

Abstract
In the study of host–pathogen interactions, vaccines and drug delivery, particulate delivery system are widely used to mimic pathogen size, pattern recognition receptor agonist presentation, and target cells or organs. However, some of the polymeric systems used in particulate delivery have inherent inflammatory properties that are variable and nonspecific. These properties enhance their adjuvant activity, but confound the analysis of signaling mechanisms. Here, we present a method for particle coating with minimal background immune activation via passivation of the surface with silica-silane. We show herein that a silica-silane shell passivates polymer particles rendering them inert to activation of innate immune cells. The method is broadly applicable and can be used to coat polymeric particles of many different compositions. This method of silica-silane coating also allows conjugation of amine-bearing agonists and provides for controlled variation of agonist loading. Finally, we demonstrate our particles maintain and enhance qualities of known pathogens, making this a potentially general method for improving immune agonist activity.
Keywords: microparticle, adjuvant, drug delivery, vaccines, biomaterials, polymers, coatings, immunology, core, shell
Graphical abstract
To better understand vaccines and immunotherapies, many researchers are elucidating the mechanisms by which the innate immune system responds to bacterial pathogens.1a–e Our group, and many others, are interested in how micrometersized pathogens stimulate multiple innate immune receptors to elicit protective responses.2a–p In our own work, we employed the current methods used in many studies to conjugate pattern recognition receptor agonists to commercial polystyrene particles to study these processes.2a–p However, as others have reported, naked polystyrene has an inherent, nonspecific background activation of innate immune cells, convoluting the results.3a–h Indeed, innate immune activation has been a recurring problem for many nanoscale materials, highlighting the need for inert particles and coatings.3a–h For us, it was difficult to differentiate the signal of the polystyrene particle from its conjugated agonists. Here we present a coating method for polymeric particles that reduces the background inflammatory response caused by nanoparticles while providing a chemical handle for conjugation of immune agonists. In addition, this coating is more “cell-like”, overcoming the intrinsic hydrophobicity of polystyrene, allowing the particles to be more uniformly dispersed in aqueous solutions without surfactant stabilization. This method provides a general route for researchers attempting to reduce nonspecific nanoparticle inflammatory properties and will find use in studies of the innate immune system. Particles with this coating will allow researchers to analyze responses solely from the agonists while mimicking the size and agonist presentation of a pathogen.
In designing an improved polymeric delivery system, we sought a general design strategy that would apply to different polymeric particles for both cellular and in vivo applications. Our design was guided by three main principles: low immunogenicity, ease of functionalization and pathogen mimicry. We further showed that this shell formation technique is generalizable to multiple polymeric cores (Figure 1).
Figure 1.
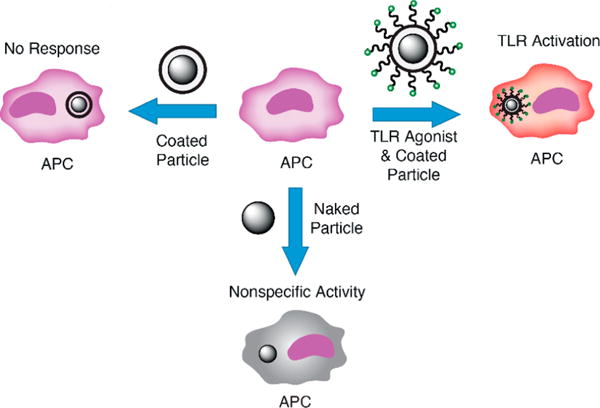
Schematic of cellular outcomes of particle exposure. Coated polystyrene particles alone do not elicit an immune response (top left). Agonist-functionalized coated polystyrene particles activate the cell’s immune response based on the agonists loaded on the surface (top right), whereas polystyrene particles cause nonspecific activation of immune cells (bottom). APC, antigen-presenting cell.
RESULTS
Synthesis of Coated Polystyrene Particles
We hypothesized that if the particles had a more biologically relevant surface potential it would reduce immunogenicity. To create a test particle, we synthesized monodisperse polystyrene (PS) particles.4 This synthesis was accomplished by first dissolving purified styrene monomer and poly(vinylpyrrolidone) MW 40 000 in ethanol and purging with dry nitrogen for 1 h. The addition of the poly(vinylpyrrolidone) increased the viscosity of the monomer solution. The stir rate was adjusted to 200 rpm to promote the formation of micronsized particles. Polymerization was initiated by the addition of 1 wt % AIBN, and the mixture was stirred at 70 °C for 24 h. To remove unreacted material, we centrifuged the reaction mixture and washed the pellet three times with ethanol. We next screened conditions to synthesize a uniform tetraethyl orthosilicate (TEOS)-mercaptosilane copolymer shell. This judicious choice of silane allowed us to graft a heterobifunctional cross-linker, maleimide-PEG6-succinimidyl ester (maleimide-PEG6-NHS) through the maleimide to the surface of the particles. The succinimidyl ester moiety on the cross-linker allowed us to attach agonists containing an amine handle. We found that optimal coatings were achieved using an emulsion polymerization approach.5 In a representative coating procedure used for all core particle types, a flask was charged with cyclohexane, n-hexanol, water, Triton X-114, and sonicated for 20 min. To this emulsion, uncoated particles were added, and the resulting mixture was sonicated for an additional 40 min to increase particle suspension. TEOS was added dropwise followed by 14 M ammonia solution and the mixture stirred for 30 min. 3-Mercaptosilane was then added, and the reaction mixture stirred for 6 h. The mixture was centrifuged and then washed three times with ethanol to provide silica-silane coated microparticles. The particles were characterized by a combination of light microscopy, dynamic light scattering (DLS), zeta potential, scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS). SEM analysis confirmed the coated particles retained relative size dispersities (Figure 2a and Figures S1–S24). High-resolution SEM analysis of the particle surface suggested that the coating was established by a Pickering-type emulsion process.6
Figure 2.
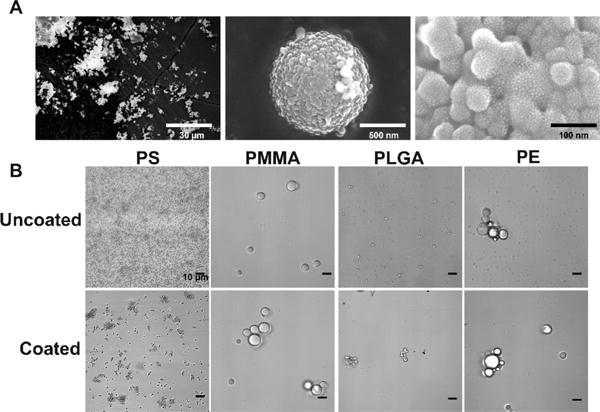
Microscopy of particles. (A) Scanning electron microscopy of PS coat particle at 1677×; 80000×; and 500000× magnification (left to right). (B) Bright-field microscopy of dispersions of uncoated and coated particles in ethanol to give accurate representations of particle size and features. Aggregation in ethanol does not reflect aggregation in aqueous solutions.
Before testing the immune activity of the particles, we assayed the physical properties of the coated particles. Using light microscopy, we examined particle morphology and bulk solution behavior (Figures 2 and 3). The particles were dispersed in ethanol to provide similar suspension and sedimentation rates across different particle types (Figure 2b).
Figure 3.
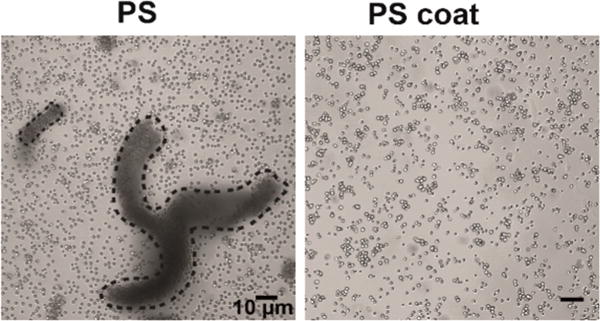
Light microscopy of particles in media. Microscopy of dispersions of PS and PS coat particles in cell culture media, demonstrating the clumping behavior of the PS particles. Clumps are denoted by dotted outlines.
We also examined dispersions of PS and coated PS particles (PS coat) in an aqueous medium, which is more relevant for biological studies. We determined that coated PS was more uniformly dispersed in aqueous solution (cell culture media) than PS (Figure 3).
Immune Response to Core–Shell Constructs
After validating uniform coating of the particles in the TEOS-mercaptosilane copolymer, we tested the immunogenicity of the particles coated with only the copolymer (before treatment with cross-linker or agonist) and compared it to their uncoated equivalents. To determine activity, we employed the transgenic RAW macrophage RAW-Blue NF-κB reporter cell line that provides a colorimetric readout of NF-κB activity.7a–d This cell line is commonly used as an assay for immunogenicity due to the central role of the transcription factor NF-κB in immune activation and cytokine production. NF-κB is a central transcription factor in immune response specifically the inflammatory response associated with innate immunity. We observed a dramatic and significant difference between PS and coated PS particles. To demonstrate generality, we also tested coated poly(methyl methacrylate) (PMMA), poly(lactic-co-glycolic acid) (PLGA) and polyethylene (PE) particles (Figure 4). All TEOS-mercaptosilane coated particles showed a significant reduction in NF-κB activity compared to their uncoated equivalents in all polymers except PE. Both coated PLGA and PMMA particles showed no significant difference in NF-κB activity compared to cells with no particles added (PBS) demonstrating that the coating passivates immune activation.
Figure 4.
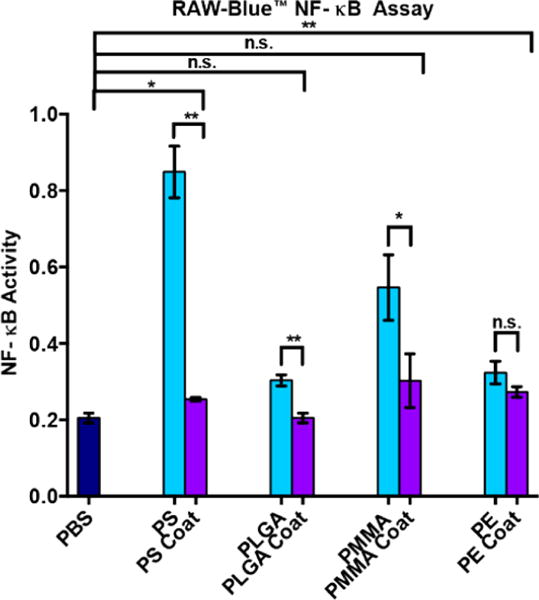
NF-κB activity of coated and uncoated particles. Blue bars, uncoated particles. Purple bars: particles with TEOS-mercaptosilane coating. RAW macrophages were stimulated for 18 h with coated or uncoated particles at 1:1 stoichiometry of particles to cells and then assayed for NF-κB activity. PBS, phosphate-buffered saline solution; PS, polystyrene particles; PS coat, TEOS-mercaptosilane-coated PS particles; PLGA, poly(lactic coglycolic acid) particles; PLGA coat, TEOS-mercaptosilane-coated PLGA particles; PMMA, poly(methyl methacrylate) particles; PMMA coat, TEOS-mercaptosilane coated PMMA particles; PE, polyethylene particles; PE coat, TEOS-mercaptosilane coated PE particles.*p < 0.05, **p < 0.01.
To further test the ability of the coating to passivate the immunogenicity of the polymer surface, we also tested immunogenicity in THP-1 cells (human APCs) by observing IL-1β secretion (Figure 5). IL-1β is a common inflammatory cytokine observed with nanoparticles.12a–c We found that the coating decreased IL-1β expression in all cases except PE which we believe is due to the low immunogenicity of uncoated PE. Further we found a statistically significant difference between PS and coated PS and PMMA and PMMA coat this reinforces our findings from the NF-κB activity assay.
Figure 5.
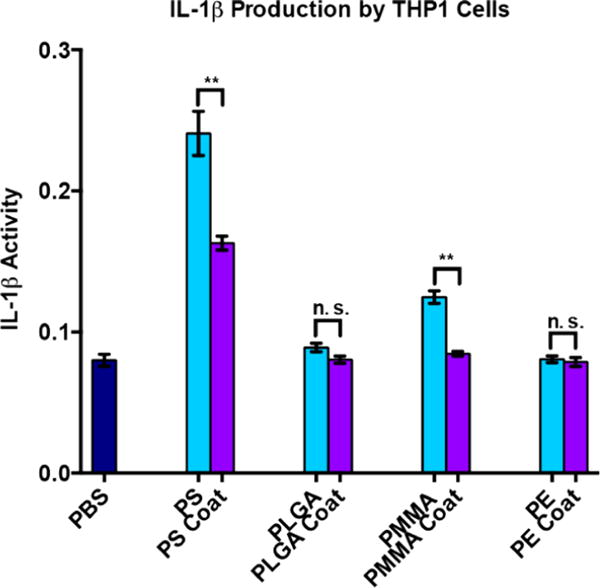
IL-1β expression in THP-1 cells. Blue bars: uncoated particles. Purple bars: particles with TEOS-mercaptosilane coating. THP-1 cells were stimulated for 18 h with coated or uncoated particles at 1:1 stoichiometry of particles to cells and then assayed for IL-1β activity. PBS, phosphate-buffered saline solution; PS, polystyrene particles; PS coat, TEOS-mercaptosilane-coated PS particles; PLGA, poly(lactic coglycolic acid) particles; PLGA coat, TEOS-mercaptosilane coated PLGA particles; PMMA, poly(methyl methacrylate) particles; PMMA coat, TEOS-mercaptosilane coated PMMA particles; PE, polyethylene particles; PE coat, TEOS-mercaptosilane coated PE particles. *p < 0.05, **p < 0.01.
For further studies we focused on polystyrene particles because they are extremely uniform in size and we intend to use them in future applications. To explore how the coating affects immune signaling and downstream effects, we performed a quantitative PCR analysis of key immune genes. This assay allowed us to monitor the expression levels of inflammatory markers and cell surface receptors involved in the innate immune response. We included TLR2 and TLR9 in our assay, as these are the target receptors for our model agonists. Changes in expression levels of these receptors from the particle itself would confound results in future experiments. We found that important immune players: CD4, TLR9, MyD88, TLR2, and TNF-α were upregulated in response to stimulation from PS particles. The coated particles did not demonstrate this degree of immunogenicity (Table 1).
Table 1.
qPCR Panel of Immune Genesa
| PS | PS coat | |
|---|---|---|
| CD4(***) | 1.132σ ± 0.18 | 0.6169σ ± 0.15 |
| IL-10(n.s) | 1σ ± 0.17 | 0.9265σ ± 0.11 |
| TLR9(*) | 1.561σ ± 0.18 | 1σ ± 0.18 |
| MyD88(***) | 1.569σ ± 0.12 | 1σ ± 0.13 |
| TLR2(***) | 1.757σ ± 0.15 | 0.9908σ ± 0.18 |
| TNF-a(**) | 1.635σ ± 0.19 | 1.089σ ± 0.19 |
RAW macrophages were stimulated for 4 h with PS or PS coat particles at 1:1 stoichiometry of particles to cells and then assayed. Values shown are fold change over unstimulated cells. Red, grey, and green signify upregulation, no change, and down regulation, respectively. Statistical significance between PS and PS coat values: CD4 (***); IL-10 (n.s.); TLR9 (*); MyD88 (***); TLR2 (***); TNF-α (**).
p < 0.05,
p < 0.01,
p < 0.001.
Agonist Loading
After demonstrating the improved immune compatibility of our TEOS-mercaptosilane copolymer coating, we wanted to ascertain if the particles could be loaded with agonists. The TLR9 agonist CpG-1826 was used as a model because an amine-bearing, highly fluorescent version is available that retains robust bioactivity.7
Uniform labeling of the core–shell particles was accomplished via treatment with a heterobifunctional cross-linker and subsequent conjugation with a fluorophore-labeled immune agonist. Due to the wide availability of amine-functionalized immune agonists, we selected a cross-linker with a succinimidyl group, maleimide-PEG6-NHS. We included a poly(ethylene glycol) spacer arm to optimize agonist presentation. Additionally, the maleimide was used to link the cross-linker to the free thiols derived from the mercaptosilane functionalization on the particle.
To control agonist loading we varied the concentration of the cross-linked particles in a solution of FAM-CpG-NH2 (Figure 6a). Flow cytometry indicated a shift in median fluorescence corresponding to an increase in the amount of FAM-CpG per particle (Figure 6b) demonstrating the ability to dose the particle with varying amounts of agonist. Using a colorimetric readout of NF-κB, we determined that NF-κB activity increased with increasing agonist loading on the particles (Figure 6c). Similarly, we increased the stoichiometry of particles to cells and observed an increase in the overall NF-κB activity. To demonstrate the generality of this method, we loaded the particles with lipoteichoic acid (LTA), a TLR2 agonist.8 Our results indicated that the increased LTA loading corresponded with increased NF-κB activation. Confocal microscopy of the particle bearing cells confirmed that they were internalized (Figure 7b and c).
Figure 6.
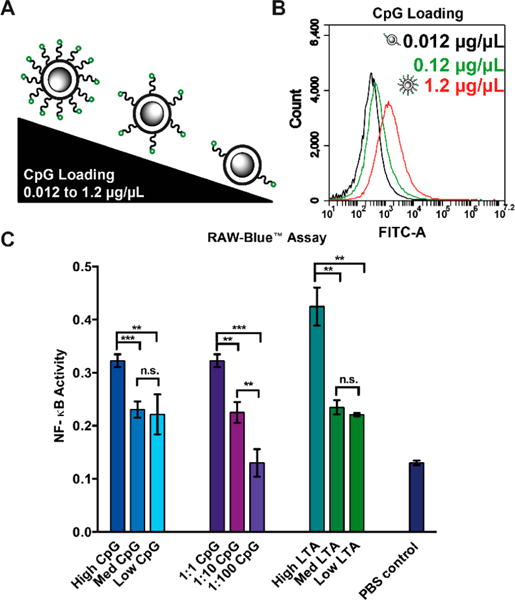
Cell activation and endocytosis of coated and uncoated particles. (A) Flow cytometry analysis of PS coat particles loaded with increasing amounts of FAM-CpG from 0.012 μg/μL to 1.2 μg/μL. (B) Agonist titration analysis using RAW macrophage RAW-Blue NF-κB activity assay. High, med, and low refer to the concentration of free agonist in the PS coat particle loading reaction (1.2, 0.12, and 0.012 μg/μL CpG, respectively, and 20, 2, and 0.2 μg/mL LTA, respectively). Ratios refer to stoichiometry of particles to cells.
Figure 7.
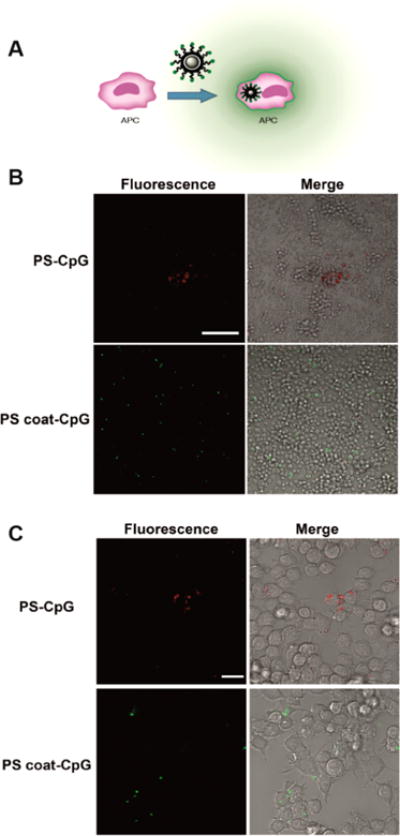
Cell internalization of particles. (A) Schematic of particle internalization. (B) Confocal microscopy (63× magnification) of RAW macrophages incubated with PS-CpG (top) or PS coat-CpG (bottom) for 4 h. Stoichiometry of particles to cells was 1:1. PS-CpG and PS coat-CpG samples were imaged using 561 or 488 nm laser excitation, respectively. Scale bar: 20 μm. (C) Confocal microscopy of samples in B at 20× magnification. Scale bar: 100 μm.
Differences in uptake were observed between PS particle and coated PS particles. Macrophages exposed to coated PS particles display internalization, while PS particles remain uninternalized (Figure 7a). Further differences were observed in the aggregation of the two particle types. PS particles tend to form large, aggregated clumps while the coated PS particles are more evenly suspended throughout the medium (Figure 7b, c). The aggregates in the PS-CpG varied in size, including some extremely large aggregates approximately 70 μm in diameter (Figure S25), lowering the effective concentration of the particles. These results indicate that the coated particles provide a more uniform bioavailability providing better control of agonist dosing.
Biomimimetic Nature of Core–Shell Particles
To study the biomimetic nature of our particles, we first examined the zeta potential of coated and uncoated particles. We found that coating the particles decreased the zeta potential by −1.2 to −20.8 mV compared to their uncoated precursors (Table S1). This negative zeta potential confers several benefits to the coated particle including: (1) reduced clumping, which increases particle dosing consistency, and (2) surface potential similar to both mammalian cells (−19.4 mV) and bacteria (−21.9 mV).9,10 We hypothesized that these advantages would increase the immune cell uptake of the particles.
To test this hypothesis, we devised a cellular uptake experiment. We incubated RAW 264.7 macrophages with fluorescein-labeled PS, coated fluorescein-labeled PS, or commercially available Alexa Fluor 488-labeled, heat-inactivated E. coli at 1:1 stoichiometry of particles to cells. After 4 h, we examined the fluorescence of the RAW cells using flow cytometry (Figure 8). We found that 1.8% of cells treated with labeled PS particles took up the particles. In contrast, 44.0% of cells showed uptake of the coated fluorescein-labeled PS particles. Further, 98.5% of cells displayed uptake of the labeled E. coli. These results showed that the uptake profile of the coated PS particles is increased compared to that of the PS particle, and therefore, closer to that of the pathogenic E. coli or a mammalian cell. We hypothesize that the reason for the lowered uptake of the coated PS particles in contrast to E. coli is due to the presence of TLR agonists on the surface of the E. coli that induce endocytosis in immune cells.11a–d
Figure 8.
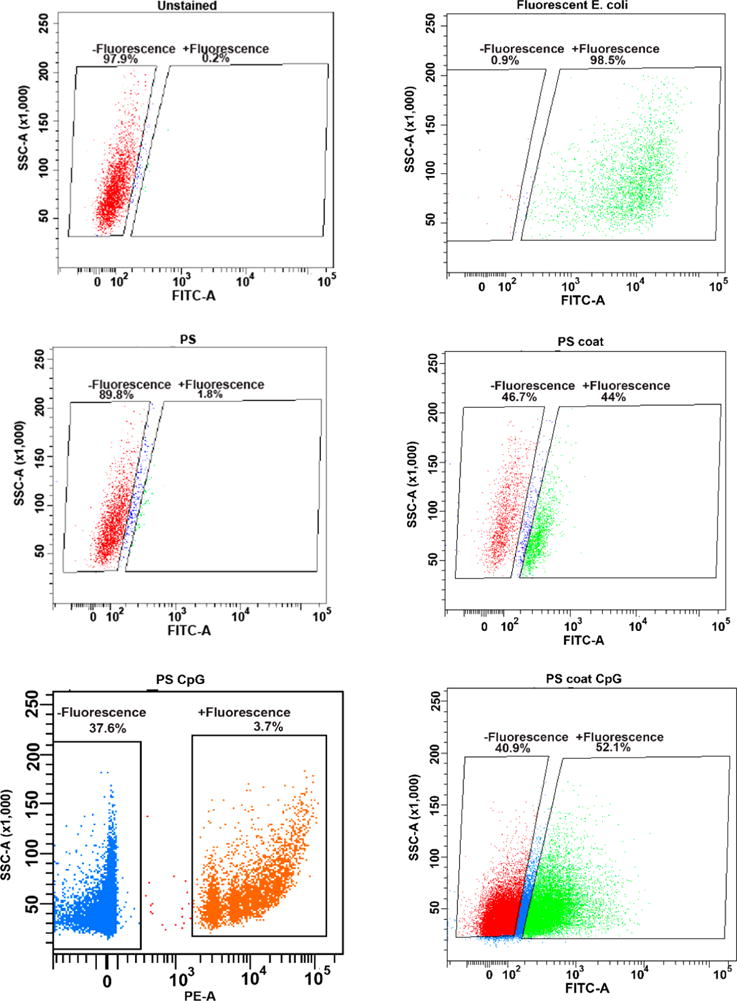
Cell uptake of particles. Flow cytometry plots of unstained cells, PS particle, PS coat particle, and fluorescent bacteria (top to bottom).
To assay the ability of our particles to recreate a pathogenic insult, we incubated RAW 264.7 cells with CpG-loaded particles. We compared coated PS particles functionalized with FAM-CpG, commercially available amine-modified PS particles that we functionalized with CpG, and Alexa Fluor 488-labeled, heat-inactivated E. coli (Figure 8). Cells were incubated with particle or E. coli for 4 h at a ratio of one particle or E. coli bacterium to one cell. We found that 3.7% of cells incubated with PS-CpG particles successfully internalized the particles. In contrast, 52.1% of cells incubated with coated PS–CpG particles displayed fluorescence, indicating successful uptake of the particles. This compares favorably with the fluorescent E. coli, which had uptake by 98.5% of the cells. We hypothesize that the coated polymer had more uptake because the zeta potential is closer to that of E. coli or a mammalian cell. Additionally, PS particles are hydrophobic and prone to clumping, lowering the effective concentration. E. coli have an additional advantage over the coated PS-CpG particles in that they display many different TLR agonists which activate the cells, potentially increasing uptake.11a–d
After sorting, the cells were analyzed by fluorescence microscopy (Figure 9). The microscopy of the cells sorted for the FITC or AlexaFluor488 marker showed punctate green fluorescence on the interior of the cells indicating uptake of the particles or bacteria. Enough cells for microscopy was only obtained for cells incubated with PS coat, PS coat-CpG, and fluorescent E. coli. This is due to the low percentage of cells positive for uptake of PS or PS-CpG particles.
Figure 9.
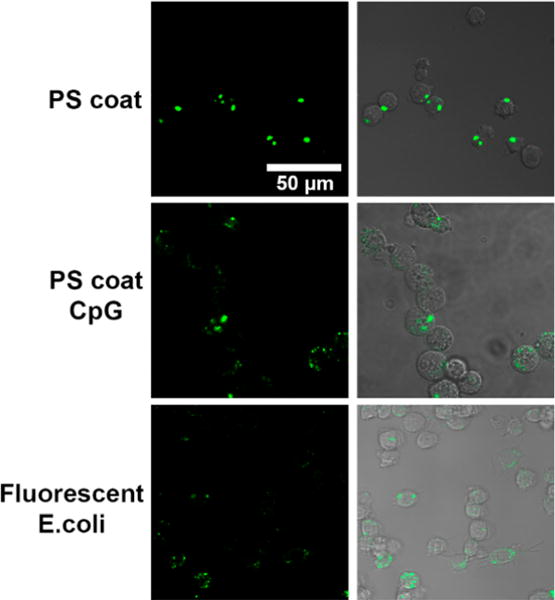
Fluorescence microscopy of sorted cells. Cells FACS sorted for green fluorescence demonstrate the presence of particle puncta.
CONCLUSION
Particulate forms of polystyrene and other common delivery polymers nonspecifically activate the immune system to varying degrees, while limiting the amount of particulate matter that is internalized. To incorporate these useful polymers into tools to study the immune system, we developed a method to passivate their surface. This technique provides a significant decrease in the immunogenicity of common polymer particles. We further showed that this easy and scalable coating technique can be applied to many polymeric particles conferring them with the same useful qualities, including ease of cross-linking and agonist loading, uniform dispersion in aqueous solutions, and increased cellular uptake. The coated particles display surface properties closely resembling that of a pathogen, opening new avenues for study of the innate immune system and adjuvant development.
MATERIALS
5′-FAM-TCCATGACGTTCCTGACGTT-3′-NH2 (FAM-CpG-amino) was purchased from IDT. All cell culture reagents were obtained from Life Technologies unless otherwise specified. Chemicals were from Sigma-Aldrich unless otherwise specified. PMMA (3–10 μm) and PE (3–16 μm) particles were purchased from Cospheric. PLGA (2 μm) particles were purchased from Sigma-Aldrich. Rhodamine-NH2 (1 μm) functionalized polystyrene particles were purchased from Fisher Scientific. AlexaFluor 488 functionalized heat inactivated E. coli were purchased from ThermoFisher Pierce.
METHODS
Synthesis of 1 μm Polystyrene Particles
Uniform, spherical polystyrene particles were synthesized via controlled styrene polymerization. Polyvinylpyrrolidone, MW 40,000 (2.0 g) and styrene (20 g), washed with NaOH and dried with MgSO4, was dissolved in EtOH (250 mL) and purged with nitrogen for 1 h. AIBN (0.2 g) was added and the mixture stirred at 70 °C and 200 rpm (IKA) for 24 h. Mixture was pelleted by centrifugation at 3400 rpm (Baxter Varifuge 3.0R) for 30 min and washed 3× in 30 mL of EtOH to remove residual monomer, initiator, and stabilizer. PS particles were stored in EtOH at 4 °C.
Synthesis of Functionalized Silica Shell on Polymer Particles
Cyclohexane (45 mL), n-hexanol (10.8 mL), endotoxinfree water (2 mL) and Triton X-114 (10.8 mL) were placed in a round-bottom flask and sonicated for 20 min. Particles (0.2 g) were added and the suspension was sonicated for 40 min. TEOS (400 μL) was added dropwise followed by 14 M aqueous ammonia (1.2 mL). Solution was stirred for 30 min RT. 3-Mercaptosilane (200 μL) was added dropwise and stirred for 6 h. TEOS-mercaptosilane copolymer coated particles were pelleted at 3400 rpm for 30 min and washed 3× with EtOH. Particles were dried at 70 °C overnight.
Agonist Attachment to Functionalized Particles
TEOS-mercaptosilane copolymer coated particles or NH2-functionalized polystyrene particles (0.4 g) and maleimide-PEG6-NHS (0.4 g) were suspended in 5 mL of DMSO and 5 mL of dPBS, sonicated for 1 h and incubated at 37 °C overnight. Resulting particles were pelleted and washed 3× in DMSO. FAM-CpG-NH2 (0.5–5 equiv) was added in 50 mM Tris-HCl, 150 mM NaCl (pH 8) and sonicated for 1 h in the dark and incubated at 37 °C for 0.5–4 h. Particles were pelleted and removed and resulting supernatant was measured using NanoDrop 2000 (ThermoScientific) to gauge DNA loading by measuring the DNA remaining in solution. Resulting particles were pelleted and washed 3× with DMSO and 3× with dPBS and stored at 4 °C.
Flow Cytometry of Agonist Labeled Particles
Particles were placed in dPBS and sonicated for 1 h. Particles were aliquoted and diluted 1:50 in a 1.5 mL microcentrifuge tube in dPBS. Particles were analyzed using Accuri C6 flow cytometer. Gating parameters were selected for size of a single particle.
SEM and EDS of Particles
Scanning electron microscopy (SEM) and energy-dispersive spectroscopy (EDS) of the particles was performed using an FEI Quanta 3D FEG dual beam (SEM/FIB) equipped with Inca EDS (Oxford Instruments). High-resolution images were taken with an FEI Magellan 400 XHR SEM particle samples were dried under vacuum for 24 h, mounted on carbon tape, and sputter coated (South Bay Technologies) with approximately 2–4 nm of Au/Pd 60:40 or Ir.
Zeta Potential
Zeta potential was performed using a Zetasizer ZS Nano DLS (Malvern). Particles were suspended at a concentration of approximately 1 mg/mL in a solution of 10 mM NaCl in nanopure water. The mixture was then sonicated (Bransonic) for 8 min, and vortexed immediately before measurement.
Dynamic Light Scattering
Dynamic light scattering (DLS) was performed using a Zetasizer ZS Nano DLS (Malvern). The particles were placed in a solution of 10 mM NaCl in nanopure water at 1 mg/mL.
Light Microscopy of Particles
1 × 105 particles were placed in 200 μL of phosphate-buffered saline (PBS) or EtOH in an 8-well coverslip bottom plate (ThermoScientific). Particles were imaged using Zeiss LSM780 confocal microscope and Zeiss Plan-Apochromat 63x/1.40 oil objective.
Microscopy of Particle Internalization
RAW 264.7 macrophages were plated at 1 × 105 cells/well in an 8-well coverslip bottom plate in 200 μL Dulbecco’s modified eagle medium (DMEM) containing 10% heat-inactivated fetal bovine serum (HIFBS) and incubated at 37 °C, 5% CO2 for 24 h. Media was changed to DMEM, 10% HIFBS and cells were incubated for 1 h. Particles were added at a 1:1 ratio of cells to particles and incubated for 4 h. Cells were imaged using a Zeiss LSM780 confocal microscope and Zeiss Plan-Apochromat 63x/1.40 oil objective and 488 nm laser.
RAW-Blue NF-κB Assay
RAW-Blue NF-κB cells (Invivogen) were passaged and plated in a 96 well plate at 100k cells/well in 180 μL DMEM containing 10% HIFBS. Cells were incubated at 37 °C and 5% CO2 for 1 h. Particles were counted using flow cytometer. 100k particles were added to each well (1 particle:1 cell). The volume of each well was brought to 200 μL and incubated at 37 °C and 5% CO2 for 18 h. After 18 h, 20 μL of the cell supernatant was placed in 180 μL freshly prepared QuantiBlue (Invivogen) solution and incubated at 37 °C/5% CO2 for up to 2 h. The plate was analyzed every hour using a Multiskan FC plate reader (Thermo Scientific) and absorbance was measured at 620 nm.
THP-1 IL-1β
THP-1 cells were cultured in Roswell Park Memorial Institute medium (RPMI) 1640 medium supplemented with 10% HIFBS. 3.6 × 105 cells were plated in each well of a 96 well plate in 180 μL DMEM supplemented with 10% HIFBS. Cells were incubated with 20 μL of LPS at 10 μg/mL was for 3 h at 37 °C/5% CO2. Supernatant was removed and 180 μL supplemented RPMI added to the cells. Twenty μL of particle suspension (1.8 × 107 particles/mL) was added to the cells. Cells were incubated overnight at 37 °C/5% CO2. Detection of IL-1β was accomplished using HEK-Blue IL-1β cells (Invivogen). HEK-Blue IL-1β cells were washed twice with prewarmed dPBS and detached using a cell scraper. Cells were resuspended in fresh prewarmed DMEM supplemented with 10% HIFBS. 5 × 105 cells were placed in each well in 150 μL of supplemented DMEM. Fifty microliters of THP-1 cell supernatant was added to each well. Cells were incubated overnight 37 °C in 5% CO2. IL-1β production was determined by adding 150 μL QuantiBlue (Invivogen) solution and incubated at 37 °C/5% CO2 for up to 2 h. to 50 μL HEK-Blue IL-1β cell supernatant. The plate was analyzed every hour using a Multiskan FC plate reader (Thermo Scientific) and absorbance was measured at 620 nm.
Particle vs E. coli Uptake Analysis
RAW 264.7 macrophages were passaged and 1 × 107 cells were placed in 15 mL conical tube. Particles or heat inactivated fluorescent bacteria were added at a ratio of 1:1. Cells were incubated at 37 °C 5% CO2 for 4 h. Cells were pelleted and washed 3× with dPBS and placed in 5 mL of FACS tubes. Cells were sorted using FACSAria Fusion flow cytometer (BD).
qPCR Analysis
RAW 264.7 (2 × 106 cells) were plated in a 6 well plate in 2 mL DMEM supplemented with 10% HIFBS. Two ×106 particles were added to each well. Cells were incubated for 4 h and RNA was extracted using RNeasy Plus Mini kit (Qiagen). RT-PCR was performed using RT2 first strand kit (Qiagen) and BioRad thermocycler according to manufacturer’s protocol. cDNA was stored at −20 °C. RT2 SYBR ROX qPCR Master mix (Qiagen) was used according to manufacturer’s protocol. qPCR amplification was performed using a Stratagene Mx3005P thermocycler.
Supplementary Material
Acknowledgments
The authors thank the Optical Biology Core at University of California, Irvine, for assistance in image acquisition, and the Institute for Immunology Flow Core at University of California, Irvine, for cell sorting assistance. We acknowledge the Laser Spectroscopy Facility at the University of California, Irvine, directed by Dr. Dmitry Fishman, for training and use of the Malvern Zetasizer. SEM and EDS work was performed at the UC Irvine Materials Research Institute (IMRI), using instrumentation funded in part by the National Science Foundation Center for Chemistry at the Space-Time Limit (CHE-082913). The authors acknowledge Dr. Qiyin Lin and William Gaieck for assistance with SEM, and IMRI facility director Dr. Jian-Guo Zheng.
Funding
We would like to acknowledge support by the NIH (1U01Al124286-01 and 1DP2Al112194-01, GM099594), Prof. Esser-Kahn thanks the Pew Scholars Program and the Cottrell Scholars Program for generous support. This work was supported, in part, by a grant from the Alfred P. Sloan foundation. The authors acknowledge the financial support provided by NIH (1U01AL124286–01 and 1DP2AL112194–01, GM0099594), B.M. thanks NSF-GRFP (DGE-1321846), Professor Esser-Kahn thanks the Pew Scholars Program, the Cottrell Scholars Program for generous support. This work was supported, in part, by the Alfred P. Sloan Foundation.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsbiomaterials.6b00473.
SEM data, zeta potential data, dynamic light scattering, and further confocal microscopy images (PDF)
ORCID
Aaron P. Esser-Kahn: 0000-0003-1273-0951
Notes
The authors declare no competing financial interest.
References
- 1.(a) Hubbell J, Thomas S, Swartz M. Materials engineering for immunomodulation. Nature. 2009;462(7272):449–460. doi: 10.1038/nature08604. [DOI] [PubMed] [Google Scholar]; (b) Moyer T, Zmolek A, Irvine D. Beyond antigens and adjuvants: formulating future vaccines. J Clin Invest. 2016;126(3):799–808. doi: 10.1172/JCI81083. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Irvine D, Swartz M, Szeto G. Engineering synthetic vaccines using cues from natural immunity. Nat Mater. 2013;12:978–990. doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pradhan P, Qin H, Leleux J, Gwak D, Sakamaki I, Kwak L, Roy K. The effect of combined IL10 siRNA and CpG ODN as pathogenmimicking microparticles on Th1/Th2 cytokine balance in dendritic cells and protective immunity against B cell lymphoma. Biomaterials. 2014;35(21):5491–5504. doi: 10.1016/j.biomaterials.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kasturi S, Skountzou I, Albrecht R, Koutsonanos D, Hua T, Nakaya H, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan R, Garcia-Sastre A, Compans R, Pulendran B. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470:543–547. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Petersen L, Ramer-Tait A, Broderick S, Kong C, Ulery B, Rajan K, Wannemuehler MJ, Narasimhan B. Activation of innate immune responses in a pathogen-mimicking manner by amphiphilic polyanhydride nanoparticle adjuvants. Biomaterials. 2011;32(28):6822. doi: 10.1016/j.biomaterials.2011.05.063. [DOI] [PubMed] [Google Scholar]; (b) Stutts L, Esser-Kahn A. A Light-Controlled TLR4 Agonist and Selectable Activation of Cell Subpopulations. ChemBioChem. 2015;16(12):1744–1748. doi: 10.1002/cbic.201500164. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mancini R, Tom J, Esser-Kahn A. Covalently Coupled Immunostimulant Heterodimers. Angew Chem, Int Ed. 2014;53(1):189–192. doi: 10.1002/anie.201306551. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mancini R, Stutts L, Moore T, Esser-Kahn A. Controlling the Origins of Inflammation with a Photoactive Lipopeptide Immunopotentiator. Angew Chem, Int Ed. 2015;54(20):5962–5965. doi: 10.1002/anie.201500416. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Fytianos K, Rodriguez- Lorenzo L, Clift M, Blank F, Vanhecke D, Von Garnier C, Petri-Fink A, Rothen-Rutishauser B. Uptake efficiency of surface modified gold nanoparticles does not correlate with functional changes and cytokine secretion in human dendritic cells in vitro. Nanomedicine. 2015;11(3):633–644. doi: 10.1016/j.nano.2014.11.004. [DOI] [PubMed] [Google Scholar]; (f) Toy R, Roy K. Engineering nanoparticles to overcome barriers to immunotherapy. Bioeng Transl Med. 2016;1(1):47–62. doi: 10.1002/btm2.10005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Leleux J, Atalis A, Roy K. Engineering immunity: Modulating dendritic cell subsets and lymph node response to direct immune-polarization and vaccine efficacy. J Controlled Release. 2015;219:610–621. doi: 10.1016/j.jconrel.2015.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Siefert A, Caplan M, Fahmy T. Artificial bacterial biomimetic nanoparticles synergize pathogen-associated molecular patterns for vaccine efficacy. Biomaterials. 2016;97:85–96. doi: 10.1016/j.biomaterials.2016.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Moon J, Suh H, Li A, Ockenhouse C, Yadava A, Irvine D. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc Natl Acad Sci U S A. 2012;109(4):1080–1085. doi: 10.1073/pnas.1112648109. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Nuhn L, Vanparijs N, de Beuckelaer A, Lybaert L, Verstraete G, Deswarte K, Lienenklaus S, Shukla N, Salyer A, Lambrecht B, Grooten J, David S, de Koker S, De Geest B. pH-degradable imidazoquinoline-ligated nanogels for lymph node-focused immune activation. Proc Natl Acad Sci U S A. 2016;113(29):8098–8103. doi: 10.1073/pnas.1600816113. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Andorko J, Gammon J, Tostanoski L, Zeng Q, Jewell C. Targeted Programming of the Lymph Node Environment Causes Evolution of Local and Systemic Immunity. Cell Mol Bioeng. 2016;9(3):418–432. doi: 10.1007/s12195-016-0455-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Irvine D, Hanson M, Rakhra K, Tokatlian T. Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem Rev. 2015;115(19):11109–11146. doi: 10.1021/acs.chemrev.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Koshy S, Mooney D. Biomaterials for enhancing anti-cancer immunity. Curr Opin Biotechnol. 2016;40:1–8. doi: 10.1016/j.copbio.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Dowling D, Levy O. Pediatric vaccine adjuvants: Components of the modern vaccinologist’s toolbox. Pediatr Infect Dis J. 2015;34(12):1395–1398. doi: 10.1097/INF.0000000000000893. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Meyer R, Sunshine J, Green J. Biomimetic particles as therapeutics. Trends Biotechnol. 2015;33(9):514–524. doi: 10.1016/j.tibtech.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Lynn G, Laga R, Darrah P, Ishizuka A, Balaci A, Dulcey A, Pechar M, Pola R, Gerner M, Yamamoto A, Buechler C, Quinn K, Smelkinson M, Vanek O, Cawood R, Hills Y, Vasalatiy O, Kastenmuller K, Francica J, Stutts L, Tom J, Ryu K, Esser-Kahn A, Etrych T, Fisher K, Seymour L, Seder R. In vivo characterization of the physiochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat Biotechnol. 2015;33(11):1201–1210. doi: 10.1038/nbt.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Seong S, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4(6):469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]; (b) Farrera C, Fadeel B. It takes two to tango: Understanding the interactions between engineered nanomaterials and the immune system. Eur J Pharm Biopharm. 2015;95:3–12. doi: 10.1016/j.ejpb.2015.03.007. [DOI] [PubMed] [Google Scholar]; (c) Gustafson H, Holt-Casper D, Grainger D, Ghandehari H. Nanoparticle uptake: The phagocyte problem. Nano Today. 2015;10(4):487–510. doi: 10.1016/j.nantod.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fadeel B. Clear and present danger? Engineered nanoparticles and the immune system. Swiss Med Wkly. 2012;142:w13609. doi: 10.4414/smw.2012.13609. [DOI] [PubMed] [Google Scholar]; (e) Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson K. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc Natl Acad Sci U S A. 2008;105(38):14265–14270. doi: 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ekdahl K, Lambris J, Elwing H, Ricklin D, Nilsson P, Teramura Y, Nicholls I, Nilsson B. Innate immunity activation on biomaterial surfaces: A mechanistic model and coping strategies. Adv Drug Delivery Rev. 2011;63(12):1042–1050. doi: 10.1016/j.addr.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Lunov O, Syrovets T, Loos C, Nienhaus G, Mailander V, Landfester K, Rouis M, Simmet T. Amino-Functionalized Polystyrene Nanoparticles Activate the NLRP3 Inflammasome in Human Macrophages. ACS Nano. 2011;5(12):9648–9657. doi: 10.1021/nn203596e. [DOI] [PubMed] [Google Scholar]; (h) Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson K. Nanoparticle size and surface properties determine the protein corona with possible implicationsfor biological impacts. Proc Natl Acad Sci U S A. 2008;105(38):14265–14270. doi: 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Many of the most effective materials, many cited in 2, avoid this inherent inflammation through molecular design, but this may not be a viable option for those lacking synthetic approaches and requires more steps to achieve basic understanding.
- 4.Hong J, Han H, Hong C, Shim S. A direct preparation of silica shell on polystyrene microspheres prepared by dispersion polymerization with polyvinylpyrrolidone. J Polym Sci, Part A: Polym Chem. 2008;46(8):2884–2890. [Google Scholar]
- 5.Thickett S, Gilbert R. Emulsion polymerization: State of the art in kinetics and mechanisms. Polymer. 2007;48(24):6965–6991. [Google Scholar]
- 6.Pickering S. CXCVI.—Emulsions. J Chem Soc, Trans. 1907;91:2001–2021. [Google Scholar]
- 7.(a) Ballas Z, Krieg A, Warren T, Rasmussen W, Davis H, Waldschmidt M, Weiner G. Divergent Therapeutic and Immunologic Effects of Oligodeoxynucleotides with Distinct CpG Motifs. J Immunol. 2001;167(9):4878–4886. doi: 10.4049/jimmunol.167.9.4878. [DOI] [PubMed] [Google Scholar]; (b) Hayden M, West A, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]; (c) Silverman N, Maniatis T. NF-kappaB signaling pathways in mammalian and insect innate immunity. Genes Dev. 2001;15:2321–2342. doi: 10.1101/gad.909001. [DOI] [PubMed] [Google Scholar]; (d) Libermann T, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10(5):2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. Peptidoglycan- and Lipoteichoic acid-induced cell activation is mediated by Toll-like receptor 2. J Biol Chem. 1999;274:17406–17409. doi: 10.1074/jbc.274.25.17406. [DOI] [PubMed] [Google Scholar]
- 9.Alves C, Melo M, Franquelim H, Ferre H, Planas M, Feliu L, Bardaji E, Kowalczyk W, Andreu D, Santos N, Fernandes M, Castanho M. Escherichia coli Cell Surface Perturbation and Disruption Induced by Antimicrobial Peptides BP100 and pepR. J Biol Chem. 2010;285(36):27536–27544. doi: 10.1074/jbc.M110.130955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bondar OV, Saifullina DV, Shakhmaeva II, Mavlyutova II, Abdullin TI. Monitoring of the Zeta Potential of Human Cells upon Reduction in Their Viability and Interaction with Polymers. Acta Naturae. 2012;4(1):78–81. [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Takeuchi T, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11(4):443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]; (b) Platt C, Ma J, Chalouni C, Ebersold M, Bou-Reslan H, Carano R, Mellman I, Delamarre L. Mature dendritic cells use endocytic receptors to capture and present antigens. Proc Natl Acad Sci U S A. 2010;107(9):4287–4292. doi: 10.1073/pnas.0910609107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol. 2009;10:609–622. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Blander J, Medzhitov R. Regulation of Phagosome Maturation by Signals from Toll-Like Receptors. Science. 2004;304(5673):1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 12.(a) Demento S, Eisenbarth S, Foellmer H, Platt C, Caplan M, Saltzman WM, Mellman I, Ledizet M, Fikrig E, Flavell R, Fahmy T. Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine. 2009;27:3013–3021. doi: 10.1016/j.vaccine.2009.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baron L, Gombault A, Fanny M, Villeret B, Savigny F, Guillou N, Panek C, Le Bert C, Lagente V, Rassendren F, Riteau N, Couillin I. The NLRP3 inflammazome is activated by nanoparticles through ATP, ADP and adenosine. Cell Death Dis. 2015;6:e1629. doi: 10.1038/cddis.2014.576. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hari A, Zhang Y, Tu Z, Detampel P, Stenner M, Ganguly A, Shi Y. Activation of NLRP3 inflammasome by crystalline structures via cell surface contact. Sci Rep. 2014;4:7281. doi: 10.1038/srep07281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.