


Abstract
APOBEC cytidine deaminases mutate cancer genomes by converting cytidines into uridines within ssDNA during replication. Although uracil DNA glycosylases limit APOBEC-induced mutation, it is unknown if subsequent base excision repair (BER) steps function on replication-associated ssDNA. Hence, we measured APOBEC3B-induced CAN1 mutation frequencies in yeast deficient in BER endonucleases or DNA damage tolerance proteins. Strains lacking Apn1, Apn2, Ntg1, Ntg2 or Rev3 displayed wild-type frequencies of APOBEC3B-induced canavanine resistance (CanR). However, strains without error-free lesion bypass proteins Ubc13, Mms2 and Mph1 displayed respective 4.9-, 2.8- and 7.8-fold higher frequency of APOBEC3B-induced CanR. These results indicate that mutations resulting from APOBEC activity are avoided by deoxyuridine conversion to abasic sites ahead of nascent lagging strand DNA synthesis and subsequent bypass by error-free template switching. We found this mechanism also functions during telomere re-synthesis, but with a diminished requirement for Ubc13. Interestingly, reduction of G to C substitutions in Ubc13-deficient strains uncovered a previously unknown role of Ubc13 in controlling the activity of the translesion synthesis polymerase, Rev1. Our results highlight a novel mechanism for error-free bypass of deoxyuridines generated within ssDNA and suggest that the APOBEC mutation signature observed in cancer genomes may under-represent the genomic damage these enzymes induce.
INTRODUCTION
Somatic mutations are the predominant cause of cell cycle dysregulation and unrestrained proliferation that occurs during the development of sporadic cancers. Recently, members of the apolipoprotein B mRNA editing enzyme catalytic polypeptide-like (APOBEC) family of cytidine deaminases have been identified as tumor-specific sources of DNA damage (1,2) occurring in ∼15% of human cancers (3–5). The catalytically active enzymes in this family, which include activation-induced cytidine deaminase (AID), APOBEC1 and the seven APOBEC3 enzymes (A through H), all deaminate cytidines in single-strand (ss) DNA substrates resulting in cytidine (C) to thymidine (T) or C to guanidine (G) substitutions (3–5). Additionally, every active APOBEC family member except AID and APOBEC3G (A3G) deaminate cytidines specifically within TCW motifs (where the underlined C is deaminated and W = adenine (A) or T) (6). This nucleotide editing capacity normally is used to tailor immunoglobulin affinities (AID) (7), alter apolipoproteins involved in dietary lipid transport (APOBEC1) (8) and as an anti-viral/retrotransposon innate immune response (APOBEC1 and APOBEC3) (6).
Initial characterization of the role APOBECs play in cancer have relied primarily on the over-representation of APOBEC signature mutations in cancer genomes and characteristic patterns among these mutations in relationship to various chromosomal features. A large portion of the experimental data supporting that these patterns are consistent with APOBEC activity has been accumulated in yeast and other model systems. Studies aimed at understanding AID's involvement in somatic hypermutation and class switch recombination uncovered a strong preference for this enzyme to deaminate cytidine in the non-transcribed strand of genes (9–12). Based on extrapolation of this specificity, other APOBEC enzymes were initially proposed to likewise edit cancer genomes by deaminating transcription bubbles. While some evidence for non-AID APOBECs editing transcription intermediates has been observed in yeast and bioinformatics analysis of some human cancers (13–15), APOBEC deamination of other ssDNA substrates has been additionally uncovered. The formation of APOBEC-induced clustered mutations, called kataegis, has been linked to the ability of these enzymes to deaminate chromosomal ssDNA intermediates formed during 5΄ to 3΄ resection at double-stranded breaks (DSBs) (16). More recently, publications have determined in yeast and Escherichia coli that the lagging strand template during DNA replication is the primary chromosomal substrate of several members of the APOBEC3 subfamily, including APOBEC3A (A3A), APOBEC3B (A3B) and A3G (17,18). Likewise, induction of DSBs by over-expression of A3A in U2OS cells requires DNA replication, implicating the lagging strand template is an APOBEC substrate in human cells (19). Multiple analyses of the distribution of APOBEC signature mutations in human tumors also indicate that deamination at replication forks is the primary means for these enzymes to mutagenize cancer genomes (20–22), and supports the usage of yeast and other organisms as a model for studying how APOBEC enzymes contribute to cancer generation. Cytidine deamination ahead of nascent DNA synthesis by the lagging strand polymerase results in deoxyuridine (dU) templating for A insertion and consequently results in C to T substitutions (or G to A substitutions if the dU is formed on the complementary DNA strand) if left unprocessed. How the mutagenic consequences of cytidine deamination in replication-associated ssDNA are mitigated is currently unclear.
In humans and yeast, dU incorporated into double-stranded (ds) DNA is removed by base excision repair (BER). In yeast, this mechanism involves the excision of the uracil base by the Ung1 uracil glycosylase, generation of a nick 5΄ of the newly formed abasic site by Apn1, strand displacement DNA synthesis by polymerase δ to replace the damaged base, flap removal and finally ligation by DNA Ligase I (23). However, during replication the ssDNA that serves as a substrate for APOBEC-induced deamination may not be ideal for repair by this mechanism as a nick in the phosphodiester backbone following conversion of dU to an abasic site would generate a DSB. Despite this, robust repair of these deamination events seems to occur, as indicated by an increase in APOBEC-induced mutagenesis as the consequence of UNG1 deletion (16,18,24,25). Furthermore, the generation of γ-H2AX foci by over-expression of A3A in human cells (19,26,27) suggests further BER activities may occur in ssDNA, namely APE1-mediated cleavage of Ung-generated abasic sites. Replication forks may stall at Ung-generated abasic sites and subsequently regress, returning the abasic site into dsDNA (23). This would then allow normal BER to repair dU lesions in a pre-replicative manner (28). Alternatively, any of three DNA damage tolerance (DDT) pathways could be utilized to bypass the Ung-generated abasic sites (23). Usage of these pathways is regulated by specific post-translational modifications to the PCNA sliding clamp in response to fork-stalling lesions at the replication fork (29,30). This includes SUMOylation by Siz1-Ubc9 to regulate recombination-mediated fork restart (23,31), monoubiquitination by Rad18-Rad6 to signal for translesion synthesis (TLS) (31–33), or polyubiquitination by Rad5-Ubc13-Mms2 to signal for error-free template switching (29,34).
Here, we show that yeast use a lesion-conversion mechanism ahead of DNA strand synthesis to avoid mutagenic A3B-induced dU during DNA replication. This conversion is done through Ung1-mediated excision of dU generating a fork-stalling abasic site, which is then circumvented primarily by the error-free bypass pathway. Deletion of UBC13, MMS2, or MPH1 (all genes critical for error-free bypass) increase the CAN1 mutation frequency induced by A3B activity to levels approaching ung1Δ strain frequencies. In contrast, deletion of individual BER components APN1, APN2, NTG1, NTG2, or other DDT components REV3 and SIZ1, had little effect on replication-associated mutagenesis. This indicates that the downstream steps of BER, TLS and recombination-mediated fork restart play more limited roles in preventing A3B-induced mutations.
Additionally, we found that error-free lesion bypass requires Ubc13 in a replication-specific manner, as ubc13Δ does not affect the frequency of CAN1 mutations incurred during the re-synthesis of uncapped telomeres, despite Mph1-mediated error-free bypass of similar abasic sites still occurring. Interestingly, analyses of the CAN1 mutation spectra in both replicative and telomere re-synthesis systems indicate that Ubc13 and Mms2 play a significant role in determining which TLS polymerase is active in bypassing abasic sites, as the ubc13Δ and mms2Δ spectra had diminished numbers of G to C substitutions indicative of the activity of the TLS polymerase, Rev1. This suggests that Ubc13 and Mms2 promote Rev1 cytidine transferase activity during TLS-mediated abasic site bypass and play a unique role in the crosstalk between error-free lesion bypass and TLS pathways. These results indicate that robust error-free lesion bypass mechanisms may significantly mitigate the deleterious effects of APOBEC-induced damage in human cancers and enable continued cell proliferation despite high levels of DNA damage occurring within the replication fork.
MATERIALS AND METHODS
Yeast culture and strains
Yeasts were grown using standard techniques (35) on standard rich media (YPDA) or synthetic complete (SC) media. All haploid strains utilized in this study were constructed by recombination-directed chromosomal integration in the CG379 genetic background, a derivative of S288C Saccharomyces cerevisiae (36). The genotypes of each strain used are listed in Supplementary Table S1. Briefly, novel gene deletion yeast strains were produced by transformation with polymerase chain reaction (PCR) fragments containing either a nourseothricin resistance cartridge (NAT-R) or a geneticin resistance cartridge (G418-R) flanked by sequences homologous to the wild-type gene. The primers used to generate these fragments are listed in Supplementary Table S2. The transformed yeast strains were selected for on appropriate drug-supplemented media. Diploid yeast heterozygous for a chromosomally integrated A3B-HygR (hygromycin resistance) expression construct and/or rad5Δ were generated by mating haploid MATα rad5Δ strains to either haploid MATa wild-type yeast or to haploid MATa strains containing A3B-HygR integrated into LEU2 on Chr III. Six independent crosses using three rad5Δ isolates and two A3B+ strains were conducted. Single colonies were isolated by streaking onto YPDA, isolates patched out onto YPDA plates and phenotype verified using selective media.
Plasmid transformation of yeast
Yeast clonal isolates were grown to saturation in liquid standard rich media (YPDA) at either 23°C for cdc13-1 strains or 30°C for all other strains, followed by a 3–5 h secondary incubation at appropriate temperatures. The yeasts were transformed with either A3B-expression or vector-control plasmids containing a hygromycin resistance cartridge (18) and were grown on YPDA overnight before replica plating to YPDA media supplemented with hygromycin (300 μg/ml) for selection of the plasmid.
Plating efficiency and growth kinetics of yeasts expressing A3B
Yeast strains used in this study (Supplementary Table S1) were transformed with either A3B-expression or vector-control plasmids and streaked to obtain clonal single cell isolates. Six independent isolates from each genotype were resuspended in water, counted and plated on YPDA supplemented with hygromycin (300 μg/ml) at a density of ∼200 cells per plate. Following a three-day incubation period at 30°C, the number of colonies growing on the plates were counted and compared to the number of cells plated to determine the median plating efficiency for each genotype. Alternatively, six single colony isolates of the wild-type yeasts were isolated following transformation with A3B-expressing or vector-control plasmid, resuspended in water and counted. 15 ml YPDA was then inoculated with 1.5 × 104 cells and incubated at 30°C. The O/D600 of these cultures was measured at 2-h intervals for 17 h.
Tetrad dissection and plating efficiency of rad5Δ yeast expressing A3B
Verified diploid strains heterozygous for an integrated A3B expression construct and/or rad5Δ were grown on sporulation media (20 g/l KAc, 1 g/l yeast extract, 20 g/l agar) for 5 days (1 day at 30°C and 4 days at room temperature). Resulting tetrads were re-suspended in water and treated with zymolyase for 10 min to digest the outer cell wall, prior to dissection on YPDA using a Singer MSM400. After 3 days of growth, tetrad dissections were replica plated to YPDA, YPDA supplemented with G418 (200 μg/ml), and YPDA supplemented with hygromycin (300 μg/ml) to assess the genotype of individual spores. The distribution of genotypes between large and small colonies was compared by two-tailed chi-squared goodness-of-fit test. Cells from 10–15 independent spores for each genotype resulting from the dissection were then re-suspended, counted and plated on YPDA media at a density of 200 cells per plate. After three days growth at 30°C the number of colonies growing on YPDA plates were counted and compared to the number of cells plated to determine the plating efficiency for each genotype.
Determination of mutation frequency
Yeast freshly transformed with either an A3B-expression or vector-control plasmid were streaked for single colonies. Independent colonies were re-suspended in water, diluted, and plated at a density of 200 cells per plate on YPDA supplemented with hygromycin. Transformed yeast containing CAN1 on Chr II were grown at 30°C for 72 h to accumulate mutations. Six or seven independent clones were selected, re-suspended, and plated to SC media lacking arginine and supplemented with 0.006% canavanine and also plated to nonselective SC media at appropriate concentrations to determine mutation frequencies. Mutation frequencies were assessed as a ratio of yeast colonies that gain canavanine resistance to the number of colonies that grow in the absence of selection, corrected by the respective dilution factors and calculated using the following formula:
 |
CanR frequencies for cdc13-1 yeast containing CAN1 in the Chr V sub-telomere were measured as previously described in detail (37). Briefly, yeast clonal isolates containing the cdc13-1 allele, the Chr V CAN1 mutation reporter and either the vector control or A3B-expression plasmid were grown at 23°C to saturation, followed by a 1:10 dilution and a second incubation at 37°C (the non-permissive temperature for the cdc13-1 allele) for 6 h to expose ssDNA through telomere uncapping and end resection. Cells were then appropriately plated as described above to determine mutation frequencies. Differences in mutation frequency between genotypes were evaluated statistically using a two-tailed Mann–Whitney rank sum test in Graphpad Prism5 (Graphpad, La Jolla, CA, USA). All mutation frequency values are listed in Supplementary Table S3.
Evaluation of mutation spectra
Yeast transformed with A3B-expressing plasmid were plated on YPDA supplemented with hygromycin (300 μg/ml) at a density of ∼200 cells per plate and grown for three days at 30°C to accumulate mutations within independent colonies, after which they were replica plated to SC-arginine media supplemented with 0.006% canavanine and grown for an additional three days to select for CanR yeast. Alternatively, following a 6 h incubation at 37°C, cultures of cdc13-1 yeast transformed with A3B were directly plated to SC-arginine media supplemented with 0.006% canavanine and allowed to grow four days at 23°C to obtain CanR isolates. Independent CanR papillae were then selected and further clonally isolated through single colony streaking. Genomic DNA was isolated by bead agitation in a LiAc/SDS/TE buffer and purified over OMEGA HiBind DNA mini columns (Omega Bio-tek, Inc., Norcross, GA, USA). The CAN1 gene in isolates from wild-type, ubc13Δ, mph1Δ, ung1Δ, ubc13Δ mph1Δ, ubc13Δ ung1Δ and mph1Δ ung1Δ genetic backgrounds were PCR amplified using primers with unique identification barcodes listed in Supplementary Table S2. These amplicons were pooled and sequenced by SMRT sequencing on a PacBio RSII as in (38). We obtained a total of 6197 consensus sequence reads, each obtained by at least three complete passages of an individual DNA fragment. This equated to 2–63× coverage per barcoded amplicon. The sequencing results were sorted into individual isolates identified by their unique identification barcode combination. Consensus sequence reads were assembled to the reference CAN1 gene and mutations called in Geneious 8.1.8 (Biomatters Limited, Newark, NJ, USA). All called mutations were supported by multiple aligned circular consensus sequences (CCS) and occurred in greater than 50% of aligned reads per unique barcode set. The CAN1 gene from mms2Δ and apn1Δ apn2Δ yeast strains were PCR amplified using alternative primers listed in Supplementary Table S2 and Sanger sequenced (Genscript Biotech Corporation, Piscataway, NJ, USA) as described in (18). Statistical significance for strand bias or nucleotide insertion preference was determined using a G-test of goodness-of-fit. Comparisons between mutation spectra of different genotypes were evaluated using a two-tailed Fisher's exact test in Graphpad Prism5 (Graphpad, La Jolla, CA, USA). The preferred nucleotide sequences mutated in CAN1 following A3B expression were generated with the mutated base +/− 1 nt using WebLogo (http://weblogo.berkeley.edu/logo.cgi) as described in (39). All mutations identified through sequencing are listed in Supplementary Table S4 and summaries of the mutation spectra are provided in Supplementary Table S5.
RESULTS
A3B-induced deaminations are converted to abasic sites by Ung1, but not repaired through the BER pathway
To address how APOBEC-induced dU that occur in the context of DNA replication are repaired or otherwise processed, we ectopically expressed A3B within strains of Saccharomyces cerevisiae and measured CAN1 forward mutations, which result in canavanine resistance (CanR), using a reporter on chromosome II located 16 kb centromere proximal of ARS216. We previously showed that A3B-induced mutations in this location are primarily due to deamination occurring in ssDNA during the synthesis of the lagging strand (Figure 1A) (18). Consistent with our previous findings, A3B expression in this system had no detectable effect on the growth kinetics of wild-type yeast (Supplementary Figure S1) but resulted in ∼200-fold increase in mutation frequency compared to vector-only controls (Figure 2A). Deletion of UNG1 in A3B-expressing strains caused an additional 8.5-fold increase in mutation frequency, indicating that A3B-induced dU is either repaired or bypassed in an error-free manner during DNA replication. To test whether abasic sites created by Ung1-catalyzed removal of dU in ssDNA at the replication fork are moved into dsDNA through fork regression and subsequently repaired by BER, we deleted the APN1, APN2, NTG1 and NTG2 genes which encode the yeast AP endonucleases and AP lyases known to participate in BER (23) (Figure 2A). The deletion of APN1 in the vector-transformed strain showed increases in mutation frequency as a result of impaired BER activity, indicating that this deletion removes the majority of AP endonuclease activity in yeast. However, the independent deletion of APN1 or any other BER endonuclease or lyase in the A3B-expressing strains failed to significantly increase the CanR frequency beyond wild-type levels. To determine whether redundant activity between these enzymes may enable efficient removal of A3B-induced lesions and thereby obscure mutagenic effects in the single deletion strains, we measured CanR frequency in apn1Δ apn2Δ and ntg1Δ ntg2Δ double deletion strains. Yeast where both NTG1 and NTG2 were deleted demonstrated only a small 1.8-fold increase in CanR frequency compared to wild-type yeast, further supporting that these lyases play little role in the repair of abasic sites in replication-associated ssDNA. In contrast, apn1Δ apn2Δ strains displayed a moderate 2.8-fold increase in CanR.
Figure 1.

Reporters used in this study. (A) The CAN1 forward mutation reporter was placed 16 kb centromere proximal to the replication origin ARS216 on Chr II. This system has been previously shown to accumulate mutations predominantly through A3B-induced deamination during replication. During DNA replication the lagging strand template is exposed to A3B, resulting in targeted dC to dU deamination. This confers a mutation in CAN1 which allows the yeast to survive on selective canavanine media. Based upon the location of CAN1 to the centromere proximal side of ARS216, the bottom DNA strand of the gene is the lagging strand template. Thus, cytidine deamination occurs on the bottom strand, resulting in mutations at G bases (sequencing reports mutations relative to the top DNA strand). (B) The CAN1 forward mutation reporter was placed 7.5 kb from the end of the telomere of Chr V. This system contains the cdc13-1 mutant allele which uncaps telomeres at nonpermissive temperatures and halts the growth of Saccharomyces cerevisiae at G2 phase following DNA replication. Following telomere uncapping, 5΄ to 3΄ resection initiates, resulting in the exposure of dC in ssDNA to A3B. When sequenced, the mutations in the re-synthesized strand are reported as mutations in dG.
Figure 2.

BER is not a predominant method of removing A3B-induced dU formed during replication. (A) The frequencies of canavanine-resistance (CanR) induced in WT, ung1Δ, apn1Δ, apn2Δ, apn1Δ apn2Δ, ntg1Δ, ntg2Δ and ntg1Δ ntg2Δ yeast following transformation with vector control plasmid, or A3B expression plasmid were assessed. CanR frequency was determined in isogenic strains harboring the CAN1 reporter on Chr II. Horizontal bars and numeric values indicate the median frequency of six independent replicates. Statistical significance was determined by a two-tailed non-parametric Mann–Whitney rank sum test. (B) The strand bias of CAN1 mutations from A3B-expressing wild-type, apn1Δ apn2Δ and ung1Δ yeast strains were assessed by Sanger sequencing. The mutation spectra frequency represents the proportion of the CanR frequency in (A) that can be attributed to the individual mutation types. The numerical values above the bars indicate the number of mutations in each strand for each strain. Statistical significance of strand bias in each genotype was determined by a two-tailed G-test of goodness-of-fit. **:P < 0.02.
To understand the cause of mutation in these strains, we sequenced the CAN1 gene in independent CanR clones. Consistent with previous results showing that A3B targets the lagging strand template during DNA replication (18), we observed predominantly G bases mutated within GA dinucleotides (complementary to TC) in the wild-type and the UNG1 deficient strains (P < 0.002) (Figure 2B and Supplementary Figure S2A–D). However, the apn1Δ apn2Δ strains displayed a significant loss of this strand bias (Figure 2B). The frequency of G mutations that potentially originate from deamination of ssDNA in the lagging strand template in apn1Δ apn2Δ strains increased only 1.3-fold compared to the G mutation frequency in wild-type yeast (Figure 2B and Supplementary Table S5B). In contrast, C mutations in apn1Δ apn2Δ strains increased 4.7-fold. This change in mutation spectrum in apn1Δ apn2Δ strains indicates that instead of the increase in mutagenesis stemming from failure to remove A3B lesions on the lagging strand template, it occurs due to the presence of additional mutagenic A3B-induced lesions formed on the leading strand template. These added lesions likely originate from increased ssDNA substrate generated as a consequence of an inability to effectively remove spontaneous base lesions. Accordingly, vector-transformed apn1Δ apn2Δ strains showed a significant (p-0.0260 versus wild-type) decrease in survival (Supplementary Figure S3A), indicating the yeast's compromised capacity to process spontaneous lesions creates difficulties during cell division. These results therefore suggest that while Ung1-mediated dU removal occurs in ssDNA, the downstream mechanisms in BER are likely not prominently involved in the processing of A3B-induced deaminations that occur during DNA replication. Thus, pre-replicative repair of these lesions by BER is not a major mechanism for avoiding APOBEC-induced mutations. The apparent inability of AP endonucleases to mediate BER of A3B-induced lesions during replication could result from ssDNA binding proteins sterically blocking the access of these enzymes to abasic sites in the lagging strand template. Alternatively, human APE1 was previously shown biochemically to have drastically reduced affinity for abasic sites in ssDNA compared to dsDNA (40), indicating that the ssDNA context of these lesions itself may make them resistant to repair.
A3B-induced deaminations are predominantly bypassed by error-free damage avoidance
Since BER processes downstream of dU excision by Ung1 do not reduce A3B-induced mutations, other mechanism(s) must act to mitigate some or all of the mutational consequences of cytidine deamination during replication. Therefore, we sought to determine if DDT pathways are utilized to bypass abasic sites generated following excision of A3B-induced dU by Ung1 (Figure 3 and Supplementary Figure S4A). To specifically assess the contribution of TLS to A3B-induced mutagenesis, we deleted the catalytic subunit of the TLS DNA polymerase ζ (REV3), which is required for all damage-induced mutagenesis in yeast (23). We observed no notable change in mutation frequency or cell survival when ectopic A3B is expressed in rev3Δ strains(Figure 3B and Supplementary Figure S3B and C), indicating that TLS does not play a major role in damage avoidance of A3B-induced dU at the replication fork.
Figure 3.

Error-free bypass is the DDT pathway used to avoid A3B-induced lesions incurred ahead of polymerase δ lagging strand DNA synthesis. (A) Possible mechanisms for processing dU and their mutagenic outcomes. If left within DNA, the A3B-induced dU will confer a G to A mutation due to templating during replication, as indicated in black. Glycolytic removal of the uracil base by Ung1, converts dU to an abasic site, which can then serve to induce DNA damage tolerance (DDT) pathways to bypass the lesion. Specific modifications on the PCNA clamp determine pathway choice. PCNA SUMOylation by Ubc9-Siz1 controls recombination-mediated fork restart, noted in red. Monoubiquitination of PCNA by Rad18-Rad6 can result in two outcomes: the initiation of TLS and the insertion of untemplated nucleotides across from the abasic site (purple) or the extension of the ubiquitin chain by Rad5-Ubc13-Mms2 (blue). The latter results in the initiation of error-free lesion bypass. (B and C) Frequencies and mutational spectra of CanR following transformation with A3B expression plasmid assessed in yeast strains harboring the CAN1 reporter on Chr II were assessed. (B) rev3Δ, siz1Δ, ubc13Δ, mms2Δ, mph1Δ and mte1Δ yeast strains were compared to discern the usage of the various DDT pathways to avoid abasic sites generated during replication. Horizontal bars and numerical values indicate the median frequency of six or seven independent replicates. Statistical significance was determined by a two-tailed non-parametric Mann-Whitney rank sum test. WT and ung1Δ values from Figure 2 are included for reference. (C) The mutation spectra of the CAN1 in independent WT, ubc13Δ, mms2Δ or ung1Δ CanR yeast were determined through PacBio and Sanger Sequencing. The mutation spectra frequency represents the proportion of the CanR frequency in (A) that can be attributed to the individual mutation types. The numerical values above the bars indicate the number of isolates with the reported mutation. Statistical significance of G to A and G to C mutation ratios in each genetic backgrounds were determined by two-tailed G-test of goodness-of-fit test. *:P < 0.05, **: P < 0.01.
Next, we deleted genes involved in post-translational modifications of the PCNA clamp used to signal for individual DDT pathway selection. While no notable change in mutation frequency or cell survival was observed in the siz1Δ strain (Figure 3B and Supplementary Figure S3B and C), the deletion of UBC13 resulted in a 4.9-fold increase in CanR over wild-type (P = 0.0022). These mutation frequencies indicate that recombination-mediated fork restart does not have a major role in lesion avoidance during replication. Instead, the mutagenic consequences of APOBEC-induced dU are primarily bypassed through an error-free template switching mechanism. Supporting this, deletion of MMS2, another member of the Rad5-Ubc13-Mms2 E3 ubiquitin ligase complex involved in the polyubiquitination of PCNA, increased CAN1 mutagenesis 2.8-fold over the wild-type (P = 0.0022). We also deleted RAD5, the third member of the Rad5-Ubc13-Mms2 E3 ligase complex, but attempts to transform this strain with A3B-expressing plasmid consistently failed, suggesting a synthetic lethal phenotype may exist in A3B-expressing rad5Δ strains. To test this, we generated rad5Δ strains containing a chromosomally integrated A3B expression construct through mating, sporulation and tetrad dissection. We observed a slow-growth phenotype specifically in rad5Δ spores expressing A3B. Dilution and plating of ∼200 individual rad5Δ A3B+ cells on rich media demonstrated a decrease in plating efficiency (Supplementary Figure S5), indicating that the combination of RAD5 loss and A3B expression does result in cell death. This severe phenotype is likely due to greater difficulty bypassing abasic sites and completing replication as Rad5 plays roles in both initiating error-free lesion bypass and stimulating Rev1-mediated TLS (41).
To further confirm Rad5-Ubc13-Mms2 signaling of error-free bypass of abasic sites as the favored mechanism for circumventing A3B-induced mutation, we also deleted MPH1, which encodes a helicase involved in the error-free template switching pathway (42) (Figure 3B). The loss of the Mph1 helicase also increased A3B-induced CanR frequency 7.8-fold over wild-type (P = 0.0034) and nearly to the level associated with ung1Δ, suggesting that in the absence of Mph1 all A3B-induced lesions become mutagenic. Mph1 is a multifunctional protein with roles in homologous recombination (43), telomere maintenance (44), Okazaki fragment maturation (45), as well as error-free lesion bypass (42,46). To determine whether inability of Mph1 to mediate its functions during homologous recombination contributes to the observed elevation in A3B-induced CanR frequency in mph1Δ strains, we deleted the Mph1-associated factor Mte1, which supports Mph1 activities during the homology-directed repair of DNA double strand breaks (47–49). No detectable difference was observed between the CanR frequency of mte1Δ strains compared to wild-type yeast (P = 0.0931). Thus, the increased mutagenicity of A3B activity in mph1Δ strains is unlikely to be due to a defect in homology-directed repair, and the increase in CanR observed with the MPH1-deficient strains are likely due the defects in the helicase's role in error-free bypass. Supporting this, the mutation frequency in strains deficient in both UBC13 and MPH1 are similar to mph1Δ single mutant strains (P = 0.0129 versus ubc13Δ), indicating that Ubc13-mediated bypass occurs as a subset of Mph1-mediated bypass (Supplementary Figure S6). No detectable difference in mutation frequency was observed between ubc13Δ ung1Δ, mms2Δ ung1Δ, or mph1Δ ung1Δ strains and ung1Δ strains (Supplementary Figure S7B), verifying that with respect to A3B-induced mutation, UBC13, MMS2 and MPH1 are epistatic with UNG1. A small, but statistically significant difference in mutation frequency was observed between mph1Δ and mph1Δ ung1Δ strains (P = 0.038) as well as between ubc13Δ and ubc13Δ ung1Δ strains (P = 0.005).
We then sequenced the CAN1 gene in independent CanR clones to assess the mutation spectra in error-free lesion bypass-deficient yeast (Figure 3C). As with the wild-type and UNG1-deficient strains, we observed predominantly G bases mutated within GA dinucleotides (complementary to TC) (Supplementary Figure S2A–C, E–J and S8), indicating that unlike A3B-induced mutations in apn1Δ apn2Δ strains, the elevated mutations in bypass-deficient yeast result from inability to tolerate lesions in the lagging strand template in an error-free manner. Additionally, all strains deficient in Ung1 had solely G to A mutations due to retention of dU in the DNA (Supplementary Figure S7C), further confirming that these mutations result from A3B-induced deamination.
Previous reports have shown CAN1 mutations induced by A3G activity on ssDNA at uncapped telomeres (37) or APOBEC-attributed mutations in human cancer (3,5) consisted of equal amounts of G to A and G to C substitutions. This substitution pattern occurs due to abasic site bypass by the TLS polymerase, Rev1, which exclusively inserts C across from abasic sites, and a second polymerase which primarily inserts A (termed A-rule insertion) (37). However, the CAN1 mutation spectra induced by A3B activity during replication of wild-type yeast in our study consisted of almost exclusively G to A mutations (P = 0.000019) (Figure 3C). This result is consistent with previous analyses of A3B-induced mutation in yeast (16,18) and is likely because the majority of mutations in wild-type yeast result from residual dU that template C to T substitutions. Accordingly, the A3B-induced CanR frequency in the rev3Δ strain is similar to the wild-type strain (Figure 3B), indicating that most A3B mutations in wild-type yeast are generated independently of TLS. However, in strains lacking error-free template switching, the majority of A3B-induced mutations are due to error-prone TLS past abasic sites. This is evident as deletion of REV3 in the ubc13Δ and mph1Δ strains decreased the APOBEC-induced mutation frequency to wild-type levels in addition to demonstrating a decreased cell survival (Figure 4, Supplementary Figures S3B, C and S4B). The A3B-induced CAN1 mutation spectrum in mph1Δ strains has nearly equal representation of G to C and G to A substitutions (Figure 3B), demonstrating the usage of Rev1 and another ‘A-rule’ polymerase in the bypass of abasic sites. The polymerase responsible for this untemplated A-rule insertion is likely either polymerase δ or ζ, as both have been previously shown biochemically to preferentially insert deoxyadenine across from an abasic site (50), the lesions are lagging strand specific, and DNA polymerase η has previously been shown to contribute little to mutagenic abasic site bypass (37). Interestingly, the ubc13Δ and mms2Δ mutation spectra consisted predominantly of G to A mutations (P = 0.000136 and P = 0.033, respectively), indicating that abasic site bypass in these strains is accomplished primarily by A-rule insertion at the abasic site followed by polymerase ζ mediated extension past the lesion. Since Rev1 has been shown to be required for damage-induced mutagenesis as a structural component (37,51,52) and the CAN1 mutation frequency increases in ubc13Δ and mms2Δ strains, this result cannot be explained by a failure to recruit Rev1 to abasic sites. Therefore, Ubc13 and Mms2 appear to directly promote nucleotide insertion by Rev1 during abasic site TLS bypass.
Figure 4.
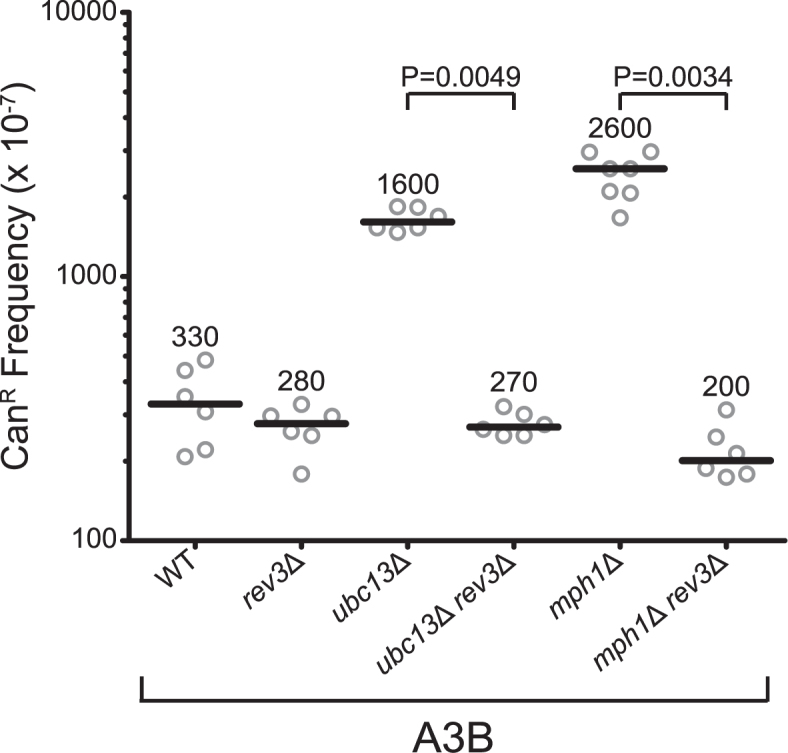
Mutagenesis in error-free bypass-deficient strains is TLS-dependent. A3B-induced CanR frequencies WT, rev3Δ, ubc13Δ, ubc13Δ rev3Δ, mph1Δ and mph1Δ rev3Δ yeast. CanR frequency was determined with CAN1 reporter on Chr II. Values for WT, rev3Δ, ubc13Δ and mph1Δ from Figure 3 are included for reference. Horizontal bars and numeric values indicate the median frequency of six or seven independent replicates. Statistical significance was determined by a two-tailed non-parametric Mann–Whitney rank sum test.
Usage of Ubc13 in error-free damage avoidance is context-specific
Although most DNA synthesis occurs during replication, gap-filling synthesis without canonical replication forks can occur during restoration of uncapped telomeres or DSB repair. If damage accumulates in the exposed ssDNA formed during these processes, usage of error-free bypass with non-allelic templates could result in genetic rearrangements as a consequence of close proximity to a DNA terminus in the template strand. To test whether error-free bypass is used to avoid damage during non-replicative DNA synthesis, we assessed the effect of Ubc13 and Mph1 deficiency on A3B-induced mutagenesis in a yeast strain with a cdc13-1 allele that results in telomere uncapping and subsequent 5΄ to 3΄ resection of the chromosome end under non-permissive temperatures. This resection renders the CAN1 gene susceptible to A3B-induced deamination due to its location 7.5 kb from the chromosome V end (Figure 1B) (37). As the result of telomere uncapping, cdc13-1 yeast grown at non-permissive temperature arrest in G2 phase of the cell cycle (53). Upon return to permissive growth temperatures, non-replicative DNA synthesis occurs to restore the resected chromosome ends, and any lesions encountered by this process could potentially utilize the available sister chromatid as a template for error-free bypass.
Within this system, we observed a 6.1-fold increase in the CanR frequency upon expressing A3B compared to the vector-only controls. Furthermore, deletion of UNG1 resulted in an additional 7.3-fold increase in CanR (P = 0.0022), showing that dU excision is involved in the avoidance of mutation during non-replicative DNA synthesis (Figure 5A) as it is during replicative DNA synthesis. In contrast to APOBEC-induced CAN1 mutagenesis during replication, rev3Δ resulted in a 95% reduction in the CanR frequency compared to the cdc13-1 strain (P = 0.0012). This result indicates that a significant amount of A3B-induced mutations during telomere re-synthesis are mediated through error-prone bypass of abasic sites by TLS, confirming previous studies assessing A3G mutagenesis in the same system (37). Additionally, we observed an increase in the CanR frequency in the cdc13-1 mph1Δ strains, demonstrating that these lesions are also bypassed in an error-free manner. Surprisingly, the cdc13-1 ubc13Δ strain failed to significantly alter the CanR frequency compared to cdc13-1 strains, indicating that the polyubiquitination of PCNA may not be required for error-free damage avoidance during gap-fill DNA synthesis. This difference in mutation frequency between the UBC13- and MPH1-deficient strains may suggest that licensing of pathway initiation may be dispensable for error-free bypass during certain types of gap-fill synthesis.
Figure 5.

Ubc13 is dispensable for error-free bypass of abasic sites in non-replicative DNA synthesis but plays a role in Rev1 usage. Frequencies and mutational spectra of CanR following transformation with vector control plasmid, or A3B expression plasmid assessed in yeast strains harboring the CAN1 reporter on Chr V. (A) cdc13-1, cdc13-1 ung1Δ, cdc13-1 mph1Δ, cdc13-1 rev3Δ and cdc13-1 ubc13Δ yeast strains were assessed to discern the usage of the error-free bypass pathway to avoid abasic sites generated during nonreplicative DNA synthesis. Horizontal bars and numerical values indicate the median frequency of six or seven independent replicates. Statistical significance was determined by a two-tailed non-parametric Mann–Whitney rank sum test. (B) The mutation spectra of the CAN1 in independent cdc13-1, cdc13-1 ung1Δ and cdc13-1 ubc13Δ CanR yeast were assessed through PacBio sequencing. The mutation spectra frequency represents the proportion of the CanR frequency in (A) that can be attributed to the individual mutation types. The numerical values above the bars indicate the number of isolates with the reported mutation. Statistical significance of G to A and G to C mutation ratios between genetic backgrounds were determined by two-tailed Fisher's exact test.
Next, we determined the roles of individual TLS polymerases in abasic site bypass by sequencing the CAN1 gene from independent CanR mutants from the cdc13-1, cdc13-1 ubc13Δ and cdc13-1 ung1Δ strains (Figure 5B). Mutations in CAN1 were strand biased consisting primarily of mutations in G bases within GA dinucleotides (Supplementary Figures S2A, K–N and S8), consistent with A3B deamination only occurring in the DNA strand remaining following 5΄ to 3΄ resection from the uncapped telomere. Additionally, cdc13-1 ung1Δ strains predominantly contained G to A substitutions (P = 0.000144) (Figure 5B), further supporting that A3B-induced dU is the primary lesion occurring in these yeast. A3B induced an equal number of G to A and G to C substitutions in cdc13-1 strains, similar to previously reported mutation spectra for A3G-induced mutations in this system and is reminiscent of APOBEC-induced human cancer mutation spectra. This contrasts with our observation in the wild-type replicative strain, which displayed a strong bias (P = 0.000019) towards G to A mutations due to dU retention. This difference may be explained by a greater efficiency of dU being converted to abasic sites in the telomere re-synthesis system due to extended holding of the yeast at non-permissive temperatures, which may give Ung1 more opportunity to excise the dU. Interestingly, the cdc13-1 ubc13Δ strain demonstrated a substantial decrease in G to C substitutions compared to the cdc13-1 strain (P = 0.0252). This result indicates that despite its dispensability for error-free lesion bypass during telomere re-synthesis, Ubc13 still promotes Rev1 usage during TLS in this system in a similar fashion as during replicative DNA synthesis.
DISCUSSION
In this study, we demonstrate in S. cerevisiae that A3B-induced dU lesions formed in ssDNA of the lagging strand template are avoided in a two-step lesion conversion and bypass mechanism (Figure 6). First, the dU lesions are efficiently converted to abasic sites by Ung1 ahead of DNA synthesis by the replicative polymerase. These abasic sites serve to stall the replicative polymerase and induce a template switching mechanism to bypass the majority of the lesions in an error-free manner (Figure 3B). Ablation of the error-free bypass pathway leads to an increase in CanR frequency, which is the result of error-prone nucleotide insertion across from the abasic site in a TLS-dependent manner (Figure 4). Surprisingly, deleting UBC13 or MMS2, traditional components promoting error-free bypass, reduced G to C substitution in the CAN1 gene mutation spectrum from an equal representation of G to A and G to C mutations(Figure 3C). This indicates that Ubc13 and Mms2 may moderate TLS polymerase usage through controlling Rev1 cytidine transferase activity. We observed a similar decrease in G to C substitution in the absence of Ubc13 during uncapped telomere re-synthesis, even though this deletion resulted in no detectable impact on mutation frequency, (Figure 5) indicating the ubiquitin ligase still directs TLS polymerase usage even when the protein is dispensable for error-free bypass.
Figure 6.

Model for avoidance of APOBEC-induced lesions ahead of lagging strand DNA synthesis. (A) Single stranded DNA in the lagging strand template contain cytidines susceptible for APOBEC-induced deamination. These dU are excised by Ung1 ahead of the replicative polymerase, generating a fork-stalling abasic site (denoted as AP). The majority of these lesions are bypassed in an error-free manner by template switching, which is dependent on Ubc13, Mms2, Rad5 and Mph1. Some abasic sites are bypassed via the error-prone TLS pathway, dependent on Rev1 and polymerase ζ. Here, Ubc13 and Rad5 influence polymerase choice by promoting Rev1-mediated dC insertion, resulting in G to C substitutions. Alternatively, A-rule insertion by a different polymerase results in G to A substitutions. (B) Following end resection, cytidines on the non-resected DNA strand are exposed to APOBEC activity. These dU are excised by Ung1, generating abasic sites. During gap-fill synthesis, the polymerase stalls at the abasic sites. At this point, the lesion is predominantly bypassed through Mph1-dependent error-free template switch, potentially using its sister chromatid as a template or through error-prone TLS, dependent on Rev1 and polymerase ζ. As with (A), Ubc13 promotes Rev1 cytidine transferase activity during TLS, however, Ubc13-mediated polyubiquitination of PCNA is unnecessary for initiating error-free template switching.
In human cells, Ung2, the human nuclear equivalent to yeast Ung1, travels with the replication fork (54), presumably to remove dU incorporated into newly synthesized DNA by replicative polymerases. Our results indicate that Ung1 can also work ahead of polymerase δ to convert spontaneous or A3B-induced dU to abasic sites prior to DNA synthesis. A consequence of this lesion conversion is the stalling of the replication fork as polymerase δ encounters these sites (55). However, contrary to previous reports in mammalian cells of glycosylases converting base lesions to abasic sites to initiate fork regression and subsequent pre-replicative DNA repair, our results in yeast indicate that the PCNA clamp is polyubiquitinated by the Rad5-Ubc13-Mms2 E3 ligase complex to initiate error-free bypass (32,56). This bypass may involve the use of an alternative template for DNA synthesis directly at the replication fork. Alternatively, DNA synthesis may reinitiate downstream of the lesion, leaving the modified PCNA clamp as a marker for error-free postreplicative repair (29) after DNA replication completes. This mechanism of lesion conversion and bypass may be used to avoid mutagenesis stemming from other ssDNA-specific DNA damage at the replication fork.
The conversion of A3B-induced dU to abasic sites ahead of DNA polymerase δ lagging strand synthesis enables us to semi-quantitatively compare the relative contribution of error-free bypass and TLS in abasic site bypass in eukaryotic chromosomal DNA. Since ubc13Δ and mph1Δ strains have similar TLS-dependent increases in CanR frequencies, we conclude that most, if not all, TLS bypass events of abasic sites derived from cytidine deamination are mutagenic. Therefore, we can utilize the CanR frequencies in strains deficient for error-free bypass and TLS as a surrogate for the relative usage of each DDT pathway. In the replicative system, we see that error-free bypass is used approximately 17.4- to 21.3-fold more than TLS (the ratio of usage between error-free bypass and TLS calculated as:  using fluctuation values) for the UBC13 and MPH1 deficient strains, respectively. Similarly, error-free bypass is predominant over TLS even during telomere re-synthesis, occurring 3.5-fold more frequently than TLS. Predominant usage of error-free bypass usage is consistent with recent findings showing RecA-dependent damage avoidance mechanisms are favored in chromosomal lesion bypass in E. coli (57). This suggests that prokaryotes and eukaryotes use similar mechanisms to avoid fork stalling DNA lesions. Whether template switching mechanisms are used to bypass lesions occurring in human cells, which restrict the use of recombination-based mechanisms of DNA repair, is unknown.
using fluctuation values) for the UBC13 and MPH1 deficient strains, respectively. Similarly, error-free bypass is predominant over TLS even during telomere re-synthesis, occurring 3.5-fold more frequently than TLS. Predominant usage of error-free bypass usage is consistent with recent findings showing RecA-dependent damage avoidance mechanisms are favored in chromosomal lesion bypass in E. coli (57). This suggests that prokaryotes and eukaryotes use similar mechanisms to avoid fork stalling DNA lesions. Whether template switching mechanisms are used to bypass lesions occurring in human cells, which restrict the use of recombination-based mechanisms of DNA repair, is unknown.
The relationship between error-free bypass and TLS appears to be more complicated than simply two alternative mechanisms that enable continued replication when DNA polymerases are confronted with fork-stalling DNA lesions. Recent studies have begun to uncover direct crosstalk between the two mechanisms, mediated by the Rad5-Ubc13-Mms2 ubiquitin ligase complex that polyubiquitinates PCNA to initiate error-free lesion bypass. We have uncovered additional evidence of this crosstalk in the A3B-induced mutation spectra of ubc13Δ and mms2Δ yeast. The deletion of UBC13 or MMS2 resulted in an under-representation of TLS-mediated G to C substitutions, indicating reduced usage of Rev1 cytidine transferase activity to bypass abasic sites in favor of an A-rule insertion polymerase. The Rad5-Ubc13-Mms2 E3 ligase complex was previously reported to associate directly with Rev1 through Rad5 and indirectly with polymerase ζ through Rev1 (41,58). Disruption of the Rev1-Rad5 binding domain results in sensitivity to DNA damaging agents and reduced damaged-induced mutagenesis, consistent with the loss of both error-free lesion bypass and TLS, presumably due to the inability to recruit Rev1 to lesions sites. A3B-expression in rad5Δ yeast produces a similar reduced cell viability (Supplementary Figure S5), indicating that Rad5 likely also promotes both error-free bypass and TLS past abasic sites ahead of polymerase δ lagging strand synthesis during replication. Supporting this interpretation, A3B expression in ubc13Δ rev3Δ and mph1Δ rev3Δ yeast which lack both TLS and error-free bypass reduces cell survival (Supplementary Figure S3B and C). In contrast, the reduction of Rev1-induced mutations in the ubc13Δ strains is unlikely to be due to failure to recruit Rev1, as this enzyme is structurally required for TLS and thus its absence should eliminate all TLS synthesis. Thus, we propose that Ubc13 directly promotes Rev1 cytidine transferase activity.
In human cancers and yeast, APOBEC enzymes target lagging strand synthesis intermediates during replication. The mutation spectra for APOBEC-mutagenized tumors show an equal representation in G to C and G to A substitutions, indicating the usage of TLS, partially attributable to Rev1, in the bypass of abasic sites as the primary mediators of APOBEC-induced mutagenesis. However, in wild-type yeast, we observed that the vast majority of A3B-induced abasic sites were bypassed in an error-free manner during replication, with remaining mutagenic lesions consisting of unexcised dU resulting in G to A substitutions. Multiple explanations could account for this difference in mutation spectra between human cancers and replicating yeast expressing A3B. The first and simplest explanation would be that human cells utilize TLS at a higher frequency to bypass abasic sites than yeast, or error-free bypass may be downregulated or dysfunctional in cancer cells. Alternatively, replication stress, which occurs in human cancers, may result in dysregulated DNA synthesis that is more akin to yeast gap-fill synthesis. Here, an increased amount of ssDNA ahead of the replicative polymerase may enable Ung to excise dU at a higher efficiency. This would act to decrease the proportion of mutations resulting from dU templating relative to those resulting from insertion by TLS. We were able to estimate the amount of dU that is retained in the DNA in each system by comparing the estimated contributions in bypassing dU by TLS and error-free bypass to the mutation frequency observed by deleting UNG1. In the replicative system, we estimate that Ung1 was ∼90.1% efficient at excising dU  and ∼99.2% efficient in the telomere re-synthesis system
and ∼99.2% efficient in the telomere re-synthesis system  . This difference in Ung1 efficiency alone between the two systems would add approximately 12-fold more G to A mutations due to dU templating to the CAN1 mutation spectrum of the replicative system in comparison to that of the telomere re-synthesis system. This would ultimately result in a strong bias towards G to A substitutions over G to C substitutions, irrespective of an altered relative usage of error-free lesion bypass and TLS. If indeed the bulk of APOBEC-induced dU in human cancer is bypassed in an error-free manner as we see in yeast, our findings suggest that the true number of APOBEC-induced damages to the DNA may be much greater than previously estimated from the amount of APOBEC signatures observed in cancer genomic sequences.
. This difference in Ung1 efficiency alone between the two systems would add approximately 12-fold more G to A mutations due to dU templating to the CAN1 mutation spectrum of the replicative system in comparison to that of the telomere re-synthesis system. This would ultimately result in a strong bias towards G to A substitutions over G to C substitutions, irrespective of an altered relative usage of error-free lesion bypass and TLS. If indeed the bulk of APOBEC-induced dU in human cancer is bypassed in an error-free manner as we see in yeast, our findings suggest that the true number of APOBEC-induced damages to the DNA may be much greater than previously estimated from the amount of APOBEC signatures observed in cancer genomic sequences.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Drs Dmitry Gordenin, Cynthia Sakofsky, John Wyrick, Jacquelyn Stone and Tony Mertz for advice and helpful comments, and the WSU Genomics Core for PacBio sequencing of CAN1 amplicons.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of Environmental Health Sciences [ES022633] and Breast Cancer Research Program Breakthrough Award from the Department of Defense [BC141727] to S.A.R. Funding for open access charge: National Institute of Environmental Health Sciences [ES022633].
Conflict of interest statement. None declared.
REFERENCES
- 1. Nik-Zainal S., Alexandrov L.B., Wedge D.C., Van Loo P., Greenman C.D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L.A. et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012; 149:979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roberts S.A., Sterling J., Thompson C., Harris S., Mav D., Shah R., Klimczak L.J., Kryukov G.V., Malc E., Mieczkowski P.A. et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell. 2012; 46:424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burns M.B., Temiz N.A., Harris R.S.. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013; 45:977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roberts S.A., Lawrence M.S., Klimczak L.J., Grimm S.A., Fargo D., Stojanov P., Kiezun A., Kryukov G.V., Carter S.L., Saksena G. et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013; 45:970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L. et al. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Refsland E.W., Harris R.S.. The APOBEC3 family of retroelement restriction factors. Curr. Top. Microbiol. Immunol. 2013; 371:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muramatsu M., Kinoshita K., Fagarasan S., Yamada S., Shinkai Y., Honjo T.. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000; 102:553–563. [DOI] [PubMed] [Google Scholar]
- 8. Teng B., Burant C.F., Davidson N.O.. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993; 260:1816–1819. [DOI] [PubMed] [Google Scholar]
- 9. Chaudhuri J., Tian M., Khuong C., Chua K., Pinaud E., Alt F.W.. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003; 422:726–730. [DOI] [PubMed] [Google Scholar]
- 10. Sohail A., Klapacz J., Samaranayake M., Ullah A., Bhagwat A.S.. Human activation-induced cytidine deaminase causes transcription-dependent, strand-biased C to U deaminations. Nucleic Acids Res. 2003; 31:2990–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ramiro A.R., Stavropoulos P., Jankovic M., Nussenzweig M.C.. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 2003; 4:452–456. [DOI] [PubMed] [Google Scholar]
- 12. Pham P., Bransteitter R., Petruska J., Goodman M.F.. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003; 424:103–107. [DOI] [PubMed] [Google Scholar]
- 13. Taylor B.J., Wu Y.L., Rada C.. Active RNAP pre-initiation sites are highly mutated by cytidine deaminases in yeast, with AID targeting small RNA genes. Elife. 2014; 3:e03553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lada A.G., Kliver S.F., Dhar A., Polev D.E., Masharsky A.E., Rogozin I.B., Pavlov Y.I.. Disruption of transcriptional coactivator Sub1 leads to genome-wide re-distribution of clustered mutations induced by APOBEC in active yeast genes. PLoS Genet. 2015; 11:e1005217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nordentoft I., Lamy P., Birkenkamp-Demtroder K., Shumansky K., Vang S., Hornshoj H., Juul M., Villesen P., Hedegaard J., Roth A. et al. Mutational context and diverse clonal development in early and late bladder cancer. Cell Rep. 2014; 7:1649–1663. [DOI] [PubMed] [Google Scholar]
- 16. Taylor B.J., Nik-Zainal S., Wu Y.L., Stebbings L.A., Raine K., Campbell P.J., Rada C., Stratton M.R., Neuberger M.S.. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife. 2013; 2:e00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bhagwat A.S., Hao W., Townes J.P., Lee H., Tang H., Foster P.L.. Strand-biased cytosine deamination at the replication fork causes cytosine to thymine mutations in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:2176–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoopes J.I., Cortez L.M., Mertz T.M., Malc E.P., Mieczkowski P.A., Roberts S.A.. APOBEC3A and APOBEC3B preferentially deaminate the lagging strand template during DNA replication. Cell Rep. 2016; 14:1273–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Green A.M., Landry S., Budagyan K., Avgousti D.C., Shalhout S., Bhagwat A.S., Weitzman M.D.. APOBEC3A damages the cellular genome during DNA replication. Cell Cycle. 2016; 15:998–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haradhvala N.J., Polak P., Stojanov P., Covington K.R., Shinbrot E., Hess J.M., Rheinbay E., Kim J., Maruvka Y.E., Braunstein L.Z. et al. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell. 2016; 164:538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morganella S., Alexandrov L.B., Glodzik D., Zou X., Davies H., Staaf J., Sieuwerts A.M., Brinkman A.B., Martin S., Ramakrishna M. et al. The topography of mutational processes in breast cancer genomes. Nat. Commun. 2016; 7:11383–11393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seplyarskiy V.B., Soldatov R.A., Popadin K.Y., Antonarakis S.E., Bazykin G.A., Nikolaev S.I.. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016; 26:174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boiteux S., Jinks-Robertson S.. DNA repair mechanisms and the bypass of DNA damage in Saccharomyces cerevisiae. Genetics. 2013; 193:1025–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Di Noia J., Neuberger M.S.. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 2002; 419:43–48. [DOI] [PubMed] [Google Scholar]
- 25. Schumacher A.J., Nissley D.V., Harris R.S.. APOBEC3G hypermutates genomic DNA and inhibits Ty1 retrotransposition in yeast. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:9854–9859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Landry S., Narvaiza I., Linfesty D.C., Weitzman M.D.. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011; 12:444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Narvaiza I., Landry S., Weitzman M.D.. APOBEC3 proteins and genomic stability: the high cost of a good defense. Cell Cycle. 2012; 11:33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hegde M.L., Hegde P.M., Bellot L.J., Mandal S.M., Hazra T.K., Li G.M., Boldogh I., Tomkinson A.E., Mitra S.. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E3090–E3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu X., Blackwell S., Lin A., Li F., Qin Z., Xiao W.. Error-free DNA-damage tolerance in Saccharomyces cerevisiae. Mutat. Res. Rev. Mutat. Res. 2015; 764:43–50. [DOI] [PubMed] [Google Scholar]
- 30. Branzei D., Foiani M.. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010; 11:208–219. [DOI] [PubMed] [Google Scholar]
- 31. Stelter P., Ulrich H.D.. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003; 425:188–191. [DOI] [PubMed] [Google Scholar]
- 32. Kannouche P.L., Wing J., Lehmann A.R.. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell. 2004; 14:491–500. [DOI] [PubMed] [Google Scholar]
- 33. Sharma N.M., Kochenova O.V., Shcherbakova P.V.. The non-canonical protein binding site at the monomer-monomer interface of yeast proliferating cell nuclear antigen (PCNA) regulates the Rev1-PCNA interaction and Polzeta/Rev1-dependent translesion DNA synthesis. J. Biol. Chem. 2011; 286:33557–33566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brusky J., Zhu Y., Xiao W.. UBC13, a DNA-damage-inducible gene, is a member of the error-free postreplication repair pathway in Saccharomyces cerevisiae. Curr. Genet. 2000; 37:168–174. [DOI] [PubMed] [Google Scholar]
- 35. Sherman F., Fink G.R., Hicks J.B.. Laboratory Course Mmanual for Methods in Yeast Genetics. 1986; NY: Cold Spring Harbor Laboratory [Google Scholar]
- 36. Morrison A., Bell J.B., Kunkel T.A., Sugino A.. Eukaryotic DNA polymerase amino acid sequence required for 3΄—-5΄ exonuclease activity. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:9473–9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chan K., Resnick M.A., Gordenin D.A.. The choice of nucleotide inserted opposite abasic sites formed within chromosomal DNA reveals the polymerase activities participating in translesion DNA synthesis. DNA Repair (Amst). 2013; 12:878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guo X., Lehner K., O’Connell K., Zhang J., Dave S.S., Jinks-Robertson S.. SMRT sequencing for parallel analysis of multiple targets and accurate SNP phasing. G3. 2015; 5:2801–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marenstein D.R., Wilson D.M. 3rd, Teebor G.W.. Human AP endonuclease (APE1) demonstrates endonucleolytic activity against AP sites in single-stranded DNA. DNA Repair (Amst). 2004; 3:527–533. [DOI] [PubMed] [Google Scholar]
- 41. Xu X., Lin A., Zhou C., Blackwell S.R., Zhang Y., Wang Z., Feng Q., Guan R., Hanna M.D., Chen Z. et al. Involvement of budding yeast Rad5 in translesion DNA synthesis through physical interaction with Rev1. Nucleic Acids Res. 2016; 44:5231–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schurer K.A., Rudolph C., Ulrich H.D., Kramer W.. Yeast MPH1 gene functions in an error-free DNA damage bypass pathway that requires genes from Homologous recombination, but not from postreplicative repair. Genetics. 2004; 166:1673–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Prakash R., Satory D., Dray E., Papusha A., Scheller J., Kramer W., Krejci L., Klein H., Haber J.E., Sung P., Ira G.. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 2009; 23:67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luke-Glaser S., Luke B.. The Mph1 helicase can promote telomere uncapping and premature senescence in budding yeast. PLoS One. 2012; 7:e42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kang Y.H., Kang M.J., Kim J.H., Lee C.H., Cho I.T., Hurwitz J., Seo Y.S.. The MPH1 gene of Saccharomyces cerevisiae functions in Okazaki fragment processing. J. Biol. Chem. 2009; 284:10376–10386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prakash R., Krejci L., Van Komen S., Anke Schurer K., Kramer W., Sung P.. Saccharomyces cerevisiae MPH1 gene, required for homologous recombination-mediated mutation avoidance, encodes a 3΄ to 5΄ DNA helicase. J. Biol. Chem. 2005; 280:7854–7860. [DOI] [PubMed] [Google Scholar]
- 47. Silva S., Altmannova V., Luke-Glaser S., Henriksen P., Gallina I., Yang X., Choudhary C., Luke B., Krejci L., Lisby M.. Mte1 interacts with Mph1 and promotes crossover recombination and telomere maintenance. Genes Dev. 2016; 30:700–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yimit A., Kim T., Anand R.P., Meister S., Ou J., Haber J.E., Zhang Z., Brown G.W.. MTE1 functions with MPH1 in double-strand break repair. Genetics. 2016; 203:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xue X., Papusha A., Choi K., Bonner J.N., Kumar S., Niu H., Kaur H., Zheng X.F., Donnianni R.A., Lu L. et al. Differential regulation of the anti-crossover and replication fork regression activities of Mph1 by Mte1. Genes Dev. 2016; 30:687–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Haracska L., Unk I., Johnson R.E., Johansson E., Burgers P.M., Prakash S., Prakash L.. Roles of yeast DNA polymerases delta and zeta and of Rev1 in the bypass of abasic sites. Genes Dev. 2001; 15:945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. D'Souza S., Walker G.C.. Novel role for the C terminus of Saccharomyces cerevisiae Rev1 in mediating protein-protein interactions. Mol. Cell. Biol. 2006; 26:8173–8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wood A., Garg P., Burgers P.M.. A ubiquitin-binding motif in the translesion DNA polymerase Rev1 mediates its essential functional interaction with ubiquitinated proliferating cell nuclear antigen in response to DNA damage. J. Biol. Chem. 2007; 282:20256–20263. [DOI] [PubMed] [Google Scholar]
- 53. Garvik B., Carson M., Hartwell L.. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol. Cell. Biol. 1995; 15:6128–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kavli B., Sundheim O., Akbari M., Otterlei M., Nilsen H., Skorpen F., Aas P.A., Hagen L., Krokan H.E., Slupphaug G.. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem. 2002; 277:39926–39936. [DOI] [PubMed] [Google Scholar]
- 55. Maga G., van Loon B., Crespan E., Villani G., Hubscher U.. The block of DNA polymerase delta strand displacement activity by an abasic site can be rescued by the concerted action of DNA polymerase beta and Flap endonuclease 1. J. Biol. Chem. 2009; 284:14267–14275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xiao W., Chow B.L., Broomfield S., Hanna M.. The Saccharomyces cerevisiae RAD6 group is composed of an error-prone and two error-free postreplication repair pathways. Genetics. 2000; 155:1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Naiman K., Pages V., Fuchs R.P.. A defect in homologous recombination leads to increased translesion synthesis in E. coli. Nucleic Acids Res. 2016; 44:7691–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pustovalova Y., Magalhaes M.T., D'Souza S., Rizzo A.A., Korza G., Walker G.C., Korzhnev D.M.. Interaction between the Rev1 C-terminal domain and the PolD3 subunit of polzeta suggests a mechanism of polymerase exchange upon Rev1/Polzeta-dependent translesion synthesis. Biochemistry. 2016; 55:2043–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.