

Abstract
Introduction
Developing biomarker tools for identification of individuals at high-risk for late-onset Alzheimer’s disease (LOAD) is important for prognosis and early treatment. This review focuses on genetic factors and their potential role for precision medicine in LOAD.
Areas covered
APOEe4 is the strongest genetic risk factor for non-Mendelian LOAD, and the APOE-linkage disequilibrium (LD) region has produced the most significant association signal in multi-center genome-wide-association-studies (GWAS). Consideration of extended haplotypes in the APOE-LD region and specifically, non-coding variants in putative enhancer elements, such as the TOMM40-polyT, in-addition to the coding variants that comprise the APOE-genotypes, may be useful for predicting subjects at high-risk of developing LOAD and estimating age-of-onset of early disease-stage symptoms. A genetic-biomarker based on APOE–TOMM40-polyT haplotypes, and age is currently applied in a clinical trial for prevention/delay of LOAD onset. Additionally, we discuss LOAD-GWAS discoveries and the development of new genetic risk scores based on LOAD-GWAS findings other than the APOE-LD region.
Expert Commentary
Deciphering the precise causal genetic-variants within LOAD-GWAS regions will advance the development of genetic-biomarkers to complement and refine the APOE-LD region based prediction model. Collectively, the genetic-biomarkers will be translational for early diagnosis and enrichment of clinical trials with subjects at high-risk.
Keywords: APOE, genetic-based LOAD prediction models, genetic biomarkers, Late-Onset Alzheimer’s disease (LOAD), precision medicine, TOMM40 poly-T
1. Introduction
An ultimate goal for precision medicine is to develop and identify personalized disease treatment and prevention approaches considering individual variability in genetics, environmental and lifestyle factors. Gaining better insight into the biological pathways and molecular basis underlying disease etiology will advance the potential role of precision medicine. For many human complex diseases (i.e. don’t exhibit Mendelian inheritance pattern) of aging, including late-onset Alzheimer’s disease (LOAD), it is clear that family history is associated with increased risk; however, understanding the precise genetic variants, target genes and specific biological pathways contributing to disease risk has greatly lagged the discovery of disease-associated Single Nucleotide Polymorphisms (SNPs) from genome-wide association studies (GWAS).
The two strongest risk factors for the most common form of Alzheimer’s disease, non-Mendelian LOAD, are age [1] followed by APOE genotype[2]. Recently, epidemiological, biological and clinical studies have pointed to sex and gender differences in cognitive decline and the progression of LOAD [3, 4, 5]. Increasingly, it is clear that the risk of LOAD and the associated genetic loci must be interpreted in a framework of age-dependent risk and sex-specific risk of the disease.
In this paper, we focus on genetic factors and their potential role for precision medicine in LOAD. Developing biomarkers for precise identification of subjects at high-risk for LOAD is important for prognosis and early intervention. Here, we review the discovery of genetic markers of LOAD risk that provide a greater level of precision and accuracy for the early prediction of LOAD and mild cognitive impairment (MCI) due to AD and the impact of these markers on precision diagnosis for LOAD. These genetic markers can be assayed using DNA from a simple blood draw and used for prediction of LOAD-risk either alone or in conjunction with neuroimaging, cerebrospinal fluid (CSF), clinical, neuropsychological, and metabolomics biomarkers. As APOE is the strongest genetic risk factor for non-Mendelian LOAD, we primarily describe a comprehensive characterization of genetic variation in the APOE linkage disequilibrium (LD) region of the human genome and in particular within the neighboring TOMM40 gene. We suggest that consideration of extended haplotypes in this region of the genome and specifically non-coding variants in putative enhancer elements, in addition to the coding variants that comprise the APOE genotype, may be useful for predicting who is at risk of developing LOAD and estimating age of onset of early disease-stage symptoms. Supporting this premise are a number of clinical studies that have examined episodic memory and brain imaging changes associated with the manifestation of early-stage LOAD. In addition, we include a discussion of the development of new genetic risk score models based on LOAD-GWAS findings other than the APOE region. We conclude with highlighting the application of genetic risk prediction tools for stratification and enrichment of clinical trials for prevention and/or delay the onset of LOAD with individuals at high-risk.
2. APOE – the first and most firmly established genetic risk locus for LOAD
Over 20 years ago, genetic linkage analysis of pedigrees with familial Alzheimer’s disease identified a disease-associated region on chromosome 19[6]. Over the ensuing years, it has been demonstrated that the e4 allele of APOE gene, within the chromosome 19 linkage region, is the strongest and most highly replicated genetic risk factor for LOAD, is associated with lower age of clinical disease onset, and with development of amyloid plaques, a pathological hallmark of AD[7, 8, 9, 10]. The APOE alleles are defined by two coding SNPs (rs429358 and rs7412) that are located in exon 4 of the APOE gene and present in humans three forms, e2, e3 and e4. The protein coding consequences of these SNPs are to change two amino acids in the protein: Cys130Arg (rs429358) and Arg176Cys (rs7412). Age of onset (AOO) Kaplan-Meier (KM) curves for the development of LOAD stratified by APOE genotype are shown in Figure 1. The APOE e3/3 genotype is often considered the reference or “neutral” genotype. APOE e4 alleles contribute a dose-dependent increase in risk and a shift of the KM curves to earlier ages of disease onset. APOE e2 containing genotypes are associated with later ages of LOAD onset compared with genotypes that carry only e3 or e4 APOE alleles.
Figure 1. Alzheimer’s disease age of onset curves by APOE genotypes.
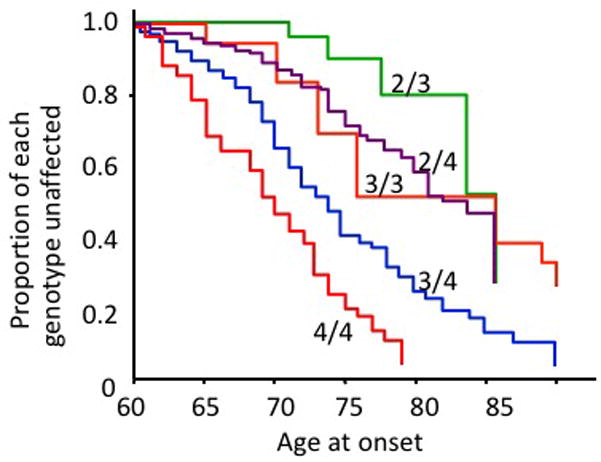
This figure is adapted from Corder et al. [7]. Kaplan-Meyer survival curves, where the Y axis shows the proportion of individuals unaffected by LOAD who have stratified by the 5 most common APOE genotypes. The X axis is the age of onset of LOAD for cases and age when last examined for unaffected individuals. N=234 subjects: 95 diagnosed with LOAD, 139 cognitively normal. N for each genotype: 2/3: 16; 3/3:77; 2/4:5; 3/4:113; 4/4: 23.
There is a body of work that explores how APOE isoforms differentially affect a number of processes that lead to development of AD-related pathologies[2, 11, 12]. While these examples directly change the nature of the encoded proteins, it is also possible that regulatory variants within the APOE LD region that affect gene expression or splicing may be directly related to the pathophysiology of LOAD [13, 14, 15]. Common variants in genomic regulatory elements that affect gene expression, transcript splicing, or epigenomic modification may also modify the phenotypic expression of LOAD (e.g. severity and AOO).
3. LOAD Genetics post APOE discovery
3.1 The GWAS era
GWAS have produced an extensive list of genetic variants associated with LOAD[16, 17, 18]. However, the translation of these data into biochemical targets for drugs development has not followed. Moreover, these studies have not provided significantly better prediction of risk or age of onset of LOAD than does APOE genotype alone [19]. The GWAS studies have provided valuable information on biological pathways involved in the etiology of AD, including cholesterol metabolism, immune response, neuroinflammation and endocytosis[20, 21].
3.2. Phylogenetic Analysis to identify TOMM40 poly-T as a risk locus for LOAD
GWAS of LOAD produced genetic variants with relatively low odds ratios (typically 1.1 – 1.3) that have been followed up by examining proximal genes, conducting computational analysis of enriched biological pathways or whole-genome/whole exome sequencing to identify rare variants. In contrast, as a consequence of the high LD present in the APOE-TOMM40 region which has an extremely high association with LOAD, the identification of genetic variants in this region that modify LOAD age of onset or risk requires a different approach that allows multiple variants within an LD region to contribute to the association with the phenotype. Such general strategy constitutes of three major steps, a search using bioinformatics tools to find and annotate highly polymorphic structural variants such as simple sequence repeats (SSRs) and homopolymers followed by phase sequencing of the region that contains these variants and phylogenetic analyses of the sequence data[22]. An important aspect of this general strategy is to fully characterize the defined genomic region and consider single variants as well as long-range haplotypes and interactions between variants. The strengths of this general approach are that sequencing captures broadly defined structural variants[23] (including insertion/deletions, inversions, homopolymers and other repeated sequences) in addition to SNPs and that haplotype phase is known with certainty. This methodology is likely to be particularly applicable to the genetics of phenotypes that result from cis-acting alleles, each with relatively small effect size and collectively adding to a larger effect size. From a mechanistic standpoint, this strategy is suitable for identification of variants that impact gene regulation or genomic structure or tag other variants that affect regulatory functions[24]. Phylogenetic approaches are used to interrogate regions of interest previously highlighted by genetic association studies (e.g. linkage analysis or GWAS) and are particularly relevant for regions that exhibit high LD. A recent review by Templeton presents the rationale, limitations and illustrative examples for using phylogenetic methods for genotype/phenotype associations[22]. Detailed examples of specific methods are covered in several publications[25, 26, 27]. For regions of weaker LD, methods based on analysis of phylogenetic network analysis are needed [28] since recombination will limit the applicability of phylogenetic analysis.
Roses et al. employed phased sequencing coupled with phylogenetic analysis to identify variants associated with LOAD-risk within the high LD region at chromosome 19 proximal to APOE, a genomic region that has repeatedly shown the most significant, by a wide margin, LOAD-GWA signal [29]. Briefly, the TOMM40-APOE region of both chromosomes from all subjects was cloned and sequenced, so that the phase of all variants was precisely known, not inferred, and all types of variants were included in the analysis [30]. The sequences, including all distinguishing variants, were then used to construct a phylogenetic, or more appropriately a genealogical, tree. A number of methods, including Bayesian and maximum parsimony, were used to estimate the possible genealogies for the TOMM40-APOE region. Combinations of variants that separate clades of the tree were identified and tested for association with the phenotype of interest. The TOMM40-APOE genomic region is a fairly unique case in that it is a region of very strong LD, is populated with relatively abundant polymorphisms, and contains large effect-size, causal alleles in APOE [29]. This study resulted in the identification of the rs10524523 (‘523) locus, a highly polymorphic poly-T repeat tract positioned in intron 6 of the TOMM40 gene[29] (hereafter TOMM40 poly-T) and subsequent work demonstrated that this TOMM40 poly-T improve the precision of age-of-onset (AOO) prediction for LOAD [31].
The use of haplotype-based methods, including clustering based on evolutionarily-informed haplotypes, is currently limited by the size of a region of the genome that can be sequenced at a reasonable cost. In the discovery of TOMM40 Poly-T, Roses et al. overcame this challenge by conducting phased sequencing by cloning long-range PCR products and sequencing multiple clones per individual using the traditional Sanger method [29]. However, this approach is very expensive to carry out on a genome wide scale. Newer next generation sequencing technologies have made it possible to sequence entire human genomes; for example the 1000 Genomes Project has sequenced and analyzed over 1000 individual human genomes and made the data publicly available. While access to this data enables analysis of rare and structural variants in phenotype association studies, the phase of the polymorphisms on each of the two chromosomes must still be statistically inferred, i.e. the content of variants unique to each of the two homologous chromosomes, or diplotype, is usually not directly observed.
4. Clinical and biological implications of the role of TOMM40 poly-T in LOAD
Three allele groups were defined for the TOMM40 poly-T polymorphism, based on the modes of the distributions of the number of ‘T’-residues: ‘Short’ (S, T≤19), ‘Long’ (L, 20≤T≤29) and ‘Very Long’ (VL, T≥30). The haplotypic relationship between the TOMM40 poly-T polymorphism and APOE SNPs in different populations [32, 33] and the association between specific TOMM40 poly-T – APOE haplotypes and age of onset of LOAD has been described in several publications[31, 34]. The Kaplan-Meier (KM) curves illustrated in Figure 2 represent a retrospective stratification by TOMM40 poly-T genotype of the age of LOAD onset. These KM curves were constructed using genotypic and AOO data for Caucasian individuals followed longitudinally at the Joseph & Kathleen Bryan Alzheimer’s Disease Research Center at Duke University [32, 33]. Subjects were tested over time with a battery of neuropsychological tests to monitor cognitive changes and to diagnose onset of cognitive impairment and probable AD dementia. The L/L curve in Figure 2 corresponds closely to APOE e4/4 since 98% of all APOE e4 alleles are linked to TOMM40 poly-T ‘L’ in Caucasians. In this particular cohort, 50% of the L/L subjects were diagnosed with probable AD by age 76. By the age of 78 years, 50% of subjects in this cohort with the L/VL genotype developed cognitive impairment whereas, in general, L/S subjects developed cognitive impairment at later ages (50% had clinical symptoms by age 81). L/S and L/VL genotypes are also APOE e3/4; therefore, stratification by TOMM40 poly-T genotype gave two, separable age-of-onset (AOO) curves for APOE e3/4 subjects and provided more granularity to the age of disease onset distributions. Similarly, instead of one curve for APOE e3 homozygotes, three distinct curves, for S/S, S/VL and VL/VL, were resolved thus creating three different AOO risk groups. As with APOE e3, APOE e2 may be linked to an S or a VL allele. Carriage of at least one allele of APOE e2 appears to result in later AOO[35], regardless of the TOMM40 poly-T allele in the haplotype, although AOO may be modified by APOE e4 (or an L allele) in the genotype. In conclusion, the combination of the TOMM40 poly-T – APOE haplotypes provides a more precise estimation of LOAD AOO compared to AOO estimations based on APOE status solely.
Figure 2. Alzheimer’s disease age of onset curves by TOMM40 poly-T and APOE haplotypes.

The figure is adapted from Roses et al.[34]. Kaplan-Meyer survival curves, where the Y axis shows the percent survival without cognitive impairment, and the X axis represents age. Data was obtained from the Duke Bryan ADRC cohort N=438 subjects: 106 diagnosed with dementia, 332 cognitively normal. N for each genotype: L,L:23; VL,L:54; S,L:72; S,S:100; S,VL:138; VL,VL:51. TOMM40 genotypes and the corresponding APOE genotypes are indicated on the figure. The red line corresponds to APOE ε4/4; the two green lines correspond to APOE e3/4, and the three blue lines correspond to APOE e3/3.
The TOMM40 poly-T polymorphism has also been associated with performance in the recall of words from the primacy portion of a long word list, a cognitive deficiency noted in the early manifestation of AD, in cognitively healthy individuals in midlife[36]. An association was also reported between the S allele and improved memory and executive function into very old age in APOE e3/3 cognitively healthy elderly people[37]. Furthermore, the TOMM40 poly-T locus was also associated with gray matter volume in regions of the brain affected early in the development of AD in APOE e3/3, cognitively normal, middle-age people [36]. A recent study based on data from the Religious Orders Study and Rush Memory and Aging Project reported an association of APOE e3/3-TOMM40 poly-T haplotypes with cognitive decline in community based older persons[38]. The large sample size (N=1170) and long-term follow up (up to 21 years) were critical factors in demonstrating that there was an APOE-independent effect of the TOMM40 poly-T locus on cognitive decline by limiting the analysis cohort to APOE e3/3 individuals thus removing the confounding effects of APOE e4 or e2 alleles. The results showed that subjects with TOMM40 poly-T-S/S had faster decline in global cognition than subjects with TOMM40 poly-T-S/VL or VL/VL (p=0.002). The same association was observed for episodic memory (p=0.0004) and semantic memory (p=0.003), but not for working memory, processing speed or visuospatial ability (all p>.05). Collectively, these studies provided genetic evidence for the contribution of TOMM40 poly-T locus to LOAD and related endophenotypes. However, other studies reported conflicting data and did not replicate the reported association of TOMM40 poly-T with LOAD risk and AOO [39, 40, 41].
The biochemical mechanism by which the TOMM40 poly-T variant affects AD pathophysiology and biochemistry is not clear, however, it is different from the effects of APOE status. The TOMM40 gene encodes the Tom40 protein, dimers of which create the pore in the outer mitochondrial membrane through which almost all proteins enter mitochondria. The protein is essential for mitochondrial biogenesis and function [42]. Structural DNA variations, especially those in intronic or intergenic regions like the TOMM40 poly-T variant, most likely exert their effects on phenotype by altering gene transcription efficiency, the timing of transcription, transcript stability, transcript splicing, or possibly by changing patterns of epigenomic modification[43, 44, 45, 46, 47, 48] [49]. There is support for the idea that short structural variants, rather than or in addition to SNPs, may be responsible for many human complex traits[50, 51, 52]. It has been demonstrated that TOMM40 poly-T affects the expression levels of APOE and TOMM40 mRNAs in the temporal and occipital cortexes of LOAD patients and normal controls[32], and the effect of the TOMM40 poly-T variation on transcription regulation was recapitulated in a cell-based luciferase reporting system[32] [53]. Gene expression studies suggest that the number of protein import channels per mitochondrion may be regulated by TOMM40 poly-T variants. Bekris et al. described a complex transcriptional regulatory region for TOMM40 and APOE expression that extends throughout both genes and is influenced by multiple polymorphisms including the TOMM40 poly-T locus[54]. It has been suggested that a relatively modest change in mRNA expression may produce pathology that accumulate over time and is expressed clinically in later age. Changes in levels of the Tom40 protein have also been detected in the brains of Parkinson’s disease patients, and in a mouse model of Parkinson’s disease[55], suggesting that expression of the Tom40 protein may have a broader impact on a spectrum of neurological diseases in aging[56].
5. Developing LOAD prediction models
Utilization of any biomarker is dependent not only on its performance characteristics (NPV, PPV) but also by the intended application (e.g. clinical trial enrichment, diagnostic use in a clinical setting companion diagnostic to a therapeutic) often referred to as “fit for purpose”. In addition to the test performance (specificity, sensitivity), features like availability of specialized reagents, instruments and/or qualified personnel, willingness of subjects to be tested, invasiveness, cost/reimbursement and consideration of the clinical utility in the practice of medicine contribute to the use of any given test. The usage of a biomarker to enrich a clinical trial in normal subjects at risk for conversion to disease symptoms needs to consider how that test would be employed in the practice of medicine in a global environment.
Combinations of biomarkers could conceivably be used to further refine selection of subjects for clinical trials, balancing cost and invasiveness of the assay with the improvement in accuracy of conversion prediction in a pre-specified time frame. There is strong precedent for the combination of APOE, CSF, imaging biomarkers and neurocognitive measures to improve predictive accuracy [57], and recent work using the Biomarkers for Older Controls at Risk for Dementia (BIOCARD) cohort [58, 59], with 20 years of longitudinal follow up has provided promising results [60]. Specifically, for prediction of the onset of MCI in a five-year time frame, the combination of six measures provided the best performance: Two memory and thinking tests (the Digit Symbol and Paired Associates Immediate Recall tests), levels of CSF amyloid beta and p-tau, two measures of MRI brain scan – one to assess the thickness of the right entorhinal cortex and another to measure the volume of the hippocampus. Accuracy of prediction was reported as: area under the receiver operating characteristic curve (AUC)=0.89, sensitivity = 0.85, specificity = 0.70 [60].
Genetics-based risk biomarkers rely on genomic DNA readily collected from a blood draw, using robust, widely available inexpensive DNA testing of an analyte that is unaffected by environmental, disease state or other conditions. A two-stage process is also possible for specific settings, where the genetic biomarkers would be used to initially screen subjects for a prevention trial using an inexpensive blood test followed by CSF and/or imaging biomarkers for further screening of individuals.
5.1 Prediction of LOAD risk based on GWAS findings
Recently, two studies used genotype data from the International Genomics of Alzheimer’s Project (IGAP) to investigate the accuracy of LOAD prediction models based on risk alleles identified in GWAS. Escott-Price et al. conducted a polygenic score analysis to test whether the alleles identified to associate with LOAD in one sample set were significantly enriched in the cases relative to the controls in an independent sample, and found a significant enrichment for a polygenic component in LOAD [61]. They further showed that best prediction accuracy, AUC = 78.2%, was achieved by a logistic regression model with APOE, the polygenic score, sex and age as predictors. Another study constructed a genetic risk score (GRS) using top 19 GWAS SNPs and evaluate its capacity to improve prediction of LOAD-risk in prospective cohorts. The GRS was associated with a 17% increase in risk of developing LOAD. This study showed a small improvement in LOAD risk prediction when adding the GRS to age, sex, APOEe4 and education [62]. Overall, these studies demonstrated that the LOAD-GWAS based genetic predictors have utility alongside other models based on traditional LOAD predictors, however, their inclusion provided only a minor improvement in LOAD-risk prediction.
5.2 Prediction of LOAD risk based on TOMM40 poly-T – APOE haplotypes and age
A simple, robust genetics-based biomarker risk algorithm (GBRA) utilizing a combination of APOE genotype, TOMM40 poly-T genotypes and age has been developed as a prognostic tool for assessing LOAD age of onset in asymptomatic people [31, 63]. Figure 3 summarizes the principles of the LOAD-GBRA algorithm. The positive predictive values (PPV) and negative predictive values (NPV) of the GBRA are in the range of 70–80%. The relatively high odds ratio (approximately 3–5) comparing the GBRA to predictive models based on APOE and age alone support the value of the GBRA in risk prediction for MCI due to LOAD. In addition, the GBRA “high” and “low” LOAD-risk categorizations correlated well with pathological CSF biomarker levels, Positron Emission Tomography (PET) amyloid burden and neurocognitive scores. For example, for the data from the Alzheimer’s Disease Neuroimaging initiative (ADNI), subjects predicted to be high risk by the GBRA showed a significantly (p < 0.0001) lower mean level of CSF Aβ1-42 (157.15±3.78) in comparison to subjects predicted to be low risk (195.59±4.67); subjects predicted to be high risk by the GBRA showed a significantly (p < 0.0001) higher (worse) Standard Uptake Value ratio (SUVR) measurement of amyloid burden (mean SUVR 1.90±0.06, n=40) in comparison to subjects predicted to be low risk (mean SUVR 1.52±0.07, n=30)[63].
Figure 3. Genetics-based biomarker risk algorithm (GBRA).
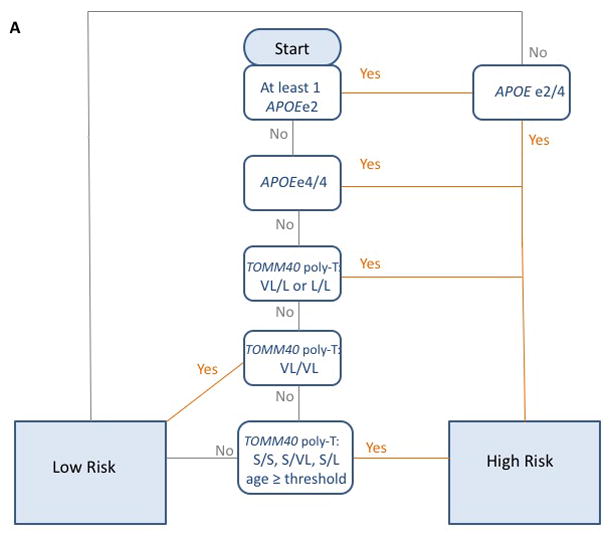
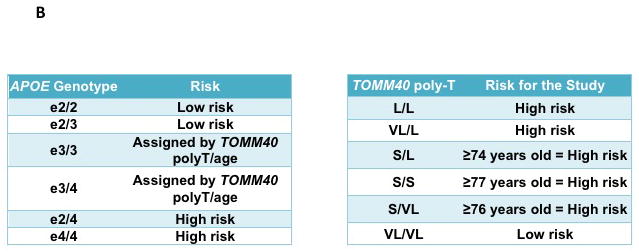
This figure is adapted from Lutz et al. [63] Flowchart (A) and Tables (B) for the process for the generation of the risk assessment for MCI due to LOAD using the GBRA. Risk of high or low is assigned based on APOE genotype, TOMM40 poly-T genotype and current age. The GBRA was developed for assessment of asymptomatic individuals and currently being used for the TOMMORROW clinical trial.
Receiver operating curves and comparative analysis of area under the curve (AUC) showed that the combinations of APOE genotype, TOMM40 poly-T genotype and age outperformed age in sensitivity and specificity for risk prediction for MCI due to LOAD. For a well-characterized cohort of individuals with LOAD, MCI and cognitively normal controls followed at the Duke Bryan Alzheimer’s Disease Research, the GBRA outperformed age and either genotype (APOE or TOMM40) in terms of sensitivity and specificity[63]. For the replication ADNI cohort, the performance of age combined with either or both genotypes was equivalent. For both cohorts, the combination of age and APOE genotype provided slightly better performance than age and TOMM40 poly-T genotype, although the difference was not statistically significant[63]. It was surprising that the AUC for the GBRA did not show a statistically-significant improvement for the ADNI cohort when compared with the combination of APOE genotype and age considering that age interaction terms with genotype were highly statistically-significant (p<0.01)[63]. Testing in additional replication cohorts from longitudinal observational studies with more precise estimation of age of onset of MCI/AD will be needed to more clearly define the performance of the GBRA.
6. Application of the TOMM40 poly-T – APOE haplotypes in Clinical Trial
The development of genetic biomarkers for precise identification of subjects at high-risk to develop LOAD will permit preclinical intervention and improve prognosis. Another important application for genetic risk prediction tools is for enrichment and stratification of clinical trials for individuals with high-risk. This approach is particularly relevant for prevention and/or delay the onset trails. The ultimate proof of the value of including TOMM40 poly-T in addition to APOE variants for risk prediction (GBRA summarizes in Figure 3) will come from the TOMMORROW clinical trial, a pharmacogenetically-enriched, double-blind, delay-of-disease-onset clinical trial of cognitively normal subjects aged 65–83, inclusive, classified as having high or low risk for development of cognitive symptoms over the course of a 5-year study (TOMMORROW trial; ClinicalTrials.gov Identifier=NCT01931566, [34]). In this study, high risk subjects are randomized to active therapy (low-dose pioglitazone) or placebo; low risk subjects are randomized to placebo only. Stratification for risk of developing MCI due to AD during the study, prior to randomization is accomplished with the GBRA at the beginning of the study (when neuropsychological testing verifies normal cognition). The GBRA will be qualified for use as a prognostic biomarker at the end of the phase 3 trial when the performance characteristics and Receiver Operating Curve (ROC) curves can be calculated from the trial data which will provide a large (n>3,000), prospectively sampled cohort. Once qualified, the biomarker can be used as a companion pharmacogenetics test for a therapeutic to delay the onset of LOAD. While the GBRA was developed as a binary predictive algorithm for a delay-of-disease-onset clinical trial of cognitively normal subjects to high and low risk groups, the algorithm could also be adapted to a continuous scale based on likelihood of conversion within a pre-specified time frame.
7. Conclusion
The APOE status was associated with LOAD age-of-onset and stratification by TOMM40 poly-T genotype provides a more precise estimation of LOAD AOO compared to AOO estimations based on APOE status solely. A genetics-based biomarker risk algorithm for LOAD based on the APOE–TOMM40 poly-T haplotypes, and age was constructed and has been applied in the TOMMORROW study, a clinical trial for prevention and/or delay the onset of LOAD, for enrichment with individuals at high-risk. The utility of the APOE–TOMM40 poly-T based genetic biomarker will be validate at the end of phase 3 of the TOMMORROW clinical trial. GWAS have identified over 20 genomic regions associated with LOAD in addition to the APOE LD region. New genetic risk scores based on LOAD-GWAS have been developed and demonstrated utility alongside other models based on traditional LOAD predictors, however, their inclusion provided only a minor improvement in LOAD-risk prediction compare to models based only on traditional LOAD predictors – age, sex, APOEe4. A better understanding of the genetic factors underpinning the etiology of LOAD may provide new informative genetic variants for integration in genetic based disease-risk models that will improve the precision of LOAD-risk prediction.
8. Expert Commentary
Lack of effective therapies for Alzheimer’s stems from poor understanding of the disease’s etiology. Multiple etiologies and age-related prodromal processes contribute to the pathophysiology of LOAD, which highlights the significance of precision medicine in LOAD. Genetics plays an important role in the risk to develop LOAD, nonetheless, deciphering the genetic factors underpinning the etiology LOAD has been a challenge. This challenge attributes, at least in part, to the facts that LOAD is a heterogeneous, multifactorial disease, and the constellation of the genetic determinants might be personalized and distinct among individuals. Large multi-center GWA studies have found associations between over 20 genomic loci and LOAD. However, the precise target genes, the defined causal genetic variants and their molecular mechanisms of action through which they exert their pathogenic effects remain largely unknown.
The large majority of the LOAD-GWAS risk SNPs are in non-coding intragenic or intergenic regulatory regions of the genome. Furthermore, changes in gene expression in LOAD vs. healthy control were described in brain tissues and previous studies reported the cis-associations of tagging SNPs with expression of nearby LOAD-risk genes, providing a strong scientific premise for the proposed study. These evidences led us to hypothesize that changes in expression profiles of critical disease genes is an important molecular mechanism underlying disease etiology and that causal variants modulate expression of these disease genes, via transcriptional and post-transcriptional regulatory events, and by that contribute to LOAD susceptibility.
Short Structural Variants (SSVs), variants other than SNPs, include short deletions, short insertions, indels, homopolymer stretches and short tandem repeats (STR). SSVs were not included in GWAS, including for LOAD, and expression traits (eQTL) association studies (eGWA). In the post-GWA era this understudied class of genetic variation is increasingly thought to play an important role in complex disease such as LOAD and it has been suggested that SSVs have regulatory functions in gene transcription and splicing. Therefore, we advocate that investigations of SSVs within the LOAD associated genomic regions will move the field forward towards identification of the functional and causal genetic variations. Moreover, with the exception of the APOE status that consists of two SNPs and the combination of APOE with TOMM40 poly-T, haplotypes have been underrepresented in genetic studies of LOAD. The example of the APOE LD region demonstrates that extended haplotypes are likely to be more informative than a single variant. Collectively, we suggest that consideration of a broad spectrum of genetic variants and of extended haplotypes is crucial for the elucidation of causal genetic risk-factors and their functional effects that contribute to the risk of developing LOAD.
Uncovering the genetic etiologies of LOAD is the fundamental milestone in order to achieve and practice precision medicine in LOAD. The prospectively identified genome-wide causal variants will be translational for the construction of refined integrative genetic biomarkers to complement and improve other LOAD-risk models based on traditional LOAD predictors.
Complex diseases including LOAD involve the interaction of numerous biological pathways and interactions between genetic, metabolic and environmental factors. Understanding these interactions is critical both to understand the pathophysiology of the disease and to identify potential targets for interventions. In this review, we showed that a region of the genome that has an extremely strong genetic association with LOAD also has two relevant pathways cholesterol metabolism and mitochondrial transport that map back to two genes, APOE and TOMM40 that are in high LD. Integrative disease modeling (IDM) is an approach will link genetic, molecular, neurophysiological and neuroimaging data across multiple physiological levels in order to more clearly elucidate the biological networks that are affected by the causal genetic variants[64, 65, 66, 67, 68, 69]. Integrative disease modeling and systems-biology approaches for understanding the interaction of complex genetic and metabolic factors and networks are increasingly being employed for LOAD research specifically [70, 71, 72, 73]. Moreover, as LOAD develops over the course of decades, IDM will be essential to understand the time course of the development of disease at the levels of the genome, epigenome, transcriptome, microRNome, proteome/peptidome, metabolome/lipidome, microbiome, lifestyle, and environmental factors that are involved in complex cellular networks.
9. Five-year view
New emerging genomic technologies and cutting-edge biological model systems will advance the identification and validation of the precise LOAD-causal variants and LOAD-risk haplotypes, and the understanding of their molecular mechanisms of action through which they exert their pathogenic effects. Subsequent studies using innovative models will establish functional genotype-phenotype relationships of the identified LOAD-risk genetic variants and will evaluate their direct functional activity and their causal links to molecular and cellular phenotypes. This knowledge will give greater insight into molecular targets contributing to the etiology of LOAD. The variants and haplotypes with direct effects will have a strong translational impact as they will be utilized in combination with APOE LD region to develop new genetic LOAD-risk prediction algorithms, i.e. integrative genetic biomarkers, that will demonstrate an improved precision of LOAD-risk prediction.
The highly informative integrative genetic biomarkers will be implemented for early diagnosis, and for enrichment of clinical trials with subjects at high risk. In depth understanding of genetic risk factors will also advance the identification of actionable targets for development of novel therapies for LOAD. Ultimately, integrative genetic biomarkers will inform regarding personalized treatment approaches.
Key issues.
APOE is the strongest genetic risk factor for non-Mendelian LOAD. APOE e4 allele contributes a dose-dependent increase in risk and is associated with earlier age-of-onset.
A highly polymorphic, poly-T in intron 6 of the TOMM40 gene that is adjacent to APOE, hereafter TOMM40 poly-T, is associated with age-of-onset of LOAD and with cognitive performance in the elderly.
It has been suggested that the TOMM40 poly-T acts as a regional transcription regulator of the TOMM40 and APOE genes, that may provide the underpinning molecular mechanism for the reported associations with LOAD and related endophenotypes.
Large multi-center GWA studies have found associations between over 20 genomic loci and LOAD. The GWAS discoveries have provided valuable information on biological pathways involved in the etiology of LOAD. However, the precise target gene and causal genetic variants have yet to be uncovered.
A genetics-based biomarker risk algorithm for LOAD based on the APOE–TOMM40 poly-T haplotypes, and age was developed and is being used for enrichment with individuals at high-risk in clinical trials for prevention and/or delay the onset of LOAD, particularly the TOMMORROW study.
New genetic risk scores based on LOAD-GWAS findings have also been developed by different groups, however, up-to-date provided only a minor improvement in LOAD-risk prediction compared to the traditional LOAD predictors.
LOAD develops over the course of decades. Integrative disease modeling and systems-biology approaches will be essential to understand the time course of the development of disease at the levels of the genome, epigenome, transcriptome, microRNome, proteome/peptidome, metabolome/lipidome, microbiome, lifestyle, and environmental factors that are involved in complex cellular networks. These approaches will help focus LOAD research on potential therapeutic approaches to apply at the earliest stages of disease pathophysiology.
Acknowledgments
Funding
This work was funded in part by the National Institute on Aging (NIA) [R01AG040370 to O.C.].
Footnotes
Declaration of interest
MW Lutz is a paid consultant to Zinfandel Pharmaceuticals. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- 1.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s & dementia: the journal of the Alzheimer’s Association. 2007;3:186–91. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- 2.Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jack CR, Jr, Wiste HJ, Weigand SD, Knopman DS, Vemuri P, Mielke MM, Lowe V, Senjem ML, Gunter JL, Machulda MM, Gregg BE, Pankratz VS, Rocca WA, Petersen RC. Age, Sex, and APOE epsilon4 Effects on Memory, Brain Structure, and beta-Amyloid Across the Adult Life Span. JAMA Neurol. 2015;72:511–9. doi: 10.1001/jamaneurol.2014.4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin Epidemiol. 2014;6:37–48. doi: 10.2147/CLEP.S37929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snyder HM, Asthana S, Bain L, Brinton R, Craft S, Dubal DB, Espeland MA, Gatz M, Mielke MM, Raber J, Rapp PR, Yaffe K, Carrillo MC. Sex biology contributions to vulnerability to Alzheimer’s disease: A think tank convened by the Women’s Alzheimer’s Research Initiative. Alzheimer’s & dementia: the journal of the Alzheimer’s Association. 2016 doi: 10.1016/j.jalz.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pericak-Vance MA, Bebout JL, Gaskell PC, Jr, Yamaoka LH, Hung WY, Alberts MJ, Walker AP, Bartlett RJ, Haynes CA, Welsh KA, et al. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet. 1991;48:1034–50. Epub 1991/06/01. [PMC free article] [PubMed] [Google Scholar]
- 7*.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science (New York, NY. 1993;261:921–3. doi: 10.1126/science.8346443. The first demonstration of the relationship between APOE genotypes and LOAD age-of-onset. [DOI] [PubMed] [Google Scholar]
- 8.Liu N, Zhang K, Zhao H, Rao DC, Gu CC. Advances in Genetics. Vol. 60. Academic Press; 2008. Haplotype-Association Analysis; pp. 335–405. [DOI] [PubMed] [Google Scholar]
- 9.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proceedings of the National Academy of Sciences. 1993;90:9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10**.Saunders AM, Strittmatter WJ, Schmechel D, St George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD. Association of apolipoprotein E allele e4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–72. doi: 10.1212/wnl.43.8.1467. The original report of APOEe4 association with LOAD-risk. [DOI] [PubMed] [Google Scholar]
- 11.Holtzman DM, Herz J, Bu G. Apolipoprotein E and Apolipoprotein E Receptors: Normal Biology and Roles in Alzheimer Disease. Cold Spring Harbor Perspectives in Medicine. 2012:2. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamazaki Y, Painter MM, Bu G, Kanekiyo T. Apolipoprotein E as a Therapeutic Target in Alzheimer’s Disease: A Review of Basic Research and Clinical Evidence. CNS Drugs. 2016;30:773–89. doi: 10.1007/s40263-016-0361-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13**.Linnertz CAL, Gottschalk W, Crenshaw D, Lutz MW, Allen J, Saith S, Mihovilovic M, Burke JR, Welsh-Bohmer KA, Roses AD, Chiba-Falek O. The cis-regulatory effect of an Alzheimer’s disease-associated poly-T locus on expression of TOMM40 and APOE genes. Alzheimer’s & Dementia. 2013 doi: 10.1016/j.jalz.2013.08.280. The first report of the functional role of the TOMM40 poly-T locus in the transcriptional regulation of TOMM40 and APOE genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allen M, Zou F, Chai HS, Younkin CS, Crook J, Pankratz VS, Carrasquillo MM, Rowley CN, Nair AA, Middha S, Maharjan S, Nguyen T, Ma L, Malphrus KG, Palusak R, Lincoln S, Bisceglio G, Georgescu C, Schultz D, Rakhshan F, Kolbert CP, Jen J, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD, Alzheimer’s Disease Genetics C. Petersen RC, Graff-Radford NR, Dickson DW, Younkin SG, Ertekin-Taner N. Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology. 2012;79:221–8. doi: 10.1212/WNL.0b013e3182605801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gottschalk WK, Mihovilovic M, Roses AD, Chiba-Falek O. The Role of Upregulated APOE in Alzheimer’s Disease Etiology. J Alzheimers Dis Parkinsonism. 2016:6. doi: 10.4172/2161-0460.1000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Medway C, Morgan K. Review: The genetics of Alzheimer’s disease; putting flesh on the bones. Neuropathol Appl Neurobiol. 2014;40:97–105. doi: 10.1111/nan.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17**.Lambert J-C, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, Jun G, DeStefano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thornton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin C-F, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau M-T, Choi S-H, Reitz C, Pasquier F, Hollingworth P, Ramirez A, Hanon O, Fitzpatrick AL, Buxbaum JD, Campion D, Crane PK, Baldwin C, Becker T, Gudnason V, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letenneur L, Moron FJ, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fievet N, Huentelman MJ, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuinness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossu P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Naranjo MCD, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannfelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O’Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH, Jr, Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RFAG, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JSK, Boerwinkle E, Riemenschneider M, Boada M, Hiltunen M, Martin ER, Schmidt R, Rujescu D, Wang L-S, Dartigues J-F, Mayeux R, Tzourio C, Hofman A, Nothen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P European Alzheimer’s Disease I, Genetic Environmental Risk in Alzheimer’s D, Alzheimer’s Disease Genetic C, Cohorts for H, Aging Research in Genomic E. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–8. doi: 10.1038/ng.2802. A large meta-analysis of multi-center GWAS that finds associations between over 20 genomic loci and LOAD,11 of which were new loci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chouraki V, Seshadri S. Genetics of Alzheimer’s disease. Adv Genet. 2014;87:245–94. doi: 10.1016/B978-0-12-800149-3.00005-6. [DOI] [PubMed] [Google Scholar]
- 19.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ER, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Ruther E, Schurmann B, Heun R, Kolsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Gallacher J, Hull M, Rujescu D, Giegling I, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, van Duijn CM, Breteler MM, Ikram MA, DeStefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S, Berr C, Campion D, Epelbaum J, Dartigues JF, Tzourio C, Alperovitch A, Lathrop M, Feulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snaedal J, Bjornsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia A, Porcellini E, Hanon O, Coto E, Alvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossu P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O’Donovan M, Amouyel P, Williams J. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 43:429–35. doi: 10.1038/ng.803. Epub 2011/04/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karch CM, Cruchaga C, Goate AM. Alzheimer’s disease genetics: from the bench to the clinic. Neuron. 2014;83:11–26. doi: 10.1016/j.neuron.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77:43–51. doi: 10.1016/j.biopsych.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Templeton AR. The diverse applications of cladistic analysis of molecular evolution, with special reference to nested clade analysis. Int J Mol Sci. 2010;11:124–39. doi: 10.3390/ijms11010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nature reviews Genetics. 2009;10:241–51. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- 24.Roses AD, Saunders AM, Lutz MW, Zhang N, Hariri AR, Asin KE, Crenshaw DG, Budur K, Burns DK, Brannan SK. New applications of disease genetics and pharmacogenetics to drug development. Current opinion in pharmacology. 2014;14:81–9. doi: 10.1016/j.coph.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 25.Bardel C, Danjean V, Hugot JP, Darlu P, Genin E. On the use of haplotype phylogeny to detect disease susceptibility loci. BMC genetics. 2005;6:24. doi: 10.1186/1471-2156-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tachmazidou I, Verzilli CJ, De Iorio M. Genetic association mapping via evolution-based clustering of haplotypes. PLoS Genet. 2007;3:e111. doi: 10.1371/journal.pgen.0030111. Epub 2007/07/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Berthier-Schaad Y, Fallin MD, Fink NE, Tracy RP, Klag MJ, Smith MW, Coresh J. IL-6 haplotypes, inflammation, and risk for cardiovascular disease in a multiethnic dialysis cohort. J Am Soc Nephrol. 2006;17:863–70. doi: 10.1681/ASN.2005050465. [DOI] [PubMed] [Google Scholar]
- 28.Huson DH, Scornavacca C. A survey of combinatorial methods for phylogenetic networks. Genome Biol Evol. 2011;3:23–35. doi: 10.1093/gbe/evq077. Epub 2010/11/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, Huentelman MJ, Welsh-Bohmer KA, Reiman EM. A TOMM40 variable length polymorphism determines the age of late-onset Alzheimers disease. Pharmacogenomics J. 2010;10:375–84. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30**.Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, Huentelman MJ, Welsh-Bohmer KA, Reiman EM. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 2010;10:375–84. doi: 10.1038/tpj.2009.69. The discovery of the TOMM40 poly-T and its effect on LOAD age-of-onset. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crenshaw DG, Gottschalk WK, Lutz MW, Grossman I, Saunders AM, Burke JR, Welsh-Bohmer KA, Brannan SK, Burns DK, Roses AD. Using genetics to enable studies on the prevention of Alzheimer’s disease. Clinical pharmacology and therapeutics. 2013;93:177–85. doi: 10.1038/clpt.2012.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Linnertz C, Saunders AM, Lutz MW, Crenshaw DM, Grossman I, Burns DK, Whitfield KE, Hauser MA, McCarthy JJ, Ulmer M, Allingham R, Welsh-Bohmer KA, Roses AD, Chiba-Falek O. Characterization of the poly-T variant in the TOMM40 gene in diverse populations. PloS one. 2012;7:e30994. doi: 10.1371/journal.pone.0030994. Illustartion of the high variability of the TOMM40 poly-T allele frequencies and range of lengths in different ethnic groups. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roses AD, Lutz MW, Saunders AM, Goldgaber D, Saul R, Sundseth SS, Akkari PA, Roses SM, Gottschalk WK, Whitfield KE, Vostrov AA, Hauser MA, Allingham RR, Burns DK, Chiba-Falek O, Welsh-Bohmer KA. African-American TOMM40’523-APOE haplotypes are admixture of West African and Caucasian alleles. Alzheimer’s & dementia: the journal of the Alzheimer’s Association. 2014;10:592–601. e2. doi: 10.1016/j.jalz.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 34.Roses AD, Lutz MW, Crenshaw DG, Grossman I, Saunders AM, Gottschalk WK. TOMM40 and APOE: Requirements for replication studies of association with age of disease onset and enrichment of a clinical trial. Alzheimers Dement. 2013;9:132–6. doi: 10.1016/j.jalz.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 35.Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Rimmler JB, Locke PA, Conneally PM, Schmader KE, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–4. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- 36.Johnson SC, La Rue A, Hermann BP, Xu G, Koscik RL, Jonaitis EM, Bendlin BB, Hogan KJ, Roses AD, Saunders AM, Lutz MW, Asthana S, Green RC, Sager MA. The effect of TOMM40 poly-T length on gray matter volume and cognition in middle-aged persons with APOE ε3/ε3 genotype. Alzheimer’s and Dementia. 2011;7:456–65. doi: 10.1016/j.jalz.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayden KM, McEvoy JM, Linnertz C, Attix D, Kuchibhatla M, Saunders AM, Lutz MW, Welsh-Bohmer KA, Roses AD, Chiba-Falek O. A homopolymer polymorphism in the TOMM40 gene contributes to cognitive performance in aging. Alzheimers Dement. 2012;8:381–8. doi: 10.1016/j.jalz.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu L, Lutz MW, Wilson RS, Burns DK, Roses AD, Saunders AM, Gaiteri C, De Jager PL, Barnes LL, Bennett DA. TOMM40’523 variant and cognitive decline in older persons with APOE ε3/3 genotype. Neurology. 2017 Jan 20; doi: 10.1212/WNL.0000000000003614. pii:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jun G, Vardarajan BN, Buros J, et al. COmprehensive search for alzheimer disease susceptibility loci in the apoe region. Archives of Neurology. 2012;69:1270–9. doi: 10.1001/archneurol.2012.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cruchaga C, Nowotny P, Kauwe JSK, Ridge PG, Mayo K, Bertelsen S, Hinrichs A, Fagan AM, Holtzman DM, Morris JC, Goate AM for the Alzheimer’s Disease Neuroimaging Initiative. Association and Expression Analyses With Single-Nucleotide Polymorphisms in TOMM40 in Alzheimer Disease. Arch Neurol. 2011;68:1013–9. doi: 10.1001/archneurol.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maruszak A, Pepłońska B, Safranow K, Chodakowska-Żebrowska M, Barcikowska M, Żekanowski C. TOMM40 rs10524523 Polymorphism’s Role in Late-Onset Alzheimer’s Disease and in Longevity. Journal of Alzheimer’s Disease. 2012;28:309–22. doi: 10.3233/JAD-2011-110743. [DOI] [PubMed] [Google Scholar]
- 42.Gottschalk WK, Lutz MW, He YT, Saunders AM, Burns DK, Roses AD, Chiba-Falek O. The Broad Impact of TOM40 on Neurodegenerative Diseases in Aging. Journal of Parkinson’s disease and Alzheimer’s disease. 2014;1:12. doi: 10.13188/2376-922X.1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akai J, Kimura A, Hata RI. Transcriptional regulation of the human type I collagen alpha2 (COL1A2) gene by the combination of two dinucleotide repeats. Gene. 1999;239:65–73. doi: 10.1016/s0378-1119(99)00380-7. [DOI] [PubMed] [Google Scholar]
- 44.Chiba-Falek O, Nussbaum RL. Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum Mol Genet. 2001;10:3101–9. doi: 10.1093/hmg/10.26.3101. Epub 2001/12/26. [DOI] [PubMed] [Google Scholar]
- 45.Okladnova O, Syagailo YV, Tranitz M, Stober G, Riederer P, Mossner R, Lesch KP. A promoter-associated polymorphic repeat modulates PAX-6 expression in human brain. Biochemical and biophysical research communications. 1998;248:402–5. doi: 10.1006/bbrc.1998.8972. [DOI] [PubMed] [Google Scholar]
- 46.Peters DG, Kassam A, St Jean PL, Yonas H, Ferrell RE. Functional polymorphism in the matrix metalloproteinase-9 promoter as a potential risk factor for intracranial aneurysm. Stroke. 1999;30:2612–6. doi: 10.1161/01.str.30.12.2612. [DOI] [PubMed] [Google Scholar]
- 47.Searle S, Blackwell JM. Evidence for a functional repeat polymorphism in the promoter of the human NRAMP1 gene that correlates with autoimmune versus infectious disease susceptibility. Journal of medical genetics. 1999;36:295–9. [PMC free article] [PubMed] [Google Scholar]
- 48.Shimajiri S, Arima N, Tanimoto A, Murata Y, Hamada T, Wang KY, Sasaguri Y. Shortened microsatellite d(CA)21 sequence down-regulates promoter activity of matrix metalloproteinase 9 gene. FEBS Lett. 1999;455:70–4. doi: 10.1016/s0014-5793(99)00863-7. [DOI] [PubMed] [Google Scholar]
- 49.Hefferon TW, Groman JD, Yurk CE, Cutting GR. A variable dinucleotide repeat in the CFTR gene contributes to phenotype diversity by forming RNA secondary structures that alter splicing. Proc Natl Acad Sci U S A. 2004;101:3504–9. doi: 10.1073/pnas.0400182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–40. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 51.Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nature reviews Genetics. 2005;6:729–42. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 52.Willems T, Gymrek M, Highnam G, Mittelman D, Erlich Y Genomes Project C. The landscape of human STR variation. Genome Res. 2014;24:1894–904. doi: 10.1101/gr.177774.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Payton A, Sindrewicz P, Pessoa V, Platt H, Horan M, Ollier W, Bubb VJ, Pendleton N, Quinn JP. A TOMM40 poly-T variant modulates gene expression and is associated with vocabulary ability and decline in nonpathologic aging. Neurobiology of aging. 2015 doi: 10.1016/j.neurobiolaging.2015.11.017. [DOI] [PubMed] [Google Scholar]
- 54.Bekris LM, Lutz F, Yu C-E. Functional analysis of APOE locus genetic variation implicates regional enhancers in the regulation of both TOMM40 and APOE. J Hum Genet. 2012;57:18–25. doi: 10.1038/jhg.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bender A, Desplats P, Spencer B, Rockenstein E, Adame A, Elstner M, Laub C, Mueller S, Koob AO, Mante M, Pham E, Klopstock T, Masliah E. TOM40 mediates mitochondrial dysfunction induced by alpha-synuclein accumulation in Parkinson’s disease. PloS one. 2013;8:e62277. doi: 10.1371/journal.pone.0062277. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Gottschalk WK, Lutz MW, He YT, Saunders AM, Burns DK, Roses AD, Chiba-Falek O. The Broad Impact of TOM40 on Neurodegenerative Diseases in Aging. J Parkinsons Dis Alzheimers Dis. 2014:1. doi: 10.13188/2376-922X.1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu P, Dean RA, Hall SD, Qi Y, Sethuraman G, Willis BA, Siemers ER, Martenyi F, Tauscher JT, Schwarz AJ. Enriching amnestic mild cognitive impairment populations for clinical trials: optimal combination of biomarkers to predict conversion to dementia. Journal of Alzheimer’s disease: JAD. 2012;32:373–85. doi: 10.3233/JAD-2012-120832. [DOI] [PubMed] [Google Scholar]
- 58.Albert M, Soldan A, Gottesman R, McKhann G, Sacktor N, Farrington L, Grega M, Turner R, Lu Y, Li S, Wang MC, Selnes O. Cognitive changes preceding clinical symptom onset of mild cognitive impairment and relationship to ApoE genotype. Curr Alzheimer Res. 2014;11:773–84. doi: 10.2174/156720501108140910121920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Soldan A, Pettigrew C, Lu Y, Wang MC, Selnes O, Albert M, Brown T, Ratnanather JT, Younes L, Miller MI, Team BR. Relationship of medial temporal lobe atrophy, APOE genotype, and cognitive reserve in preclinical Alzheimer’s disease. Hum Brain Mapp. 2015;36:2826–41. doi: 10.1002/hbm.22810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Albert M. Using Combinations of Variables to Identify Individuals with Preclinical AD (F1-03-02) Alzheimer’s Association; 2015. Available from: https://www.alz.org/aaic/releases_2015/Sun-8amET.asp. [Google Scholar]
- 61*.Escott-Price V, Sims R, Bannister C, Harold D, Vronskaya M, Majounie E, Badarinarayan N, Morgan K, Passmore P, Holmes C, Powell J, Brayne C, Gill M, Mead S, Goate A, Cruchaga C, Lambert JC, van Duijn C, Maier W, Ramirez A, Holmans P, Jones L, Hardy J, Seshadri S, Schellenberg GD, Amouyel P, Williams J Gerad/Perades, consortia I. Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain. 2015;138:3673–84. doi: 10.1093/brain/awv268. The first LOAD prediction model based on risk alleles identified in GWAS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chouraki V, Reitz C, Maury F, Bis JC, Bellenguez C, Yu L, Jakobsdottir J, Mukherjee S, Adams HH, Choi SH, Larson EB, Fitzpatrick A, Uitterlinden AG, de Jager PL, Hofman A, Gudnason V, Vardarajan B, Ibrahim-Verbaas C, van der Lee SJ, Lopez O, Dartigues JF, Berr C, Amouyel P, Bennett DA, van Duijn C, DeStefano AL, Launer LJ, Ikram MA, Crane PK, Lambert JC, Mayeux R, Seshadri S International Genomics of Alzheimer’s P. Evaluation of a Genetic Risk Score to Improve Risk Prediction for Alzheimer’s Disease. J Alzheimers Dis. 2016;53:921–32. doi: 10.3233/JAD-150749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63**.Lutz MW, Sundseth S, Burns DK, Saunders AM, Hayden KM, Burke JR, Welsh-Bohmer KA, Roses AD. A Genetics-based Biomarker Risk Algorithm for Predicting Risk of Alzheimer’s Disease Alzheimer’s and Dementia: Translational Research and Clinical Interventions. 2016;2:30–44. doi: 10.1016/j.trci.2015.12.002. Epub 2016 Jan 1. The development of LOAD-GBRA utilizing a combination of APOE genotype, TOMM40 poly-T genotypes and age. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moreno-Moral A, Pesce F, Behmoaras J, Petretto E. Systems Genetics as a Tool to Identify Master Genetic Regulators in Complex Disease. Methods in molecular biology. 2017;1488:337–62. doi: 10.1007/978-1-4939-6427-7_16. [DOI] [PubMed] [Google Scholar]
- 65.Mungall CJ, McMurry JA, Kohler S, Balhoff JP, Borromeo C, Brush M, Carbon S, Conlin T, Dunn N, Engelstad M, Foster E, Gourdine JP, Jacobsen JO, Keith D, Laraway B, Lewis SE, NguyenXuan J, Shefchek K, Vasilevsky N, Yuan Z, Washington N, Hochheiser H, Groza T, Smedley D, Robinson PN, Haendel MA. The Monarch Initiative: an integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic acids research. 2017;45:D712–D22. doi: 10.1093/nar/gkw1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Y, Maxwell S, Feng T, Zhu X, Elston RC, Koyuturk M, Chance MR. Gene, pathway and network frameworks to identify epistatic interactions of single nucleotide polymorphisms derived from GWAS data. BMC systems biology. 2012;6(Suppl 3):S15. doi: 10.1186/1752-0509-6-S3-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schadt EE, Lamb J, Yang X, Zhu J, Edwards S, Guhathakurta D, Sieberts SK, Monks S, Reitman M, Zhang C, Lum PY, Leonardson A, Thieringer R, Metzger JM, Yang L, Castle J, Zhu H, Kash SF, Drake TA, Sachs A, Lusis AJ. An integrative genomics approach to infer causal associations between gene expression and disease. Nature genetics. 2005;37:710–7. doi: 10.1038/ng1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HY, Chen R, Miriami E, Karczewski KJ, Hariharan M, Dewey FE, Cheng Y, Clark MJ, Im H, Habegger L, Balasubramanian S, O’Huallachain M, Dudley JT, Hillenmeyer S, Haraksingh R, Sharon D, Euskirchen G, Lacroute P, Bettinger K, Boyle AP, Kasowski M, Grubert F, Seki S, Garcia M, Whirl-Carrillo M, Gallardo M, Blasco MA, Greenberg PL, Snyder P, Klein TE, Altman RB, Butte AJ, Ashley EA, Gerstein M, Nadeau KC, Tang H, Snyder M. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. 2012;148:1293–307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karczewski KJ, Dudley JT, Kukurba KR, Chen R, Butte AJ, Montgomery SB, Snyder M. Systematic functional regulatory assessment of disease-associated variants. Proc Natl Acad Sci U S A. 2013;110:9607–12. doi: 10.1073/pnas.1219099110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, Lamb JR, Bennett DA, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–20. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gaiteri C, Mostafavi S, Honey CJ, De Jager PL, Bennett DA. Genetic variants in Alzheimer disease - molecular and brain network approaches. Nature reviews Neurology. 2016;12:413–27. doi: 10.1038/nrneurol.2016.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rollo JL, Banihashemi N, Vafaee F, Crawford JW, Kuncic Z, Holsinger RM. Unraveling the mechanistic complexity of Alzheimer’s disease through systems biology. Alzheimers Dement. 2016;12:708–18. doi: 10.1016/j.jalz.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 73.Geerts H, Dacks PA, Devanarayan V, Haas M, Khachaturian ZS, Gordon MF, Maudsley S, Romero K, Stephenson D Brain Health Modeling I. Big data to smart data in Alzheimer’s disease: The brain health modeling initiative to foster actionable knowledge. Alzheimers Dement. 2016;12:1014–21. doi: 10.1016/j.jalz.2016.04.008. [DOI] [PubMed] [Google Scholar]