

Abstract
Until recently, the genetic basis of neuroblastoma, a heterogeneous neoplasm arising from the developing sympathetic nervous system, remained undefined. The discovery of gain-of-function mutations in the ALK receptor tyrosine kinase gene as the major cause of familial neuroblastoma led to the discovery of identical somatic mutations and rapid advancement of ALK as a tractable therapeutic target. Inactivating mutations in a master regulator of neural crest development, PHOX2B, have also been identified in a subset of familial neuroblastomas. Other high penetrance susceptibility alleles likely exist, but together these heritable mutations account for less than 10% of neuroblastoma cases. A genome-wide association study of a large neuroblastoma cohort identified common and rare polymorphisms highly associated with the disease. Ongoing resequencing efforts aim to further define the genetic landscape of neuroblastoma.
Keywords: Genome-wide association study, Cancer, Pediatrics, Hereditary
Introduction
Neuroblastoma is the most common solid extracranial malignancy of childhood, accounting for about 7% of all cancers in children under the age of 15.[1] It is the most common cancer in the first year of life, with a median age of diagnosis of 17 months.[1,2] It is a cancer of the developing sympathetic nervous system, arising in the adrenal medulla or paraspinal ganglia.[3] Approximately 65% of these tumors present in the abdomen, along with the neck, pelvis and chest.[4] Clinical course can vary widely, with infants often having spontaneous regression of the tumor without chemotherapy,[5–8] while older children generally have a poor prognosis despite highly intensive chemotherapy, radiation therapy, and immunotherapy.[4] Demonstrating the phenotypic heterogeneity of neuroblastoma, low-risk patients have a greater than 95% survival probability whereas high-risk patients have a 40–50% probability of long-term survival. [9,10] It has been known for some time that MYCN amplification in tumors portends a poor prognosis,[11–13] and thus is used as a biomarker for treatment stratification. Recently, there has been significant effort made to better classify subgroups of patients based on age, and tumor spread, genomics and differentiation.[14–18] The International Neuroblastoma Risk Group (INRG) classification has led to 16 statistically distinct risk groups based on clinical and molecular features which has made prognosis more accurate for patients and helps guide physicians on treatment regimens.[15]
Significant progress has been made recently in the understanding of the heritability of neuroblastoma through linkage scans of families with the disease and genome-wide association studies (GWAS) of sporadic cases (Table 1). The primary advantages of GWAS over previous methods are that no assumptions about candidate genes are necessary, variations can be localized precisely, and no testing in families or family members is required.[19] From a clinical standpoint, it is clear that improvement must continue to be made in defining novel therapeutic approaches to neuroblastoma as it continues to account for 12% of childhood cancer mortality,[10] with advancement especially crucial in high-risk patients.[20] One starting point to develop optimal treatments is to understand the underlying genetic alterations that initiate tumorigenesis. We review here the current understanding of the genetic susceptibility of neuroblastoma.
Table 1.
Summary of neuroblastoma susceptibility loci. A majority of this cohort of genomic loci are significantly associated with distinct neuroblastoma phenotypes, while some remain to be characterized. P values and Odds Ratios (ORs) are combined values between discovery and replication studies from the original publication. Predicted mechanisms on protein function are indicated as loss of function, gain of function, or currently unknown. MAF = minor allele frequency.
| Genomic Locus | Phenotype Association | Top SNP | P-Value (combined) | MAF cases | Odds Ratio (OR) | Proposed Mechanism |
|---|---|---|---|---|---|---|
| 6p22 | High-risk | rs6939340 | 9.33 × 10−15 | 56% | 1.37 | Loss of function |
| 2q35 | High-risk | rs6435862 | 5.20 × 10−18 | 40% | 1.68 | Gain of function |
| 11p15 | High-risk | rs110419 | 5.20 × 10−16 | 55% | 1.34 | Gain of function |
| 6q16 | High-risk | rs17065417 | 1.20 × 10−8 | 8% | 1.38 | Gain of function |
| 6q16 | High-risk | rs4336470 | 2.70 × 10−7 | 30% | 1.26 | Loss of function |
| 1q23 | Low-risk | rs1027702 | 2.07 × 10−6 | 31% | 2.01 | unknown |
| 5q11 | Low-risk | rs2619046 | 2.94 × 10−6 | 32% | 1.47 | unknown |
| 5q11 | Low-risk | rs10055201 | 6.54 × 10−7 | 29% | 1.49 | unknown |
| 11p11 | Low-risk | rs11037575 | 4.20 × 10−7 | 39% | 1.67 | unknown |
| 8p21 | rs11994014 | 0.005 | 20% | 0.88 | Loss of function | |
| 17p13 | rs35850753 | 3.34 × 10−14 | 3.6% | 2.7 | Loss of function | |
| 1q21 | CNV | 2.97 × 10−17 | 15% | 2.49 | unknown |
Familial Neuroblastoma
About 1–2% of neuroblastoma is inherited in an autosomal dominant fashion within families.[21–24] As with many cancer predisposition syndromes, patients often have multiple primary tumor sites and an earlier age of onset. The disease is typically highly penetrant, but there is variability and unaffected obligate carriers are often observed.[21,22,25] Neuroblastoma families often show significant clinical variability in severity of disease, with low- and high-risk cases observed in the same pedigrees.[26–30] While rare, these families provide a unique opportunity to learn about genetic drivers of neuroblastoma.
The first gene found to predispose to neuroblastoma was identified in families affected with neuroblastoma along with Hirschsprung disease and/or congenital central hypoventilation syndrome (also known as “Ondine’s Curse”). These disorders of neural crest-derived cells are known as neurocristopathies and are occasionally seen coincident with neuroblastoma.[31–35] Amiel and colleagues identified loss of function mutations in the paired-like homeobox 2B (PHOX2B) gene in the majority of patients with congenital central hypoventilation syndrome after sequencing this candidate gene.[36,37] This gene was of interest because the PHOX2B transcription factor is essential during development of the autonomic nervous system. Germline mutations in PHOX2B were subsequently found in a small proportion (~10%) of pedigrees with familial neuroblastoma, making this the first bone fide neuroblastoma predisposition gene.[38,39] As expected, the families with PHOX2B mutations also had variable penetrance of each of the component neurocristopathies, with non-polyalanine repeat expansion mutations (NPARM) typically lead to the most severe phenotype. [40,41]
In order to identify additional hereditary predisposition genes in the familial neuroblastoma cases, a genome-wide linkage scan at 6,000 single nucleotide polymorphisms (SNPs) was undertaken in 20 neuroblastoma families.[25] A linkage signal was found and narrowed down to chromosome bands 2p23–p24, which contained 104 genes including MYCN. This known neuroblastoma oncogene was resequenced in all probands, but no mutation was found. The anaplastic lymphoma kinase (ALK) is also in this region and had been previously identified as a potential oncogene in this malignancy[42,43] as well as in other cancers through active translocations and point mutations.[44–51] When ALK was resequenced, three distinct mutations were found in this gene in eight discrete families.[25] Subsequent studies have confirmed that about 80% of families with neuroblastoma harbor mutations in ALK. Mutations in ALK were also found to be somatically acquired in about 10% of all cases of neuroblastoma.[25,52–54] ALK is a receptor tyrosine kinase, and all of these were activating mutations in the tyrosine kinase domain that caused constitutive phosphorylation and were predicted to be oncogenic drivers.[25] While Knudson and Strong’s prediction of a “two-hit” model has held true for most hereditary cancers,[21] these susceptibility genes are usually tumor suppressor genes. In contrast, ALK was the first oncogene mutation shown to cause a familial pediatric cancer. The Mosse lab has subsequently biochemically characterized each of the germline and somatic mutations, and there is a correlation between penetrance and mutation type. [55,56] For example, the R1275Q mutation leads to near complete penetrance in families and was shown to be one of the most activating mutations tolerated in the germline, whereas the G1128A is more weakly activating and is correlated with an approximate 25% likelihood of developing neuroblastoma. Interestingly, the two most highly activating hotspot mutations acquired somatically (F1174* and F1245*) were each observed in the germline once, but in the setting of neuroblastoma with severe neurocognitive defects and brain stem abnormalities, further emphasizing the genotype-phenotype relationship as well as the critical role plays in normal neurodevelopment.[57] Genetic testing for both ALK and PHOX2B are currently available for identifiying genetic susceptibility and informing decisions about screening other family members (http://www.ncbi.nlm.nih.gov/sites/GeneTests/).
ALK was quickly identified as a potential pharmacologic target in neuroblastoma when knockdown of ALK resulted in growth inhibition in all neuroblastoma cell lines with ALK mutations and some with wild-type ALK.[25] Further testing with an ALK small molecule inhibitor, crizotinib, showed profound sensitivity in vitro and in vivo to the drug in a panel of neuroblastoma cell lines and xenografts, respectively, with certain mutations and ALK amplification.[56,58–60] Based on these data, only 18 months after ALK was discovered as a neuroblastoma oncogene, the Children’s Oncology Group initiated a Phase I/II clinical trial testing crizotinib in patients with relapsed pediatric solid tumors and anaplastic large cell lymphoma (ALCL) (www.clinicaltrials.gov, Identifier: NCT00939770). Toxicity has remained low, and seven patients with ALCL and two patients with neuroblastoma have had complete responses as the trial continues.[61] This is a hallmark example of how identifying genetic susceptibility can be quickly advanced for clinical benefit.
However, there are some families that do not show mutations in ALK or PHOX2B, thus the search for additional familial neuroblastoma gene continues. Whole exome analysis of one family with two affected cousins and two healthy members showed a mutation in GALNT14 predicted to be functionally damaging, but continued efforts are necessary to further define this familial variant.[113] In parallel, germline mutations in TP53, SDHB, PTPN11, APC, and NF1 have been reported to occur rarely in neuroblastoma patients (Figure 1).[62–68,69] Neuroblastoma has also been reported to arise in complex congenital malformation syndromes, such as the subtelomeric 1p36.3 or 11q23 deletions.[70,71] The heritability of neuroblastoma remains only partially understood, yet continued investigation is expected to reveal new insights into familial neuroblastoma predisposition, including gene-gene and gene-environment interactions.
Figure 1.
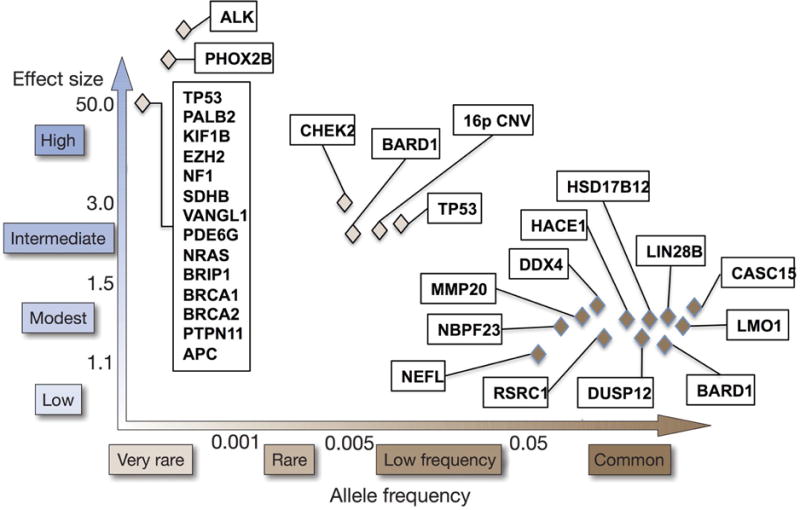
Graphical representation of genetic predisposition to neuroblastoma. Known familial and sporadic predisposition genes have been compiled into one summary figure across multiple studies. The familial mutations are shown in the top left of the graph representing a very rare allele frequency and high effect size. GWAS-discovered variations are in the bottom right corner representing a higher allele frequency with a lower effect size. Continued sequencing efforts are likely to uncover additional rare susceptibility variants along this spectrum, of which dozens are predicted to be discovered to explain the heritability of neuroblastoma.
Genetic susceptibility to sporadic neuroblastoma
In familial neuroblastoma, there are rare mutations that lead to a high probability of disease. For the 99% of cases that occur sporadically, a common variant hypothesis proposes that common germline variations influence the probability of disease occurrence, each with a low relative risk, but presumably acting in concert. A large GWAS consisting of 720 neuroblastoma cases and 2,128 controls was undertaken in neuroblastoma as an unbiased method for discovering these polymorphisms (Figure 2).[89] This original GWAS has been expanded and replicated as additional patient samples have been accrued, leading to the identification of DNA alleles significantly associated with high-risk and low-risk neuroblastoma predisposition, including CASC15, BARD1, LMO1, LIN28B, HACE1, DUSP12, DDX4, IL31RA, HSD17B12, NEFL, TP53, AND NBPF23 (Table 1).[72, 74, 75, 79, 81, 88, 89, 102–104, 107] The discovery of these susceptibility loci demonstrates the utility of interrogating GWAS signals for clues into the underlying biology driving neuroblastoma genesis.
Figure 2.
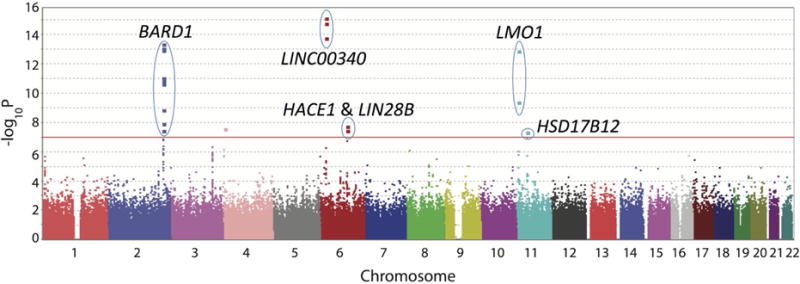
Manhattan plot of high-risk neuroblastoma GWAS results across multiple studies. Level of significance (−log10 transformed p values) for each SNP along the genome in chromosomal order is plotted, and the corresponding genes are labeled. Red line: genome-wide significance threshold based on Bonferroni adjustment. Adapted from Diskin, et al. 2012.
Results from the initial GWAS identified three SNPs at chromosome 6p22 within a newly identified long noncoding RNA (lncRNA) annotated as CASC15.[72] Homozygosity for the risk alleles was significantly associated with metastatic disease, amplification of MYCN oncogene in the tumors, and patient relapse. Recently, decreased expression of the truncated isoform CASC15-S was associated with more advanced disease. [73] Another lncRNA, NBAT-1 (CASC14), was shown to be located at the 6p22 susceptibility locus as well, and functional studies have shown that loss of NBAT-1 promotes proliferation and invasion. [74] Subsequently, a GWAS restricted to high-risk neuroblastoma identified the BRCA-associated ring domain-1 gene (BARD1) at chromosome 2q35 was identified as a susceptibility locus.[75] Six SNPs were discovered in three different N-terminal introns of this gene. BARD1, along with its binding partner, breast cancer 1, early onset (BRCA1), had been previously implicated in breast and other cancers, but genetic variants in BARD1 had not been shown to lead to cancer susceptibility, even in breast cancer.[76–78] Continuing efforts in BARD1 have found that an isoform, BARD1β, which lacks the RING domain necessary for BRCA1 binding, is preferentially expressed in neuroblastoma cell lines that are homozygous for the risk alleles.[79] Consistent with oncogenic behavior, knockdown of this isoform inhibits cell growth, while overexpression leads to increased proliferation. Additionally, BARD1β was found to stabilize the Aurora family of kinases in neuroblastoma cell lines, suggesting a possible mechanism of action and potential therapeutic strategy as Aurora kinase inhibitors are in clinical development for cancer.[79,80]
This GWAS was expanded (2,251 neuroblastoma cases and 6,097 controls) and the gene LMO1 was shown to be significantly associated with high-risk neuroblastoma, which had previously been implicated in human cancer, but not neuroblastoma. Four SNPs that were significantly associated with neuroblastoma at chromosome 11p15.4 were within the LIM domain only 1 (LMO1) gene.[81] This gene, along with LMO2, LMO3 and LMO4, encodes a cysteine-rich transcriptional cofactor that is preferentially expressed in the nervous system.[82] This family of genes has been found to be critically involved in leukemia (reviewed in ref. [83]) and breast cancers,[84–86] while LMO3 has been shown to be oncogenic in neuroblastoma through its interaction with a neuronal-restricted transcription factor.[87] These common variations in LMO1 were found to be associated with high-risk disease and decreased survival.[81] Neuroblastoma tumors with LMO1 risk alleles were found to have increased expression of LMO1, and depletion of LMO1 in cell lines decreased growth while forced over-expression increased growth.[81] This is consistent with a gain-of-function role in tumor progression. Recent investigation showed that the causal SNP resides in a super enhancer element within the first intron, with the G>A transversion ablating a canonical GATA transcription factor binding site. [88] Investigators showed that the A allele was “protective”, as there was no GATA binding, and not cis-mediated LMO1 transcription, providing one of the first clear mechanistic insights into a genetic association.
By further expanding this GWAS to 2,817 neuroblastoma cases and 7,473 controls, two new association signals emerged at 6q16 in two different genes, HACE1 and LIN28B.[89] HACE1 encodes an E3 ubiquitin ligase and has been identified as a tumor suppressor gene silenced in Wilms’ tumors, colorectal cancer, and gastric carcinoma.[90–92] It has also been shown to suppress cell growth in human cancer cells, including a neuroblastoma cell line, by inhibiting cell cycle progression during stress.[93] LIN28B, a known oncogene, encodes an RNA-binding protein that is developmentally regulated and blocks the expression of the let-7 family of microRNAs.[94] High expression of LIN28B and correlated low levels of let-7 have been observed in many human cancers.[95,96] LIN28B and let-7 are involved in stem cell differentiation, as overexpressing the former or inhibiting the latter leads to the reprogramming of human and mouse fibroblasts into pluripotent stem cells.[97,98] In the GWAS, LIN28B was expressed at significantly higher levels in neuroblastoma cell lines homozygous for the risk allele, and this correlated with lower levels of let-7 and growth inhibition following knockdown of LIN28B.[89] In tumor samples, HACE1 expression was significantly lower and LIN28B significantly higher in high-risk neuroblastomas and were correlated similarly with worse overall survival. Mechanistic studies have shown that LIN28B promotes increased expression of the oncogenic protein RAN, which both converge on Aurora Kinase A.[99] Increased activity was shown to drive tumorigenesis, providing further evidence that targeting Aurora kinases may provide a benefit to neuroblastoma patients.[100,101]
In an integrated proteomic-GWAS approach, Capasso identified three SNPs significantly associated with neuroblastoma in the NEFL gene, encoding the light chain neurofilament protein in which mutations are known in disorders of the peripheral nervous system.[102] Overexpression of NEFL in cells with a protective allele caused cells to adopt a more differentiated phenotype and to have reduced proliferative capacity. The authors suggested that decreased expression of NEFL alters the differentiation state of sympathetic neurons and may predispose neuroblastoma.[102]
After enriching the GWAS for patients with low-risk neuroblastoma, SNPs in four genes, DUSP12, DDX4, IL31RA and HSD17B12, were discovered to be significantly associated with this phenotypic subset.[103] These genes are different than those found in high-risk neuroblastoma, suggesting these subtypes are likely genetically distinct and emphasizing the importance of robust phenotypic information in GWAS efforts. These data further support the notion that widely divergent neuroblastoma phenotypes are genetically predetermined.
A genome wide SNP scan for copy number variation (CNV) identified a novel CNV at 1q21.1 that is associated with neuroblastoma, and they were able to confirm deletions in this region by quantitative PCR and FISH.[104] A new neuroblastoma breakpoint family gene, NBPF23, was identified at this location by a transcript that was similar to other genes in the family. This transcript is most commonly expressed in fetal brain and sympathetic nervous system tissues, and in neuroblastoma, its expression was correlated with this CNV. NBPF1 was identified originally at the translocation breakpoint in the germline of a child with neuroblastoma,[68] and research continues to elucidate the role of this family of genes in disease development.
The prevalence of GWAS-associated genes has been further interrogated among different ethnic groups. A follow up study to the previously described BARD1 GWAS was carried out in African American children with neuroblastoma looking at SNPs in the gene regions identified by the GWAS in Caucasians.[106] Two of the six SNPs found in BARD1 were also significantly associated with neuroblastoma in the African-American cohort, validating the original GWAS. Due to different patterns of linkage disequilibrium in the two ethnicities, this effort narrowed the potential location of the causal variant. Another study in patients of African descent identified an allele in a new gene, sperm associated antigen 16 (SPAG16), associated with high-risk neuroblastoma in patients of both African and European ancestry showing the potential of discovering new associations by studying specific ethnic groups.[107]
In the Oldridge manuscript noted above defining a mechanistic basis for the LMO1 association, the protective T-allele was noted to be common in people of European ancestry, but is largely absent in African and African-American populations, which retain the G-allele.[102] This may provide a partial explanation for the more aggressive forms of neuroblastoma observed in African-American patients. Altogether, these results indicate that ethnic background may play a role in genetic predisposition and that therapeutic approaches may require requisite tailoring.
Collectively, these GWAS-discovered genes account for only a small portion of neuroblastoma heritability, which remains poorly understood. It is likely that further expansion of GWAS efforts will continue to uncover more susceptibility genes that will confer risk in an additive manner. No epistasis was found when the most significant SNPs from 2q35, 6p22, 11p15.4 and 1q21.1 CNV were studied together;[81] however, specific clusters of combinations of these SNPs were significantly associated with neuroblastoma.[105] Mechanistic insights are being discovered, but the underlying basis for most statistical associations remain unknown. Neuroblastoma GWASs were expected to discover genes that affect development of the sympathetic nervous system, showing that common variants can lead to missteps in development and therefore malignancies. Investigators are pursuing ongoing studies to model GWAS variants and heritability in zebrafish and induced pluripotent stem cell models to understand the biological consequences in neuroblastoma and investigate potential therapeutic interventions.
Rare Variants
There are currently two main groups of germline DNA variations that predispose to neuroblastoma: very rare genetic mutations leading to Mendelian inheritance of familial neuroblastoma with a high penetrance, and common variations that only increase risk of disease in small increments. These discoveries thus far have only explained a small proportion of the heritability of neuroblastoma. While further expansion of the GWAS will continue to uncover more common variants and genes important in the development of neuroblastoma, we suggest that these discoveries lie on a spectrum with the middle ground only beginning to be realized (Figure 1). These are rare germline variations or mutations with a lower penetrance than familial disease but with a larger effect on predisposition than the common SNPs. Owing to their rarity and the relatively small number of patients with neuroblastoma, it has been difficult to identify these rare variants. Recently, two rare germline variants in TP53 were found to be robustly associated with neuroblastoma using the 1000 Genomes Project[108] and an advanced imputation process elucidating associations with SNPs not directly assayed on the limited arrays.[109] Likewise, germline sequencing has identified putative damaging mutations in ALK, CHEK2, PINK1, BARD1 and APC1 in small percentages of patients with neuroblastoma.[69,110]. As sequencing technology improves and costs decrease, discoveries of additional rare variants are on the horizon to define and characterize further the heritability of neuroblastoma. The influence of germline mosaicism and epistatic interaction of de novo or inherited mutations with GWAS-defined polymorphisms remains undefined.
Summary and Future Directions
Significant progress has been made in the last six years in describing the genetic landscape of neuroblastoma and continuing studies will aim to further identify Mendelian susceptibility genes. This is already influencing clinical care as genetic testing is available, and there are noninvasive screening methods to surveil for disease in young children. Current recommendations suggest that children with a known damaging germline mutation in ALK or PHOX2B based on familial pedigrees should undergo surveillance with every 3-month ultrasonography and urinary catecholamines until a minimum of age 5, if not beyond.[111] The main impact of GWAS studies to date is in identifying genes critical to neuroblastoma progression and maintenance, thus uncovering potential oncogenic vulnerabilities. With the discovery of ALK as an example, it is important that translational approaches related to these genes be prioritized, as additional targeted therapies for patients with neuroblastoma are essential to improving survival. Future work to extend the discovery of germline polymorphisms to those that influence response to therapy and impact co-morbidities such as hearing loss also has the potential to improve patient survival and quality of life. The ultimate goal of genomic studies in neuroblastoma is to inform precision medicine with genetic evaluations to tailor clinical treatments and extend survival.[113] As additional patient samples are accrued over time, future GWAS endeavors will be required to continue the discovery of additional susceptibility alleles. Extensive further investigation, both computationally and in designing better models for these rare genetically defined subsets, will be required to translate these genomic discoveries into actionable targets for diagnosis and treatment.
Acknowledgments
This work was supported by the Institutional Clinical and Translational Science Award, grant number TL1 RR024133, and John Maris’ grant R01 CA124709: The Genetic Basis of Neuroblastoma Tumorigenesis.
Grant support: This work was supported by the Institutional Clinical and Translational Science Award, grant number TL1 RR024133, and John Maris’ grant R01 CA124709: The Genetic Basis of Neuroblastoma Tumorigenesis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that there are no actual or perceived conflicts of interest.
References
- 1.Howlader N, Noone A, Krapcho M, Neyman N, Aminou R, Waldron W, Altekruse S, Kosary C, Ruhl J, Tatalovich Z, et al. SEER Cancer Statistics Review1975–2008based on November 2010 SEER data submission. National Cancer Institute; BethesdaMD: 2011. Edited by. [Google Scholar]
- 2.London WB, Castleberry RP, Matthay KK, Look AT, Seeger RC, Shimada H, Thorner P, Brodeur G, Maris JM, Reynolds CP, et al. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children’s Oncology Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23:6459–6465. doi: 10.1200/JCO.2005.05.571. [DOI] [PubMed] [Google Scholar]
- 3.Hoehner JC, Gestblom C, Hedborg F, Sandstedt B, Olsen L, Pahlman S. A developmental model of neuroblastoma: differentiating stroma-poor tumors’ progress along an extra-adrenal chromaffin lineage. Lab Invest. 1996;75:659–675. [PubMed] [Google Scholar]
- 4.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 5.Carlsen NL. The new International Neuroblastoma Staging System: some critical notes. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1990;8:935–936. doi: 10.1200/JCO.1990.8.5.935. [DOI] [PubMed] [Google Scholar]
- 6.Cole WH, Everson TC. Spontaneous regression of cancer: preliminary report. Ann Surg. 1956;144:366–383. doi: 10.1097/00000658-195609000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamamoto K, Hanada R, Kikuchi A, Ichikawa M, Aihara T, Oguma E, Moritani T, Shimanuki Y, Tanimura M, Hayashi Y. Spontaneous regression of localized neuroblastoma detected by mass screening. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1998;16:1265–1269. doi: 10.1200/JCO.1998.16.4.1265. [DOI] [PubMed] [Google Scholar]
- 8.Hero B, Simon T, Spitz R, Ernestus K, Gnekow AK, Scheel-Walter HG, Schwabe D, Schilling FH, Benz-Bohm G, Berthold F. Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:1504–1510. doi: 10.1200/JCO.2007.12.3349. [DOI] [PubMed] [Google Scholar]
- 9.Oberthuer A, Juraeva D, Hero B, Volland R, Sterz C, Schmidt R, Faldum A, Kahlert Y, Engesser A, Asgharzadeh S, et al. Revised risk estimation and treatment stratification of low- and intermediate-risk neuroblastoma patients by integrating clinical and molecular prognostic markers. Clin Cancer Res. 2015;21:1904–1915. doi: 10.1158/1078-0432.CCR-14-0817. *Event free survival and overall survival of low- and intermediate-risk neuroblastoma patients were stratified and utilized to propose revised courses of treatment based on risk group. [DOI] [PubMed] [Google Scholar]
- 10.Maris JM. Recent advances in neuroblastoma. The New England Journal of Medicine. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, Brodeur G, Goldstein M, Trent J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. 1983;305:245–248. doi: 10.1038/305245a0. [DOI] [PubMed] [Google Scholar]
- 12.Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- 13.Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, Hammond D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. New England Journal of Medicine. 1985;313:1111–1116. doi: 10.1056/NEJM198510313131802. [DOI] [PubMed] [Google Scholar]
- 14.Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, De Bernardi B, Evans AE, Favrot M, Hedborg F, et al. Revisions of the international criteria for neuroblastoma diagnosisstagingand response to treatment. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1993;11:1466–1477. doi: 10.1200/JCO.1993.11.8.1466. [DOI] [PubMed] [Google Scholar]
- 15.Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:289–297. doi: 10.1200/JCO.2008.16.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cecchetto G, Mosseri V, De Bernardi B, Helardot P, Monclair T, Costa E, Horcher E, Neuenschwander S, Toma P, Rizzo A, et al. Surgical risk factors in primary surgery for localized neuroblastoma: the LNESG1 study of the European International Society of Pediatric Oncology Neuroblastoma Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23:8483–8489. doi: 10.1200/JCO.2005.02.4661. [DOI] [PubMed] [Google Scholar]
- 17.Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, Kaneko M, London WB, Matthay KK, Nuchtern JG, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:298–303. doi: 10.1200/JCO.2008.16.6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deyell RJ, Attiyeh EF. Advances in the understanding of constitutional and somatic genomic alterations in neuroblastoma. Cancer Genet. 2011;204:113–121. doi: 10.1016/j.cancergen.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 20.Tonini GP, Nakagawara A, Berthold F. Towards a turning point of neuroblastoma therapy. Cancer Lett. 2012;326:128–134. doi: 10.1016/j.canlet.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 21.Knudson AG, Jr, Strong LC. Mutation and cancer: neuroblastoma and pheochromocytoma. Am J Hum Genet. 1972;24:514–532. [PMC free article] [PubMed] [Google Scholar]
- 22.Kushner BH, Gilbert F, Helson L. Familial neuroblastoma. Case reports literature review and etiologic considerations. Cancer. 1986;57:1887–1893. doi: 10.1002/1097-0142(19860501)57:9<1887::aid-cncr2820570931>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 23.Dodge HJ. BM: Neuroblastoma of the adrenal medulla in siblings. Rocky Mt Med. 1945;42:35–38. [Google Scholar]
- 24.Chompret A, dVF, Brugieres L, Abel A, Raquin MA, Hartmann O, et al. Excess of cancers in relatives of patients with neuroblastoma. Med Pediatr Oncol. 1998;31:211. [Google Scholar]
- 25.Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–935. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardy PC, Nesbit ME., Jr Familial neuroblastoma: report of a kindred with a high incidence of infantile tumors. The Journal of pediatrics. 1972;80:74–77. doi: 10.1016/s0022-3476(72)80456-6. [DOI] [PubMed] [Google Scholar]
- 27.Gerson JM, Chatten J, Eisman S. Letter: Familial neuroblastoma: a follow-up. N Engl J Med. 1974(290):1487. doi: 10.1056/NEJM197406272902615. [DOI] [PubMed] [Google Scholar]
- 28.Wong KY, Hanenson IB, Lampkin BC. Familial neuroblastoma. Am J Dis Child. 1971;121:415–416. doi: 10.1001/archpedi.1971.02100160085010. [DOI] [PubMed] [Google Scholar]
- 29.Bergstrom JF, Long JM. Familial occurrence of ganglioneuromas. Tex Med. 1974;70:62–65. [PubMed] [Google Scholar]
- 30.Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nature reviews Cancer. 2003;3:203–216. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 31.Bolande RP. Neurocristopathy: its growth and development in 20 years. Pediatr Pathol Lab Med. 1997;17:1–25. [PubMed] [Google Scholar]
- 32.Bower RJ, Adkins JC. Ondine’s curse and neurocristopathy. Clinical pediatrics. 1980;19:665–668. doi: 10.1177/000992288001901004. [DOI] [PubMed] [Google Scholar]
- 33.Michna BA, McWilliams NB, Krummel TM, Hartenberg MA, Salzberg AM. Multifocal ganglioneuroblastoma coexistent with total colonic aganglionosis. J Pediatr Surg. 1988;23:57–59. doi: 10.1016/s0022-3468(88)80541-4. [DOI] [PubMed] [Google Scholar]
- 34.Roshkow JE, Haller JO, Berdon WE, Sane SM. Hirschsprung’s disase, Ondine’s curse, and neuroblastoma–manifestations of neurocristopathy. Pediatr Radiol. 1988;19:45–49. doi: 10.1007/BF02388410. [DOI] [PubMed] [Google Scholar]
- 35.Stovroff M, Dykes F, Teague WG. The complete spectrum of neurocristopathy in an infant with congenital hypoventilation, Hirschsprung’s disease, and neuroblastoma. J Pediatr Surg. 1995;30:1218–1221. doi: 10.1016/0022-3468(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 36.Amiel J, Laudier B, Attie-Bitach T, Trang H, de Pontual L, Gener B, Trochet D, Etchevers H, Ray P, Simonneau M, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nature genetics. 2003;33:459–461. doi: 10.1038/ng1130. [DOI] [PubMed] [Google Scholar]
- 37.Weese-Mayer DE, Berry-Kravis EM, Zhou L, Maher BS, Silvestri JM, Curran ME, Marazita ML. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2b. Am J Med Genet A. 2003;123A:267–278. doi: 10.1002/ajmg.a.20527. [DOI] [PubMed] [Google Scholar]
- 38.Trochet D, Bourdeaut F, Janoueix-Lerosey I, Deville A, de Pontual L, Schleiermacher G, Coze C, Philip N, Frebourg T, Munnich A, et al. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet. 2004;74:761–764. doi: 10.1086/383253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mosse YP, Laudenslager M, Khazi D, Carlisle AJ, Winter CL, Rappaport E, Maris JM. Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet. 2004;75:727–730. doi: 10.1086/424530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heide S, Masliah-Planchon J, Isidor B, Guimier A, Bodet D, Coze C, Deville A, Thebault E, Pasquier CJ, Cassagnau E, et al. Oncologic Phenotype of Peripheral Neuroblastic Tumors Associated With PHOX2B Non-Polyalanine Repeat Expansion Mutations. Pediatr Blood Cancer. 2016;63:71–77. doi: 10.1002/pbc.25723. *This study identified four patients with poorly differentiated nueroblastomas with PHOX2B NPARM, indicating that this predisposition can be associated with more aggressive cancer phenotypes. [DOI] [PubMed] [Google Scholar]
- 41.Nagashimada M, Ohta H, Li C, Nakao K, Uesaka T, Brunet JF, Amiel J, Trochet D, Wakayama T, Enomoto H. Autonomic neurocristopathy-associated mutations in PHOX2B dysregulate Sox10 expression. J Clin Invest. 2012;122:3145–3158. doi: 10.1172/JCI63401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osajima-Hakomori Y, Miyake I, Ohira M, Nakagawara A, Nakagawa A, Sakai R. Biological role of anaplastic lymphoma kinase in neuroblastoma. Am J Pathol. 2005;167:213–222. doi: 10.1016/S0002-9440(10)62966-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.George RE, Attiyeh EF, Li S, Moreau LA, Neuberg D, Li C, Fox EA, Meyerson M, Diller L, Fortina P, et al. Genome-wide analysis of neuroblastomas using high-density single nucleotide polymorphism arrays. PLoS One. 2007;2:e255. doi: 10.1371/journal.pone.0000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T, Perlman EJ. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res. 1999;59:2776–2780. [PubMed] [Google Scholar]
- 45.Jazii FR, Najafi Z, Malekzadeh R, Conrads TP, Ziaee AA, Abnet C, Yazdznbod M, Karkhane AA, Salekdeh GH. Identification of squamous cell carcinoma associated proteins by proteomics and loss of beta tropomyosin expression in esophageal cancer. World J Gastroenterol. 2006;12:7104–7112. doi: 10.3748/wjg.v12.i44.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 47.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 48.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 49.Inamura K, Takeuchi K, Togashi Y, Nomura K, Ninomiya H, Okui M, Satoh Y, Okumura S, Nakagawa K, Soda M, et al. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J Thorac Oncol. 2008;3:13–17. doi: 10.1097/JTO.0b013e31815e8b60. [DOI] [PubMed] [Google Scholar]
- 50.Wang YW, Tu PH, Lin KT, Lin SC, Ko JY, Jou YS. Identification of oncogenic point mutations and hyperphosphorylation of anaplastic lymphoma kinase in lung cancer. Neoplasia. 2011;13:704–715. doi: 10.1593/neo.11222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murugan AK, Xing M. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res. 2011;71:4403–4411. doi: 10.1158/0008-5472.CAN-10-4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–970. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- 53.George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–978. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–974. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- 55.Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, Laudenslager M, Rappaport EF, Wood AC, McGrady PW, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. 2014;26:682–694. doi: 10.1016/j.ccell.2014.09.019. *Analyses of ALK mutations in over 1,500 neuroblastoma tumor samples characterized oncogenic and passenger mutations using biochemical assays and computational prediction studies. Crizotinib sensitivity varied among the ALK mutant variants suggesting differential clinical outcomes based on ALK genomic status. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bresler SC, Wood AC, Haglund EA, Courtright J, Belcastro LT, Plegaria JS, Cole K, Toporovskaya Y, Zhao H, Carpenter EL, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci Transl Med. 2011;3:108ra114. doi: 10.1126/scitranslmed.3002950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Pontual L, Kettaneh D, Gordon CT, Oufadem M, Boddaert N, Lees M, Balu L, Lachassinne E, Petros A, Mollet J, et al. Germline gain-of-function mutations of ALK disrupt central nervous system development. Hum Mutat. 2011;32:272–276. doi: 10.1002/humu.21442. [DOI] [PubMed] [Google Scholar]
- 58.Schonherr C, Ruuth K, Yamazaki Y, Eriksson T, Christensen J, Palmer RH, Hallberg B. Activating ALK mutations found in neuroblastoma are inhibited by Crizotinib and NVP-TAE684. Biochem J. 2011;440:405–413. doi: 10.1042/BJ20101796. [DOI] [PubMed] [Google Scholar]
- 59.Heuckmann JM, Holzel M, Sos ML, Heynck S, Balke-Want H, Koker M, Peifer M, Weiss J, Lovly CM, Grutter C, et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin Cancer Res. 2011;17:7394–7401. doi: 10.1158/1078-0432.CCR-11-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carpenter EL, Mosse YP. Targeting ALK in neuroblastoma–preclinical and clinical advancements. Nat Rev Clin Oncol. 2012;9:391–399. doi: 10.1038/nrclinonc.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, Rolland D, Balis FM, Maris JM, Weigel BJ, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14:472–480. doi: 10.1016/S1470-2045(13)70095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, Eden OB, Varley JM. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene. 2001;20:4621–4628. doi: 10.1038/sj.onc.1204621. [DOI] [PubMed] [Google Scholar]
- 63.Hasle H. Malignant diseases in Noonan syndrome and related disorders. Horm Res. 2009;72(Suppl 2):8–14. doi: 10.1159/000243773. [DOI] [PubMed] [Google Scholar]
- 64.Mutesa L, Pierquin G, Janin N, Segers K, Thomee C, Provenzi M, Bours V. Germline PTPN11 missense mutation in a case of Noonan syndrome associated with mediastinal and retroperitoneal neuroblastic tumors. Cancer Genet Cytogenet. 2008;182:40–42. doi: 10.1016/j.cancergencyto.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 65.Chantrain CF, Jijon P, De Raedt T, Vermylen C, Poirel HA, Legius E, Brichard B. Therapy-related acute myeloid leukemia in a child with Noonan syndrome and clonal duplication of the germline PTPN11 mutation. Pediatr Blood Cancer. 2007;48:101–104. doi: 10.1002/pbc.20527. [DOI] [PubMed] [Google Scholar]
- 66.Schimke RN, Collins DL, Stolle CA. Paraganglioma, neuroblastoma, and a SDHB mutation: Resolution of a 30-year-old mystery. Am J Med Genet A. 2010;152A:1531–1535. doi: 10.1002/ajmg.a.33384. [DOI] [PubMed] [Google Scholar]
- 67.Cascon A, Landa I, Lopez-Jimenez E, Diez-Hernandez A, Buchta M, Montero-Conde C, Leskela S, Leandro-Garcia LJ, Leton R, Rodriguez-Antona C, et al. Molecular characterisation of a common SDHB deletion in paraganglioma patients. J Med Genet. 2008;45:233–238. doi: 10.1136/jmg.2007.054965. [DOI] [PubMed] [Google Scholar]
- 68.Vandepoele K, Andries V, Van Roy N, Staes K, Vandesompele J, Laureys G, De Smet E, Berx G, Speleman F, van Roy F. A constitutional translocation t(1;17)(p36.2;q11.2) in a neuroblastoma patient disrupts the human NBPF1 and ACCN1 genes. PLoS One. 2008;3:e2207. doi: 10.1371/journal.pone.0002207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, Hedges D, Ma X, Zhou X, Yergeau DA, et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med. 2015;373:2336–2346. doi: 10.1056/NEJMoa1508054. *This next-generation sequencing effort identified a new association of germline APC and SDHB mutations in neuroblastoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Isidor B, Le Cunff M, Boceno M, Boisseau P, Thomas C, Rival JM, David A, Le Caignec C. Complex constitutional subtelomeric 1p36.3 deletion/duplication in a mentally retarded child with neonatal neuroblastoma. Eur J Med Genet. 2008;51:679–684. doi: 10.1016/j.ejmg.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 71.Mosse Y, Greshock J, King A, Khazi D, Weber BL, Maris JM. Identification and high-resolution mapping of a constitutional 11q deletion in an infant with multifocal neuroblastoma. Lancet Oncol. 2003;4:769–771. doi: 10.1016/s1470-2045(03)01283-x. [DOI] [PubMed] [Google Scholar]
- 72.Maris JM, Mosse YP, Bradfield JP, Hou C, Monni S, Scott RH, Asgharzadeh S, Attiyeh EF, Diskin SJ, Laudenslager M, et al. Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. N Engl J Med. 2008;358:2585–2593. doi: 10.1056/NEJMoa0708698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Russell MR, Penikis A, Oldridge DA, Alvarez-Dominguez JR, McDaniel L, Diamond M, Padovan O, Raman P, Li Y, Wei JS, et al. CASC15-S Is a Tumor Suppressor lncRNA at the 6p22 Neuroblastoma Susceptibility Locus. Cancer Res. 2015;75:3155–3166. doi: 10.1158/0008-5472.CAN-14-3613. *This study identified CASC15 as the tumor supppressor lncRNA located at a known susceptibility locus on chromosome 6p22 and showed that reduced expression of the shortened form, CASC15-S, increased cellular migration and was associated with poor patient outcome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pandey GK. The Risk-Associated Long Noncoding RNA NBAT-1 Controls Neuroblastoma Progression by Regulating Cell Proliferation and Neuronal Differentiation. Cancer Cell. 2014 doi: 10.1016/j.ccell.2014.09.014. *Differential expression of NBAT-1, a lncRNA located at the 6p22 susceptibility locus, was identified as a predictive biomarker of patient outcome in neuroblastoma. Decrease or loss of NBAT-1 conferred increased proliferative and invasive properties in vitro via epigenetic silencing. [DOI] [PubMed] [Google Scholar]
- 75.Capasso M, Devoto M, Hou C, Asgharzadeh S, Glessner JT, Attiyeh EF, Mosse YP, Kim C, Diskin SJ, Cole KA, et al. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nat Genet. 2009;41:718–723. doi: 10.1038/ng.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, Yang MC, Hwang LY, Bowcock AM, Baer R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- 77.Irminger-Finger I, Jefford CE. Is there more to BARD1 than BRCA1? Nat Rev Cancer. 2006;6:382–391. doi: 10.1038/nrc1878. [DOI] [PubMed] [Google Scholar]
- 78.Hosking FJ, Dobbins SE, Houlston RS. Genome-wide association studies for detecting cancer susceptibility. Br Med Bull. 2011;97:27–46. doi: 10.1093/bmb/ldq038. [DOI] [PubMed] [Google Scholar]
- 79.Bosse KR, Diskin SJ, Cole KA, Wood AC, Schnepp RW, Norris G, Nguyen le B, Jagannathan J, Laquaglia M, Winter C, et al. Common variation at BARD1 results in the expression of an oncogenic isoform that influences neuroblastoma susceptibility and oncogenicity. Cancer Res. 2012;72:2068–2078. doi: 10.1158/0008-5472.CAN-11-3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ryser S, Dizin E, Jefford CE, Delaval B, Gagos S, Christodoulidou A, Krause KH, Birnbaum D, Irminger-Finger I. Distinct roles of BARD1 isoforms in mitosis: full-length BARD1 mediates Aurora B degradation, cancer-associated BARD1beta scaffolds Aurora B and BRCA2. Cancer Res. 2009;69:1125–1134. doi: 10.1158/0008-5472.CAN-08-2134. [DOI] [PubMed] [Google Scholar]
- 81.Wang K, Diskin SJ, Zhang H, Attiyeh EF, Winter C, Hou C, Schnepp RW, Diamond M, Bosse K, Mayes PA, et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature. 2011;469:216–220. doi: 10.1038/nature09609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Curtis DJ, McCormack MP. The molecular basis of Lmo2-induced T-cell acute lymphoblastic leukemia. Clin Cancer Res. 2010;16:5618–5623. doi: 10.1158/1078-0432.CCR-10-0440. [DOI] [PubMed] [Google Scholar]
- 84.Sum EY, Peng B, Yu X, Chen J, Byrne J, Lindeman GJ, Visvader JE. The LIM domain protein LMO4 interacts with the cofactor CtIP and the tumor suppressor BRCA1 and inhibits BRCA1 activity. J Biol Chem. 2002;277:7849–7856. doi: 10.1074/jbc.M110603200. [DOI] [PubMed] [Google Scholar]
- 85.Visvader JE, Venter D, Hahm K, Santamaria M, Sum EY, O’Reilly L, White D, Williams R, Armes J, Lindeman GJ. The LIM domain gene LMO4 inhibits differentiation of mammary epithelial cells in vitro and is overexpressed in breast cancer. Proc Natl Acad Sci U S A. 2001;98:14452–14457. doi: 10.1073/pnas.251547698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Montanez-Wiscovich ME, Seachrist DD, Landis MD, Visvader J, Andersen B, Keri RA. LMO4 is an essential mediator of ErbB2/HER2/Neu-induced breast cancer cell cycle progression. Oncogene. 2009;28:3608–3618. doi: 10.1038/onc.2009.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aoyama M, Ozaki T, Inuzuka H, Tomotsune D, Hirato J, Okamoto Y, Tokita H, Ohira M, Nakagawara A. LMO3 interacts with neuronal transcription factor, HEN2, and acts as an oncogene in neuroblastoma. Cancer Res. 2005;65:4587–4597. doi: 10.1158/0008-5472.CAN-04-4630. [DOI] [PubMed] [Google Scholar]
- 88.Oldridge DA, Wood AC, Weichert-Leahey N, Crimmins I, Sussman R, Winter C, McDaniel LD, Diamond M, Hart LS, Zhu S, et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature. 2015;528:418–421. doi: 10.1038/nature15540. **This study identified the G-allele of the GATA binding site for transcription factor GATA3 as a SNP highly associated with neuroblastoma and found more commonly in African and African-American populations. The more recently evolved T-allele (TATA), shown to prevent GATA3 binding in association with decreased LMO1 expression, was found more commonly in European populations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Diskin SJ, Capasso M, Schnepp RW, Cole KA, Attiyeh EF, Hou C, Diamond M, Carpenter EL, Winter C, Lee H, et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat Genet. 2012;44:1126–1130. doi: 10.1038/ng.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Anglesio MS, Evdokimova V, Melnyk N, Zhang L, Fernandez CV, Grundy PE, Leach S, Marra MA, Brooks-Wilson AR, Penninger J, et al. Differential expression of a novel ankyrin containing E3 ubiquitin-protein ligase, Hace1, in sporadic Wilms’ tumor versus normal kidney. Hum Mol Genet. 2004;13:2061–2074. doi: 10.1093/hmg/ddh215. [DOI] [PubMed] [Google Scholar]
- 91.Hibi K, Sakata M, Sakuraba K, Shirahata A, Goto T, Mizukami H, Saito M, Ishibashi K, Kigawa G, Nemoto H, et al. Aberrant methylation of the HACE1 gene is frequently detected in advanced colorectal cancer. Anticancer Res. 2008;28:1581–1584. [PubMed] [Google Scholar]
- 92.Sakata M, Kitamura YH, Sakuraba K, Goto T, Mizukami H, Saito M, Ishibashi K, Kigawa G, Nemoto H, Sanada Y, et al. Methylation of HACE1 in gastric carcinoma. Anticancer Res. 2009;29:2231–2233. [PubMed] [Google Scholar]
- 93.Zhang L, Anglesio MS, O’Sullivan M, Zhang F, Yang G, Sarao R, Mai PN, Cronin S, Hara H, Melnyk N, et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat Med. 2007;13:1060–1069. doi: 10.1038/nm1621. [DOI] [PubMed] [Google Scholar]
- 94.Piskounova E, Polytarchou C, Thornton JE, LaPierre RJ, Pothoulakis C, Hagan JP, Iliopoulos D, Gregory RI. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell. 2011;147:1066–1079. doi: 10.1016/j.cell.2011.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, O’Sullivan M, Lu J, Phillips LA, Lockhart VL, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41:843–848. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Melton C, Judson RL, Blelloch R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature. 2010;463:621–626. doi: 10.1038/nature08725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 99.Schnepp RW, Khurana P, Attiyeh EF, Raman P, Chodosh SE, Oldridge DA, Gagliardi ME, Conkrite KL, Asgharzadeh S, Seeger RC, et al. A LIN28B-RAN-AURKA Signaling Network Promotes Neuroblastoma Tumorigenesis. Cancer Cell. 2015;28:599–609. doi: 10.1016/j.ccell.2015.09.012. **This study identified RAN as a target of the neuroblastoma oncogenic driver LIN28B and characterized the downstream effects of both proteins on increased Aurora Kinase A activity to further drive oncogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Carol H, Boehm I, Reynolds CP, Kang MH, Maris JM, Morton CL, Gorlick R, Kolb EA, Keir ST, Wu J, et al. Efficacy and pharmacokinetic/pharmacodynamic evaluation of the Aurora kinase A inhibitor MLN8237 against preclinical models of pediatric cancer. Cancer chemotherapy and pharmacology. 2011;68:1291–1304. doi: 10.1007/s00280-011-1618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mosse YP, Lipsitz E, Fox E, Teachey DT, Maris JM, Weigel B, Adamson PC, Ingle MA, Ahern CH, Blaney SM. Pediatric Phase I Trial and Pharmacokinetic Study of MLN8237, an Investigational Oral Selective Small-Molecule Inhibitor of Aurora Kinase A: A Children’s Oncology Group Phase I Consortium Study. Clinical Cancer Research. 2012;18:6058–6064. doi: 10.1158/1078-0432.CCR-11-3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Capasso M, Diskin S, Cimmino F, Acierno G, Totaro F, Petrosino G, Pezone L, Diamond M, McDaniel L, Hakonarson H, et al. Common genetic variants in NEFL influence gene expression and neuroblastoma risk. Cancer Res. 2014;74:6913–6924. doi: 10.1158/0008-5472.CAN-14-0431. *GWAS efforts identified three SNPs in the neurofilament gene NEFL shown to associated with neuroblastoma. High levels of NEFL were shown to promote differentiation and limit proliferation and are association with improved overal survival in neuroblastoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nguyen le B, Diskin SJ, Capasso M, Wang K, Diamond MA, Glessner J, Kim C, Attiyeh EF, Mosse YP, Cole K, et al. Phenotype restricted genome-wide association study using a gene-centric approach identifies three low-risk neuroblastoma susceptibility Loci. PLoS Genet. 2011;7:e1002026. doi: 10.1371/journal.pgen.1002026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, Cole K, Mosse YP, Wood A, Lynch JE, et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 2009;459:987–991. doi: 10.1038/nature08035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Capasso M, Calabrese FM, Iolascon A, Mellerup E. Combinations of genetic data in a study of neuroblastoma risk genotypes. Cancer Genet. 2014;207:94–97. doi: 10.1016/j.cancergen.2014.02.004. *In this study, combinations of genotypes were investigated for susceptibility between neuroblastoma patients and controls, finding 24 combinations with a significant association to neuroblastoma. [DOI] [PubMed] [Google Scholar]
- 106.Latorre V, Diskin SJ, Diamond MA, Zhang H, Hakonarson H, Maris JM, Devoto M. Replication of neuroblastoma SNP association at the BARD1 locus in African-Americans. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2012;21:658–663. doi: 10.1158/1055-9965.EPI-11-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gamazon ER, Pinto N, Konkashbaev A, Im HK, Diskin SJ, London WB, Maris JM, Dolan ME, Cox NJ, Cohn SL. Trans-population analysis of genetic mechanisms of ethnic disparities in neuroblastoma survival. Journal of the National Cancer Institute. 2013;105:302–309. doi: 10.1093/jnci/djs503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Diskin SJ, Capasso M, Diamond M, Oldridge DA, Conkrite K, Bosse KR, Russell MR, Iolascon A, Hakonarson H, Devoto M, et al. Rare variants in TP53 and susceptibility to neuroblastoma. Journal of the National Cancer Institute. 2014;106:dju047. doi: 10.1093/jnci/dju047. *Two rare germline variants in TP53 were discovered to be associated with neuroblastoma, despite the rarity of TP53 mutations in the majority of cases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–284. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Laug WE, Siegel SE, Shaw KN, Landing B, Baptista J, Gutenstein M. Initial urinary catecholamine metabolite concentrations and prognosis in neuroblastoma. Pediatrics. 1978;62:77–83. [PubMed] [Google Scholar]
- 112.Schnepp RW, Bosse KR, Maris JM. Improving patient outcomes with cancer genomics: unique opportunities and challenges in pediatric oncology. JAMA. 2015;314:881–883. doi: 10.1001/jama.2015.9794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.De Mariano M, Gallesio R, Chierici M, Furlanello C, Conte M, Garaventa A, Croce M, Ferrini S, Tonini GP, Longo L. Identification of GALNT14 as a novel neuroblastoma predisposition gene. Oncotarget. 2015;6:26335–26346. doi: 10.18632/oncotarget.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]