

Abstract
Background and Aims Endoparasitic root-knot nematodes (RKNs) (Meloidogyne spp.) cause considerable losses in banana (Musa spp.), with Meloidogyne incognita a predominant species in Cavendish sub-group bananas. This study investigates the root transcriptome in Musa acuminata genotypes 4297-06 (AA) and Cavendish Grande Naine (CAV; AAA) during early compatible interactions with M. incognita.
Methods Roots were analysed by brightfield light microscopy over a 35 d period to examine nematode penetration and morphological cell transformation. RNA samples were extracted 3, 7 and 10 days after inoculation (DAI) with nematode J2 juveniles, and cDNA libraries were sequenced using lllumina HiSeq technology. Sequences were mapped to the M. acuminata ssp. malaccensis var. Pahang genome sequence, differentially expressed genes (DEGs) identified and transcript representation determined by gene set enrichment and pathway mapping.
Key Results Microscopic analysis revealed a life cycle of M. incognita completing in 24 d in CAV and 27 d in 4279-06. Comparable numbers of DEGs were up- and downregulated in each genotype, with potential involvement of many in early host defence responses involving reactive oxygen species and jasmonate/ethylene signalling. DEGs revealed concomitant auxin metabolism and cell wall modification processes likely to be involved in giant cell formation. Notable transcripts related to host defence included those coding for leucine-rich repeat receptor-like serine/threonine-protein kinases, peroxidases, thaumatin-like pathogenesis-related proteins, and DREB, ERF, MYB, NAC and WRKY transcription factors. Transcripts related to giant cell development included indole acetic acid-amido synthetase GH3.8 genes, involved in auxin metabolism, as well as genes encoding expansins and hydrolases, involved in cell wall modification.
Conclusions Expression analysis in M. acuminata during compatible interactions with RKNs provides insights into genes modulated during infection and giant cell formation. Increased understanding of both defence responses to limit parasitism during compatible interactions and effector-targeted host genes in this complex interaction will facilitate the development of genetic improvement measures for RKNs.
Keywords: Musa acuminata, Meloidogyne incognita, root-knot nematode, biotic stress, transcriptome, monocotyledons
INTRODUCTION
Banana (Musa spp.) is a major monocotyledonous crop in > 100 tropical and sub-tropical countries in sub-Saharan Africa, Central and South America and Asia, contributing towards global food security, nutrition and poverty alleviation as an important source of carbohydrates, fibre, vitamins and minerals, and livelihood.
In contrast to fertile wild diploid genotypes, many commercial diploid and triploid cultivars are seedless and parthenocarpic. As evolution of such plants is limited to asexual reproduction, via clonal vegetative micropropagation or suckers, today’s commercial crop possesses a narrow genetic base, with many cultivars lacking resistance to pests and diseases. Given the predominant global consumption of susceptible genotypes such as the sterile triploids of the Musa acuminata Cavendish sub-group, advances in crop improvement for the development of new cultivars that offer high quality, yield and resistance to biotic stresses is of paramount importance for the Musa industry.
Root-knot nematodes (RKNs) (Meloidogyne spp.) are obligate sedentary endoparasites that infect root tissues and develop biotrophic interactions with susceptible host plants. With worldwide distribution on a wide range of monocotyledonous and dicotyledonous plant species, they cause significant losses to the global agricultural economy. RKN larvae complete their life cycles through infection of plant roots, inducing the development of specialized feeding cells (giant cells), which are the nutritive source for all stages of the nematode life cycle. Subsequent hyperplasy and hypertrophy of the neighbouring root cells results in the formation of visible galls on infected root tissues. While RKN infection of young plants can be lethal, damage and deformation of roots and rhizomes in mature plants can result in increased crop cycle duration and reduced yield (Perry et al., 2009; Jones et al., 2013).
While the migratory endoparasitic nematode Radopholus similis is recognized as perhaps the most damaging nematode on banana globally (Gowen et al., 2005), RKN species M. incognita, M. javanica, M. arenaria and M. hapla also cause considerable losses across Musa-growing regions (Stoffelen et al., 2000; Gowen et al., 2005). Meloidogyne incognita is a predominant species in the important edible Cavendish sub-group bananas, which are predominantly destined for export markets (Quénehervé et al., 2009). The life cycle of M. incognita on susceptible Musa genotypes typically completes in 4–6 weeks, depending on environmental conditions. Following embryonic development, second-stage infective juveniles (J2) emerge from eggs. These then penetrate the root apex region, migrate through the root and establish feeding sites in vascular parenchyma cells. This involves the puncture of host cells by the nematode stylet, followed by injection of substances secreted from the oesophageal glands. Nematodes then ingest the cytoplasmic content of giant cells, which acts as a metabolic drain that diverts nutrients from the plant to the nematodes. Nematode secretions induce hypertrophy and hyperplasia of cells, resulting in an enlargement of infected roots with the presence of galls, which are composed of giant cells and nematode larvae. During this process, nematode juveniles pass through successive moults to J3, J4 and finally to male or female adult stages. Males typically occur either during adverse conditions or when population densities are high (Perry et al., 2009). Adult females each produce several hundred eggs in a gelatinous matrix that forms an egg mass in gall tissues (De Waele and Davide, 1998). Damage to root vascular tissues can also compromise plant development in aerial plant tissues, with a reduction in leaf size and number, reduced yield, and delay and unevenness in fruit ripening.
Given the increasing prohibition of chemical nematicides, efforts are increasing to develop integrated approaches for RKN management, which include cultural practices, biological control and introgression of resistance alleles into cultivated genotypes (Molinari, 2011). In banana, however, sources of genetic resistance to RKNs are limited, with very few reports of inclusion of promising genotypes with effective resistance or tolerance in Musa breeding programmes (Davide and Marasigan, 1985; Vilas Boas et al., 2002; Quénéhervé et al., 2009).
Two interconnected tiers of the plant immune system are currently recognized (Jones and Dangl, 2006). In the first, pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI) is activated following the interaction between host pattern recognition receptors (PRRs), which include receptor-like kinases (RLKs) and receptor-like proteins (RLPs) (Monaghan and Zipfel, 2012), and diverse conserved PAMPs, which are essential in microbial fitness or survival (Medzhitov and Janeway, 1997). Transient production of reactive oxygen species (ROS) at the cell surface is associated with NADPH oxidases and peroxidases (Zipfel et al., 2006; Daudi et al., 2012), with ROS acting as antimicrobials, in cell wall strengthening or in intracellular signalling (Lamb and Dixon, 1997). PTI involves the activation of a mitogen-activated protein kinase (MAPK) cascade and activation of WRKY transcription factors (TFs), with a rapid calcium cytoplasmic influx and converse efflux of K+ and Cl–. Callose deposition between the plant cell wall and plasma membrane is also characteristic of PTI. The second tier of the plant immune system, known as effector-triggered immunity (ETI), involves interaction between plant resistance R protein receptors, such as the nucleotide-binding domain leucine-rich repeat (NB-LRR) proteins, and cognate pathogen effector molecules that target PTI or other key host functions. Downstream defence responses of ETI can overlap with those of PTI, with activation of MAPK cascades and involvement of WRKY TFs, and a long-lasting accumulation of ROS. Upregulation of pathogenesis-related (PR) genes is associated with biosynthesis of salicylic acid (SA), jasmonic acid (JA) and ethylene (ET), together with production of antimicrobial secondary metabolites. SA accumulation, which typically regulates defence responses to biotrophs, can lead to a hypersensitive response (HR), as well as to systemic acquired resistance (SAR) (Durrant and Dong, 2004), with the expression of genes coding for acidic PR proteins (van Loon et al., 2005; Park et al., 2007). JA and ET signalling can also be activated, typically, although not exclusively, against necrotrophic pathogens and insect pests (Glazebrook, 2005; Pieterse et al., 2012). JA is known to be involved in biosynthesis of PR proteins and proteinase inhibitors, with ET acting in synergy with JA signalling, with involvement in PR protein production and enhancement of the SA-mediated NPR1 pathway in SAR (Lorenzo et al., 2003; Leon-Reyes et al., 2009).
Analysis of the plant response to RKN infection can provide insights into the host pathways both manipulated by the parasite and activated or suppressed by the plant during the defence response. In contrast to the considerable data published in recent years for important plant–fungal and plant–bacterial pathosystems, analysis of molecular interactions between plants and nematodes has been relatively limited. Most research to date on host gene expression in response to RKNs has focused on interactions in dicotyledonous host plants (e.g. Jammes et al., 2005; Alkharouf et al., 2006; Ithal et al., 2007a; Klink et al., 2007; Szakasits et al., 2009; Barcala et al., 2010; Bagnaresi et al., 2013; Guimaraes et al., 2015), with relatively few studies on monocotyledonous hosts (Kyndt et al., 2012a; Ji et al., 2013; Nguyên et al., 2014). Analysis of the transcriptome of compatible interactions between nematodes and monocotyledonous hosts has so far been limited to studies only on rice. Kyndt and colleagues (2012a) compared gene expression following susceptible host challenge with the sedentary RKN Meloidogyne graminicola and the migratory root rot nematode Hirschmanniella oryzae, with Ji et al. (2013) characterizing the transcriptome of giant cells induced by M. graminicola. In these studies, plant root necrosis following infection with the migratory nematode H. oryzae resulted in induction of plant immune responses that included the jasmonate biosynthesis pathway, the production of PR proteins and activation of WRKY TFs (Kyndt et al., 2012a). In contrast, gene expression data for galls and giant cells formed as a result of RKN infection revealed suppression of plant immune responses and activation of primary metabolism. Similar suppression of plant defence pathways due to RKNs, involving PR proteins and flavonoid, phenylpropanoid and jasmonate biosynthesis pathways, was also observed by Ji et al. (2013).
In this study, a whole-genome transcriptome analysis of root tissues in two RKN-susceptible M. acuminata genotypes was conducted during early stages of infection with M. incognita. The characterization of both defence responses that may limit RKN parasitism during compatible interactions and the identification of potential effector-targeted host genes in this complex interaction will facilitate the downstream development of novel genetic improvement-based control measures for RKNs.
MATERIALS AND METHODS
Nematode infection
Plantlets of M. acuminata Cavendish sub-group cultivar ‘Grande Naine’ (CAV) (AAA) and a M. acuminata breeding genitor genotype, accession code 4279-06 (4279-06) (AA) were provided by Embrapa Cassava and Tropical Fruits. Following regeneration from tissue culture, in vitro rooting was achieved following 30 d growth in Murashige and Skoog (MS) medium. Plantlets were transplanted into 1·5 L pots containing sterilized substrate composed of a mixture of soil and sand (1:1), fertilizer and lime, then adapted for a period of 45 d under glasshouse conditions at an average temperature of 25 °C prior to inoculation with M. incognita.
Nematode inoculum was provided by Embrapa Genetic Resources and Biotechnology, originating from a population isolated from infected banana root tissues. Stock cultures were maintained on tomato (Solanum lycopersicum ‘Santa Cruz’), under glasshouse conditions (20–30 °C, 12 h light period). Nematode eggs were extracted from tomato roots according to Hussey and Barker (1973). Roots were cut into portions of 1–2 cm, placed in a sodium hypochlorite solution (10 %) and agitated for 2 min. The suspension was then sieved through a series of 45, 140 and 500 mesh, with eggs recovered at the latter size. Retained eggs were washed thoroughly to remove sodium hypochlorite. For the onset of second-stage juveniles (J2), the suspension was placed in a Baerman funnel and incubated at 28 °C for 8 d. Recovered J2 juveniles were quantified and inoculum calibrated to 1000 J2 mL–1 suspension. Musa acuminata CAV and M. acuminata 4279-06 plantlets at the 45 d stage after transplantation were individually inoculated with 5000 M. incognita J2s. For inoculation, nematode suspension aliquots were pipetted uniformly around the pseudostem of each plant. Non-inoculated control plants were treated identically, with sterile distilled water as inoculum. For each genotype, plants were arranged in a randomized block design, with three replicate plants for each evaluation time point. Plants were maintained at an average temperature of 25 °C and watered at 3 d intervals. Root tip samples (1 g) from each of the three replicate plants for each genotype were collected at 3, 7 and 10 days after inoculation (DAI), immediately snap-frozen in liquid nitrogen and stored at − 80 °C until further processing. Bioassays were performed in duplicate.
Histopathological analysis
Inoculated root samples for each genotype were harvested at 3 d intervals over a period of 35 d, using three plants of each genotype per time point. In order to observe RKN penetration, localization and subsequent development within the roots, fine root tips were stained with acid fuchsin, according to Byrd et al. (1983) and Hooper et al. (2005), then observed by stereomicroscopy (Leica MZ75) and bright-field light microscopy (Zeiss Axiophoto). Root segments were also embedded in resin for sectioning (4 μm). Root fragments were excised, fixed, and embedded in Technovit 7100 epoxy resin (Kulzer Friedrichsdorf), as described by Pegard et al. (2005). Sections were subsequently stained (1 min at 60 °C) with 0·05 % toluidine blue in 0·1 m sodium phosphate buffer, pH 5·5 and observed under bright-field light microscopy (Zeiss Axiophoto).
RNA-Seq library construction and Illumina sequencing
Total RNA was extracted from inoculated root tissues and corresponding root tip regions in non-inoculated control plants. A total of 72 plant samples were processed, representing infected and non-inoculated control root tissues for each genotype, at each evaluation time point, and considering duplicate bioassays. Extraction was performed using Concert® Plant RNA Reagent (Invitrogen, Carlsbad, CA, USA) and purification with the INVISORB Spin Plant RNA Mini Kit (Invitek, Hayward, CA, USA), according to the manufacturer’s instructions. RNA concentration and integrity were assessed using the Agilent 2100 Bioanalyzer and RNA LabChip® kit (Agilent Technologies, Santa Clara, CA, USA). Construction of cDNA libraries and Illumina RNA-Seq sequencing were conducted by Eurofins (Eurofins MWG Operon, Huntsville, AL, USA). Paired-end (2 × 125 bases) cDNA libraries were prepared from 5 μg of total RNA of each plant root sample (comprising a pool of RNA from all three replicate plants) using a KAPA stranded mRNA-Seq kit (Kapa Biosystems, Wilmington, MA, USA). All multiplexed libraries from the duplicate bioassays were sequenced on two flow cell channels of an Illumina HiSeq 2500 sequencing system (Illumina, San Diego, CA, USA), to provide technical replicates too.
Bioinformatics
Mapping of reads to reference M. acuminata genome sequence
High quality sequence reads (Fastq QC > 20) were identified and selected using the program Trimmomatic (Bolger et al., 2014). Alignment was conducted to gene regions of the reference genome sequence for M. acuminata DH-Pahang, publicly available on the Banana Genome HUB platform (http://banana-genome-hub.southgreen.fr/download). In order to enable analysis of individual data sets, alignment was conducted with sequences from each cDNA library in batch mode, using the program Novoalign/Useq (http://www.novocraft.com/documentation/novoalign-2/novoalign-user-guide/rnaseq-analysis-mrna-and-the-spliceosome/).
Normalization and gene expression profiling.
In silico analysis of statistically significant differential gene expression between the evaluated treatments for aligned sequences was conducted using the Python script HTseq-count, which computes read counts mapped per gene (Anders et al., 2015). This was followed by analysis with EdgeR (Robinson et al., 2010) for identification of statistically significant differentially expressed genes (DEGs). EdgeR is employed to model data as a negative binomial distribution, allowing normalization of biological and technical variation using a generalization of the Poisson distribution model. Raw counts for each gene were normalized in relation to different sequence depths between duplicate bioassay samples.
Genes were considered differentially expressed in the infected libraries when compared with the respective non-inoculated controls, for each evaluated time point, following employment of the Benjamini–Hochberg algorithm for estimation of fold change (FC). DEGs were considered significant when relative gene expression between inoculated and non-inoculated control treatments displayed at least a 2-fold FC (log2FC threshold >1·0 or < –1·0), considering a false discovery rate (FDR)-adjusted P-value (padj) of 0·1. The terms upregulation and downregulation were employed throughout the text to indicate, respectively, transcript levels that were higher or lower than observed in non-inoculated controls.
Gene Ontology and analysis of enrichment.
A hypergeometric test within the program FUNC (Prüfer et al., 2007) was employed to analyse under- and over-representation of DEGs on the basis of gene function classification within Gene Ontology (GO) categories, with redundant category terms eliminated using REVIGO (http://revigo.irb.hr/).
Pathway analysis.
Gene expression data were analysed in terms of metabolic functionalities using the Mercator pipeline (Lohse et al., 2014) to ascribe potential gene function and MapMan BINs (Usadel et al., 2005). Fisher’s exact test, adjusted with Bonferroni correction for multiple testing, was employed for identification of MapMan BIN categories that were considered significantly enriched (adjusted P-value <0·05).
Quantitative real-time PCR validation of RNA-Seq-derived gene expression data
Validation of expression for a total of 13 selected DEGs was conducted via quantitative real-time PCR (qRT-PCR) analysis, using a Platinum SYBR Green qPCR Super Mix-UDG w/ROX kit (Invitrogen) and amplification on an ABI StepOne® Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). RNA material was selected from samples that were analysed via RNA-Seq, with three independent biological replicates analysed for each genotype and time point and three technical replicates per amplification. A total of 3 μg of each total RNA pool was treated with 2 U of Amplification Grade DNase I (Invitrogen) to digest residual DNA. RNA was reverse transcribed to cDNA using Super Script IV reverse transcriptase and oligo(dT)20 primers (Invitrogen). Specific primers (Supplementary Data Table S2) were designed using Primer Express software (Applied Biosystems), with expected amplicon sizes of approx. 100 bp. Primers were initially tested for specificity and efficiency against a mix of cDNAs originating from roots of M. acuminata CAV and 4279-06 (data not shown). Primer sequences that resulted in specific amplification are listed in Supplementary Data Table S1. The PCR mixtures contained 1 μL of template cDNA, primers (175 nm) and Platinum® SYBR® Green qPCR Super Mix-UDG w/ROX kit (Invitrogen). Thermal cycling was conducted using 40 cycles of denaturation at 95 °C for 15 s, primer annealing and extension at 60 °C for 30 s. Data were analysed using SDS 2.2.2 software (Applied Biosystems) to determine cycle threshold (Ct) values. The specificity of the PCR products generated for each set of primers was verified by analysing the Tm (dissociation) of amplified products. Gene expression levels were normalized against the M. acuminata DH Pahang v1 reference genes GSMUA_Achr1G20200_001 and GSMUA_Achr10g22980 (Table S1). For each genotype, expression profiles of mRNA transcripts at 3, 7 and 10 DAI were obtained as the ratio ΔRn (Treated)/ΔRn (Control) using the program SDS 2.2.2.
RESULTS
Histopathology
Observation of acid fuchsin- and toluidine blue-stained root sections confirmed a similar life cycle of M. incognita in both susceptible Musa genotypes in terms of penetration and nematode development in the plant, completing in 24 d in CAV and 27 d in 4279-06 (Fig. 1). At 3 DAI, J2s had penetrated root tips in both genotypes, migrating along the elongation zone. At 6 DAI, J2s were present in the root cortex of each genotype, migrating in parenchyma tissues towards the vascular cylinder. Between 9 and 12 DAI, J2 had established feeding sites of giant cells in the vascular cylinder, with J3 present in CAV and J2a in 4279-06. In the case of both genotypes, J3 and J4 were observed at 18 DAI, with J4 individuals associated with giant cells in the central cylinder appearing at 21 DAI. Multinucleation and enlargement of giant cells continued to develop in both genotypes to 21 DAI, with no evidence of cell or nematode degeneration. By 24 DAI, adult stage egg-laying females had developed in CAV, with gelatinous egg masses visible at this time point on root surface tissues. Egg mass presence was observed in 4279-06 at 27 DAI. Enlarged and damaged vascular cylinders were apparent in both genotypes at 24 DAI, with parenchyma, and xylem and phloem vessels transformed into active feeding sites harbouring numerous intact giant cells (Fig. 2).
Fig. 1.

Histopathological examination of root sections of Musa acuminata genotypes CAV (left) and 4279-06 (right) infected with Meloidogyne incognita. Root sections (10 μm) were observed under brightfield microscopy following staining with acid fuchsin. (A, B) Second-stage juveniles (J2) migrating inside the root tip. (C) Adult females with egg masses released in the cortical zone. (D) Adult female attached to the central cylinder. (E, F) Adult females with egg masses released in the cortical zone. DAI, days after inoculation; cc, central cylinder; co, cortex; em, egg mass; e, egg; F, female; gc, giant cell.
Fig. 2.

Histopathological examination of root sections of Musa acuminata genotypes CAV (left) and 4279-06 (right) infected with Meloidogyne incognita. Root sections (10 μm) were observed under brightfield microscopy following staining with toluidine blue. (A, B) Adult females surrounding giant cells which completely occupy the central cylinder. (C, D) Non-inoculated control root sections. DAI, days after inoculation; cc, central cylinder; co, cortex; f, female; gc, giant cell; N, nematode; vc, vacuole.
Sequence statistics
For each Musa genotype, root samples from plants inoculated with M. incognita and from non-inoculated controls were collected at 3, 7 and 10 DAI, to enable host gene expression analysis during early phases of the interaction. Illumina Hi-seq 2500 RNA-Seq generated 290 308 118 raw paired-end sequence reads from cDNA libraries prepared in bioassay 1 and 220 629 858 from libraries prepared in bioassay 2. Sequence reads averaged 150 bp in length. A total of 195 053 336 high-quality reads were produced from the 12 multiplexed cDNA libraries for CAV and 163 798 990 for an equal number of libraries for 4279-06. Reads from CAV that mapped to the M. acuminata genome sequence (http://banana-genome.cirad.fr/) totalled >28 billion bases, representing a 54·69-fold coverage of the transcriptome. Similar numbers were observed with data for 4279-06, with 24 billion bases mapped, representing a 45·92-fold coverage of the M. acuminata transcriptome. As such, sequence depth and coverage were sufficient for analysis of differential gene expression for gene sequences from each cDNA library. The Pearson correlation coefficient was employed to determine the correlation between data obtained from the two biological replicate bioassays, with an average Pearson R value of 0·80. A summary of sequence statistics and read mapping to the reference M. acuminata genome sequence is shown in Table 1. Raw sequence data from this study were deposited at the NCBI Sequence Read Archive (SRA) database (BioProject ID PRJNA349155). Sequence data generated from the cDNA libraries for each time point and for both Musa genotypes contained reads aligning to the total 37 604 M. acuminata ssp. malaccensis var. Pahang (DH Pahang) WGS gene models (Table S2), with mapping to > 22 000 gene models in the case of CAV data, and to > 24 000 in the case of 4279-06.
Table 1.
Overview of Illumina Hiseq 2500 sequence data
| Musa acuminata genotype | cDNA library | High quality reads (Q30)* | Reads mapped to Musa acuminata reference genome (%) | |
|---|---|---|---|---|
| Bioassay 1 | ||||
| CAV | Uninfected roots 3 DAI | 16 769 217 | 97·55% | |
| Uninfected roots 7 DAI | 23 209 664 | 98·03% | ||
| Uninfected roots 10 DAI | 8 696 880 | 97·57% | ||
| M. incognita-infected roots 3 DAI | 20 073 517 | 97·57% | ||
| M. incognita-infected roots 7 DAI | 14 708 226 | 97·75% | ||
| M. incognita-infected roots 10 DAI | 17 827 098 | 97·87% | ||
| 4279-06 | Uninfected roots 3 DAI | 26 583 657 | 97·30% | |
| Uninfected roots 7 DAI | 20 175 582 | 97·67% | ||
| Uninfected roots 10 DAI | 10 800 357 | 97·74% | ||
| M. incognita-infected roots 3 DAI | 15 023 526 | 97·84% | ||
| M. incognita-infected roots 7 DAI | 15 475 299 | 97·94% | ||
| M. incognita-infected roots 10 DAI | 14 907 813 | 98·03% | ||
| Total | 204 250 836 | |||
| Bioassay 2 | ||||
| CAV | Uninfected roots 3 DAI | 26 551 739 | 97·78% | |
| Uninfected roots 7 DAI | 14 064 646 | 97·72% | ||
| Uninfected roots 10 DAI | 12 970 992 | 97·45% | ||
| M. incognita-infected roots 3 DAI | 16 667 993 | 97·74% | ||
| M. incognita-infected roots 7 DAI | 12 960 975 | 98·05% | ||
| M. incognita-infected roots 10 DAI | 10 552 389 | 97·79% | ||
| 4279-06 | Uninfected roots 3 DAI | 9 835 864 | 98·00% | |
| Uninfected roots 7 DAI | 11 447 367 | 97·89% | ||
| Uninfected roots 10 DAI | 8 376 730 | 97·86% | ||
| M. incognita-infected roots 3 DAI | 5 025 640 | 97·79% | ||
| M. incognita-infected roots 7 DAI | 13 795 865 | 97·76% | ||
| M. incognita-infected roots 10 DAI | 12 351 290 | 97·70% | ||
| Total | 154 601 490 | |||
(Q30) = 1 in 103 base calling errors.
Transcriptome modulation
Analysis of fold change in gene expression was conducted using HTseq-count and EdgeR on mapped read counts originating from all inoculated and respective non-inoculated control samples, at each investigated time point during the M. acuminata–M. incognita interaction. Statistically significant DEGs were identified among inoculated samples, when compared with respective non-inoculated controls, across the three investigated time points. A total of 680 DEGs were identified across the sampling points of 3, 7 and 10 DAI in the two susceptible genotypes, with 479 of these up- or downregulated genes observed in 4279-06 and 238 in CAV (Table 2; Table S2). When comparing DEG numbers between the two genotypes, greater numbers of DEGs were observed at 3 DAI in 4279-06, in contrast to at 7 DAI for CAV. A total of 29 DEGs were common to both genotypes (Fig. 3; Table S2) Of these, 11 were commonly upregulated and six commonly downregulated between the two genotypes over the evaluated time course.
Table 2.
Summary of differentially expressed genes (DEGs) identified in Musa acuminata genotypes CAV and 4279-06, based on comparison of read counts per gene between Meloidogyne incognita-infected root samples and non-inoculated controls
| M. acuminata genotype | Time point after inoculation | Total number of DEGS | No. of up-regulated DEGs | No. of down-regulated DEGs |
|---|---|---|---|---|
| CAV | 3 DAI | 54 | 25 | 29 |
| 7 DAI | 170 | 143 | 27 | |
| 10 DAI | 14 | 1 | 13 | |
| 4279-06 | 3 DAI | 394 | 217 | 177 |
| 7 DAI | 59 | 31 | 28 | |
| 10 DAI | 26 | 25 | 1 |
Fig. 3.
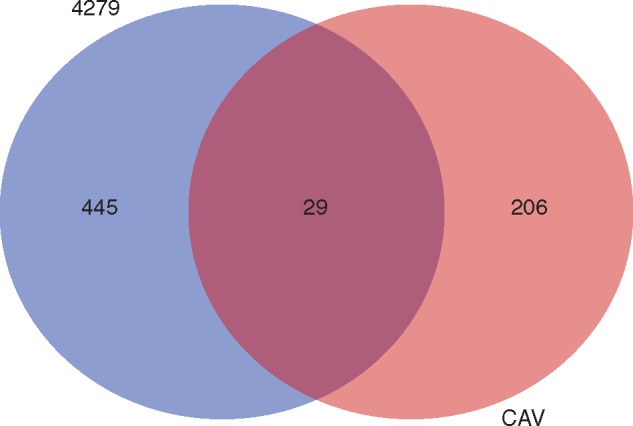
Global Venn diagram summarizing numbers of distinct differentially expressed genes (DEGs) observed in Musa acuminata genotypes CAV and 4279-06 following infection by Meloidogyne incognita over a 10 d period. DEGs were considered significant when relative gene expression between inoculated and non-inoculated treatments showed at least a 2-fold FC (log2FC threshold >1·0 or < –1·0), considering a false discovery rate (FDR)-adjusted P-value (padj) of 0·01. The overlapping region of the diagram represents DEGs common to both Musa genotypes during the interaction with M. incognita. Annotation and expression data for all DEGs are listed in Table S1.
GO enrichment analysis
The relative abundance of DEGs was analysed according to GO classifications (Supplementary Data Figs S1–S3). Analysis for CAV at 3 DAI showed upregulated genes classified in biological process sub-categories related to recognition and signalling pathways. At this time point, cellular component terms cell wall, extracellular region and external encapsulating structure were also represented. At 7 DAI, enrichment was considerable for terms involved in biological process sub-categories related to response to stress or stimulus, defence responses, phytohormone signalling [ET, abscisic acid (ABA), JA and SA] and signal transduction. Enriched terms related to molecular function were also more apparent than at 3 DAI, related to TF binding activity, nuclease activity, transferase activity, receptor protein kinase binding and oxidoreductase activity. Cell component terms were enriched for cell wall/apoplast and extracellular components. By 10 DAI, enrichment was again less pronounced, with biological process terms relating to cell processes and cell wall, molecular function terms relating to protein binding and transferase activity, and cell components relating to cell wall or extracellular region.
In contrast to CAV, considerable gene set enrichment was observed in 4279-06 at 3 DAI, with enrichment for biological process terms related to response to stress or stimulus, signalling, metabolic processes, cell growth, organization, development and cycle, apoptosis and gene silencing. Molecular functions assigned to DEGs were related to hydroxylase, transferase, oxidoreductase, peroxidase, transaminase, and 1,3-β-d-glucan synthase activities. Cell component terms enriched comprised cell wall, and extracellular and nuclear regions. At 7 DAI, fewer terms were observed in DEGs in comparison with CAV, with biological process categories enriched in relation to stress or stimulus, defence responses, phytohormone signalling and signal transduction. Enriched molecular function terms related to TF binding and nuclease activity, and cell component terms for intracellular organelles and the CCR4–NOT complex. At 10 DAI, as with CAV, fewer terms were enriched amongst the DEGs. Biological process terms enriched were related to cell wall, vascular histogenesis, oxidation–reduction processes, response to oxygen, SA signalling and SAR. Molecular function terms comprised only dioxygenase and oxidoreductase activities, with cell components enriched for respiratory chain, mitochondria and cytoplasm.
MapMan ontology
Analysis of DEGs using MapMan provided a pictorial representation of gene expression across selected metabolic pathways (Fig. 4). Genes were identified with homology to Oryza sativa genome probes and allocated to MapMan bins. Significant upregulation of genes involved in signalling, biotic signalling, cell wall, protein degradation and secondary metabolism were observed initially at 3 DAI in 4279-06. These were followed by genes related to signalling, TFs and ET upregulated at 7 DAI. In contrast to earlier time points, at 10 DAI genes related to signalling, biotic signalling, cell wall, protein degradation, secondary metabolism and TFs were generally downregulated.
Fig. 4.

MapMan-derived visualization of expression profiles of differentially expressed genes (DEGs). Differential expression patterns are based on log2FCs of mRNA transcripts from inoculated host cells vs. non-inoculated controls. Each dot represents the presence of a paralogue gene that encodes a particular enzyme within a metabolic pathway. A blue colour indicates an upregulated gene in infected tissue vs. the non-inoculated control, while a red colour indicates a downregulated gene.
For CAV, most apparent upregulated genes at 3 DAI were related to signalling and TFs. At 7 DAI, in contrast, the greatest upregulation was observed relating to signalling, cell wall, protein degradation, secondary metabolism, TFs and ET At 10 DAI, as in the case of 4279-06, most represented pathways appeared downregulated, with the exception of biotic stress signalling, appearing upregulated only at this later stage.
Global representation of differential gene expression
Representation of differential gene expression for each genotype over the time course of the interaction with M. incognita, based on expression levels (log2FC) of transcripts in relation to non-inoculated controls, is presented graphically as volcano scatter plots in Fig. 5 and as heat maps with hierarchical cluster analysis of expression patterns over time in Supplementary Data Figs S4 and S5. Expression varied considerably over the evaluated time period of the interaction in each genotype, with six heat map cluster groups for DEGs observed in CAV and eight in 4279-06. Different gene expression modulation patterns were also apparent between the two susceptible genotypes, probably reflecting their different genetic backgrounds. All DEGs relative to controls that were identified across the sampling points of 3, 7 and 10 DAI with M. incognita are listed for each of the two susceptible M. acuminata genotypes in Table S2.
Fig. 5.

Volcano scatter plots for differentially expressed genes (DEGs) in Musa acuminata genotypes CAV and 4279-06 following infection by Meloidogyne incognita over a 10 d period. Genes that were highly modulated following infection, in relation to non-inoculated controls, appear farther to the left and right sides, with highly significant changes appearing higher on the plot. Significant DEGs are represented in blue, with up-regulated genes following infection represented on the right-hand side of each plot and downregulated genes on the left-hand side. Non-significant DEGs are represented in red.
Transcriptome changes in CAV over the time course
Data summarizing transcriptome changes in CAV are presented in Table 2, Fig. 5 and Table S2. At 3 DAI, gene expression was relatively unchanged for the majority of genes during early interaction with M. incognita, with only 25 genes significantly upregulated and 29 downregulated, relative to non-inoculated controls. At 7 DAI, in contrast, 143 genes appeared upregulated, with a total of 27 downregulated. At 10 DAI, expression patterns were similar to those observed at 3 DAI, with low numbers of genes modulated in response to M. incognita. Only one gene was significantly upregulated, with a total of 13 genes downregulated at this time point.
Among the most up-regulated transcripts at 3 DAI (Table S2), genes potentially involved in defence responses were identified, coding for an LRR receptor-like serine/threonine-protein kinase and for peroxidase enzymes, involved in response to auxin catabolism and biotic and oxidative stresses. Additional upregulated transcripts included those coding for a proline-rich DC2.15 protein, typically involved in metabolic changes due to auxin depletion; for a cyclin-U2-1 protein, involved in the cell cycle and cell division; and an expansin-A15 protein, potentially involved in loosening plant cell wall cellulose microfibrils and matrix glucans. Downregulated transcripts at 3 DAI included a number potentially involved in defence responses such as those coding for an LRR receptor-like serine/threonine-protein kinase, a xylanase inhibitor protein, a wound-induced protein WIN1 and a thaumatin-like PR protein. Further downregulated transcripts included a homeobox protein knotted-1-like 2 protein, involved in cytokinin-activated signalling and meristematic state cell maintenance, and a probable xyloglucan endotransglucosylase/hydrolase protein, potentially involved in cell wall organization.
At 7 DAI, among the transcripts most upregulated, numerous genes associated with stress responses were observed (Table S2). These included DREB1D, DREB1E and DREB1F TFs, associated with gibberellin biosynthesis and dehydration-inducible transcription; a TIFY 5A protein, involved in repression of JA-mediated signaling; ET-responsive TFs ERF024 and ERF112, which are activators in ET defence signalling; an ERF4 transcriptional repressor, potentially involved in negative regulation of gene expression in response to stress, ET- and JA-mediated signalling; an E3 ubiquitin-protein ligase PUB22, associated with negative regulation of water stress responses; lipoxygenase (LOX) genes, potentially involved in JA biosynthesis; a gibberellin-responsive protein 1 CIGR1; a Myb4 transcription repressor, potentially involved in phenylpropanoid metabolism regulation and JA/ET signalling; a Zinc finger A20 and AN1 domain-containing stress-associated protein 1 SAP1, associated with environmental stress response; and a WRKY41 TF. Numerous transcripts were also observed that code for GDSL esterase/lipases, potentially involved in response to abiotic stress, pathogen defence, seed development and lipid metabolism. Strongly downregulated genes at 7 DAI included a number associated with defence response, such as those coding for an l-ascorbate peroxidase 4; an LRR receptor-like serine/threonine-protein kinase; and an 1-aminocyclopropane-1-carboxylate oxidase (ACO), involved in ET biosynthesis. Other downregulated transcripts included those coding for an expansin-B15 protein, involved in plant cell wall loosening; an endoglucanase 8, involved in cellulose catabolism; and two indole-3-acetic acid-amido synthetase GH3.8 genes, associated with auxin homeostasis.
At 10 DAI, only a single upregulated gene was identified amongst the DEGs (Table S2), coding for a metallothionein-like protein 4A, which is typically involved in oxidative stress responses in plants. Significant downregulated DEGs involved in defence included those coding for caffeic acid 3-O-methyltransferase (COMT) proteins, involved in phenylpropanoid biosynthesis; a thaumatin-like PR protein; and a (+)-delta-cadinene synthase isozyme A, involved in biosynthesis of terpenoid secondary metabolites. Other downregulated genes observed included those coding for a NAC098 protein TF, associated with plant development and inhibition of cell division; and expansin proteins B18 and A15, which, like other expansins observed, may play roles in plant cell wall modification.
Transcriptome changes in 4279-06 over the time course
Data for transcriptome changes in 4279-06 are also summarized in Table 2, Fig. 5 and Table S2. In the case of this genotype, similar numbers of genes were up- and downregulated during early phases of the interaction with M. incognita, relative to non-inoculated controls, with considerable numbers of genes modulated at 3 DAI, in contrast to CAV. Although the same modulation trend was observed at 7 DAI, low numbers of genes were differentially expressed at this time point. Analysis of gene expression at 10 DAI revealed low numbers of DEGs, almost exclusively upregulated.
In addition to numerous hypothetical proteins, amongst the most upregulated genes at 3 DAI (Table S2) were those coding for proteins potentially involved in defence responses, including a subtilisin-like protease, potentially involved in programmed cell death; a putative LRR receptor-like serine/threonine-protein kinase; a NAC domain-TF protein, associated with stress response or phytohormone regulation; a MYB family TF; callose synthases; and a mannose-binding plant lectin. Upregulated genes also included those coding for a pectate lyase and putative expansins, potentially indicative of cell modification involved in giant cell development in infected root tissues. Genes most downregulated at 3 DAI comprised a number potentially involved in defence responses, including those coding for a putative LRR receptor-like serine/threonine-protein kinase; a PER7 peroxidase; a gibberellin-regulated protein GASA4, associated with cell division and downregulated by ABA, SA and JA; a bHLH135 TF involved in brassinosteroid (BR) and gibberellin signalling; snakin-1 and -2 antimicrobial proteins; WRKY TFs; and a GDSL esterase/lipase. Repressed genes probably involved in cell division and cell wall modification associated with feeding site development comprised a serine/threonine-protein kinase Haspin, involved in chromosome cohesion during mitosis; a transcriptional regulator gene SUP, involved in repression expression of genes with a role in cell division; a xyloglucan endotransglucosylase/hydrolase protein XTH28; a polygalacturonase gene; and a putative pectinmethylesterase PME1 gene.
Similar findings were observed for the most up-regulated genes at 7 DAI (Table S2), with gene expression again appearing to reflect both host defence responses and cell modification during disease development. Upregulated genes potentially involved in stress responses included DREB1D and DREB1E TFs; a SAP1 protein; ET-responsive TFs ERF112, ERF105, ERF5 and ERF4; an ll-diaminopimelate aminotransferase, chloroplastic (DAP), associated with systemic acquired resistance via SA-mediated signalling; a peroxidase 4; and a pathogenesis-related protein 1 (PR1). A phytosulphokines 3 (PSK3) gene, typically involved in promotion of cell differentiation was also significantly upregulated. Significantly downregulated genes at this time point included a number potentially involved in defence responses, comprising a MYB family TF; an LRR protein kinase family protein; a putative protein TIME FOR COFFEE TIC, involved in JA signalling; an AP2/ERF and B3 domain-containing TF RAV1, involved in ET signalling; and a BBTI8 Bowman–Birk type bran trypsin inhibitor precursor. Downregulation of this protease inhibitor may be indicative of defence response, given that proteolysis is known to be involved in the plant immune response. Downregulated genes potentially involved in cell modification comprised a proline-rich protein DC2.15, typically associated with metabolic changes following auxin removal; together with two GH3.8 genes.
Differentially exporessed genes identified at 10 DAI were almost exclusively upregulated (Table S2). Highly expressed genes included a number potentially involved in defence responses, including an ACO2 gene encoding an ACO2, involved in ET biosynthesis; an ERF071 ethylene-responsive TF activator; and a wound-induced protein. Additional upregulated DEGs included those coding for two genes associated with chloroplasts, namely an ATJ20 gene, coding for a putative chloroplastic chaperone protein dnaJ 20; together with a CPX gene, which codes for a chloroplastic coproporphyrinogen-III oxidase. Upregulated genes potentially associated with feeding site development comprised a nicotianamine synthase 3, involved in iron sensing and transport; and an EXPB15 gene coding for a B15 expansin. A GA2OX1 gene encoding a gibberellin 2-β-dioxygenase 1, involved in gibberellin biosynthesis and homeostasis, was also amongst highly upregulated genes. A single highly downregulated gene was identified among the significant DEGs, coding for a subtilisin-like protease ARA12 enzyme. Subtilases are abundant serine proteases in plants, with functions in cell development, activation of cell wall loosening enzymes, and defence and stress responses associated with programmed cell death (Vartapetian et al., 2011; Figueiredo et al., 2014).
Comparison of gene expression between M. acuminata genotypes
Of the 29 DEGs observed in both genotypes across the investigated time points (Fig. 3; Table S2), 11 were commonly upregulated. These included genes coding for proteins potentially involved in defence responses, comprising a stress-response A/B barrel domain-containing protein HS1; a Prx protein, associated with peroxidise activity and cell redox homeostasis; a CCR4-associated factor 1 homologue 11, involved in PR protein expression; and ET-responsive TFs involved in ET-activated signalling, namely the activators ERF5 and ERF112 and repressor ERF4. A number of common upregulated genes were also observed where function was less clear, either because of potential roles in cell defence or giant cell formation, or unknown function. These comprised DREB1D and 1E TFs, associated with dehydration-inducible transcription, wound responses and gibberellin biosynthesis; a HARBI1 protein nuclease; and a number of uncharacterized proteins.
Six genes were commonly downregulated in the two genotypes over the evaluated time course. In addition to proteins with unknown function, these comprised genes potentially related to giant cell development and activity, namely a At3g43660 pcl1 iron transporter; a C24B11·05 SPAC24B11·05 hydrolase; a high affinity sulphate transporter 2 ST2; and a probable indole-3-acetic acid-amido synthetase GH3.8, involved in indole-3-acetic acid (IAA) synthesis and auxin homeostasis.
Real-time PCR validation of gene expression
A total of 13 genes were analysed for expression profile by qRT-PCR, to enable comparison with Illumina RNA-Seq-derived host gene expression data. Although the selected genes were not necessarily significantly differentially expressed in silico, in relation to non-inoculated controls, the analysis across all three time points for each Musa genotype showed that expression pattern tendencies to up- or downregulation for each genotype, and at each evaluated time point, were in broad agreement between RNA-Seq and qRT-PCR data for ten of the analysed genes (Fig. 6). Differences in log2FC values may occur due to the different algorithms applied for estimation of fold change as well as due to variation in biological replicates.
Fig. 6.

Validation of differential expression profiles (log2FC) based on RNA-Seq and qRT-PCR for selected genes in Musa acuminata CAV and 4279-06 in infected vascular root tissues vs. non-infected controls. qRT-PCR data are presented on the left y-axis, with RNA-Seq data on the right y-axis. Bars indicating standard error values are based upon three independent biological replicates per genotype and time point and three technical replicates per amplification.
DISCUSSION
Although a number of studies have been conducted to examine resistance or tolerance of Musa genotypes to Meloidogyne spp. (e.g. Stoffelen et al., 2000; Van den Bergh et al., 2002a, b; Quénéhervé et al., 2009), effective sources of resistance remain lacking in breeding programmes. Complementing recent characterization of gene expression during a Musa–nematode incompatible interaction, namely with the migratory endoparasitic nematode, Pratylenchus coffeae (Backiyarani et al., 2014), this data set for the M. acuminata–M. incognita compatible interaction also represents an important addition to existing transcriptome resources for M. acuminata–pathogen interactions (Li et al., 2012; Bai et al., 2013; Passos et al., 2013). Here, we investigated gene expression modulation at early stages of the compatible interaction between the biotrophic RKN M. incognita and two M. acuminata genotypes. CAV was included as a susceptible reference (Van den Bergh et al., 2002a), with 4279-06, a breeding genitor diploid hybrid that, whilst serving as a source of resistance to the fungal pathogen Pseudocercospora musae (Silva et al., 2001), also displays susceptibility to RKNs, despite this different genetic background. While there has been considerable focus on elucidation of host mechanisms activated during incompatible plant–nematode interactions, our understanding of how RKN modulates host genes during infection and giant cell development, as well as defence mechanisms active during the compatible interaction, remains limited, especially in monocotyledonous plants.
Modulation of plant immune responses in compatible interactions with RKNs
In the case of RKNs, compatible interactions and feeding site development in plants, nematodes appear to modulate both host development and immune responses. Recent transcriptome analysis of gall tissues induced by the biotrophic RKN M. graminicola has revealed suppression of ETI, SA and ET/JA defence pathways, together with activation of gibberellic acid and BR biosynthesis and signalling (Kyndt et al., 2012a; Ji et al., 2013; Nahar et al., 2013; Nguyên et al., 2014). The phytohormone ABA, although more typically associated with abiotic stress responses, together with auxins, BRs, cytokinins and gibberellins, which are known regulators of plant developmental, also all appear to play overlapping roles in plant immune responses to RKNs (Kyndt et al., 2014). Within giant cells, repression of genes involved in SA and JA biosynthesis has been confirmed (Ji et al., 2013). Secondary metabolism also appears to be altered during compatible responses to RKNs in monocots and dicots with examples of repression of flavonoid and phenylpropanoid biosynthetic pathway genes, together with genes involved in biosynthesis of pathogenesis-related proteins (Jammes et al., 2005; Barcala et al., 2010; Kyndt et al., 2012a; Ji et al., 2013).
In our study, evidence of potential plant immune responses to infection existed amongst the gene enrichment, pathway mapping and DEG data sets. In the case of 4279-06, DEG data for transcripts at early time points provide evidence for upregulation of genes encoding LRR receptor-like serine/threonine-protein kinases, peroxidases, programmed cell death-related proteins, NAC and MYB TF proteins, and callose synthases. At later time points, these included peroxidases, stress-associated proteins, ET biosynthesis and ERF TF proteins, PR proteins and wound-induced proteins. In the case of CAV, although fewer DEGs appeared related to defence responses than in 4279-06, similar trends were apparent, with genes encoding peroxidases up-regulated at early time points, followed later by ERF TF protein activators of ET-activated defence signalling. DEG data also provided evidence for repression of immune responses during RKN–Musa interaction. For 4279-06, these included downregulated transcripts at early time points for LRR receptor-like serine/threonine-protein kinases, peroxidases, WRKY TFs and snakins, later extending to MYB TF proteins, JA and ET signalling-related proteins and TFs, and programmed cell death-related proteins. In the case of CAV, similar groups of downregulated genes were observed, including early downregulation of LRR receptor-like serine/threonine-protein kinases and thaumatin-like PR proteins, and later downregulation of genes involved in JA and ET signalling, as well as in phenylpropanoid and terpenoid biosynthesis.
Phytohormone-related genes
The role of ET appears complex in both incompatible and compatible plant–nematode interactions. Although RKN infection has been associated with suppression of ET responses (Barcala et al., 2010; Nahar et al., 2011; Kyndt et al, 2012b), patterns of repression or activation have also been observed in both incompatible and compatible interactions between rice and M. graminicola (Petitot et al., 2017). Our data revealed similar patterns, with numerous ET signalling-related proteins and ERF activators up- or downregulated amongst the DEGs. A number of such plant ERFs are also involved in regulation of JA-mediated resistance (McGrath et al., 2005). The common upregulation of the ET-responsive TF repressor ERF4 at 7 DAI in both genotypes may indicate participation in negative regulation of biotic stress responses during nematode infection.
As gibberellins are involved in cell division and elongation (Richards et al., 2001), their biosynthesis may favour giant cell and feeding site development, as evidenced in tomato galls (Klink et al., 2007) and arabidopsis (Jammes et al., 2005). Constant expression of genes involved in their biosynthesis has been reported in giant cells and galls (Kyndt et al., 2012b). In our study, a number of genes associated with gibberellin biosynthesis and signalling were modulated, with significant downregulation at 3 DAI in 4269-06, and later upregulation in both genotypes. While these hormones have also been shown to interact negatively with JA signalling in arabidopsis (Navarro et al., 2008), thus increasing disease susceptibility, a report of increased induction of gibberellin pathways during incompatible soybean–RKN interaction shows that the role of this hormone in defence is still unresolved (Beneventi et al., 2013).
In addition to control of plant growth and development (Woodward and Bartel, 2005), auxin signalling occurs during compatible interactions with RKNs, with increased transport and hormone levels reported in feeding sites (Doyle et al., 2003; Grunewald et al., 2009). IAA, which is the main auxin in plants, is known to induce expression of expansin proteins, which although typically involved in cell wall loosening during plant growth, can also render a plant susceptible to biotic stress (Ding et al., 2008). In our study, downregulation of the indole-3-acetic acid-amido synthetase GH3.8 genes was observed in DEGs for both genotypes, at 3 and 7 DAI in 4279-06, and at 7 DAI in CAV. As this gene is involved in auxin homeostasis through preventing free IAA accumulation, downregulation is associated with increased auxin concentration. As activation of this gene can suppress expansin expression and activate basal immunity in rice (Ding et al., 2008), the frequent downregulation in our data sets may indicate a potential role in RKN establishment through allowing increased concentration of auxins and expansins, together with potential inhibition of basal immune responses.
Cytokinins are involved in the control of root development, with a reciprocal relationship to auxins, inhibiting root meristem enlargement and lateral root development (Chapman and Estelle, 2009). In observations in rice and Lotus, both suppression and activation of cytokinin pathway genes have been noted in root gall tissues (Lohar et al., 2004). In our study, mapman ontology of DEGs indicated early upregulation of genes relating to auxin metabolism and downregulation of genes relating to cytokinins in both CAV and 4279-06. Such contrasting induction and suppression of these phytohormones has been hypothesized to be associated with the induction of lateral root growth. This commonly occurs during RKN development in root tissues (Kyndt et al., 2012b) and was observed in both evaluated genotypes in this study.
Genes involved in cell metabolism and development
As RKN is a sendentary endoparasite, feeding sites in infected root tissues are the exclusive nutrient source for these pathogens. As such, considerable host gene expression modulation in relation to the cell cycle, primary cellular metabolism, protein synthesis and transport is expected, with movement of nutrients across plasma membranes from the phloem to giant cells. Gene expression data for giant cells and galls in both monocotyledonous and dicotyledonous plants has confirmed such activities (e.g. Jammes et al., 2005; Kyndt et al., 2012a, 2014; Ji et al., 2013). In our study, while DEGs did not indicate increased metabolic activity, which probably reflects the fact that giant cells were under development during the time course evaluated via RNA-Seq, regulation of genes associated with cell development was observed amongst the DEGs. These included, for example, upregulation of genes involved in cell division, such as a cyclin-U2-1 protein in CAV and a phytosulphokines 3 gene in 4279-06. Downregulation of a transcriptional regulator gene SUP was also observed in 4279-06, which typically represses gene expression involved in cell division.
Cell wall-related genes
Root-knot nematodes are known to secrete enzymes into infected plant root tissues that are involved in cell wall degradation or modification during giant cell formation (Haegeman et al., 2012). Similarly, plant genes encoding hydrolases such as endoglucanases, pectinases and expansins are also commonly upregulated following nematode infection, facilitating the establishment of feeding sites (Gheysen and Mitchum, 2009). Within our data sets, MapMan pathway analysis generally indicated an upregulation of genes associated with the cell wall at early time points of the interaction in both genotypes, diminishing over time. Amongst the individual DEGs, similar trends were apparent, with upregulated genes for expansins and pectate lyases observed at early time points, as well as downregulation throughout of expansins, pectin methylesterases, endoglucanases and xyloglucan endotransglucosylase hydrolases. The latter family of enzymes, while documented to be upregulated in giant cells and syncytia, has also been shown to be downregulated in surrounding root tissues in galls (Ithal et al., 2007a, b; Barcala et al., 2010; Kyndt et al., 2012b). Such an observation may be relevant to our findings, given that analysis of gene expression was based on infected root tissues rather than individual giant cells.
Conclusions and future directions
In conclusion, this first study into the dynamics of gene expression in root tissues in two M. acuminata genotypes during early stages of compatible interactions with M. incognita reveals, among DEGs, both host genes associated with defence response to infection and those modulated during giant cell formation. The results provide evidence for early host defence responses that involve ROS and JA/ET signalling events which are later suppressed, with concomitant auxin metabolism and cell wall modification processes likely to be involved in giant cell formation. This examination also highlights that a single RKN population may induce genotype-specific host gene modulation patterns during the compatible interaction in order to cause disease. Complementing work on characterization of R-gene-mediated resistance, increased understanding of both activated defence responses to limit parasitism during compatible interactions and the identification of potential host targets of nematode effectors in this complex interaction will facilitate the development of novel genetic improvement-based control measures for RKNs. Many of the modulated genes described in this study represent interesting candidates for further analysis of function in host defence or susceptibility, through either knock-out or gain-of-function analysis using genetic modification or gene editing approaches such as the CRISPR–Cas9 system (Lozano-Juste and Cutler, 2014). Host genes that can facilitate pathogen infection and compatible responses can be considered as susceptibility genes (van Schie and Takken, 2014). As mutation or loss of such a gene may impair pathogenicity, modification of candidate susceptibility genes from compatible interaction data sets which are unlikely, based on predicted function, to result in fitness penalties to the plant, offers considerable potential in disease resistance breeding.
SUPPLEMENTARY DATA
Supplementary data are available online at https://academic.oup.com/aob and consist of the following. Table S1: list of target genes, specific primer sequences and supporting information in qRT-PCR validation of differential gene expression. Table S2: global gene expression data showing DEGs identified across the sampling points of 3, 7 and 10 DAI with Meloidogyne incognita in the two susceptible Musa acuminata genotypes. Figure S1: Gene Ontology terms enriched in differentially expressed genes in Musa acuminata genotypes CAV and 4279-06 at 3 DAI with Meloidogyne incognita. Figure S2: Gene Ontology terms enriched in differentially expressed genes in Musa acuminata genotypes CAV and 4279-06 at 7 DAI with Meloidogyne incognita. Figure S3: Gene Ontology terms enriched in differentially expressed genes in Musa acuminata genotypes CAV and 4279-06 at 10 DAI with Meloidogyne incognita. Figure S4: hierarchical cluster analysis of expression changes among Musa acuminata CAV DEGs (A), categorized into six groups according to their expression pattern over the time course of the interaction with Meloidogyne incognita (B). Figures S5: hierarchical cluster analysis of expression changes among Musa acuminata 4279-06 DEGs (A), categorized into eight groups according to their expression pattern over the time course of the interaction with Meloidogyne incognita (B).
Supplementary Material
ACKNOWLEDGEMENTS
This work was partially funded by the Fundação de Amparo à Pesquisa do Estado de Distrito Federal (FAPDF) (project 193.000.983/2015) and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (project 473437/2012-0). N.E.N.C. was supported by a fellowship from CAPES. R.N.G.M. was supported by a fellowship from CNPq. We thank anonymous reviewers for their useful comments on the manuscript.
LITERATURE CITED
- Alkharouf NW, Klink VP, Chouikha IB, et al. 2006. Timecourse microarray analyses reveal global changes in gene expression of susceptible Glycine max (soybean) roots during infection by Heterodera glycines (soybean cyst nematode). Planta 224: 838–852. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W.. 2015. HTSeq – a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backiyarani S, Uma S, Arunkumar G, Saraswathi MS, Sundararaju P.. 2014. Differentially expressed genes in incompatible interactions of Pratylenchus coffeae with Musa using suppression subtractive hybridization. Physiological and Molecular Plant Pathology 86: 11–18. [Google Scholar]
- Bagnaresi P, Sala T, Irdani T, et al. 2013. Solanum torvum responses to the root-knot nematode Meloidogyne incognita. BMC Genomics 14: 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai TT, Xie WB, Zhou PP, et al. 2013. Transcriptome and expression profile analysis of highly resistant and susceptible banana roots challenged with Fusarium oxysporum f. sp. cubense tropical race 4. PLoS One 8: e73945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcala M, Garcia A, Cabrera J, et al. 2010. Early transcriptomic events in microdissected Arabidopsis nematode-induced giant cells. The Plant Journal 61: 698–712. [DOI] [PubMed] [Google Scholar]
- Beneventi MA, da Silva OB Jr, de Sá ME, et al. 2013. Transcription profile of soybean–root-knot nematode interaction reveals a key role of phytohormones in the resistance reaction. BMC Genomics 14: 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd DW, Kirkpatrick T, Barker KR.. 1983. An improved technique for clearing and staining plant tissues for detection of nematodes. Journal of Nematology 15: 142–143. [PMC free article] [PubMed] [Google Scholar]
- Chapman EJ, Estelle M.. 2009. Cytokinin and auxin intersection in root meristems. Genome Biology 10: 210. [Google Scholar]
- Daudi A, Cheng Z, O’Brien JA, et al. 2012. The apoplastic oxidative burst peroxidase in Arabidopsis is a major component of pattern triggered immunity. The Plant Cell 24: 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davide RG, Marasigan LQ.. 1985. Yield loss assessment and evaluation of resistance of banana cultivars to the nematode Radopholus similis Thorne and Meloidogyne incognita Chitwood. Phillipin Agricultural 63: 335–349. [Google Scholar]
- De Waele D, Davide RG.. 1998. The root-knot nematodes of banana Musa Pest Fact Sheet No. 3. Montpellier, France: INIBAP. [Google Scholar]
- Ding X, Cao Y, Huang L, et al. 2008. Activation of the indole-3-acetic acid-amido synthetase GH3-8 suppresses expansin expression and promotes salicylate- and jasmonate-independent basal immunity in rice. The Plant Cell 20: 228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle EA, Lambert KN.. 2003. Meloidogyne javanica chorismate mutase 1 alters plant cell development. Molecular Plant-Microbe Interactions 16: 123–131. [DOI] [PubMed] [Google Scholar]
- Durrant WE, Dong X.. 2004. Systemic acquired resistance. Annual Review of Phytopathology 42: 185–209. [DOI] [PubMed] [Google Scholar]
- Figueiredo A, Monteiro F, Sebastiana M.. 2014. Subtilisin-like proteases in plant–pathogen recognition and immune priming: a perspective. Frontiers in Plant Science 5: 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheysen G, Mitchum MG.. 2009. Molecular insights in the susceptible plant response to nematode infection. Plant Cell Monographs 15: 45–81. [Google Scholar]
- Glazebrook J. 2005. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annual Review of Phytopathology 43: 205–227. [DOI] [PubMed] [Google Scholar]
- Gowen SR, Quénéhervé P, Fogain R.. 2005. Nematode parasites of banana and plantains In: Luc M, Sikora RA, Bridge J, eds. Plant-parasitic nematodes in subtropical and tropical agriculture, 2nd edn.Wallingford, UK: CAB International, 611–643. [Google Scholar]
- Grunewald W, van Noorden G, van Isterdael G, Beeckman T, Gheysen G, Mathesius U.. 2009. Manipulation of auxin transport in plant roots during Rhizobium symbiosis and nematode parasitism. The Plant Cell 21: 2553–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes PM, Guimaraes LA, Morgante CV, et al.2015. Root transcriptome analysis of wild peanut reveals candidate genes for nematode resistance. PLoS One 10: e0140937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haegeman A, Mantelin S, Jones JT, Gheysen Y.. 2012. Functional roles of effectors of plant-parasitic nematodes. Gene 492: 19–31. [DOI] [PubMed] [Google Scholar]
- Hooper DJ, Hallmann J, Subbotin AS.. 2005. Methods for extraction, processing and detection of plant and soil nematodes In: M Luc, RA Sikora, J Bridge, eds. Plant-parasitic nematodes in subtropical and tropical agriculture, 2nd edn.Wallingford, UK: CAB International, 611–643. [Google Scholar]
- Hussey RS, Barker KR.. 1973. A comparison of methods of collecting inocula of Meloidogyne spp., including a new technique. Plant Disease Reporter 57: 1025–1028. [Google Scholar]
- Ithal N, Recknor J, Nettleton D, et al. 2007a. Parallel genome-wide expression profiling of host and pathogen during soybean cyst nematode infection of soybean. Molecular Plant–Microbe Interactions 20: 293–305. [DOI] [PubMed] [Google Scholar]
- Ithal N, Recknor J, Nettleton D, Maier T, Baum TJ, Mitchum MG.. 2007b. Developmental transcript profiling of cyst nematode feeding cells in soybean roots. Molecular Plant–Microbe Interactions 20: 510–525. [DOI] [PubMed] [Google Scholar]
- Jammes F, Lecomte P, Almeida-Engler J, et al.2005. Genome-wide expression profiling of the host response to root-knot nematode infection in Arabidopsis. The Plant Journal 44: 447–458. [DOI] [PubMed] [Google Scholar]
- Ji HL, Gheysen G, Denil S, et al. 2013. Transcriptional analysis through RNA sequencing of giant cells induced by Meloidogyne graminicola in rice roots. Journal of Experimental Botany 64: 3885–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JDG, Dangl JL.. 2006. The plant immune system. Nature 444: 323–329. [DOI] [PubMed] [Google Scholar]
- Jones JT, Haegeman A, Danchin EGJ, et al. 2013. Top 10 plant-parasitic nematodes in molecular plant pathology. Molecular Plant Pathology 14: 946–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klink VP, Overall CC, Alkharouf NW, MacDonald MH, Matthews BF.. 2007. A time-course comparative microarray analysis of an incompatible and compatible response by Glycine max (soybean) to Heterodera glycines (soybean cyst nematode) infection. Planta 226: 1423–1447. [DOI] [PubMed] [Google Scholar]
- Kyndt T, Nahar K, Haegeman A, De Vleesschauwer D, Höfte M, Gheysen G.. 2012a. Comparing the defence-related gene expression changes upon migratory and sedentary nematode attack in rice. Plant Biology 14: 73–82. [DOI] [PubMed] [Google Scholar]
- Kyndt T, Denil S, Haegeman A, et al. 2012b. Transcriptional reprogramming by root knot and migratory nematode infection in rice. New Phytologist 196: 887–900. [DOI] [PubMed] [Google Scholar]
- Kyndt T, Fernandez D, Gheysen G.. 2014. Plant-parasitic nematode infections in rice: molecular and cellular insights. Annual Review of Phytopathology 52: 135–153. [DOI] [PubMed] [Google Scholar]
- Lamb C, Dixon RA.. 1997. The oxidative burst in plant disease resistance. Annual Review of Plant Physiology and Plant Molecular Biology 48: 251–275. [DOI] [PubMed] [Google Scholar]
- Leon-Reyes A, Spoel SH, De Lange ES, et al. 2009. Ethylene modulates the role of NONEXPRESSOR OF PATHOGENESIS-RELATED GENE1 in cross talk between salicylate and jasmonate signaling. Plant Physiology 149: 1797–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CY, Deng GM, Yang J, et al. 2012. Transcriptome profiling of resistant and susceptible Cavendish banana roots following inoculation with Fusarium oxysporum f. sp. cubense tropical race 4. BMC Genomics 13: 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohar DP, Schaff JE, Laskey JG, Kieber JJ, Bilyeu KD, Bird DM.. 2004. Cytokinins play opposite roles in lateral root formation, and nematode and rhizobial symbioses. The Plant Journal 38: 203–214. [DOI] [PubMed] [Google Scholar]
- Lohse M, Nagel A, Herter T, et al. 2014. Mercator: a fast and simple web server for genome scale functional annotation of plant sequence data. Plant, Cell & Environment 37: 1250–1258. [DOI] [PubMed] [Google Scholar]
- Lorenzo O, Piqueras R, Sánchez-Serrano JJ, Solano R.. 2003. ETHYLENE RESPONSE FACTOR1 integrates signaling from ethylene and jasmonate pathways in plant defense. The Plant Cell 15: 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano-Juste J, Cutler SR.. 2014. Plant genome engineering in full bloom. Trends in Plant Science 19: 284–287. [DOI] [PubMed] [Google Scholar]
- McGrath KC, Dombrecht B, Manners JM, et al. 2005. Repressor- and activator-type ethylene response factors functioning in jasmonate signaling and disease resistance identified via a genome-wide screen of Arabidopsis transcription factor gene expression. Plant Physiology 139: 949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, Janeway CA Jr.. 1997. Innate immunity: the virtues of a nonclonal system of recognition. Cell 91: 295–298. [DOI] [PubMed] [Google Scholar]
- Molinari S. 2011. Natural genetic and induced plant resistance, as a control strategy to plant-parasitic nematodes alternative to pesticides. Plant Cell Reports 30: 311–323. [DOI] [PubMed] [Google Scholar]
- Monaghan J, Zipfel C.. 2012. Plant pattern recognition receptor complexes at the plasma membrane. Current Opinion in Plant Biology 15: 349–357. [DOI] [PubMed] [Google Scholar]
- Nahar K, Kyndt T, De Vleesschauwer D, Hofte M, Gheysen G.. 2011. The jasmonate pathway is a key player in systemically induced defense against root knot nematodes in rice. Plant Physiology 157: 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahar K, Kyndt T, Hause B, Hofte M, Gheysen G.. 2013. Brassinosteroids suppress rice defense against root-knot nematodes through antagonism with the jasmonate pathway. Molecular Plant-Microbe Interactions 26: 106–115. [DOI] [PubMed] [Google Scholar]
- Navarro L, Bari R, Achard P, et al. 2008. DELLAs control plant immune responses by modulating the balance of jasmonic acid and salicylic acid signaling. Current Biology 18: 650–655. [DOI] [PubMed] [Google Scholar]
- Nguyên VP, Bellafiore S, Petitot AS, et al. 2014. Meloidogyne incognita–rice (Oryza sativa) interaction: a new model system to study plant-root knot nematode interactions in monocotyledons. Rice 7: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-W, Kaimoyo E, Kumar D, Mosher S, Klessig DF.. 2007. Methyl salicylate is a critical mobile signal for plant systemic acquired resistance. Science 318: 113–116. [DOI] [PubMed] [Google Scholar]
- Passos MAN, de Cruz VO, Emediato FL, et al. 2013. Analysis of the leaf transcriptome of Musa acuminata during interaction with Mycosphaerella musicola: gene assembly, annotation and marker development. BMC Genomics 14: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegard A, Brizzard G, Fazari A, Soucaze O, Abad P, Djan-Caparolino C.. 2005. Histological characterization of resistance to different root-knot nematode species related to phenolics accumulation in Capsicum annuum. Phytopathology 95: 158–165. [DOI] [PubMed] [Google Scholar]
- Perry RN, Moens M, Starr JL.. 2009. Root-knot nematodes. Wallingford, UK: CAB International. [Google Scholar]
- Petitot A-S, Kyndt T, Haidar R. 2017. Transcriptomic and histological responses of African rice (Oryza glaberrima) to Meloidogyne graminicola provide new insights into root-knot nematode resistance in monocots. Annals of Botany119: 885–899. [DOI] [PMC free article] [PubMed]
- Pieterse CMJ, Van der Does D, Zamioudis C, Leon-Reyes A, Van Wees SCM.. 2012. Hormonal modulation of plant immunity. Annual Review of Cell and Developmental Biology 28: 489–521. [DOI] [PubMed] [Google Scholar]
- Prüfer K., Muetzel B, do HH, et al.2007. FUNC: a package for detecting significant associations between gene sets and ontological annotations. BMC Bioinformatics 8: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quénéhervé P, Valette C, Topart P, Tezenas DU, Montcel H, Salmon F.. 2009. Nematode resistance in bananas: screening results on some wild and cultivated accessions of Musa spp. Euphytica 165: 123–136. [Google Scholar]
- Richards DE, King KE, Ait-Ali T, Harberd NP.. 2001. How gibberellin regulates plant growth and development: a molecular genetic analysis of gibberellin signaling. Annual Review of Plant Physiology and Plant Molecular Biology 52: 67–88. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK.. 2010. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schie1 CCN, Takken FLW.. 2014. Susceptibility genes 101: how to be a good host. Annual Review of Phytopathology 52: 551–581. [DOI] [PubMed] [Google Scholar]
- Silva S de O, Souza Júnior MT, Alves EJ, Silveira JRS, Lima MB.. 2001. Banana breeding program at Embrapa. Crop Breeding and Applied Biotechnology 1: 399–436. [Google Scholar]
- Stoffelen R, Verlinden R, Xuyen NT, Swennen R, De Waele D.. 2000. Host plant response of Eumusa and Australimusa bananas (Musa spp) to migratory endoparasitic and root-knot nematodes. Nematology 2: 907–916. [Google Scholar]
- Szakasits D, Heinen P, Wieczorek K, et al. 2009. The transcriptome of syncytia induced by the cyst nematode Heterodera schachtii in Arabidopsis roots. The Plant Journal 57: 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usadel B, Nagel A, Thimm O, et al. 2005. Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiology 138: 1195–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bergh I, Nguyet DTM, Tuyet NT, Tuyet Nhi HH, De Waele D.. 2002a. Screening of Vietnamese Musa germplasm for resistance to root knot and root lesion nematodes in the greenhouse. Australasian Plant Pathology 31: 363. [Google Scholar]
- Van den Bergh I, Nguyet DTM, Tuyet NT, Tuyet Nhi HH, De Waele D.. 2002b. Responses of Vietnamese Musa genotypes to Meloidogyne spp. under field conditions. Nematology 4: 917–923. [Google Scholar]
- Van Loon LC, Rep M, Pieterse CMJ.. 2005. Significance of inducible defense-related proteins in infected plants. Annual Review of Phytopathology 44: 135–162. [DOI] [PubMed] [Google Scholar]
- Vartapetian A, Tuzhikov A, Chichkova N, Taliansky M, Wolpert T.. 2011. A plant alternative to animal caspases: subtilisin-like proteases. Cell Death and Differentiation 18: 1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilas Boas LC, Tenente RCV, Gonzaga V, Silva Neto SP, Rocha HS.. 2002. Reação de clones de bananeira (Musa spp.) ao nematóide Meloidogyne incognita (Kofoid & White, 1919) Chitwood, 1949, Raça 2. Revista Brasileira de Fruticultura 24: 690–693. [Google Scholar]
- Woodward AW, Bartel B.. 2005. Auxin: regulation, action, and interaction. Annals of Botany 95: 707–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel C, Kunze G, Chinchilla D, et al. 2006. Perception of the bacterial PAMP EF-Tu by the receptor EFR restricts Agrobacterium-mediated transformation. Cell 125: 749–760. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.