

Abstract
We investigated the effect of Single Nucleotide Polymorphisms (SNPs) spanning 10 methotrexate (MTX) pathway genes, namely AMPD1, ATIC, DHFR, FPGS, GGH, ITPA, MTHFD1, SHMT1, SLC19A1 (RFC) and TYMS on the outcome of MTX treatment in a UK rheumatoid arthritis (RA) patient cohort. Tagging SNPs were selected and genotyping performed in 309 patients with predefined outcomes to MTX treatment. Of the 129 SNPs tested, 11 associations were detected with efficacy (p-trend ≤ 0.05) including four SNPs in the ATIC gene (rs12995526, rs3821353, rs7563206 and rs16853834), 6 SNPs in the SLC19A1 gene region (rs11702425, rs2838956, rs7499, rs2274808, rs9977268, rs7279445) and a single SNP within the GGH gene (rs12681874). Five SNPs were significantly associated with adverse events; three in the DHFR gene (rs12517451, rs10072026, and rs1643657) and two of borderline significance in the FPGS gene. The results suggest that genetic variations in several key MTX pathway genes may influence response to MTX in RA patients. Further studies will be required to validate these findings and if confirmed these results could contribute towards a better understanding of and ability to predict MTX response in RA.
Keywords: Rheumatoid Arthritis, Methotrexate, polymorphism, pharmacogenetics, ATIC, RFC
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic disabling disease, requiring long-term treatment. Drug therapy is a key component of the treatment pathway and disease modifying anti rheumatic drugs (DMARDs) provide the mainstay of therapy, with mounting evidence suggesting that earlier treatment with DMARDS offers benefits in the longer term [1, 2]. There are several DMARDs available, but in clinical practice methotrexate (MTX) is increasingly recognised as the anchor drug for the treatment of RA [3-6]. This is because of substantial clinical experience, established efficacy, superior continuation rates, affordability and the fact that treatment with MTX not only reduces disease activity in the short term, but can also delay or stabilise the development of bone erosions in some patients over the longer term [7, 8]. Nevertheless, there is still significant variability in patient responses to treatment, with an estimated one third of patients failing to respond to MTX either due to lack of efficacy or adverse events (AE) [9-11]. As a result of this inter-patient variability in response and the fact that no predictive tests are available, routine blood and liver function testing are required in clinical practice that can be costly and inconvenient for patients [12]. For these reasons MTX represents an interesting target for pharmacogenetic testing, to identify response predictors that could maximise response and minimise toxicity.
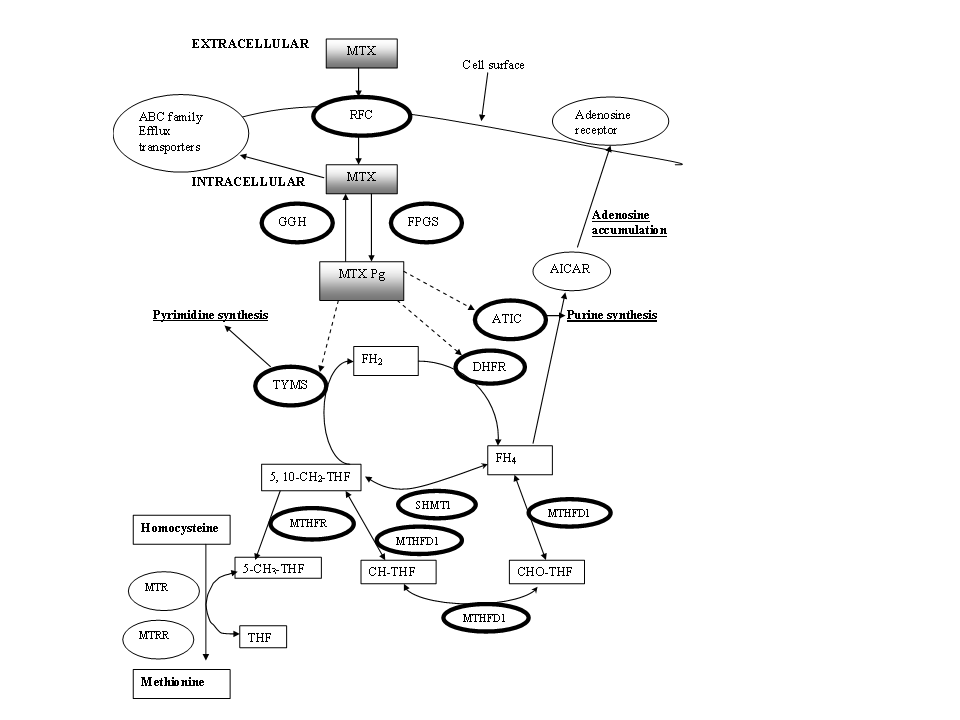
Treatment response is a complex multi-factorial trait with various contributing factors, including individual patient factors (age, sex, ethnicity, co-morbidities), disease specific factors (disease duration, severity, activity) and genetic factors [1, 13-15] A complex interplay of several genes encoding proteins involved in drug uptake and disposal, absorption, retention, distribution, metabolism and interaction with cellular targets can influence drug actions and thus these genes represent logical targets for pharmaocogenetic testing. The actual mechanism of action of low-dose MTX, used in the treatment of RA, is still not fully understood, but it is thought that the anti-inflammatory effects, mediated by adenosine release, are more important than the anti-proliferative effects [16-18]. In order to enter the cell, MTX is internalised by the reduced folate carrier (SLC19A1/RFC), with impaired transport correlated with MTX resistance [19, 20]. Once internalised, MTX requires intracellular polyglutamation, controlled by the polyglutumation-deconjugation cycle which is instigated by the enzymes γ- folypolyglutamate synthetase (FPGS) and glutamyl hydrolase (GGH), respectively [21]. It is these active MTX-polyglutamates (MTX-Pg) that determine MTX functional status and play an important role in directly suppressing various enzymes, such as dihydrofolate reductase (DHFR), thymidylate synthase (TYMS), 5-aminoimidazole 4-carboximide ribonucleotide (AICAR) transformylase (ATIC) and having an indirect effect on methylenetetrahydrofolate reductase (MTHFR).
The association of polymorphisms in these genes with MTX response have been described by various groups, although many of these studies have tested isolated polymorphisms within a few genes relevant to MTX metabolism. Our aim was to examine several important MTX pathway genes to determine if single nucleotide polymorphism (SNP) markers, selected to comprehensively cover the genes, were associated with MTX treatment response outcomes in a well-characterised group of patients with established RA.
PATIENTS AND METHODS
Study subjects and outcomes
Details of the patients included in this study and the methods by which they were recruited are outlined elsewhere [22]. In brief, all subjects were considered eligible for inclusion if they had taken MTX monotherapy for at least 3 months for RA, aged over 18 years, of white Caucasian ethnic origin and classified as having RA according to the ACR 1987 criteria [23]. The patient cohort was recruited retrospectively from two hospitals: The University Hospital of North Staffordshire (UHNS) and Central Manchester NHS Foundation Trust (CMFT). Patients were identified either via an electronic database (UHNS) or case note review (CMFT) (Table 1). Eligible patients had to fulfill one of three defined outcomes to MTX: (i) good responder (physician statement of good response plus a stable dose of MTX for at least 6 months, with an ESR <20 and/or normal CRP); (ii) inefficacy failure (physician statement of inefficacy plus failure to reduce ESR and/or CRP by at least 20% with MTX therapy for at least 3 months at a minimum dose of 15mg/wk) or (iii) AE failure (AE had to be persistent or serious and lead to treatment cessation: verified by medical record review. Furthermore, the AE had to resolve on treatment cessation and, in the case of GI AE, recur after MTX re-challenge). Individuals that did not meet one of these defined outcomes were not included in the study. Ethical approval for the study was obtained from North Staffordshire LREC (Ref 03/20) and Central Manchester LREC (Ref 03/CM/315) and subjects written consent was obtained according to the Declaration of Helsinki.
Table 1.
Patients demographics and baseline characteristics
| Responders | IE failure | AE failure | |
|---|---|---|---|
| Number patients (%) | 147 (48) | 101 (33) | 61 (19) |
| Age at diagnosis in years median (range) |
50.1 (41.2-58.8) | 46.4 (35.9-53.5) | 44.9 (39-56.8) |
| Gender: female (%) | 103 (70): | 77 (76.2): | 50 (82) |
| Age at MTX start in years median (range) |
57.4 (49.6-64.6) | 52.8 (46.3-59.3) | 52.2 (46.5-60.7) |
| RF +ve status *(%) | 99 (67.4) | 76 (75.3) | 45 (73) |
| Erosions § (%) | 115 (78) | 93 (92) | 52 (87) |
| Copies SE *(%) 0 |
27 (19) | 15 (15) | 13 (22) |
| 1 | 66 (47) | 40 (41) | 24 (41) |
| 2 | 48 (34) | 43 (44) | 22 (37) |
| No previous DMARDs median (range) |
2 (0-3) | 2 (1-3) | 2 (2-4) |
| MTX first DMARD (%) | 40 (27.2) | 21(21.8) | 7 (11.5) |
SE= Shared epitope, RF= rheumatoid factor, DMARD= disease modifying anti-rheumatic drug, MTX= methotrexate,, AE= adverse event, IE= inefficacy,
n= 297 with information,
n=301 with information
Selection of single-nucleotide polymorphisms (SNPs) and genotyping
Ten candidate genes were selected for study on the basis of putative involvement in the MTX metabolic pathway and previous evidence from the literature. They included genes involved in MTX cellular influx (SLC19A/RFC), polyglutamation (GGH, FPGS), folate pathway (DHFR, SHMT1, MTHFD1), purine synthesis (ATIC), pyrimidine synthesis (TYMS), adenosine pathway (AMPD1, ATIC) and ITPA (Supplementary Figure 1).
For each gene SNPs were selected based on a pair wise tagging SNP approach, supplemented with other commonly investigated SNPs from the literature. Marker coverage of each gene was extended to include the 10-kb upstream and downstream flanking region. Tag SNPs for each gene were selected from the CEPH/CEU Hapmap dataset (release 22) (http://www.hapmap.org) and this downloaded SNP data was then filtered through the Haploview software (http://www.broad.mit.edu/haploview/ [24] and pair-wise tagging SNPs (r2 cut off > 0.8 and MAF >5%) were selected for genotyping. In addition to the tag SNPs identified, we also included additional SNPs in each gene in case of SNP failure, 7 duplicate SNPs for quality control purposes and one 28 base pair variable number tandem repeat (VNTR) located in the 5′UTR of the TYMS gene.
Genotyping
SNP genotyping was performed using the Sequenom iPLEX® MASS ARRAY platform according to the manufacturer’s instructions (Sequenom San Diego, CA http://www.sequenom.com). Genotyping for the VNTR was performed in a 5 μl reaction volume, using primer sequences as described by Zhang et al [25]. Amplicons were electrophoresed through a 3% agarose gel and visualised with ethidium bromide staining. Quality control (QC) procedures before analysis were used such that 80% sample and polymorphism genotyping success rate was required and any samples and polymorphisms failing to meet this threshold were removed from further analysis.
Statistical analysis
Each polymorphism was tested for association with MTX efficacy and AE. Genotype and allele frequencies were compared between the groups and analysed as a nested case-control study with the responders as the referent category, using STATA version 9.2 software (StataCorp, Texas, USA) and the χ2 test for trend implemented in PLINK [26]. Two different analyses were conducted; 1) ‘responders vs inefficacy failures’ and 2) ‘responders vs AE failures’. The threshold for significance was defined at trend p≤ 0.05 and associations were expressed as trend p values, allelic odds ratios (ORs) and their 95% confidence intervals (CIs). Where variants were significantly associated with a trend p ≤ 0.05, tests of inheritance of the minor allele under dominant, recessive and multiplicative models were also conducted. Deviation from the Hardy–Weinberg equilibrium (HWE) in the responders group was tested using a chi-squared test with a threshold of p≤ 0.05.
Haplotype analysis and association was performed using Haploview software and conditional logistic regression was used to determine whether independent genetic effects existed [24].
RESULTS
Patient characteristics
The clinical and demographic features of the subjects included in the analysis have been described previously and are also presented in table 1 [22]. Subjects with established RA treated with MTX monotherapy were included in the study (median age at MTX onset=54.2 years); folic acid supplementation was taken by all patients and NSAIDs were allowed. Analysis of response to MTX was based on 309 patients comprising a maximum 147 good responders (48 %), 101 inefficacy failures (33 %) and 61 adverse event failures (19 %). The observed AEs included; gastrointestinal (n=24), abnormal liver function (n=20), and other AE’s (n=17) (Other AE’s comprising: haematological (n=7), skin rashes (n=6), renal (n=1), headaches and pneumonitis (n=3)).
Genetic association
145 SNPs were selected for testing within 10 genes. 7 duplicate SNPs were shown to be concordant; these SNPs were removed from analysis along with a further 9 SNPs, which failed QC, based on a genotyping success of <80% and deviation from HWE (p<0.05). This left 129 SNPs for further analysis. Of the 129 SNPs analysed, eleven SNPs in 3 gene regions (ATIC, GGH, SLC19A1/RFC) were found to be significantly associated (trend p≤ 0.05) with MTX efficacy and five SNPs in two genes (DHFR and FPGS) with AE (table 2 and 3). Overall no significant associations were detected with the genes, SHMT1, MTHFD1, AMPD1, ITPA and TYMS and MTX related efficacy and toxicity (supplementary table 1)
Table 2.
SNPs significantly associated with MTX efficacy (p trend ≤ 0.05)
| MAF (%) | Genotype frequencies (%) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| Gene | SNP | Base Pairs |
SNP position |
R | IE | Responders | Inefficacy failures | Trend p |
*Allelic OR (95%CI) |
||||
| (1/2) | 1_1 | 1_2 | 2_2 | 1_1 | 1_2 | 2_2 | R vs IE | ||||||
| ATIC | rs7563206 | C/T | Intron | 42.4 | 54.3 | 45 (32.4) | 70 (50.4) | 24 (17.3) | 20 (21.3) | 46 (48.9) | 28 (29.8) | 0.01 | 1.60 (1.10-2.33) |
| rs3821353 | G/T | Intron | 24.4 | 14.4 | 83 (59.7) | 44 (31.7) | 12 (8.6) | 67 (71.3) | 27 (28.7) | 0 (0.0) | 0.009 | 0.51 (0.31-0.84) | |
| rs12995526 | C/T | Intron | 42.4 | 54.9 | 45 (34.1) | 62 (47.0) | 25 (18.9) | 20 (22.0) | 42 (46.2) | 29 (31.9) | 0.01 | 1.65 (1.13-2.42) | |
| rs16853834 | C/T | Exonic 5′UTR |
13.9 | 21.5 | 96 (73.8) | 32 (24.6) | 2 (1.5) | 54 (62.8) | 27 (31.4) | 5 (5.8) | 0.04 | 1.70 (1.02-2.82) | |
| GGH | rs12681874 | C/T | Intron | 16.9 | 10.0 | 104 (78.2) | 25 (18.8) | 4 (3.0) | 77 (86.5) | 9 (10.1) | 3 (3.4) | 0.04 | 0.54 (0.30-1.00) |
| SLC19A1 | rs11702425# | T/C | Exon | 26.9 | 40.7 | 75 (53.2) | 56 (39.7) | 10 (7.1) | 34 (35.1) | 47 (48.5) | 16 (16.5) | 0.001 | 1.86 (1.26-2.74) |
| rs2838956 | A/G | Intron | 38.3 | 47.4 | 52 (36.9) | 70 (49.6) | 19 (13.5) | 24 (24.7) | 54 (55.7) | 19 (19.6) | 0.04 | 1.45 (1.00-2.10) | |
| rs7499 | G/A | Exonic 5′ UTR |
35.4 | 45.3 | 59 (41.8) | 64 (45.4) | 18 (12.8) | 27 (28.1) | 51 (53.1) | 18 (18.8) | 0.02 | 1.50 (1.03-2.19) | |
| rs2274808# | C/T | Intron | 21.8 | 33.0 | 88 (62.9) | 43 (30.7) | 9 (6.4) | 45 (46.4) | 40 (41.2) | 12 (12.4) | 0.009 | 1.76 (1.17-2.67) | |
| rs9977268# | C/T | Intron | 18.9 | 28.1 | 95 (67.9) | 37 (26.4) | 8 (5.7) | 52 (54.2) | 34 (35.4) | 10 (10.4) | 0.02 | 1.67 (1.08-2.58) | |
| rs7279445# | C/T | Intron | 45.0 | 54.2 | 43 (30.5) | 69 (48.9) | 29 (20.6) | 18 (18.8) | 52 (54.2) | 26 (27.1) | 0.05 | 1.44 (0.99-2.08) | |
based on carriage of the minor (rare) allele;
= In An overlapping gene COL18A1 MAF= minor allele frequency, R= Responder, IE= Inefficacy failure to MTX, CI= Confidence Interval 1_1= major allele homozygote 1_2= heterozygote 2_2= minor allele homozygote
Table 3.
SNPs significantly associated with MTX related AE (p trend ≤ 0.05)
| MAF (%) | Genotype frequencies (%) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| Gene | SNP | Base pairs |
SNP Position |
R | AE | Responders | Adverse events | Trend p |
*Allelic OR (95% CI) |
||||
| (1/2) | 1_1 | 1_2 | 2_2 | 1_1 | 1_2 | 2_2 | R vs AE | ||||||
| DHFR | rs12517451 | C/T | Intron | 20.0 | 29.7 | 91 (65.0) | 42 (30.0) | 7 (5.0) | 32 (54.2) | 19 (32.2) | 8 (13.6) | 0.04 | 1.68 (1.03-2.75) |
| rs1643657 | A/G | Intron | 31.0 | 21.2 | 66 (48.2) | 57 (41.6) | 14 (10.2) | 36 (61.0) | 21 (35.6) | 2 (3.4) | 0.04 | 0.60 (0.39-0.99) | |
| rs10072026 | T/C | Exonic 3′UTR |
12.7 | 5.9 | 107 (75.9) | 32 (22.7) | 2 (1.4) | 53 (89.8) | 5 (8.5) | 1 (1.7) | 0.04 | 0.43 (0.19-0.99) | |
| FPGS | rs1054774 | A/C | 5′ gene | 39.6 | 49.0 | 48 (35.8) | 67 (50.0) | 19 (14.2) | 16 (30.8) | 21 (40.4) | 15 (28.8) | 0.06 | 1.52 (0.98-2.34) |
| rs4451422 | T/A | Exonic 5′ UTR |
40.0 | 49.0 | 46 (34.3) | 69 (51.5) | 19 (14.2) | 15 (28.8) | 23 (44.2) | 14 (26.9) | 0.06 | 1.49 (0.97-2.30) | |
based on carriage of the minor (rare) allele, MAF= minor allele frequency, R= Responders AE= Adverse events to MTX, CI= Confidence Interval 1_1= major allele homozygote 1_2= heterozygote 2_2= minor allele homozygote
Association with MTX efficacy
Four of the SNPs associated with MTX inefficacy (trend p≤ 0.05) lie within the ATIC gene: Two SNPs in high LD using genotype data from our cohort (rs12995526 & rs7563206, r2 = 0.98) and a single SNP (rs16853834) were associated with a poor response to MTX (OR 1.65, 95%CI 1.13-2.42, OR 1.60, 95%CI 1.10-2.33 and OR 1.70 95%CI 1.02-2.82 respectively) and one SNP (rs3821353) with a good response to MTX (OR 0.51, 95%CI 0.31-0.84) (Table 2). Six SNPs within the SLC19A1/RFC gene region were associated with a poor response to MTX: two SNPs in high LD (rs2274808 & rs9977268, r2=0.86), rs2838956 & rs7499 (r2=0.74) and rs11702425 and one with borderline significance, rs7279445 (Table 2). Finally one SNP within the GGH gene (rs12681874) was associated with a good response to MTX (OR 0.54 95% CI 0.30-1.00).
In the ATIC and SLC19A1 (RFC) genes, as some SNPs were in high LD, we used conditional logistic regression to determine if the associations were independent of each other. The significantly associated markers were conditioned against the effect of the most significant marker in the gene but there was no evidence of independence demonstrated in this sample (data not shown). Similarly, haplotype analysis did not reveal haplotypic effects (data not shown).
Association with MTX toxicity
With regard to MTX related toxicity, two SNPs (rs10072026 and rs1643657) within the DHFR gene region were associated with a reduced risk of MTX related AE (OR 0.43 95%CI 0.19-0.99 & OR 0.60 95%CI 0.39-0.99) and another (rs12517451) showed evidence of association with an increased risk of AE (OR 1.68, 95%CI 1.03-2.75). In addition two SNPs in high LD (r2=0.96) within the FPGS gene (rs1054774 & rs44511422) showed borderline evidence of association with an increased risk of AE (Table 3). Both SNPs in the FPGS gene (rs1054774 & rs4451422) were highly significant under a recessive model of inheritance (OR 3.03, 95% CI 1.14-7.99 and OR 3.60, 95% CI 1.39-9.33 respectively) (Table 3).
Replication of previous pharmacogenetic results
Twelve of the 145 SNPs were included in order to validate previously reported associations from the literature (Table 4). Of these, one SNP in the DHFR gene (rs1650697) for which a proxy SNP (rs12517451) (r2 =1.0) was genotyped provided evidence of association with AEs (OR= 1.68, 95%CI 1.03-2.75) and two SNPs in the FPGS gene (rs1544105 & rs10106) for which proxies were genotyped showed borderline evidence of association (trend p = 0.06) with an increased risk of AE, with a further increased risk with carriage of two copies of the minor allele under a recessive model (Table 4). No significant associations were revealed with any of the other previously reported SNPs (Table 4).
Table 4.
Results for SNPs previously found to show evidence of association with MTX response
| Gene (ref) |
SNP from literature | Reported assoc |
Position | Proxy SNP typed |
MAF | R vs AE | R vs IE | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| R | AE | IE | Trend p |
*Allelic OR (95% CI) |
Trend p |
*Allelic OR (95% CI) |
|||||
|
ITPA [44, 45, 47] |
rs1127354 | Efficacy | 94 C/A | 9.6 | 9.3 | 8.8 | 0.93 | 0.97 0.47-2.02 | 0.76 | 0.90 0.48-1.71 | |
|
ATIC [31-33, 35, 39, 45, 47, 48] |
rs2372536 | AE/Efficacy | 347 C/G | 32.8 | 34.6 | 32 | 0.74 | 1.08 0.67-1.74 | 0.86 | 0.94 0.63-1.39 | |
| DHFR [36] | rs1650697 | AE (Hepatitis) | −473 G/A | rs12517451 | 20.0 | 29.7 | 23.7 | 0.04 | 1.68 1.03-2.75 | 0.35 | 1.24 0.80-1.93 |
|
GGH [49, 50] |
rs11545078 | Efficacy | 452 C/T | 10.7 | 11.9 | 8.6 | 0.74 | 1.12 0.57- 2.20 | 0.49 | 0.80 0.42-1.50 | |
|
AMPD1 [35, 45, 47] |
rs17602729 | Efficacy | 34 C/T | 13.2 | 19.6 | 12.1 | 0.13 | 1.61 0.88-2.94 | 0.72 | 0.91 0.51-1.61 | |
|
MTHFD1 [47, 51] |
rs17850560 | Efficacy | 1958 G/A | rs2236225 | 46.8 | 50.8 | 49.0 | 0.44 | 1.18 0.75-1.81 | 0.64 | 1.09 0.76-1.56 |
|
SLC19A1 [31, 33, 34, 36-40, 42, 43, 52] |
rs1051266 | AE /Efficacy | 80 G/A | 41.4 | 42.0 | 47.8 | 0.91 | 1.02 0.66-1.59 | 0.17 | 1.29 0.89-1.89 | |
|
FPGS [34, 35, 49] |
rs1544105 | Efficacy | G/A | rs1054774 | 39.2 | 49.0 | 37.6 | 0.06 | 1.52 0.98-2.34 | 0.48 | 088 0.60-1.29 |
| rs10106 | 1994 A/G | rs4451422 | 39.9 | 49.0 | 39.9 | 0.06 | 1.49 0.97-2.30 | 0.66 | 0.99 0.68-1.47 | ||
|
TYMS [31-33, 41, 43, 53-55] |
28bp VNTR | AE/Efficacy | N/A | § 45.0 | 46.5 | 47.9 | 0.80 | 1.06 0.69-1.64 | 0.53 | 1.12 0.78-1.62 | |
| 6bp deletion | N/A | 33.5 | 29.3 | 29.0 | 0.43 | 0.83 0.51-1.32 | 0.31 | 0.81 0.54-1.28 | |||
|
SHMT1 [32, 43, 48, 51] |
rs1979277 | AE/Efficacy | 1420 C/T | 31.9 | 29.7 | 35.6 | 0.67 | 0.89 0.56-1.43 | 0.41 | 1.17 0.80-1.73 | |
based on carriage of the minor (rare) allele; emboldened SNP significant or approaching statistical significance
= 2R2R genotype MAF= minor allele frequency, AE= Adverse event failure IE=Inefficacy failure, OR= odds ratio, CI= Confidence Interval, VNTR= Variable number tandem repeat
DISCUSSION
The ability to individually tailor MTX treatment to meet individual patient’s needs remains an important goal and would be valuable if applied in clinical practice. Our study has identified 16 SNPs, some novel and others replicating previous findings, in 5 key MTX metabolic pathway genes which show evidence for association with MTX efficacy or AE’s in this cohort of RA patients. No significant associations or replications of previous associations of SNPs within the genes, SHMT1, MTHFD1, AMPD1, ITPA, and TYMS with MTX efficacy or AE’s were found.
There have been a number of studies conducted to determine MTX response in RA patients, with the majority adopting a candidate gene approach, genotyping isolated SNPs within the gene and testing for association. Our study also focused on candidate genes in the MTX metabolic pathway, partly because this approach has proven successful for a number of other common treatments. One of the best examples is the identification of polymorphisms in the vitamin K (VKORC1) and cytochrome p450 (CYP2C9) genes which influence response to warfarin where findings have since been validated in independent studies [27-30]. Our study had the additional strength that, rather than testing single SNPs in specific genes, we systematically screened selected MTX pathway genes ensuring gene coverage following quality control measures exceeded 85% when compared to the HapMap data. In this way, we can confidently exclude association with a number of genes in our cohort for effect sizes >1.5. This was a retrospective study and patients were recruited to the study based on phenotypes defined using the data available in the patients notes. We set out to define phenotypes for both inefficacy and AE’s in order to minimise variation and maximise the power to detect significant genetic effects. Furthermore, all of the patients were recruited within a well-defined geographical area and comprised an ethnically homogenous patient population.
Despite these strengths, our study has several limitations: Firstly, incomplete knowledge of the MTX metabolic pathway means that we may have failed to screen some important genes and although we have found several SNPs to be associated with either efficacy or AE in this study; it is likely that combinations of risk SNPs will be more predictive of response to MTX than individual SNP effects, as shown in previous studies [31-33]. It would be interesting to look at combinations of the polymorphisms reported here in an independent patient cohort. Secondly, the sample size was modest (n=309); although this represents one of the largest cohorts studied to date in the context of MTX pharmacogenetics in RA, the power to detect effect sizes less than 1.5 was consequently limited. Thirdly, retrospective data collection can introduce biases: this can be due to limitations of missing information on important clinical variables that are thought to be predictive of response to MTX, for example, disease specific factors, co-morbidities or folate. Thus, apart from folic acid, which all patients received at a dose of at least 5mg/week, we did not adjust for clinical factors in the analysis. Phenotypes were pre-defined using the data available in the medical notes and a broad term was used to define good responders and inefficacy failures that took into account physician statement, inflammatory markers (CRP and ESR), dose of MTX and time on treatment. Whilst these criteria were selected to identify those with well-validated response measures, many researchers now advocate using the DAS score, because it is used clinically to base treatment decisions. Unfortunately, the DAS information was not available for many patients in the current cohort. This remains an area of ongoing debate for pharmacogenetic studies, with a need to standardise outcome measures and should be a consideration for future prospective studies, including other important patient reported outcomes, measures of disability and psychological outcome measures. Finally ensuring good gene coverage inevitably results in multiple testing and a consequent increased likelihood of false positive findings. Given that 129 SNPs have been tested for both response and inefficacy, a Bonferroni corrected p-value threshold of p < 2 × 10−4 could be used as the threshold for claims of significance. At this threshold, none of the SNPs would have remained statistically associated with response. We present the uncorrected p-values because, due to the limited sample size, the fact that corrective procedures may unnecessarily reduce power and a proportion of our positive findings do support those from previous studies suggesting that the effects seen may be true. For example, we report borderline association with a SNP (rs1054774) within the FPGS gene with MTX AE. Interestingly a SNP in high LD with our associated SNP, namely rs1544105, has been associated with response in a previous study [34, 35]. Also, a SNP mapping to the DHFR gene (rs1650697) and perfectly correlated (r2= 1) with rs12517451, which we report to be associated with AE, specifically liver AE (data not shown), was associated with the occurrence of hepatitis in RA patients in an independent study [36]. Finally, a SNP mapping to the ATIC gene and tested in this RA cohort for the first time has been reported to be associated with MTX in a JIA cohort as described later. In terms of MTX efficacy, several studies have investigated the role of the SLC19A1 (RFC) gene, focusing on the RFC 80A/C non-synonymous SNP (rs1051266), which results in a substitution of arginine to histidine at codon 27 in the first transmembrane domain of the RFC protein [34, 36-43]. Our results did not replicate previous findings of association with this particular SNP, in keeping with the results from some studies [34, 36, 39, 42]. We did however find six other SNPs both in the RFC gene and a neighbouring gene COL18A1 which associated with MTX efficacy. As the RFC gene is highly polymorphic in humans and shows strong LD across the gene, some of these variants were correlated with other less commonly reported polymorphisms in the RA literature [42] . This may well suggest that the overall contribution of polymorphisms on phenotype cannot be explained by the most commonly reported SNP (rs1051266) alone, but that other variants within the gene may also be important influencing response to MTX.
Several previous RA studies have reported a SNP (rs2372536) located in exon 5 of the ATIC gene, leading to a threonine to serine substitution, to be associated with both response and AE to MTX [31, 32, 36, 44]. Our finding of no association with this SNP and MTX response is in keeping with those of Takatori et al and Sharma et al [35, 39]. Differences seen with the present study and others could be explained by inter study variability; small sample sizes; comparing patients with different stages of disease (early onset [45] vs established RA [44], different outcomes to measure efficacy and AE and different treatment regimes However, we have found several other SNPs in the ATIC gene to be associated with response (efficacy) to MTX in this study and one of these (rs12995526) has also been found to be associated with MTX response in two independent patient cohorts with juvenile idiopathic arthritis (JIA) with a similar magnitude of effect [46]. Increasing evidence suggests that the anti-inflammatory role mediated by adenosine is key to MTXs mode of action and furthermore previous work reported from our group has demonstrated the importance of adenosine receptors and response to MTX [22]. The fact that SNPs in the same gene are showing evidence for association with MTX response in different cohorts suggest that this gene may play a role in determining efficacy. It is possible that multiple variants within the gene contribute or there may be a single causal variant but different LD patterns in different populations may mean that different SNPs show evidence for association.
In summary, results from this study replicate some previous findings reported in the literature and at the same time we report associations between several MTX pathway genes, namely GGH, ATIC, DHFR and SLC19A1 (RFC) and either efficacy or AE in MTX-treated RA patients. In particular there is growing evidence to support the role of the ATIC gene in the response to MTX treatment. Many of the SNPs reported here reside in non-coding regions, therefore impact on functional properties of the gene are unknown and thus no specific biological mechanisms can be proposed. Further investigation in a larger, prospectively collected cohort with well-defined outcome and clinical measures is required to confidently confirm the association of these SNPs with MTX treatment response. Once confirmed, fine mapping will be needed to determine the actual causal variant. One of the most important applications will be in providing biological insight into the mechanisms by which some patients respond to MTX whilst others do not.
Supplementary Material
{kind=link}
Acknowledgements
We thank Arthritis Research UK for their support, Arthritis Research UK grant reference no 17552, SH, P M, I N B, A.B, W.T, funded by Arthritis Research-UK and S.A.O salary is funded by Pfizer. We acknowledge the NIHR Manchester Biomedical Research Centre for support.
Footnotes
Conflicts of Interest: The authors declare no conflicts of interest
Supplementary information is available at The Pharmacogenomics Journal’s website
REFERENCES
- [1].Anderson JJ, Wells G, Verhoeven AC, Felson DT. Factors predicting response to treatment in rheumatoid arthritis: the importance of disease duration. Arthritis Rheum. 2000 Jan;43(1):22–9. doi: 10.1002/1529-0131(200001)43:1<22::AID-ANR4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- [2].Finckh A, Liang MH, van Herckenrode CM, de Pablo P. Long-term impact of early treatment on radiographic progression in rheumatoid arthritis: A meta-analysis. Arthritis Rheum. 2006 Dec 15;55(6):864–72. doi: 10.1002/art.22353. [DOI] [PubMed] [Google Scholar]
- [3].Weinblatt ME, Coblyn JS, Fox DA, Fraser PA, Holdsworth DE, Glass DN, et al. Efficacy of low-dose methotrexate in rheumatoid arthritis. N Engl J Med. 1985 Mar 28;312(13):818–22. doi: 10.1056/NEJM198503283121303. [DOI] [PubMed] [Google Scholar]
- [4].Williams HJ, Willkens RF, Samuelson CO, Jr., Alarcon GS, Guttadauria M, Yarboro C, et al. Comparison of low-dose oral pulse methotrexate and placebo in the treatment of rheumatoid arthritis. A controlled clinical trial. Arthritis Rheum. 1985 Jul;28(7):721–30. doi: 10.1002/art.1780280702. [DOI] [PubMed] [Google Scholar]
- [5].Le Loet X, Berthelot JM, Cantagrel A, Combe B, De Bandt M, Fautrel B, et al. Clinical practice decision tree for the choice of the first disease modifying antirheumatic drug for very early rheumatoid arthritis: a 2004 proposal of the French Society of Rheumatology. Ann Rheum Dis. 2006 Jan;65(1):45–50. doi: 10.1136/ard.2005.035436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sokka T, Kautiainen H, Toloza S, Makinen H, Verstappen SM, Lund Hetland M, et al. QUEST-RA: quantitative clinical assessment of patients with rheumatoid arthritis seen in standard rheumatology care in 15 countries. Ann Rheum Dis. 2007 Nov;66(11):1491–6. doi: 10.1136/ard.2006.069252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jones G, Halbert J, Crotty M, Shanahan EM, Batterham M, Ahern M. The effect of treatment on radiological progression in rheumatoid arthritis: a systematic review of randomized placebo-controlled trials. Rheumatology (Oxford) 2003 Jan;42(1):6–13. doi: 10.1093/rheumatology/keg036. [DOI] [PubMed] [Google Scholar]
- [8].Pincus T, Ferraccioli G, Sokka T, Larsen A, Rau R, Kushner I, et al. Evidence from clinical trials and long-term observational studies that disease-modifying anti-rheumatic drugs slow radiographic progression in rheumatoid arthritis: updating a 1983 review. Rheumatology (Oxford) 2002 Dec;41(12):1346–56. doi: 10.1093/rheumatology/41.12.1346. [DOI] [PubMed] [Google Scholar]
- [9].Alarcon GS, Tracy IC, Blackburn WD., Jr Methotrexate in rheumatoid arthritis. Toxic effects as the major factor in limiting long-term treatment. Arthritis Rheum. 1989 Jun;32(6):671–6. doi: 10.1002/anr.1780320603. [DOI] [PubMed] [Google Scholar]
- [10].Gispen JG, Alarcon GS, Johnson JJ, Acton RT, Barger BO, Koopman WJ. Toxicity of methotrexate in rheumatoid arthritis. J Rheumatol. 1987 Feb;14(1):74–9. [PubMed] [Google Scholar]
- [11].Salliot C, van der Heijde D. Long-term safety of methotrexate monotherapy in patients with rheumatoid arthritis: a systematic literature research. Ann Rheum Dis. 2009 Jul;68(7):1100–4. doi: 10.1136/ard.2008.093690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chakravarty K, McDonald H, Pullar T, Taggart A, Chalmers R, Oliver S, et al. BSR/BHPR guideline for disease-modifying anti-rheumatic drug (DMARD) therapy in consultation with the British Association of Dermatologists. Rheumatology (Oxford) 2008 Jun;47(6):924–5. doi: 10.1093/rheumatology/kel216a. [DOI] [PubMed] [Google Scholar]
- [13].Hoekstra M, van Ede AE, Haagsma CJ, van de Laar MA, Huizinga TW, Kruijsen MW, et al. Factors associated with toxicity, final dose, and efficacy of methotrexate in patients with rheumatoid arthritis. Ann Rheum Dis. 2003 May;62(5):423–6. doi: 10.1136/ard.62.5.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hider SL, Silman AJ, Thomson W, Lunt M, Bunn D, Symmons DP. Can clinical factors at presentation be used to predict outcome of treatment with methotrexate in patients with early inflammatory polyarthritis? Ann Rheum Dis. 2009 Jan;68(1):57–62. doi: 10.1136/ard.2008.088237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].van Ede AE, Laan RF, Rood MJ, Huizinga TW, van de Laar MA, van Denderen CJ, et al. Effect of folic or folinic acid supplementation on the toxicity and efficacy of methotrexate in rheumatoid arthritis: a forty-eight week, multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2001 Jul;44(7):1515–24. doi: 10.1002/1529-0131(200107)44:7<1515::AID-ART273>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- [16].Cronstein BN. Going with the flow: methotrexate, adenosine, and blood flow. Ann Rheum Dis. 2006 Apr;65(4):421–2. doi: 10.1136/ard.2005.049601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cutolo M, Sulli A, Pizzorni C, Seriolo B, Straub RH. Anti-inflammatory mechanisms of methotrexate in rheumatoid arthritis. Ann Rheum Dis. 2001 Aug;60(8):729–35. doi: 10.1136/ard.60.8.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Riksen NP, Barrera P, van den Broek PH, van Riel PL, Smits P, Rongen GA. Methotrexate modulates the kinetics of adenosine in humans in vivo. Ann Rheum Dis. 2006 Apr;65(4):465–70. doi: 10.1136/ard.2005.048637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jansen G, Mauritz R, Drori S, Sprecher H, Kathmann I, Bunni M, et al. A structurally altered human reduced folate carrier with increased folic acid transport mediates a novel mechanism of antifolate resistance. J Biol Chem. 1998 Nov 13;273(46):30189–98. doi: 10.1074/jbc.273.46.30189. [DOI] [PubMed] [Google Scholar]
- [20].Laverdiere C, Chiasson S, Costea I, Moghrabi A, Krajinovic M. Polymorphism G80A in the reduced folate carrier gene and its relationship to methotrexate plasma levels and outcome of childhood acute lymphoblastic leukemia. Blood. 2002 Nov 15;100(10):3832–4. doi: 10.1182/blood.V100.10.3832. [DOI] [PubMed] [Google Scholar]
- [21].McGuire JJ, Hsieh P, Bertino JR. Enzymatic synthesis of polyglutamate derivatives of 7-hydroxymethotrexate. Biochemical pharmacology. 1984 Apr 15;33(8):1355–61. doi: 10.1016/0006-2952(84)90192-8. [DOI] [PubMed] [Google Scholar]
- [22].Hider SL, Thomson W, Mack LF, Armstrong DJ, Shadforth M, Bruce IN. Polymorphisms within the adenosine receptor 2a gene are associated with adverse events in RA patients treated with MTX. Rheumatology (Oxford) 2008 Aug;47(8):1156–9. doi: 10.1093/rheumatology/ken182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988 Mar;31(3):315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- [24].Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics (Oxford, England) 2005 Jan 15;21(2):263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- [25].Zhang Z, Shi Q, Sturgis EM, Spitz MR, Hong WK, Wei Q. Thymidylate synthase 5′- and 3′-untranslated region polymorphisms associated with risk and progression of squamous cell carcinoma of the head and neck. Clin Cancer Res. 2004 Dec 1;10(23):7903–10. doi: 10.1158/1078-0432.CCR-04-0923. [DOI] [PubMed] [Google Scholar]
- [26].Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007 Sep;81(3):559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wadelius M, Chen LY, Downes K, Ghori J, Hunt S, Eriksson N, et al. Common VKORC1 and GGCX polymorphisms associated with warfarin dose. Pharmacogenomics J. 2005;5(4):262–70. doi: 10.1038/sj.tpj.6500313. [DOI] [PubMed] [Google Scholar]
- [28].Eichelbaum M, Ingelman-Sundberg M, Evans WE. Pharmacogenomics and individualized drug therapy. Annual review of medicine. 2006;57:119–37. doi: 10.1146/annurev.med.56.082103.104724. [DOI] [PubMed] [Google Scholar]
- [29].Cooper GM, Johnson JA, Langaee TY, Feng H, Stanaway IB, Schwarz UI, et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood. 2008 Aug 15;112(4):1022–7. doi: 10.1182/blood-2008-01-134247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Takeuchi F, McGinnis R, Bourgeois S, Barnes C, Eriksson N, Soranzo N, et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS genetics. 2009 Mar;5(3):e1000433. doi: 10.1371/journal.pgen.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dervieux T, Furst D, Lein DO, Capps R, Smith K, Walsh M, et al. Polyglutamation of methotrexate with common polymorphisms in reduced folate carrier, aminoimidazole carboxamide ribonucleotide transformylase, and thymidylate synthase are associated with methotrexate effects in rheumatoid arthritis. Arthritis Rheum. 2004 Sep;50(9):2766–74. doi: 10.1002/art.20460. [DOI] [PubMed] [Google Scholar]
- [32].Weisman MH, Furst DE, Park GS, Kremer JM, Smith KM, Wallace DJ, et al. Risk genotypes in folate-dependent enzymes and their association with methotrexate-related side effects in rheumatoid arthritis. Arthritis Rheum. 2006 Feb;54(2):607–12. doi: 10.1002/art.21573. [DOI] [PubMed] [Google Scholar]
- [33].Dervieux T, Furst D, Lein DO, Capps R, Smith K, Caldwell J, et al. Pharmacogenetic and metabolite measurements are associated with clinical status in patients with rheumatoid arthritis treated with methotrexate: results of a multicentred cross sectional observational study. Ann Rheum Dis. 2005 Aug 1;64(8):1180–5. doi: 10.1136/ard.2004.033399. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sharma S, Das M, Kumar A, Marwaha V, Shankar S, Aneja R, et al. Interaction of genes from influx-metabolism-efflux pathway and their influence on methotrexate efficacy in rheumatoid arthritis patients among Indians. Pharmacogenet Genomics. 2008 Dec;18(12):1041–9. doi: 10.1097/fpc.0b013e328311a8fd. [DOI] [PubMed] [Google Scholar]
- [35].Sharma S, Das M, Kumar A, Marwaha V, Shankar S, Singh P, et al. Purine biosynthetic pathway genes and methotrexate response in rheumatoid arthritis patients among north Indians. Pharmacogenet Genomics. 2009 Oct;19(10):823–8. doi: 10.1097/fpc.0b013e328331b53e. [DOI] [PubMed] [Google Scholar]
- [36].Wessels JA, de Vries-Bouwstra JK, Heijmans BT, Slagboom PE, Goekoop-Ruiterman YP, Allaart CF, et al. Efficacy and toxicity of methotrexate in early rheumatoid arthritis are associated with single-nucleotide polymorphisms in genes coding for folate pathway enzymes. Arthritis Rheum. 2006 Apr;54(4):1087–95. doi: 10.1002/art.21726. [DOI] [PubMed] [Google Scholar]
- [37].Drozdzik M, Rudas T, Pawlik A, Gornik W, Kurzawski M, Herczynska M. Reduced folate carrier-1 80G>A polymorphism affects methotrexate treatment outcome in rheumatoid arthritis. Pharmacogenomics J. 2007 Dec;7(6):404–7. doi: 10.1038/sj.tpj.6500438. [DOI] [PubMed] [Google Scholar]
- [38].Hayashi H, Fujimaki C, Daimon T, Tsuboi S, Matsuyama T, Itoh K. Genetic polymorphisms in folate pathway enzymes as a possible marker for predicting the outcome of methotrexate therapy in Japanese patients with rheumatoid arthritis. Journal of clinical pharmacy and therapeutics. 2009 Jun;34(3):355–61. doi: 10.1111/j.1365-2710.2009.01046.x. [DOI] [PubMed] [Google Scholar]
- [39].Takatori R, Takahashi KA, Tokunaga D, Hojo T, Fujioka M, Asano T, et al. ABCB1 C3435T polymorphism influences methotrexate sensitivity in rheumatoid arthritis patients. Clin Exp Rheumatol. 2006 Sep-Oct;24(5):546–54. [PubMed] [Google Scholar]
- [40].Fukino K, Kawashima T, Suzuki M, Ueno K. Methylenetetrahydrofolate reductase and reduced folate carrier-1 genotypes and methotrexate serum concentrations in patients with rheumatoid arthritis. The Journal of toxicological sciences. 2007 Oct;32(4):449–52. doi: 10.2131/jts.32.449. [DOI] [PubMed] [Google Scholar]
- [41].Grabar P Bohanec, Logar D, Lestan B, Dolzan V. Genetic determinants of methotrexate toxicity in rheumatoid arthritis patients: a study of polymorphisms affecting methotrexate transport and folate metabolism. European journal of clinical pharmacology. 2008 Nov;64(11):1057–68. doi: 10.1007/s00228-008-0521-7. [DOI] [PubMed] [Google Scholar]
- [42].Chatzikyriakidou A, Georgiou I, Voulgari PV, Papadopoulos CG, Tzavaras T, Drosos AA. Transcription regulatory polymorphism -43T>C in the 5′-flanking region of SLC19A1 gene could affect rheumatoid arthritis patient response to methotrexate therapy. Rheumatol Int. 2007 Sep;27(11):1057–61. doi: 10.1007/s00296-007-0339-0. [DOI] [PubMed] [Google Scholar]
- [43].James HM, Gillis D, Hissaria P, Lester S, Somogyi AA, Cleland LG, et al. Common polymorphisms in the folate pathway predict efficacy of combination regimens containing methotrexate and sulfasalazine in early rheumatoid arthritis. J Rheumatol. 2008 Apr;35(4):562–71. [PubMed] [Google Scholar]
- [44].Lee YC, Cui J, Costenbader KH, Shadick NA, Weinblatt ME, Karlson EW. Investigation of candidate polymorphisms and disease activity in rheumatoid arthritis patients on methotrexate. Rheumatology (Oxford) 2009 Jun;48(6):613–7. doi: 10.1093/rheumatology/ken513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wessels JA, Kooloos WM, De Jonge R, De Vries-Bouwstra JK, Allaart CF, Linssen A, et al. Relationship between genetic variants in the adenosine pathway and outcome of methotrexate treatment in patients with recent-onset rheumatoid arthritis. Arthritis Rheum. 2006 Sep;54(9):2830–9. doi: 10.1002/art.22032. [DOI] [PubMed] [Google Scholar]
- [46].Hinks A, Moncrieffe H, Martin P, Ursu S, Lal S, Kassoumeri L, et al. Association of the 5-aminoimidazole-4-carboxamide ribonucleotide transformylase gene with response to methotrexate in juvenile idiopathic arthritis. Ann Rheum Dis. 2011 Aug;70(8):1395–400. doi: 10.1136/ard.2010.146191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wessels JA, van der Kooij SM, le Cessie S, Kievit W, Barerra P, Allaart CF, et al. A clinical pharmacogenetic model to predict the efficacy of methotrexate monotherapy in recent-onset rheumatoid arthritis. Arthritis Rheum. 2007 Jun;56(6):1765–75. doi: 10.1002/art.22640. [DOI] [PubMed] [Google Scholar]
- [48].Dervieux T, Greenstein N, Kremer J. Pharmacogenomic and metabolic biomarkers in the folate pathway and their association with methotrexate effects during dosage escalation in rheumatoid arthritis. Arthritis Rheum. 2006 Oct;54(10):3095–103. doi: 10.1002/art.22129. [DOI] [PubMed] [Google Scholar]
- [49].van der Straaten R, Wessels JA, de Vries-Bouwstra JK, Goekoop-Ruiterman YP, Allaart CF, Bogaartz J, et al. Exploratory analysis of four polymorphisms in human GGH and FPGS genes and their effect in methotrexate-treated rheumatoid arthritis patients. Pharmacogenomics. 2007 Feb;8(2):141–50. doi: 10.2217/14622416.8.2.141. [DOI] [PubMed] [Google Scholar]
- [50].Kato T, Hamada A, Mori S, Saito H. Genetic polymorphisms in metabolic and cellular transport pathway of methotrexate impact clinical outcome of methotrexate monotherapy in Japanese patients with rheumatoid arthritis. Drug metabolism and pharmacokinetics. 2011 Nov 22; doi: 10.2133/dmpk.dmpk-11-rg-066. [DOI] [PubMed] [Google Scholar]
- [51].Stamp LK, Chapman PT, O’Donnell JL, Zhang M, James J, Frampton C, et al. Polymorphisms within the folate pathway predict folate concentrations but are not associated with disease activity in rheumatoid arthritis patients on methotrexate. Pharmacogenet Genomics. 2010 Jun;20(6):367–76. doi: 10.1097/FPC.0b013e3283398a71. [DOI] [PubMed] [Google Scholar]
- [52].Dervieux T, Kremer J, Lein DO, Capps R, Barham R, Meyer G, et al. Contribution of common polymorphisms in reduced folate carrier and gamma-glutamylhydrolase to methotrexate polyglutamate levels in patients with rheumatoid arthritis. Pharmacogenetics. 2004 Nov;14(11):733–9. doi: 10.1097/00008571-200411000-00004. [DOI] [PubMed] [Google Scholar]
- [53].Zeng QY, Wang YK, Xiao ZY, Chen SB. Pharmacogenetic study of 5,10-methylenetetrahydrofolate reductase C677T and thymidylate synthase 3R/2R gene polymorphisms and methotrexate-related toxicity in Chinese Han patients with inflammatory arthritis. Ann Rheum Dis. 2008 Aug;67(8):1193–4. doi: 10.1136/ard.2007.085266. [DOI] [PubMed] [Google Scholar]
- [54].Ghodke Y, Chopra A, Joshi K, Patwardhan B. Are Thymidylate synthase and Methylene tetrahydrofolate reductase genes linked with methotrexate response (efficacy, toxicity) in Indian (Asian) rheumatoid arthritis patients? Clinical rheumatology. 2008 Jun;27(6):787–9. doi: 10.1007/s10067-008-0852-x. [DOI] [PubMed] [Google Scholar]
- [55].Kumagai K, Hiyama K, Oyama T, Maeda H, Kohno N. Polymorphisms in the thymidylate synthase and methylenetetrahydrofolate reductase genes and sensitivity to the low-dose methotrexate therapy in patients with rheumatoid arthritis. International journal of molecular medicine. 2003 May;11(5):593–600. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.