it is logical to increase the immunogenicity of radiation therapy via the exogenous delivery of adjuvant.
Toll-like receptors (TLR) are pattern recognition receptors capable of recognizing microbial products39. Signaling through distinct TLR can share downstream pathways such as MyD88 and TRIF, but TLR expression varies across cell types (Figure 1). Therefore, the consequence of TLR ligation can vary according to the cell type and their differentiation40,41. More recently characterized STING and RIG-I-like receptors have also been shown to trigger the release of key innate cytokines such as type I IFN and TNFα. This review will focus on the synergy between activation of innate immune receptors and radiation therapy.
Figure 1. Distribution of innate receptors across cell types in the ImmGen dataset.
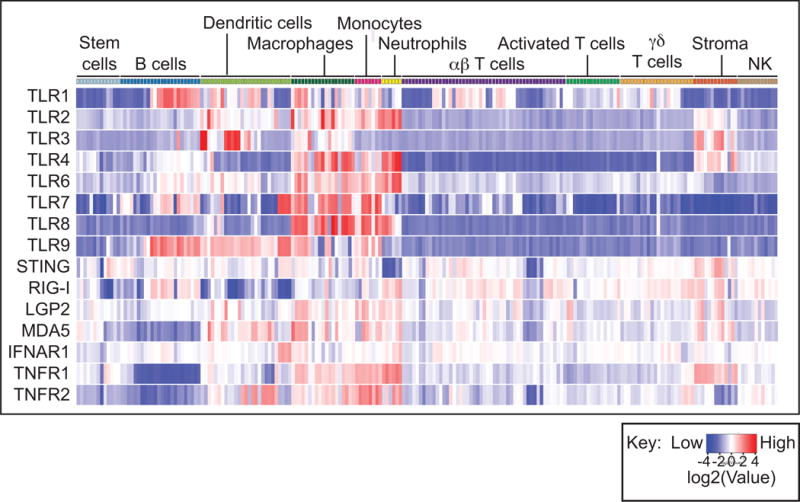
The graph shows gene expression of innate receptors in sorted cell types clustered into broad immune populations. Expression of each gene is normalized across cell populations and color-coded according to the key. There is significant variation in expression of innate sensors across immune cells. While receptors such as TLR4 are most highly expressed by macrophages and neutrophils, TLR9 shows extended expression into dendritic cells and B cells. Broadly, T cells exhibit low expression of TLR. By contrast, the innate sensor STING and the type I IFN receptor are very evenly expressed across many cell populations. This analysis is a result of data assembled by the ImmGen Consortium. Full analysis of gene expression patterns can be visualized at www.immgen.org.
TLR3
TLR3 is particularly expressed in DC and macrophage subsets (Figure 1) and mediates the host response to double stranded RNA in infectious agents42. Interestingly, the infected cell does not need to respond to the presence of double stranded RNA, but it is critical that dendritic cells express TLR3 in order to efficiently cross-present antigens from infected cells43. This is applicable to our goal to generate adaptive immune priming following radiation therapy, as the CD8+ DC population is critical for cross-presentation of cell-associated antigens to generate T cell responses44, and dying cells are efficiently tolerogenic without adjuvant45,46. TLR3 is unusual amongst TLRs because it uses TRIF rather than MyD88 to signal41,47.
The synthetic TLR3 ligand poly(I:C) has been used as a cancer immunotherapy for over 45 years. Intraperitoneal administration of poly(I:C) first showed clinical promise in treating a mouse model of melanoma48. Poly(I:C) has been widely used in vaccines, providing adjuvant signals to a range of antigen sources, including free peptides and tumor-derived apoptotic cells (reviewed in49). Early studies with poly(I:C) showed limited benefit due to its short half-life50, but a modified, degradation-resistant poly(ICLC)51 has shown increased cytokine induction, but also has produced increased toxicity in patients (reviewed in49,50). Poly(I:C) can also activate RIG-I-like receptors when poly(I:C) gains access to the cytosol. For this reason, as will be discussed below, some of the activities formerly ascribed to TLR3 are also dependent on RIG-I like receptors such as MDA552–54.
A few studies have examined poly(ICLC) in combination with radiation therapy. A phase II study of intramuscular poly(ICLC) and fractionated radiation following surgery in patients with glioblastoma showed improved survival compared to historical controls55. Similarly, a phase II study combining fractionated chemoradiation with intramuscular poly(ICLC) reported that the combination was well tolerated and that median survival was longer than prior studies with chemoradiation alone56. However, thus far there are no reports demonstrating efficacy of the combination of radiation therapy with poly(ICLC) in randomized clinical trials.
TLR4
TLR4 is expressed in neutrophils and macrophages (Figure 1), and ligation of TLR4 by lipopolysaccharide (LPS) or endogenous damage associated molecular patterns (DAMPs) such as hyaluronan, heat shock proteins (HSP), and HMGB141 results in signaling through MyD88 dependent and independent pathways57,58. Many of these factors can be released after radiotherapy and pre-clinical studies have demonstrated the importance of this endogenous TLR signaling to the success of radiotherapy. Apetoh et al. demonstrated that the release of HMBG1, resulting from radiotherapy- or chemotherapy -induced cell death, triggered TLR4, which in turn increased the processing and presentation of tumor antigens21. This process of “immunogenic cell death” was found to be critical to the anti-tumor effects of cytotoxic therapy because TLR4 knockout or blocking HMGB1 drastically abrogated the efficacy of therapy. Additionally, endogenous activation of TLR by microbiota may also play a role in the anti-tumor effects of radiotherapy since translocation of gut microbiota and subsequent TLR4 activation is critical to the efficacy of total body irradiation and adoptive transfer in mouse models59.
Early experiments demonstrating the efficacy of LPS as a therapy were performed in the late 1960s, and these early experiments recognized a tight balance between toxicity and efficacy60. A range of purified LPS preparations have been tested in clinical studies for cancer (reviewed in61) with moderate efficacy, however, the systemic toxicities of LPS are often due to the systemic consequences of IL-1 and TNFα production. Intratumoral administration of LPS has been shown to permit complete regression of tumors62. However, few studies have tested the addition of exogenous TLR4 ligands as partners for radiation therapy. BCG can be used to trigger TLR4 and increase the radiosensitivity of HCT-116 colon carcinoma cells by increasing autophagy63; however, many cancer cells cannot directly respond to TLR4 and as with other TLR ligands, it is more important that the stromal and immune cells can respond to the TLR ligand than the cancer cell64.
While LPS can induce potent pro-inflammatory responses from macrophages in particular contexts, as discussed above LPS treatment of tumor macrophages following radiation therapy results in secretion of cytokines that are anti-inflammatory and support tumor growth and tissue repair27–29,65. These data suggest that while LPS is a potent immune adjuvant, and while endogenous TLR4 adjuvants contribute to tumor control following radiation therapy, TLR4 ligation can have anti-inflammatory as well as proinflammatory effects, depending on the differentiation of the responding cells.
TLR7
TLR7 is highly expressed in monocyte and macrophage lineages (Figure 1), and the synthetic TLR7 ligand Imiquimod has been used successfully as an immunotherapy for dermatological malignancies and pre-malignancies66. Imiquimod has been shown to have direct cytotoxic effects on squamous cell carcinoma cell lines in vitro67, but when administered in vivo, the control of squamous cells is not direct and is dependent on the cytokines and inflammation generated by immune cells68–71. Topical administration of Imiquimod can alter the immune environment of skin metastases of breast cancer with some evidence of local tumor response72. In a preclinical mammary carcinoma model, topical administration of Imiquimod changed the immune environment in underlying subcutaneous tumors, and led to growth delay of both the primary treated tumor and a distant tumor70. In addition, delivery of radiation (8Gy × 3) significantly increased the number of complete local responses. Regression of distant unirradiated tumors was also observed if they were also treated with topical Imiquimod70.
However, the solubility profile of Imiquimod has limited its clinical application. There are TLR7 ligands that can be applied systemically73. Dovedi et al. demonstrated that systemic application of a novel TLR7 ligands synergized with RT for control of preclinical B and T cell lymphoma models74. This effect was entirely dependent on CD8 T cells and resulted in antigen-specific T cell immunity. Following from this work, Adlard et al. demonstrated that systemically delivered TLR7 agonist significantly improved survival in combination with RT in preclinical models of solid tumors75 while systemic delivery without RT did not impact tumor growth or progression. Improved survival was observed both with a fractionated dose of 2Gy × 5 and a single fraction of 15Gy.75. By contrast, in the T cell lymphoma model, systemic administration of the TLR7 agonist was more effective with fractionated radiation (2Gy × 5) than with a single fraction of 10Gy74. These data show that novel TLR7 ligands have significant therapeutic potential to treat cancer, particularly in conjunction with radiation therapy.
TLR9
TLR9 has a broad expression profile, in B cells, dendritic cells, macrophages and monocytes, and signals primarily through MyD88- dependent pathways. TLR9 recognizes unmethylated CpG oligonucleotide sequences that are present in microbial DNA but not mammalian DNA, as well as endogenous DAMPs including chromatinDNA complexes and ribonucleoproteins41,76,77. Synthetic CpGs that activate TLR9 have been demonstrated to be superior to bacterially derived products in tumor therapy78. A side-by-side comparison demonstrated that CpG oligonucleotides were the most effective single-agent cancer therapeutic compared to other TLR ligands64. In general, CpGs induce the activation and maturation of DCs resulting in secretion of type I interferon and up-regulation of co-stimulatory molecules such as CD80 and CD8679, leading to the activation of natural killer (NK) cells79 and the expansion of cytotoxic T lymphocytes. CpGs also enhance the differentiation of B cells into antibody-secreting plasma cells which can eradicate tumor cells through antibody dependent cellular cytotoxicity80–82.
A number of studies have shown that TLR9 agonists have anti-tumor effects in murine models when initiated while tumors are small76,83–90. Direct intratumoral or local injection of CpG appears to be more effective and less toxic than systemic administration91. Certain cancers can be refractory to single agent CpG due to low expression of MHC-I and the abundant expression of immunosuppressive TGFβ92. Overall, the most potent effects of CpG therapy are seen when used in combinatorial strategies. CpGs have been tested in combination with ionizing radiotherapy using in vitro models, animal models and in clinical trials91–101 and found to be superior in anti-tumor activity compared to either modality alone. Initial studies performed by Milas and colleagues demonstrated significant local synergy for single fraction and fractionated radiotherapy regimens in combination with CpG95,96. In a mouse fibrosarcoma model they demonstrate that the TCD50 for fractionated radiotherapy is reduced almost 4-fold (from 83.1 Gy to 23 Gy) when combined with CpG. The local synergy of radiotherapy and CpG was independently verified by a second group in a glioma model97. Although the exact mechanism of CpG-mediated radiosensitization is not known, studies have suggested that CpG increases both mTOR activation and autophagy102, decreases expression of Oct-4- mediated renewal103, and increases NF-kB signaling and nitric oxide production94.
In vivo, the efficacy of radiation in combination with CpG caused both improved local control and the generation of systemic immunity. Guha and colleagues demonstrated synergistic effects of CpG and radiation against both local irradiated tumor and systemic lung metastases in a 3LL tumor model98. This correlated with an increase in a humoral anti-tumor immune response and increased activation of dendritic cells. Clinical studies combining intralesional CpG with local radiotherapy have confirmed the safety and efficacy of this combinatorial approach in humans. A series of clinical trials by Levy and colleagues in low-grade lymphomas and demonstrated that this combinatorial approach is capable of inducing objective responses outside of the irradiated and injected lesion in about 20% of patients and disease stability in about another 20% of patients with heavily pretreated systemic91 or cutaneous lymphoma99. Many of the responses were durable and lasted for months or years.
Despite the potent immune stimulation of TLRs, studies using CpG as a therapeutic have highlighted that the increased immune effect may be a doubled edged sword. TLR activation combined with radiotherapy can generate immune responses, but as acute inflammation becomes chronic, immune suppression will ensue. Thus, chronic endogenous TLR9 signaling instigated by radiotherapy induces chronic inflammation and increases tumor recurrence104. In the clinical studies of radiotherapy combined with CpG described above, patients whose tumors induced immunosuppressive regulatory Tregs responded poorly to therapy and had a poor prognosis107. Radiotherapy combined with CpG was found to upregulate indolamine 2,3-dioxygenase (IDO) expression, which was termed “rebound immune suppression”105. This upregulation of IDO occured in response to the inflammation induced by radiotherapy and TLR activation, generating an immune suppression that included Tregs. The addition of IDO blockade reversed immune suppression and significantly improved the local and systemic efficacy of radiotherapy combined with CpG in murine models as well as companion canines with late stage metastatic spontaneous melanomas or sarcomas105. As discussed below, this rebound immune suppression may be a common feature of potent IFN-inducers106–108; thus a triple therapy approach which includes blockade of immune suppression to achieve maximal efficacy may be required to increase the clinical impact of these combinatorial approaches.
RIG-I like receptors
RIG-I, MDA5 and LGP2 are cytoplasmic sensors of viral RNA and signal though MAVS to activate type I IFN responses in infected cells. RIG-I recognizes dsRNA containing 5′ triphosphates and biphosphates109, which are present in the nucleus of cells early following transcription, but are removed before entry into the cytoplasm. During infection, the entry of unmodified RNA into the cytoplasm, triggers these sensors and activates the production of type I IFN110. RIG-I111 and LGP2112, but not MDA5,113 also can detect endogenous nuclear material that has translocated to the cytoplasm following radiation therapy. Irradiated cells activate type I IFN production in a RIG-I dependent manner and mice lacking RIG-I were protected against gastrointestinal epithelial cell death following total body radiation111. By contrast, LGP2 has an opposite effect and suppresses type I IFN induced by radiation therapy112.
While we now know that specific sequences activate RIG-I like receptors, these sequences are present in poly(I:C), which was initially thought to be exclusively a TLR3 ligand. Knockout studies using a range of synthetic ligands on dendritic cells demonstrated that MDA5 activates type I IFN responses to poly(I:C) but RIG-1 does not53. The receptor that recognizes poly(I:C) can be influenced by the route of administration – for example, RIG-I like receptors may require that the ligand access the cytoplasm. However, the cell types and pattern of receptor expression also will influence the use of these various ligands. Thus, conventional dendritic cells rely on RIG-I like receptors while plasmacytoid DCs rely on TLR3 binding to the same ligand54. Therefore, many of the studies discussed above that used poly(I:C) as a therapeutic agent may have activated a TLR3 pathway, a RIG-I like receptor pathway, or both.
Although differential activation of the two receptors (RIG-I and TLR3) may lead to different outcomes, it seems that the presence of both pathways leads to maximal response to poly(I:C). In the TRAMP murine model of prostate adenocarcinoma, poly(I:C) administered systemically resulted in complete control of tumor growth, and although much of this was lost in TLR3 knockout mice, some activity remained114. The authors attribute this to direct activity of the systemic poly(I:C) on the cancer cells, but this could also represent the activity of poly(I:C) on RIG-I like receptors. Similarly, in a model of poly(I:C)-induced lung pathology, TLR3 knockout mice exhibited similar responses with a reduced magnitude, and this may represent the activity of RIG-I like receptors115. Interestingly, a non-hematopoeitic stromal MDA5 response was shown to be required for the full efficacy of poly(I:C) as a vaccine adjuvant52, so it is possible that local inflammation driven by stromal cells is critical to support antigen-specific responses. Further studies will be necessary to determine the relative value of selective targeting of TLR3 and RIG-I like receptors, but at the moment, the data suggests that the dual response to poly(I:C) is advantageous in cancer therapy.
STING
Recently, there has been a surge of interest in STING (STimulator of INterferon Genes) for its role as a cytosolic sensor of DNA. Initially, the functions carried out by STING were attributed to TLR9, which can recognize CpG DNA and drive type I IFN responses to microbial DNA. However, double stranded DNA also activated type I IFN responses in cells lacking TLR signaling pathways and RIG-I, and this suggested that there was an unrecognized DNA-sensing mechanism that remained to be discovered116. The STING-dependent cytosolic DNA sensing pathway was discovered by two independent groups using a cDNA screen to identify proteins that induced type I IFN or IRF117,118. STING is widely expressed both among hematopoetic cells (Figure 1) and non-hematopoetic cells including cancer cells119.
The STING pathway likely evolved as an intracellular sensor of pathogen DNA such as bacterial cyclic-di-nucleotides (CDN)120,121. Mice deficient in STING also show impaired clearance of DNA viruses due to impaired generation of a type I IFN-driven immune response122. However, as has been described for RIG-I like receptors and unmodified RNA, STING has also been shown to sense the presence of endogenous DNA introduced into the cytoplasm123. These data suggest that STING may be able to sense endogenous DNA released within irradiated cancer cells; however Deng et al. demonstrated that the major mechanism of STING activation following radiation of cancer cells resulted following with cross-presentation of cell-associated antigens to dendritic cells124. In these experiments, expression of STING by the cancer cells was not required for radiation therapy-induced tumor cure, which instead was entirely dependent on STING expression in host dendritic cells124. In highly immunogenic tumors, host expression of STING is necessary for spontaneous tumor regression125. In these models, cancer cells killed by a range of methods including radiation and freeze thaw were unable to active type I IFN activation in dendritic cells; however, tumor-derived DNA transfected into the cytoplasm of DC was a potent STING-mediated activator of type I IFN production. Although it is unclear how tumor DNA is transferred to the DC cytoplasm in vivo, mice deficient in STING were unable to generate type I IFN following tumor challenge, failed to generate effective anti-tumor immunity, and were less responsive to immunotherapy125. These data suggest that the inflammatory component of cross presentation at tumor challenge was critical to generate T cell responses to tumor antigens. In a different model system using less immunogenic tumors, STING activation in the host following tumor challenge was shown to inhibit endogenous anti-tumor immunity106. The mechanisms was again via induction of type I IFN but this time also resulted in subsequent induction of the immune suppressive enzyme IDO, resulting in increased tumor growth rates. However, other studies have not found a change in tumor growth rate in wild type versus STING knockout animals that are otherwise untreated119,124,126. It is possible that some feature of the cultured cancer cells, their preparation, or their route of delivery at transfer differentially affects the likelihood and consequence of STING activation in the host.
Since CDN activation of STING results in potent induction of type I IFN, CDN have been shown to be effective vaccine adjuvants127,128. In addition, direct injection of CDN into tumors has been shown to cause dramatic regression in a range of tumor models126. Interestingly, STING was recently found to be the gene activated by DMXAA129, which is a highly active vascular disrupting agent resulting in hemorrhagic necrosis of tumors, though with variable results depending on the tumor model130. DMXAA activates mouse, but not human STING131, potentially explaining the failure in clinical translation of DMXAA. Novel CDN have been engineered for increased potency against mouse STING and human STING isoforms126,128, and novel small molecule agonists of the STING pathway have been identified132. While cancer cells can express STING119, tumor therapy with CDN is ineffective in mice lacking STING126, indicating that host STING is critical for anti-tumor activity. This also explains why DMXAA was able to cause vascular disruption in xenografts of human tumors in immunodeficient mice133, since cancer cell expression of STING was not relevant to treatment outcome. These data from immunodeficient mice also suggest that the anti-tumor efficacy of STING ligands is not necessarily dependent on functional adaptive immunity.
Both DMXAA and CDN have shown efficacy in combination with radiation therapy. DMXAA administered systemically resulted in increased tumor control by RT in a radiation and DMXAA dose-dependent manner134,135, and with fractionated radiation135. In the MC38 colorectal model that was not affected by intratumoral injection of CDN, the combination of CDN and RT resulted in significantly increased tumor control over RT alone124. Similarly, in a range of tumor models and mouse strains the combination of CDN and RT was shown to result in therapeutic synergy119. These effects were dependent on STING expression in the host, and resulted in TNFα-induced hemorrhagic necrosis in the tumor119. In these models, radiation therapy was necessary to produce CD8 T cell immunity and control distant disease, but optimal tumor control was dependent on both an early T cell independent, TNFα-dependent rapid tumor regression and a later, CD8 T cell dependent mechanism that contributed to the durable response of treated tumors119. These data demonstrate that STING ligands create synergy with radiation therapy for immune-mediated control of cancer.
Type I IFN
Type I IFN is induced downstream of many of the innate immune activators mentioned above. Type I IFN includes IFNα and IFNβ, which signal through the receptors IFNAR1 and IFNAR2136,137 (Figure 1). This contrasts with type II IFN, or IFNγ, which signals through IFNGR1 and IFNGR2, and type III IFN, that includes IFN-lambda and IL-10136,137 and signals through two α subunits and two β subunits. The ability to produce and respond to type I IFN is shared by almost all cell types, though plasmacytoid dendritic cells are able to secrete higher levels of type I IFN than any other cell type130,139. Activation of IFN receptors leads to a multifaceted response. They generate an innate anti-viral and anti-microbial state among infected and bystander cells, limiting the spread of pathogens, and also promote NK cell function and antigen presentation by dendritic cells to activate T cells (reviewed in136,137). However, type I IFN can also induce immune suppressive mechanisms, such as induction of the immune suppressive enzyme IDO106. IDO tolerizes adaptive immune responses140 and can be harnessed to treat T cell-mediated autoimmunity141. Thus, as with many immune stimuli, there is a contextual and dose-related consequence of type I IFN activation in vivo. As has been shown for IFNγ, type I IFNs also result in upregulation of PDL1 by stromal cells and cancer cells142, and therefore a role in feedback regulation of adaptive immunity.
The expression of type I IFN and IFN-responsive genes have been correlated with favorable outcome in cancer patients. A type I IFN gene signature is induced by chemotherapy and radiation therapy in some patients. This type I IFN gene signature is associated with improved outcome following neoadjuvant chemotherapy in breast cancer patients111. However, this interferon signature is expressed in a broad array of cancer cell lines, and predicts a poor response to chemotherapy and radiation therapy in patients with breast cancer143 and glioblastoma144. In preclinical models, the efficacy of radiation therapy was shown to be dependent on induction of type I IFN in murine tumor models145. Using bone marrow chimeras it was demonstrated that a functional response to radiation was dependent on IFNAR1 expression in hematopoietic cells, but not T cells, and IFNAR1 knockout mice lacked functional cross-presentation of tumor antigens by dendritic cells145. Type I IFN has been shown to be required for priming of T cell responses and recruitment of tumor-specific T cells to treated sites146.
Type I IFN has been well studied as an adjuvant therapy for melanoma, resulting in improved recurrence-free survival147,140, but no improvement on overall survival 149. Type I IFN has long been known to radiosensitize cancer cells in vitro150, and some of the earliest studies of immunotherapy combined with radiation therapy involved local or systemic application of type I IFN. This has shown mixed signs of efficacy in a range of tumor types, but toxicities have limited application of this therapy. Preclinical studies in pancreatic cancer have shown improved outcome with type I IFN and chemotherapy in pancreatic cancer151,152, and early phase studies suggested improved outcome in patients receiving type I IFN along with adjuvant chemoradiation153. Multicenter studies suggested an improved outcome; however, they also resulted in grade III or IV toxicity in 90% of patients154. A multicenter, randomized phase III trial of adjuvant chemoradiation plus type I IFN did not demonstrate a benefit of combinatorial therapy versus 5-FU alone and resulted in ≥ grade III toxicity in 85% of patients155. These data indicate that systemic delivery of type I IFN is significantly limited by toxicities and does not provide a favorable therapeutic ratio.
To minimize systemic consequences, type I IFN can be injected in the local tumor environment to generate tumor control156, and intratumoral injection of an adenoviral vector expressing murine IFNβ can produce T cell-dependent control of a murine melanoma model145. Alternatively, IFN can be engineered to accumulate in the vicinity of cancer cells using immune-conjugates. Anti-CD20 coupled to type I IFN was shown to be an effective therapy against lymphoma157 but hematological malignancies therapies require direct action of type I IFN on the cancer cells157. An anti-EGFR-IFNβ conjugate was effective against murine solid tumors expressing human EGFR, and in these models tumor clearance was dependent on functional adaptive immunity158. Further refinement of type I IFN delivery systems could potentially expand the use of this cytokine for cancer therapy.
TNFα
TNFα was identified as the factor induced by Coley’s toxins responsible for necrosis of tumors159,160. Sufficient TNFα can produce rapid hemorrhagic necrosis as a result of vascular endothelia activation and death. In addition, TNFα can induce cancer cell apoptosis. As described above, TNFα-mediated hemorrhagic necrosis of tumors has been described following administration of STING ligands, LPS, poly(I:C) and CpG119,130,161,162. However, the lower levels of endogenous TNFα produced by macrophages in the tumor environment has been shown to promote tumor regrowth following radiation therapy163. In addition, TNFα produced downstream of pattern recognition receptor ligation is the main factor contributing to systemic shock164,165 and can be causative of other toxicities. For example, pulmonary administration of CpG resulted in TLR9-dependent alveolitis and pneumonitis in mice, and this toxicity was lost in TNFα knockout mice166.
To minimize systemic toxicity, TNFα has been engineered into an adenoviral vector with expression controlled by a radiation-responsive promoter (reviewed in167). This promoter has been optimized to generate up to a 3-fold induction of gene expression following radiation therapy168 and this construct has been successfully used to induce vascular thrombosis in human tumor xenografts in immunodeficient mice169. Similarly, cancer cells modified to express TNFα constitutively or in response to radiation therapy have been shown to improve the clinical response to implanted radioactive seeds170. In immunocompetent mice, gene delivery of TNFα using an adenoviral vector resulted in improved tumor control following a single fraction of 20Gy, with a large portion of the effect due to CD8 T cells171. These data suggest that responses incorporate both a direct effect of TNFα on vascular cells, and a CD8-mediated adaptive immune response.
In the clinic, radiation-inducible TNFα gene therapy showed evidence of efficacy in preclinical and Phase II studies (reviewed in167); however, randomized phase III studies in locally advanced pancreatic cancer failed to show efficacy172. These data suggest that while TNFα is an extremely important effector cytokine that can have dramatic effects on tumors, it is very difficult to target TNFα clinically because of its pleiotrophic effects within the immune system, and subsequent toxicities.
Conclusions
Though cancer cells contain endogenous adjuvants with the potential to stimulate innate sensors, radiation therapy is not an effective stimulator of systemic immunity without the addition of immunotherapy2. It is possible that irradiated cells are only able to generate type I IFN responses in some patients, explaining variation in outcome according to the IFN gene signature143,144. This may be due to genetic variation in the endogenous DNA sensors, as has been proposed for TLR421, or due to the particular immune tumor environment. Nevertheless, the variable response presents an important opportunity to deliver the appropriate exogenous adjuvants to deliver missing signals and tap into the potential of radiation therapy as a patient-specific endogenous cancer vaccine (Figure 2).
Figure 2. Effect of adjuvant on immune cell relationships in the tumor and draining lymph nodes.
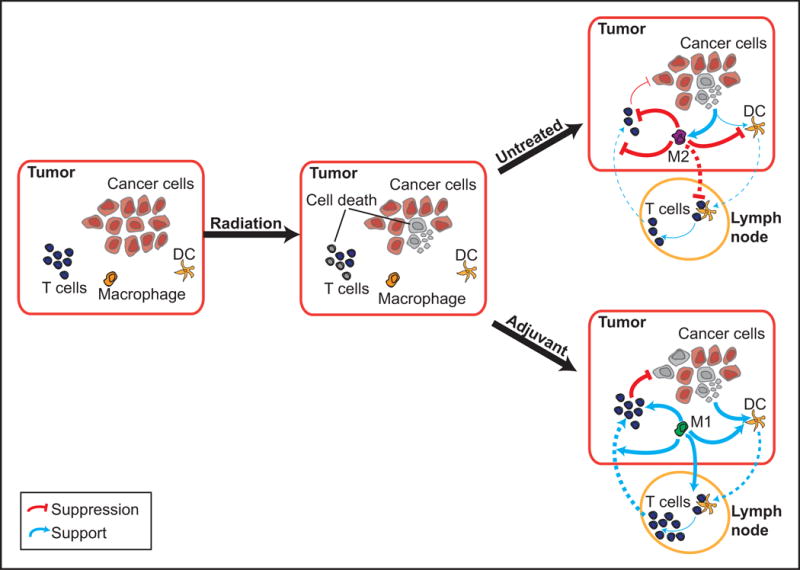
In the absence of adjuvant, cell death mediated by radiation therapy drives M2 responses that suppress DC maturation and effector T cell function. Innate immune adjuvants can drive proinflammatory M1 responses, enhance DC cross presentation of tumor antigens to T cells in the lymph node in a supportive cytokine environment, and support effector T cell control of residual disease.
When using adjuvants in addition to RT, we must dedicate efforts to learning how to best balance efficacy and toxicity. In addition, we must address the problem of local versus systemic delivery. Local delivery can maximize local tumor control while minimizing systemic toxicity, but this may also limit its clinical application. In addition, we must always consider how these therapies have the potential to interfere/suppress the adaptive immune response. As we have discussed, over-activation of inflammatory mechanisms by adjuvants can lead to adaptive immune suppression140,141, requiring additional intervention such as inhibition of IDO105. Adaptive immunity requires a critical sequence of signals and any deviation in the appropriate timing and degree of inflammation has the potential to limit antigen-specific immunity173. The appropriate timing of adjuvant delivery in relation to radiation needs to be determined in order to ensure the success of this approach174. While adjuvant might be the oldest cancer immunotherapy, the therapeutic potential of adjuvant remains to be fully exploited.
Acknowledgments
This work was supported by an American Cancer Society Postdoctoral Fellowship award (JRB), NCI R01 CA182311 (MJG), NCI R01 CA208644 (MRC), NIAID R21AI126151 (MRC), and NCI R03 CA198208 (MRC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Honscheid P, Datta K, Muders MH. Autophagy: detection, regulation and its role in cancer and therapy response. International journal of radiation biology. 2014;90:628–635. doi: 10.3109/09553002.2014.907932. [DOI] [PubMed] [Google Scholar]
- 2.Abuodeh Y, Venkat P, Kim S. Systematic review of case reports on the abscopal effect. Curr Probl Cancer. 2015 doi: 10.1016/j.currproblcancer.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Twyman-Saint Victor C, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015 doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belcaid Z, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PloS one. 2014;9:e101764. doi: 10.1371/journal.pone.0101764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Demaria S, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:728–734. [PubMed] [Google Scholar]
- 6.Sharabi AB, et al. Stereotactic Radiation Therapy Augments Antigen-Specific PD-1 Mediated Anti-Tumor Immune Responses via Cross-Presentation of Tumor Antigen. Cancer Immunol Res. 2014 doi: 10.1158/2326-6066.CIR-14-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng L, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. The Journal of clinical investigation. 2014;124:687–695. doi: 10.1172/JCI67313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. International journal of radiation oncology, biology, physics. 2013;86:343–349. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Redmond WL, Ruby CE, Weinberg AD. The role of OX40-mediated costimulation in T-cell activation and survival. Crit Rev Immunol. 2009;29:187–201. doi: 10.1615/critrevimmunol.v29.i3.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruby CE, Redmond WL, Haley D, Weinberg AD. Anti-OX40 stimulation in vivo enhances CD8+ memory T cell survival and significantly increases recall responses. Eur J Immunol. 2007;37:157–166. doi: 10.1002/eji.200636428. [DOI] [PubMed] [Google Scholar]
- 11.Evans DE, Prell RA, Thalhofer CJ, Hurwitz AA, Weinberg AD. Engagement of OX40 enhances antigen-specific CD4(+) T cell mobilization/memory development and humoral immunity: comparison of alphaOX-40 with alphaCTLA-4. Journal of immunology. 2001;167:6804–6811. doi: 10.4049/jimmunol.167.12.6804. [DOI] [PubMed] [Google Scholar]
- 12.Wen T, Bukczynski J, Watts TH. 4-1BB ligand-mediated costimulation of human T cells induces CD4 and CD8 T cell expansion, cytokine production, and the development of cytolytic effector function. Journal of immunology. 2002;168:4897–4906. doi: 10.4049/jimmunol.168.10.4897. [DOI] [PubMed] [Google Scholar]
- 13.Gramaglia I, Cooper D, Miner KT, Kwon BS, Croft M. Co-stimulation of antigen-specific CD4 T cells by 4-1BB ligand. Eur J Immunol. 2000;30:392–402. doi: 10.1002/1521-4141(200002)30:2<392::AID-IMMU392>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 14.Vinay DS, Kwon BS. Differential expression and costimulatory effect of 4-1BB (CD137) and CD28 molecules on cytokine-induced murine CD8(+) Tc1 and Tc2 cells. Cell Immunol. 1999;192:63–71. doi: 10.1006/cimm.1998.1433. [DOI] [PubMed] [Google Scholar]
- 15.Melero I, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nature medicine. 1997;3:682–685. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- 16.Weinberg AD, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. Journal of immunology. 2000;164:2160–2169. doi: 10.4049/jimmunol.164.4.2160. [DOI] [PubMed] [Google Scholar]
- 17.Gough MJ, et al. OX40 agonist therapy enhances CD8 infiltration and decreases immune suppression in the tumor. Cancer Res. 2008;68:5206–5215. doi: 10.1158/0008-5472.CAN-07-6484. [DOI] [PubMed] [Google Scholar]
- 18.Gough MJ, et al. Induction of cell stress through gene transfer of an engineered heat shock transcription factor enhances tumor immunogenicity. Gene Ther. 2004;11:1099–1104. doi: 10.1038/sj.gt.3302274. [DOI] [PubMed] [Google Scholar]
- 19.Daniels GA, et al. A simple method to cure established tumors by inflammatory killing of normal cells. Nat Biotechnol. 2004;22:1125–1132. doi: 10.1038/nbt1007. [DOI] [PubMed] [Google Scholar]
- 20.Todryk S, et al. Heat shock protein 70 induced during tumor cell killing induces Th1 cytokines and targets immature dendritic cell precursors to enhance antigen uptake. Journal of immunology. 1999;163:1398–1408. [PubMed] [Google Scholar]
- 21.Apetoh L, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nature medicine. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 22.Telusma G, et al. Dendritic cell activating peptides induce distinct cytokine profiles. Int Immunol. 2006;18:1563–1573. doi: 10.1093/intimm/dxl089. [DOI] [PubMed] [Google Scholar]
- 23.Obeid M, et al. Leveraging the immune system during chemotherapy: moving calreticulin to the cell surface converts apoptotic death from “silent” to immunogenic. Cancer Res. 2007;67:7941–7944. doi: 10.1158/0008-5472.CAN-07-1622. [DOI] [PubMed] [Google Scholar]
- 24.Luthi AU, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31:84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 25.Kono H, Chen CJ, Ontiveros F, Rock KL. Uric acid promotes an acute inflammatory response to sterile cell death in mice. The Journal of clinical investigation. 2010;120:1939–1949. doi: 10.1172/JCI40124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–521. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- 27.Crittenden MR, et al. Expression of NF-kappaB p50 in tumor stroma limits the control of tumors by radiation therapy. PloS one. 2012;7:e39295. doi: 10.1371/journal.pone.0039295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 29.Saccani A, et al. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006;66:11432–11440. doi: 10.1158/0008-5472.CAN-06-1867. [DOI] [PubMed] [Google Scholar]
- 30.Golpon HA, et al. Life after corpse engulfment: phagocytosis of apoptotic cells leads to VEGF secretion and cell growth. Faseb J. 2004;18:1716–1718. doi: 10.1096/fj.04-1853fje. [DOI] [PubMed] [Google Scholar]
- 31.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. The Journal of clinical investigation. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fadok VA, et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. The Journal of clinical investigation. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kozin SV, et al. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 2010;70:5679–5685. doi: 10.1158/0008-5472.CAN-09-4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahn GO, et al. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:8363–8368. doi: 10.1073/pnas.0911378107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiao SL, et al. TH2-Polarized CD4+ T Cells and Macrophages Limit Efficacy of Radiotherapy. Cancer Immunol Res. 2015 doi: 10.1158/2326-6066.CIR-14-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu J, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73:2782–2794. doi: 10.1158/0008-5472.CAN-12-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stafford JH, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro-oncology. 2015 doi: 10.1093/neuonc/nov272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crittenden MR, et al. Mertk on tumor macrophages is a therapeutic target to prevent tumor recurrence following radiation therapy. Oncotarget. 2016 doi: 10.18632/oncotarget.11823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 40.Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature immunology. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 42.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of doublestranded RNA and activation of NF-[kappa]B by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 43.Schulz O, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433:887–892. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- 44.Iyoda T, et al. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. The Journal of experimental medicine. 2002;195:1289–1302. doi: 10.1084/jem.20020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Belz GT, et al. The CD8alpha(+) dendritic cell is responsible for inducing peripheral self-tolerance to tissue-associated antigens. The Journal of experimental medicine. 2002;196:1099–1104. doi: 10.1084/jem.20020861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu K, et al. Immune tolerance after delivery of dying cells to dendritic cells in situ. The Journal of experimental medicine. 2002;196:1091–1097. doi: 10.1084/jem.20021215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamamoto M, et al. Role of Adaptor TRIF in the MyD88-Independent Toll-Like Receptor Signaling Pathway. Science. 2003;301:640. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 48.Bart RS, Kopf AW, Silagi S. Inhibition of the Growth of Murine Malignant Melanoma by Polyinosinic-Polycytidylic Acid. Journal of Investigative Dermatology. 1971;56:33–38. doi: 10.1111/1523-1747.ep12291892. [DOI] [PubMed] [Google Scholar]
- 49.Ammi R, et al. Poly(I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharmacol Ther. 2015;146:120–131. doi: 10.1016/j.pharmthera.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 50.Levine AS, Sivulich M, Wiernik PH, Levy HB. Initial Clinical Trials in Cancer Patients of Polyriboinosinic-Polyribocytidylic Acid Stabilized with Poly-L-lysine, in Carboxymethylcellulose [Poly(ICLC)], a Highly Effective Interferon Inducer. Cancer research. 1979;39:1645–1650. [PubMed] [Google Scholar]
- 51.Levy HB, et al. A Modified Polyriboinosinic-Polyribocytidylic Acid Complex That Induces Interferon in Primates. Journal of Infectious Diseases. 1975;132:434–439. doi: 10.1093/infdis/132.4.434. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Cella M, Gilfillan S, Colonna M. Cutting edge: polyinosinic: polycytidylic acid boosts the generation of memory CD8 T cells through melanoma differentiation-associated protein 5 expressed in stromal cells. Journal of immunology. 2010;184:2751–2755. doi: 10.4049/jimmunol.0903201. [DOI] [PubMed] [Google Scholar]
- 53.Kato H, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 54.Kato H, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 55.Butowski N, et al. A phase II clinical trial of poly-ICLC with radiation for adult patients with newly diagnosed supratentorial glioblastoma: a North American Brain Tumor Consortium (NABTC01-05) Journal of neuro-oncology. 2009;91:175–182. doi: 10.1007/s11060-008-9693-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosenfeld MR, et al. A multi-institution phase II study of poly-ICLC and radiotherapy with concurrent and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro-oncology. 2010;12:1071–1077. doi: 10.1093/neuonc/noq071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawai T, et al. Lipopolysaccharide Stimulates the MyD88-Independent Pathway and Results in Activation of IFN-Regulatory Factor 3 and the Expression of a Subset of Lipopolysaccharide-Inducible Genes. The Journal of Immunology. 2001;167:5887–5894. doi: 10.4049/jimmunol.167.10.5887. [DOI] [PubMed] [Google Scholar]
- 58.Yamamoto M, et al. Cutting Edge: A Novel Toll/IL-1 Receptor Domain- Containing Adapter That Preferentially Activates the IFN- Promoter in the TollLike Receptor Signaling. The Journal of Immunology. 2002;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 59.Paulos CM, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. The Journal of clinical investigation. 2007;117:2197–2204. doi: 10.1172/JCI32205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mizuno D, Yoshioka O, Akamatu M, Kataoka T. Antitumor effect of intracutaneous injection of bacterial lipopolysaccharide. Cancer Res. 1968;28:1531–1537. [PubMed] [Google Scholar]
- 61.Galluzzi L, et al. Trial Watch: Experimental Toll-like receptor agonists for cancer therapy. Oncoimmunology. 2012;1:699–716. doi: 10.4161/onci.20696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chicoine MR, Won EK, Zahner MC. Intratumoral injection of lipopolysaccharide causes regression of subcutaneously implanted mouse glioblastoma multiforme. Neurosurgery. 2001;48:607–614. doi: 10.1097/00006123-200103000-00032. discussion 614–605. [DOI] [PubMed] [Google Scholar]
- 63.Yuk JM, et al. Bacillus calmette-guerin cell wall cytoskeleton enhances colon cancer radiosensitivity through autophagy. Autophagy. 2010;6:46–60. doi: 10.4161/auto.6.1.10325. [DOI] [PubMed] [Google Scholar]
- 64.Grauer OM, et al. TLR ligands in the local treatment of established intracerebral murine gliomas. Journal of immunology. 2008;181:6720–6729. doi: 10.4049/jimmunol.181.10.6720. [DOI] [PubMed] [Google Scholar]
- 65.Lacave-Lapalun JV, Benderitter M, Linard C. Flagellin or lipopolysaccharide treatment modified macrophage populations after colorectal radiation of rats. The Journal of pharmacology and experimental therapeutics. 2013;346:75–85. doi: 10.1124/jpet.113.204040. [DOI] [PubMed] [Google Scholar]
- 66.Huang SJ, et al. Imiquimod enhances IFN-gamma production and effector function of T cells infiltrating human squamous cell carcinomas of the skin. The Journal of investigative dermatology. 2009;129:2676–2685. doi: 10.1038/jid.2009.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schon M, et al. Tumor-Selective Induction of Apoptosis and the Small-Molecule Immune Response Modifier Imiquimod. JNCI Journal of the National Cancer Institute. 2003;95:1138–1149. doi: 10.1093/jnci/djg016. [DOI] [PubMed] [Google Scholar]
- 68.Hemmi H, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nature immunology. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 69.Gibson SJ, et al. Cellular requirements for cytokine production in response to the immunomodulators imiquimod and S-27609. J Interferon Cytokine Res. 1995;15:537–545. doi: 10.1089/jir.1995.15.537. [DOI] [PubMed] [Google Scholar]
- 70.Dewan MZ, et al. Synergy of topical toll-like receptor 7 agonist with radiation and low-dose cyclophosphamide in a mouse model of cutaneous breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:6668–6678. doi: 10.1158/1078-0432.CCR-12-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ulrich C, et al. Topical immunomodulation under systemic immunosuppression: results of a multicentre, randomized, placebo-controlled safety and efficacy study of imiquimod 5% cream for the treatment of actinic keratoses in kidney, heart, and liver transplant patients. The British journal of dermatology. 2007;157(Suppl 2):25–31. doi: 10.1111/j.1365-2133.2007.08269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Adams S, et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:6748–6757. doi: 10.1158/1078-0432.CCR-12-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dudek AZ, et al. First in human phase I trial of 852A, a novel systemic toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:7119–7125. doi: 10.1158/1078-0432.CCR-07-1443. [DOI] [PubMed] [Google Scholar]
- 74.Dovedi SJ, et al. Systemic delivery of a TLR7 agonist in combination with radiation primes durable antitumor immune responses in mouse models of lymphoma. Blood. 2013;121:251–259. doi: 10.1182/blood-2012-05-432393. [DOI] [PubMed] [Google Scholar]
- 75.Adlard AL, et al. A novel systemically administered Toll-like receptor 7 agonist potentiates the effect of ionizing radiation in murine solid tumor models. International journal of cancer. Journal international du cancer. 2014;135:820–829. doi: 10.1002/ijc.28711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krieg AM, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 77.Hemmi H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 78.Rayburn ER, Wang W, Zhang R, Wang H. Experimental therapy for colon cancer: anti-cancer effects of TLR9 agonism, combination with other therapeutic modalities, and dependence upon p53. Int J Oncol. 2007;30:1511–1519. [PubMed] [Google Scholar]
- 79.Chaudhry UI, et al. Combined stimulation with interleukin-18 and CpG induces murine natural killer dendritic cells to produce IFN-gamma and inhibit tumor growth. Cancer Res. 2006;66:10497–10504. doi: 10.1158/0008-5472.CAN-06-1908. [DOI] [PubMed] [Google Scholar]
- 80.Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5:471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 81.Aurisicchio L, et al. Treatment of mammary carcinomas in HER-2 transgenic mice through combination of genetic vaccine and an agonist of Toll-like receptor 9. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:1575–1584. doi: 10.1158/1078-0432.CCR-08-2628. [DOI] [PubMed] [Google Scholar]
- 82.Chiron D, Bekeredjian-Ding I, Pellat-Deceunynck C, Bataille R, Jego G. Toll-like receptors: lessons to learn from normal and malignant human B cells. Blood. 2008;112:2205–2213. doi: 10.1182/blood-2008-02-140673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baines J, Celis E. Immune-mediated tumor regression induced by CpG-containing oligodeoxynucleotides. Clinical cancer research: an official journal of the American Association for Cancer Research. 2003;9:2693–2700. [PubMed] [Google Scholar]
- 84.Ballas ZK, et al. Divergent Therapeutic and Immunologic Effects of Oligodeoxynucleotides with Distinct CpG Motifs. The Journal of Immunology. 2001;167:4878–4886. doi: 10.4049/jimmunol.167.9.4878. [DOI] [PubMed] [Google Scholar]
- 85.Blazar BR, Krieg AM, Taylor PA. Synthetic unmethylated cytosine-phosphate-guanosine oligodeoxynucleotides are potent stimulators of antileukemia responses in naive and bone marrow transplant recipients. Blood. 2001;98:1217. doi: 10.1182/blood.v98.4.1217. [DOI] [PubMed] [Google Scholar]
- 86.Chu RS, Targoni OS, Krieg AM, Lehmann PV, Harding CV. CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. The Journal of experimental medicine. 1997;186:1623–1631. doi: 10.1084/jem.186.10.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Heckelsmiller K, et al. Peritumoral CpG DNA elicits a coordinated response of CD8 T cells and innate effectors to cure established tumors in a murine colon carcinoma model. Journal of immunology. 2002;169:3892–3899. doi: 10.4049/jimmunol.169.7.3892. [DOI] [PubMed] [Google Scholar]
- 88.Kawarada Y, et al. NK- and CD8(+) T cell-mediated eradication of established tumors by peritumoral injection of CpG-containing oligodeoxynucleotides. Journal of immunology. 2001;167:5247–5253. doi: 10.4049/jimmunol.167.9.5247. [DOI] [PubMed] [Google Scholar]
- 89.Lonsdorf AS, et al. Intratumor CpG-Oligodeoxynucleotide Injection Induces Protective Antitumor T Cell Immunity. The Journal of Immunology. 2003;171:3941–3946. doi: 10.4049/jimmunol.171.8.3941. [DOI] [PubMed] [Google Scholar]
- 90.Weigel BJ, Rodeberg DA, Krieg AM, Blazar BR. CpG oligodeoxynucleotides potentiate the antitumor effects of chemotherapy or tumor resection in an orthotopic murine model of rhabdomyosarcoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2003;9:3105–3114. [PubMed] [Google Scholar]
- 91.Brody JD, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28:4324–4332. doi: 10.1200/JCO.2010.28.9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chamoto K, et al. Combination immunotherapy with radiation and CpG-based tumor vaccination for the eradication of radio- and immuno-resistant lung carcinoma cells. Cancer science. 2009;100:934–939. doi: 10.1111/j.1349-7006.2009.01114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li R, Song Y, Chen W. Enhancing radiosensitivity of human pulmonary adenocarcinoma cell line A549 by CpG ODN1826. Cancer biotherapy & radiopharmaceuticals. 2011;26:69–76. doi: 10.1089/cbr.2010.0849. [DOI] [PubMed] [Google Scholar]
- 94.Li X, et al. CpG ODN107 potentiates radiosensitivity of human glioma cells via TLR9-mediated NF-kappaB activation and NO production. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2012;33:1607–1618. doi: 10.1007/s13277-012-0416-1. [DOI] [PubMed] [Google Scholar]
- 95.Mason KA, et al. CpG oligodeoxynucleotides are potent enhancers of radio- and chemoresponses of murine tumors. Radiotherapy and oncology: journal of the European Society for Therapeutic Radiology and Oncology. 2006;80:192–198. doi: 10.1016/j.radonc.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 96.Mason KA, et al. Targeting toll-like receptor 9 with CpG oligodeoxynucleotides enhances tumor response to fractionated radiotherapy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:361–369. [PubMed] [Google Scholar]
- 97.Meng Y, et al. Successful combination of local CpG-ODN and radiotherapy in malignant glioma. International journal of cancer. Journal international du cancer. 2005;116:992–997. doi: 10.1002/ijc.21131. [DOI] [PubMed] [Google Scholar]
- 98.Zhang H, et al. An in situ autologous tumor vaccination with combined radiation therapy and TLR9 agonist therapy. PloS one. 2012;7:e38111. doi: 10.1371/journal.pone.0038111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim YH, et al. In situ vaccination against mycosis fungoides by intratumoral injection of a TLR9 agonist combined with radiation: a phase 1/2 study. Blood. 2012;119:355–363. doi: 10.1182/blood-2011-05-355222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cerkovnik P, Novakovic BJ, Stegel V, Novakovic S. Tumor vaccine composed of C-class CpG oligodeoxynucleotides and irradiated tumor cells induces long-term antitumor immunity. BMC immunology. 2010;11:45. doi: 10.1186/1471-2172-11-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Milas L, et al. CpG oligodeoxynucleotide enhances tumor response to radiation. Cancer Res. 2004;64:5074–5077. doi: 10.1158/0008-5472.CAN-04-0926. [DOI] [PubMed] [Google Scholar]
- 102.Li X, et al. TLR9-ERK-mTOR signaling is critical for autophagic cell death induced by CpG oligodeoxynucleotide 107 combined with irradiation in glioma cells. Scientific reports. 2016;6:27104. doi: 10.1038/srep27104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xing N, et al. CpG oligodeoxyribonucleotide 7909 enhances radiosensitivity via downregulating Oct-4 expression in radioresistant lung cancer cells. OncoTargets and therapy. 2015;8:1443–1449. doi: 10.2147/OTT.S84467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gao C, et al. TLR9 signaling in the tumor microenvironment initiates cancer recurrence after radiotherapy. Cancer Res. 2013;73:7211–7221. doi: 10.1158/0008-5472.CAN-13-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Monjazeb AM, et al. Blocking Indolamine-2,3-Dioxygenase Rebound Immune Suppression Boosts Antitumor Effects of Radio-Immunotherapy in Murine Models and Spontaneous Canine Malignancies. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016 doi: 10.1158/1078-0432.CCR-15-3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lemos H, et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 2016 doi: 10.1158/0008-5472.CAN-15-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carlin JM, Borden EC, Sondel PM, Byrne GI. Interferon-induced indoleamine 2,3-dioxygenase activity in human mononuclear phagocytes. Journal of leukocyte biology. 1989;45:29–34. doi: 10.1002/jlb.45.1.29. [DOI] [PubMed] [Google Scholar]
- 108.Baban B, et al. A minor population of splenic dendritic cells expressing CD19 mediates IDO-dependent T cell suppression via type I IFN signaling following B7 ligation. Int Immunol. 2005;17:909–919. doi: 10.1093/intimm/dxh271. [DOI] [PubMed] [Google Scholar]
- 109.Goubau D, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature. 2014;514:372–375. doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity. 2013;38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ranoa DR, et al. Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget. 2016 doi: 10.18632/oncotarget.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Widau RC, et al. RIG-I-like receptor LGP2 protects tumor cells from ionizing radiation. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E484–491. doi: 10.1073/pnas.1323253111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sun Q, et al. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633–642. doi: 10.1016/j.immuni.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 114.Chin AI, et al. Toll-like receptor 3-mediated suppression of TRAMP prostate cancer shows the critical role of type I interferons in tumor immune surveillance. Cancer Res. 2010;70:2595–2603. doi: 10.1158/0008-5472.CAN-09-1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stowell NC, et al. Long-term activation of TLR3 by poly(I:C) induces inflammation and impairs lung function in mice. Respir Res. 2009;10:43. doi: 10.1186/1465-9921-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ishii KJ, et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nature immunology. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- 117.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhong B, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 119.Baird JR, et al. Radiotherapy Combined with Novel STING-Targeting Oligonucleotides Results in Regression of Established Tumors. Cancer Res. 2016;76:50–61. doi: 10.1158/0008-5472.CAN-14-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ishikawa H, Barber GN. The STING pathway and regulation of innate immune signaling in response to DNA pathogens. Cellular and molecular life sciences: CMLS. 2011;68:1157–1165. doi: 10.1007/s00018-010-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sauer JD, et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun. 2011;79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Abe T, et al. STING recognition of cytoplasmic DNA instigates cellular defense. Mol Cell. 2013;50:5–15. doi: 10.1016/j.molcel.2013.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Deng L, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41:843–852. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Woo SR, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Corrales L, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11:1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dubensky TW, Jr, Kanne DB, Leong ML. Rationale, progress and development of vaccines utilizing STING-activating cyclic dinucleotide adjuvants. Therapeutic advances in vaccines. 2013;1:131–143. doi: 10.1177/2051013613501988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fu J, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Science translational medicine. 2015;7:283ra252. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Prantner D, et al. 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J Biol Chem. 2012;287:39776–39788. doi: 10.1074/jbc.M112.382986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Downey CM, Aghaei M, Schwendener RA, Jirik FR. DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2′3′-cGAMP, induces M2 macrophage repolarization. PloS one. 2014;9:e99988. doi: 10.1371/journal.pone.0099988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Conlon J, et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. Journal of immunology. 2013;190:5216–5225. doi: 10.4049/jimmunol.1300097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sali TM, et al. Characterization of a Novel Human-Specific STING Agonist that Elicits Antiviral Activity Against Emerging Alphaviruses. PLoS Pathog. 2015;11:e1005324. doi: 10.1371/journal.ppat.1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Seshadri M, Toth K. Acute vascular disruption by 5,6-dimethylxanthenone-4-acetic Acid in an orthotopic model of human head and neck cancer. Transl Oncol. 2009;2:121–127. doi: 10.1593/tlo.09103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Murata R, Siemann DW, Overgaard J, Horsman MR. Improved tumor response by combining radiation and the vascular-damaging drug 5,6-dimethylxanthenone-4-acetic acid. Radiation research. 2001;156:503–509. doi: 10.1667/0033-7587(2001)156[0503:itrbcr]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 135.Wilson W, Li A, Cowan DM, Siim B. Enhancement of tumor radiation response by the antivascular agent 5,6-dimethylxanthenone-4-acetic acid. International Journal of Radiation Oncology*Biology*Physics. 1998;42:905–908. doi: 10.1016/s0360-3016(98)00358-7. [DOI] [PubMed] [Google Scholar]
- 136.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 138.Siegal FP, et al. The Nature of the Principal Type 1 Interferon-Producing Cells in Human Blood. Science. 1999;284:1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 139.Cella M, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nature medicine. 1999;5:919–923. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- 140.Huang L, et al. Cutting edge: DNA sensing via the STING adaptor in myeloid dendritic cells induces potent tolerogenic responses. Journal of immunology. 2013;191:3509–3513. doi: 10.4049/jimmunol.1301419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lemos H, et al. Activation of the STING adaptor attenuates experimental autoimmune encephalitis. Journal of immunology. 2014;192:5571–5578. doi: 10.4049/jimmunol.1303258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Terawaki S, et al. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. Journal of immunology. 2011;186:2772–2779. doi: 10.4049/jimmunol.1003208. [DOI] [PubMed] [Google Scholar]
- 143.Weichselbaum RR, et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proceedings of the National Academy of Sciences. 2008;105:18490–18495. doi: 10.1073/pnas.0809242105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Duarte CW, et al. Expression signature of IFN/STAT1 signaling genes predicts poor survival outcome in glioblastoma multiforme in a subtype-specific manner. PloS one. 2012;7:e29653. doi: 10.1371/journal.pone.0029653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Burnette BC, et al. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011;71:2488–2496. doi: 10.1158/0008-5472.CAN-10-2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lim JY, Gerber SA, Murphy SP, Lord EM. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8(+) T cells. Cancer immunology, immunotherapy: CII. 2014;63:259–271. doi: 10.1007/s00262-013-1506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Grob JJ, et al. Randomised trial of interferon α-2a as adjuvant therapy in resected primary melanoma thicker than 15 mm without clinically detectable node metastases. The Lancet. 1998;351:1905–1910. doi: 10.1016/s0140-6736(97)12445-x. [DOI] [PubMed] [Google Scholar]
- 148.Eggermont AMM, et al. Post-surgery adjuvant therapy with intermediate doses of interferon alfa 2b versus observation in patients with stage Ilb/III melanoma (EORTC 18952): randomised controlled trial. The Lancet. 2005;366:1189–1196. doi: 10.1016/S0140-6736(05)67482-X. [DOI] [PubMed] [Google Scholar]
- 149.Wheatley K, et al. Does adjuvant interferon-α for high-risk melanoma provide a worthwhile benefit? A meta-analysis of the randomised trials. Cancer Treatment Reviews. 2003;29:241–252. doi: 10.1016/s0305-7372(03)00074-4. [DOI] [PubMed] [Google Scholar]
- 150.Dritschilo A, Mossman K, Gray M, Sreevalsan T. Potentiation of radiation injury by interferon. American journal of clinical oncology. 1982;5:79–82. [PubMed] [Google Scholar]
- 151.Zhu Y, Tibensky I, Schmidt J, Ryschich E, Marten A. Interferon-alpha enhances antitumor effect of chemotherapy in an orthotopic mouse model for pancreatic adenocarcinoma. Journal of Immunotherapy. 2008;31:599–606. doi: 10.1097/CJI.0b013e3181818769. [DOI] [PubMed] [Google Scholar]
- 152.Zhu Y, et al. Interferon-alpha in combination with chemotherapy has potent antiangiogenic properties in an orthotopic mouse model for pancreatic adenocarcinoma. Journal of Immunotherapy. 2008;31:28–33. doi: 10.1097/CJI.0b013e318157c682. [DOI] [PubMed] [Google Scholar]
- 153.Linehan DC, et al. Adjuvant interferon-based chemoradiation followed by gemcitabine for resected pancreatic adenocarcinoma: a single-institution phase II study. Annals of surgery. 2008;248:145–151. doi: 10.1097/SLA.0b013e318181e4e9. [DOI] [PubMed] [Google Scholar]
- 154.Picozzi VJ, et al. Multicenter phase II trial of adjuvant therapy for resected pancreatic cancer using cisplatin, 5-fluorouracil, and interferon-alfa-2b-based chemoradiation: ACOSOG Trial Z05031. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2011;22:348–354. doi: 10.1093/annonc/mdq384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Schmidt J, et al. Open-label, multicenter, randomized phase III trial of adjuvant chemoradiation plus interferon Alfa-2b versus fluorouracil and folinic acid for patients with resected pancreatic adenocarcinoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:4077–4083. doi: 10.1200/JCO.2011.38.2960. [DOI] [PubMed] [Google Scholar]
- 156.Spaapen RM, et al. Therapeutic activity of high-dose intratumoral IFN-beta requires direct effect on the tumor vasculature. Journal of immunology. 2014;193:4254–4260. doi: 10.4049/jimmunol.1401109. [DOI] [PubMed] [Google Scholar]
- 157.Xuan C, Steward KK, Timmerman JM, Morrison SL. Targeted delivery of interferon-alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma. Blood. 2010;115:2864–2871. doi: 10.1182/blood-2009-10-250555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yang X, et al. Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer cell. 2014;25:37–48. doi: 10.1016/j.ccr.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Carswell EA, et al. An endotoxin-induced serum factor that causes necrosis of tumors. Proceedings of the National Academy of Sciences. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pennica D, et al. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature. 1984;312:724–729. doi: 10.1038/312724a0. [DOI] [PubMed] [Google Scholar]
- 161.Vicari AP, et al. Reversal of tumor-induced dendritic cell paralysis by CpG immunostimulatory oligonucleotide and anti-interleukin 10 receptor antibody. The Journal of experimental medicine. 2002;196:541–549. doi: 10.1084/jem.20020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.North RJ, Dunn PL, Havell EA. A role for tumor necrosis factor in poly(I:C)-induced hemorrhagic necrosis and T-cell-dependent regression of a murine sarcoma. Journal of interferon research. 1991;11:333–340. doi: 10.1089/jir.1991.11.333. [DOI] [PubMed] [Google Scholar]
- 163.Meng Y, et al. Cancer research. 2010;70:1534–1543. doi: 10.1158/0008-5472.CAN-09-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sparwasser T, et al. Macrophages sense pathogens via DNA motifs: induction of tumor necrosis factor-a-mediated shock. European Journal of Immunology. 1997;27:1671–1679. doi: 10.1002/eji.1830270712. [DOI] [PubMed] [Google Scholar]
- 165.Dinarello CA. Proinflammatory and Anti-inflammatory Cytokines as Mediators in the Pathogenesis of Septic Shock. Chest. 1997;112:321S–329S. doi: 10.1378/chest.112.6_supplement.321s. [DOI] [PubMed] [Google Scholar]
- 166.Campbell JD, et al. CpG-containing immunostimulatory DNA sequences elicit TNF-alpha-dependent toxicity in rodents but not in humans. The Journal of clinical investigation. 2009;119:2564–2576. doi: 10.1172/JCI38294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Weichselbaum RR, Kufe D. Translation of the radio- and chemo-inducible TNFerade vector to the treatment of human cancers. Cancer Gene Ther. 2009 doi: 10.1038/cgt.2009.37. [DOI] [PubMed] [Google Scholar]
- 168.Scott SD, Joiner MC, Marples B. Optimizing radiation-responsive gene promoters for radiogenetic cancer therapy. Gene Ther. 2002;9:1396–1402. doi: 10.1038/sj.gt.3301822. [DOI] [PubMed] [Google Scholar]
- 169.Mauceri HJ, et al. Tumor Necrosis Factor a (TNF-α) Gene Therapy Targeted by Ionizing Radiation Selectively Damages Tumor Vasculature. Cancer research. 1996;56:4311–4314. [PubMed] [Google Scholar]
- 170.Jung M, Dimtchev A, Velena A, Dritschilo A. Combining radiation therapy with interstitial radiation-inducible TNF-alpha expression for locoregional cancer treatment. Cancer Gene Ther. 2011;18:189–195. doi: 10.1038/cgt.2010.69. [DOI] [PubMed] [Google Scholar]
- 171.Meng Y, et al. Ad.Egr-TNF and local ionizing radiation suppress metastases by interferon-beta-dependent activation of antigen-specific CD8+ T cells. Mol Ther. 2010;18:912–920. doi: 10.1038/mt.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Herman JM, et al. Randomized phase III multi-institutional study of TNFerade biologic with fluorouracil and radiotherapy for locally advanced pancreatic cancer: final results. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:886–894. doi: 10.1200/JCO.2012.44.7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sckisel GD, et al. Out-of-Sequence Signal 3 Paralyzes Primary CD4(+) T-Cell-Dependent Immunity. Immunity. 2015;43:240–250. doi: 10.1016/j.immuni.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Young KH, et al. Optimizing Timing of Immunotherapy Improves Control of Tumors by Hypofractionated Radiation Therapy. PloS one. 2016;11:e0157164. doi: 10.1371/journal.pone.0157164. [DOI] [PMC free article] [PubMed] [Google Scholar]