

Abstract
In addition to the primary symptoms arising from inflammatory processes in the joints, muscle weakness is commonly reported by patients with rheumatoid arthritis (RA). Muscle weakness not only reduces the quality of life for the affected patients, but also dramatically increases the burden on society since patients' work ability decreases. A 25–70% reduction in muscular strength has been observed in pateints with RA when compared with age-matched healthy controls. The reduction in muscle strength is often larger than what could be explained by the reduction in muscle size in patients with RA, which indicates that intracellular (intrinsic) muscle dysfunction plays an important role in the underlying mechanism of muscle weakness associated with RA. In this review, we highlight the present understanding of RA-associated muscle weakness with special focus on how enhanced Ca2 + release from the ryanodine receptor and free radicals (reactive oxygen/nitrogen species) contributes to muscle weakness, and recent developments of novel therapeutic interventions.
Keywords: Rheumatoid arthritis, Muscle weakness, Ca2 +, Ryanodine receptor, Nitrosative stress, Peroxynitrite
Highlights
-
•
Muscle weakness is commonly reported by patients with rheumatoid arthritis (RA).
-
•
Intrinsic muscle weakness is important in the underlying mechanisms of muscle weakness associated with rheumatoid arthritis.
-
•
Enhanced Ca2 + release and peroxynitrite-induced stress contributes to RA-induced muscle weakness.
1. Introduction
Rheumatoid arthritis (RA) is one of the most prevalent chronic inflammatory diseases (Alamanos et al., 2006, van Vilsteren et al., 2015). In addition to the primary symptoms arising from inflammatory processes in the joints, muscle weakness is commonly reported by patients with RA (Sokka et al., 2008). Muscle weakness not only reduces the quality of life for the affected patients, but since patients' ability to work decreases it will also dramatically increase the burden on society (e.g. increased costs for long-term sick leave). Thus, RA severely affects both the individual and the society (Sokka et al., 2008, van Vilsteren et al., 2015).
In patients with RA, a 25–70% reduction in muscular strength (including both grip strength and isometric and isokinetic knee muscle strength) has been observed when compared with age-matched healthy controls (Ekdahl and Broman, 1992, Fraser et al., 1999, Helliwell and Jackson, 1994, Stenström and Minor, 2003). Reduced muscle strength is usually considered to be a result of decreased muscle mass due to disuse atrophy. Rheumatoid cachexia, a term used in RA, is defined as a loss of skeletal muscle mass and with no, or little weight loss in fat mass (Londhe and Guttridge, 2015, Walsmith and Roubenoff, 2002). However, Helliwell and Jackson stated already in 1994 that the reduction in grip strength of patients with RA is larger than what could be explained by the reduction in muscle size (Helliwell and Jackson, 1994), and this phenomenon has been reported later by others (Fraser et al., 1999, Lemmey et al., 2016). Structural analysis using electron microscopy of muscle biopsies (from e.g. vastus medialis, gluteus maximus, extensor digitorum communis) from patients with RA display dilated sarcotubular system, pleomorphic mitochondria and myofibril flaking, all signs of altered intramuscular function (de Palma et al., 2000). Thus, intracellular (intrinsic) contractile dysfunction appears to play an important role in the underlying mechanism of muscle weakness associated with RA. Indeed, force production normalized to the cross-sectional area of the muscle (i.e. the specific force, which assesses the muscle fibre's intrinsic capacity to generate force) has been shown to be markedly reduced (~ 30%) in both fast-twitch and slow-twitch skeletal muscle from two widely used rodent models of RA; collagen-induced arthritis (CIA) in mice and adjuvant-induced arthritis (AIA) in rats (Yamada et al., 2015a, Yamada et al., 2015b, Yamada et al., 2009). In the first part of this review we will discuss intrinsic muscle weakness and how altered Ca2 + and free radical signaling (reactive oxygen/reactive nitrogen species, ROS/RNS) contribute to RA-induced muscle weakness. In the latter part, we discuss therapeutic interventions to counteract RA-induced muscle weakness.
2. Intrinsic Muscle Dysfunction
The events leading to contraction of skeletal muscle fibers (excitation-contraction coupling, EC coupling) start with action potentials travelling down the transverse tubular (t-tubular) system and activating the volatage-sensitive dihydropyridine receptors (DHPR or Cav1.1). Activated DHPR mechanically trigger the ryanodine receptor type 1 (RyR1), which is the major intracellular Ca2 + release channel that releases Ca2 + from the sarcoplasmic reticulum (SR). Ca2 + then binds to the troponin complex, which moves the position of the tropomyosin filaments. This uncovers the active sites of actin for myosin binding, hence enabling and turning on myofibrillar cross-bridge cycling and contraction. Uptake of intracellular Ca2 + into SR by the SR-Ca2 + ATPase (Serca) leads to dissociation of Ca2 + from troponin, detachment of cross-bridges and muscle relaxation (Fig. 1) (Gordon et al., 2000). Regarding force generation, in simple terms one can say that the higher Ca2 + concentration, the greater force can be generated. This is valid until maximal force is reached and all ‘motors’ (the actin-myosin interaction, also known as cross-bridges) are activated. Thereby, decreased force production in skeletal muscle can be caused by; i) reduced RyR1 Ca2 + release from the SR, ii) decreased myofibrillar Ca2 + sensitivity, and/or iii) impaired ability of cross-bridges to generate force (Cheng et al., 2016, Gordon et al., 2000).
Fig. 1.
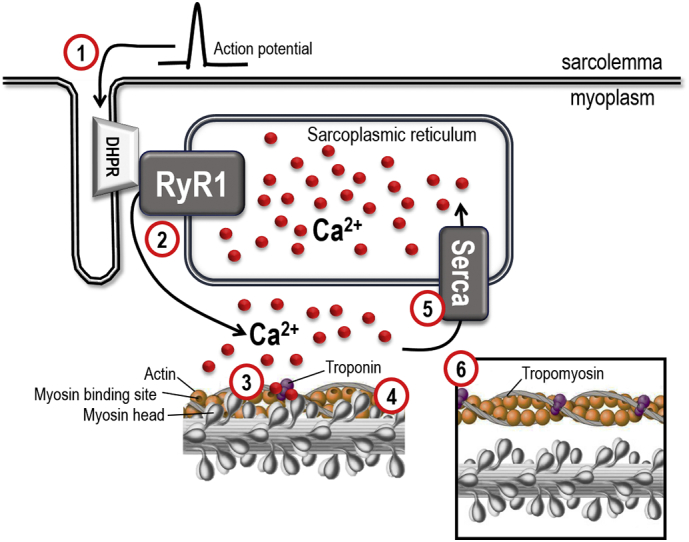
Illustration of the intracellular events leading to contraction of skeletal muscle fibers. The dihydropyridine receptor (DHPR or Cav1.1) is voltage-sensite and activated by actionpotentials (1). The DHPR opens RyR1 by mechanical interaction resulting in release of Ca2 + (2) from the sarcoplasmic reticulum (SR) and a transient increase in intracellular (myoplasmic) Ca2 + (~ 1–5 μM). Ca2 + binds to the troponin complex (3), which moves the position of the tropomyosin filaments. This uncovers the active sites of actin for myosin binding (4), which enables actin and myosin interaction and force production. The SR Ca2 + ATPase (Serca) pumps Ca2 + back into SR (5) and [Ca2 +]i returns to resting levels and the contraction ceases. At rest, when the intracellular Ca2 + concentration is low (~ 50 nM), the tropomyosin filaments hide the myosin binding sites on actin (6), hence no force can be produced.
3. Altered RyR1 Ca2 + Release and the Progression Towards Arthritis-induced Muscle Weakness
The reduction in specific force (~ 30%) observed in both fast-twitch and slow-twitch skeletal muscle from rodents with arthritis, was preceeded by a substantial and significant increase in Ca2 + release over the whole range of stimulation frequencies in muscle from mice with arthritis as compared with control muscle (Fig. 2) (Yamada et al., 2015b, Yamada et al., 2009). In fact, the free intracellular Ca2 + concentration ([Ca2 +]i) was almost twice as high in muscle fibers from CIA mice than in control fibers at the higher stimulation frequencies (50–120 Hz) (Yamada et al., 2015b). Caffeine is a potent RyR1 agonist, which is widely used in muscle research as an agent which increases Ca2 + release from SR and thereby increases myoplasmic free Ca2 + concentrations (Allen and Westerblad, 1995, Shirokova and Rios, 1996). In the presence of caffeine (5 mM), there was no longer a difference in the Ca2 + release between muscle fibers from control mice or mice with arthritis (Yamada et al., 2015b). This indicates that the increased Ca2 + release was caused by facilitated RyR1 Ca2 + release and was not the result of more Ca2 + stored in SR in muscles from mice with RA.
Fig. 2.

Increased tetanic Ca2 + accompanied by muscle weakness in muscle fibers from mice with RA (collagen-induced arthritis, CIA). (A) The intracellular Ca2 + concentration ([Ca2 +]i) was significantly increased over a wide range of stimulation frequencies in muscle fibers from RA mice (red circles) compared with controls (white circles). (B) The increased tetanic Ca2 + was accompanied by decreased force per cross-sectional area in muscle fibers from mice with RA (n = 9–10). Single intact flexor digitorium brevis (FDB) fibers (fast-twitch, type II) were used for this set of experiements. The FDB fibers were obtained by dissection and mounted in a chamber between a force transducer and an adjustable holder. The fibre length was adjusted to obtain maximum tetanic force. The diameter of the fibre at this length was used to calculate the cross-sectional area. Experiments were performed at room temperature (∼ 24 °C). The fibre was stimulated with supramaximal electrical pulses (0.5 ms in duration, 1–120 Hz) delivered via platinum electrodes placed along the long axis of the fibre. [Ca2 +]i was measured with the fluorescent Ca2 + indicator indo-1. Indo-1 was microinjected into the isolated fibre, which was then allowed to rest for at least 20 min. The mean fluorescence of indo-1 at rest and during tetanic contractions was measured and converted to [Ca2 +]i using an intracellularly established calibration curve (Andrade et al., 1998). Data are mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001 versus controls.
Adapted from Yamada et al. (2015b).
Altered RyR1 Ca2 + release has been linked to muscle weakness in several clinical aspects, e.g. bone cancer (Waning et al., 2015), muscle dystrophies (Bellinger et al., 2009, Hernández-Ochoa et al., 2015, Lanner et al., 2012), heart failure (Rullman et al., 2013) and even in normal ageing (Andersson et al., 2011). Altered RyR1 Ca2 + release often manifests as low-grade basal Ca2 + release (Ca2 + leak), i.e. Ca2 + leaking out from the RyR1 channel under basal (non-stimulated) conditions when the channel supposedly should be in its closed confirmation. RyR1 Ca2 + leak is associated with reduced Ca2 + release upon stimulation, decreased SR Ca2 + load and impaired contractility in both cardiac and skeletal muscle (Andronache et al., 2009, Aydin et al., 2008, Lanner et al., 2012, Santulli et al., 2017, Tong et al., 1999). However in Yamada et al. (2015b), a significant reduction in force production was preceeded by enhanced/increased tetanic Ca2 + release and unaltered SR Ca2 + store content in muscles from mice with arthritis (Yamada et al., 2015b).
Altered gating properties of RyR1, hence altered Ca2 + release, is thought to be the result of post-translational modifications of the channel (Aydin et al., 2008, Durham et al., 2008, Lanner et al., 2012, Waning et al., 2015). For instance, phosphorylation of RyR1Ser2843 induced by resistance exericse or β-adrenergic stimulation have been shown to activate the channel (Andersson et al., 2012, Gehlert et al., 2012, Reiken et al., 2003). On the other hand, phosphorylation of cardiac RyR2 (S2030 and S2808) has been associated with enhanced SR Ca2 + leak and reduced SR Ca2 + load, which may contribute to arrhythmias and contractile dysfunction in heart failure (Lanner et al., 2010, Marx et al., 2000). Moreover, RyR1 is known to be sensitive to ROS/RNS-induced post-translational modifications. For example, protein carbonylation (oxidations yielding reactive carbonyl groups, DNP (Fedorova et al., 2014)) and nitrosylation (nitric oxide (NO) covalently bound to cysteines (i.e., S-nitrosylation, CysNO)) are known to alter the gating properties of RyR1 (Lanner et al., 2010). For instance, S-nitrosylation of Cys3635 on RyR1 has been shown to activate the channel (Sun et al., 2001). High-levels of DNP and CysNO on RyR1 have been found in age- and disease-induced muscle dysfunction (Andersson et al., 2011, Bellinger et al., 2009, Durham et al., 2008, Lanner et al., 2012, Rullman et al., 2013, Waning et al., 2015).
Elevated levels of several ROS/RNS-induced modifications including, CysNO, DNP, malonialdehyde (MDA, highly reactive aldehydes formed by lipid peroxidation), and 3-nitrotyrosine (3-NT, a marker of peroxynitrite (ONOO•−)) have been oberved in serum, synovial fluid and synovial tissue from patients with RA (Grönwall et al., 2017, Hilliquin et al., 1997, Kaur and Halliwell, 1994). To our knowledge, the level of DNP, CysNO, MDA on RyR1 have not yet been studied in skeletal muscle from mouse-models of RA or patients with RA. However, we have previously reported that the enhanced RyR1-mediated Ca2 + release was associated with a three-fold increase in the 3-NT levels on the RyR1 complex in skeletal muscle from mice with arthritis (Yamada et al., 2015b). Thus, the facilitated RyR1 Ca2 + release seems to be linked to the eleveted levels of 3-NT in skeletal muscle from mice with RA. In summary, the arthritis-induced muscle weakness appears not to be a direct result of reduced Ca2 + release (i, discussed above), but must instead be due to decreased myofibrillar Ca2 + sensitivity (ii), and/or impaired ability of cross-bridges to generate force (iii). Nevertheless, the higher RyR3-NT levels and increased Ca2 + release could contibute to the progession towards a muscle weakning state in muscles with RA, e.g. by enhancing the NOS1 activity (see below).
4. Impaired Myofibrillar Force Production Contributes to Intrinsic Muscle Weakness in RA
Selective loss of the force producing myofibrillar proteins, including myosin heavy chain (MyHC), is linked to myofibrillar dysfunction and force loss in pathological conditions (Ochala et al., 2011). Decreased muscle mass and reduced cross-sectional area has been reported to various degrees in patients with RA (de Oliveira Nunes Teixeira et al., 2013, Hartog et al., 2009, Walsmith and Roubenoff, 2002). However, to our knowledge, only Yamada et al., 2009, have quantified the amount of myofibrillar proteins in RA subjects. A small but significant reduction (~ 7%) of the MyHC content was observed, with no loss in actin content in CIA muscles (Yamada et al., 2009). However, it is unlikely that this minor loss of MyHC could explain the overall contractile deficit (~ 30% force depression) in CIA muscles (Yamada et al., 2015b, Yamada et al., 2009). Instead, attention has been directed towards the myofibrillar function and the capacity of the actin-myosin interaction to generate force in rodents with RA (Yamada et al., 2015a, Yamada et al., 2015b, Yamada et al., 2009). Actin-myosin interaction and function can be studied in detail by quantifying myofibrillar force production using atomic force cantilevers on activated myofibrils (Yamada et al., 2015b). Impaired ability of cross-bridges to generate force can be the result of a decrease in the average force produced by the attached cross-bridges (active force) and/or a decrease in the number of myosins attached to actin in a given time (i.e. decreased cross-bridge attachment rate or increased cross-bridge detachment rate) (Cheng et al., 2016, Gordon et al., 2000).
The active force (both submaximal and maximal Ca2 +-activated force) was markedly lower (~ 33%) in myofibrils from mice with arthritis than in myofibrils from control mice. The [Ca2 +]i required to produce 50% of the maximum force (Ca50) was increased in muscles from arthritis mice, suggesting a reduced myofibrillar Ca2 + sensitivity (Yamada et al., 2015b). Moreover, myofibrils from arthritis mice showed a decreased rate of force redevelopment after shortening the fully activated myofibrils, which is consistent with slower cross-bridge attachment, reduced Ca2 + sensitivity and overall lower force generating capacity (Gordon et al., 2000, Yamada et al., 2015b). Thus, the observed force depression appears as a result of reduced myofibrillar force generating capacity and decreased myofibrillar Ca2 + sensitivity. In line with this, skeletal muscle actomyosin ATPase activity was shown to be reduced in rodents with RA (Yamada et al., 2015a). Moreover, myofibrillar irregularities, e.g. wider separation of myofibrils, dilated t-tubular system, pleomorphic mitochondria and myofibril flaking, have also been observed in muscle biopsies from patients with RA, and were correlated with muscle weakness (de Palma et al., 2000, Russell and Hanna, 1988). Thus, muscles from rodent models of RA and biopsies from patients with RA suggests that impaired myofibrillar function is a prominent factor in RA-induced muscle weakness and muscle dysfunction.
5. ROS/RNS Interfere with the Contractile Machinery in RA Muscles
Free radicals (ROS/RNS) are believed to be involved in the pathogenesis of chronic arthritis and RA, but how and to which extent is not fully understood (Datta et al., 2014, Khojah et al., 2016, Kurien et al., 2006, Mateen et al., 2016, Ozkan et al., 2007, Pacher et al., 2007). Intriguingly, extensive amount of data supports both positive (e.g. involved in gene expression, cell growth and remodeling) and negative effects (DNA damage and protein dysfunction) of ROS/RNS on cell function (Cheng et al., 2016, Kurien et al., 2006, Lambeth, 2004, Ristow, 2014, Supinski and Callahan, 2007). However, whether ROS/RNS have a protective and modulatory role or lead to damaging effects most probably depends on several factors, e.g. the type and amount of ROS/RNS as well as its localization. In skeletal muscle, several ROS/RNS species (superoxide (O2•−), hydrogen peroxide (H2O2), hydroxyl radicals (•OH), nitric oxide (NO), peroxynitrite (ONOO•−)) have been shown to directly alter contractile protein function, and data suggests that ROS/RNS also have important effects on SR function, mitochondrial function and on sarcolemmal integrity (Cheng et al., 2015, Cheng et al., 2016, Dutka et al., 2011, Murphy et al., 2008, Supinski et al., 1999, Supinski and Callahan, 2007). For example, exposure of skinned muscle fibre to ONOO•− or ONOO•− donors has been shown to cause a marked decline in maximal Ca2 +-activated force (Dutka et al., 2011, Supinski et al., 1999). Furthermore, ROS/RNS appear to play a key role in modulating inflammation-induced alterations in skeletal muscle function (Pinniger et al., 2012, Supinski and Callahan, 2007, Yamada et al., 2015a, Yamada et al., 2015b, Yamada et al., 2009). As mentioned, increased levels of several ROS/RNS markers (e.g. CysNO, DNP, MDA and 3-NT have been oberved in serum, synovial fluid and synovial tissue from patients with RA and may contribute to tissue damage and hence to the chronicity of the disease (Grönwall et al., 2017, Hilliquin et al., 1997, Kaur and Halliwell, 1994, Mateen et al., 2016, Ozkan et al., 2007). Thus, accelerating ROS/RNS formation in skeletal muscle, could play an important role in the development of myofibrillar dysfunction and muscle weakness. In fact, a two-fold increase in the ONOO•−-marker 3-NT has been observed in muscle homogenates from aged, weak animals (mice 26–28 months old) as compared with younger healthy animals (5–7 months old) (Pearson et al., 2015). Moreover, a four-fold increase in 3-NT on the contractile protein actin was shown in skeletal muscles from rodents with RA (Yamada et al., 2015a, Yamada et al., 2015b). In addition, actin aggregates, which are associated with reduced actomyosin ATPase activity and lower force production, were detected in skeletal muscles from rats with arthritis (Fedorova et al., 2009, Tiago et al., 2006, Yamada et al., 2015a). These actin aggregates contained high amounts of 3-NT, which indicates an increased ONOO•− production in skeletal muscle from rodents with RA (Yamada et al., 2015a). ONOO•− can cause oxidation of cysteine residues and nitration of tyrosine residues (Szabó et al., 2007, Tiago et al., 2006). The detailed functional significance of the increased 3-NT formation on actin in skeletal muscle from subjects with RA has not yet been determined. However, ONOO•−-induced nitration of actin has been shown to contribute to the inhibition of actin polymerization, which may have an impact on force generating capacity (Clements et al., 2003). Thus, ONOO•−-induced modifications of actin might to contribute to the decreased force generating capacity of myofibrils present in RA muscles. However, the molecular mechanisms of action remain to be determined. Furthermore, the possible effects of ROS/RNS-induced modifications on additional contractile proteins (e.g. myosin, troponin, titin, etc.) should also be investigated in skeletal muscle associated with RA-induced muscle weakness.
6. Possible Sources of ROS/RNS in RA Muscles
Increased levels of ONOO•−-induced 3-NT footprints has been consistently shown in skeletal muscles from different animal models of RA (Yamada et al., 2015a, Yamada et al., 2015b, Yamada et al., 2009). ONOO•− is a potent oxidizing and nitrating agent able to react with a wide range of cellular targets within ~ 5–20 μm (Carballal et al., 2014, Radi, 2004, Szabó et al., 2007). ONOO•− is formed by the reaction between NO and superoxide (O2•−), with a fast formation rate constant of ~ 4–16 × 109 M− 1 s− 1 (Botti et al., 2010). The rate constant for ONOO•− is ~ six times faster than the rate constant for superoxide dismutase (SOD) to convert O2•− to H2O2 (~ 1–2 × 109 M− 1 s− 1). Thus, when NO is produced at a high rate, it will rapidly react with O2•− to produce significant amounts of ONOO•− even in the presence of the high physiological concentrations of superoxide dismutase (SOD, ~ 10 to 20 μM) (Hsu et al., 1996). However, which intracellular sources of ROS/RNS are responsible for the increased redox stress that has been observed in skeletal muscle associated with RA-induced muscle weakness?
6.1. Nitric Oxide Synthase
NO is synthesized by NO synthase (NOS) from L-arginine, NADPH and O2. In addition to NO, NOS has been found to produce O2•−, hence NOS by itself can generate ONOO•−. This NOS phenomena is termed uncoupling, as O2•− production primarily occurs when NOS is not associated with cofactor or substrate (e.g. reduced L-arginine or tetrahydrobiopterin (BH4) levels) (Luo et al., 2014, Stuehr et al., 2001). Three types of NOS and several different splice isoforms have been identified in skeletal muscle; constitutively expressed NOS1 (neuronal NOS), NOS2 (inducible NOS) and NOS3 (endothelial NOS) (Knowles and Moncada, 1994). Increased levels of NOS1 have been detected in skeletal muscle both from rodents with RA (three-fold increase) and in patients with RA (two-fold increase) (Yamada et al., 2015a, Yamada et al., 2015b). Altered levels of NOS2 and NOS3 have not been reported in skeletal muscle from subjects with RA (Yamada et al., 2015a, Yamada et al., 2015b, Yamada et al., 2009). Under normal conditions, a majority of NOS1 is compartmentalized to sub-membrane scaffolds, which are part of the dystrophin glycoprotein complex (Molza et al., 2015). In addition, a small fraction of NOS1 was detectable in association with the SR and with mitochondria (Buchwalow et al., 2005). Moreover, NOS1 has been shown to co-localize with the RyR1 in skeletal and cardiac muscle from mouse and human subjects (Lee et al., 2014, Salanova et al., 2008, Yamada et al., 2015b). Interestingly, the amount NOS1 co-localized with RyR1 was increased five-fold in muscles from mice with arthritis (Yamada et al., 2015b) (Fig. 3).
Fig. 3.
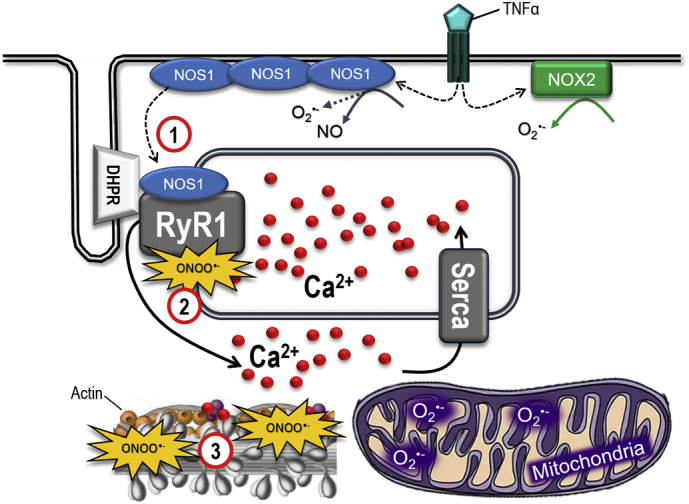
ROS/RNS sources and the tentative vicious cycle in RA-associated muscle weakness. Summary of potential sources to O2•−, NO and ONOO•− in RA muscles, including NOS1, NOX2, and mitochondria. This model suggests that NOS1 is globally increased and more NOS1 is bound to the RyR1 protein complex in arthritic muscle than in control muscle (1). This leads to ONOO•−-induced modifications of the RyR1 protein complex and the SR Ca2 + release during contractions increases, which further activates the Ca2 +-sensitive NOS1 and amplifies the ROS/RNS production (2). The increased amounts of ONOO•− attack myofibrillar proteins, e.g. actin (3), which results in contractile dysfunction and muscle weakness.
The free cytosolic Ca2 + concentration ([Ca2 +]i) evoked by electrical stimulation (tetanic [Ca2 +]i), but not resting [Ca2 +]i, has been shown to be almost two-fold higher in muscle fibers from mice with RA than healthy controls (Yamada et al., 2015b, Yamada et al., 2009). NOS1 is a Ca2 +-calmodulin (CaM)-dependent enzyme and its activity increases with [Ca2 +]i via calmodulin (CaM) (Förstermann et al., 1994). Km[Ca2 +]i for the activation of NOS1 is ~ 200 nM in the presence of 500 nM CaM (Bredt et al., 1990). However, the free accessible CaM concentration is ~ 50 nM under physiological conditions in muscle (Wu and Bers, 2007), hence the Km [Ca2 +]i for the activation of NOS1 is most probably higher than ~ 200 nM in muscle. Nevertheless, a [Ca2 +]i > 1 μM (see Fig. 1 for example) is readily reached when skeletal muscles contract, hence NOS1 becomes activated during contractile activities. Thus, not only is there more NOS1 in the muscle cells, the increased tetanic [Ca2 +]i could allow for even higher NOS1 activity and contribute to increased ROS/RNS load in RA muscles (Fig. 3).
6.2. Pro-inflammatory Cytokines and NADPH Oxidase
A number of pro-inflammatory cytokines, including TNFα, IL-1, IL-6 and IL-23 are recognized as important mediators in the processes that cause inflammation and comorbidities (e.g. bone erosion, cartilage destruction) associated with RA (Brennan and McInnes, 2008, Londhe and Guttridge, 2015, Noack and Miossec, 2017). For instance, pro-inflammatory cytokines are thought to induce ROS/RNS and apoptosis in the synovial joints and thereby contribute to the pathogenesis of RA (Croft and Siegel, 2017, Li et al., 2012). Recently Huffman et al., reported that patients with RA have 75% greater muscle concentration of IL-6 protein than healthy controls (Huffman et al., 2017). In addition to IL-6, increased levels of IL-1β, IL-8, TNF-α, and toll-like receptor (TLR)-4 has been observed in skeletal muscle from patients with RA (Huffman et al., 2017, Mikkelsen et al., 2015). Interestingly, the muscle concentration of these inflammatory markers were positively associated the disease activity, disability, pain and physical inactivity in patients with RA (Huffman et al., 2017). Furthermore, TNFα and IL-6 have been shown to promote ROS/RNS stress in skeletal muscle (Landskron et al., 2014, Reid and Moylan, 2011). In turn, ROS/RNS are suggested inducers of e.g. TNFα and IL-6 expression in various inflammatory conditions (Blaser et al., 2016, Pacher et al., 2007). Thus, a complex, and far from fully understood, crosstalk is present between cytokines and ROS/RNS in inflammation. The ROS/RNS source(s) behind cytokine-induced ROS/RNS stress are not fully established, but both NOS and NADPH oxidases type 1, 2 and 4 (NOX1, NOX2, NOX4) are suggested as downstream targets of TNFα in inflammatory processes (Blaser et al., 2016, Cangemi et al., 2014, Moe et al., 2011). Brief exposure of skeletal muscle to TNFα has been shown to increase ROS/RNS stress and reduce the force generation capacity (Alloatti et al., 2000, Reid and Moylan, 2011, Stasko et al., 2013). In these studies, the TNFα effect was blunted by addition of the NOS1 inhibitor L-NG-Nitroarginine methyl ester (L-NAME) in skeletal muscle from both guinea pig and mouse (Alloatti et al., 2000, Stasko et al., 2013), hence NOS1 was identified as the source for TNFα-induced ROS/RNS production.
Activated NOXs produce O2•− and skeletal muscle is known to express NOX2 and NOX4 (as well as the dual oxidase enzymes DUOX1, DUOX2) (Sakellariou et al., 2014). Increased levels of TNFα and NOX2 were observed in skeletal muscle from rats with arthritis, i.e. TNFα (and other not yet identified cytokines) could be a potential upstream activator of NOS1 and/or NOX2. Increased NOS1 can produce both NO and O2•− and may directly increase ONOO•− formation. Alternatively, NOS1-induced NO and NOX2-induced O2•− may contribute to the ONOO•− formation observed in RA muscles (Fig. 3). In support of this notion, an enhanced expression of NOX subunits and NOS isoforms were associated with elevated levels of 3-NT in the ischemic hemisphere following cerebral ischemia-reperfusion injury in mice (Lu et al., 2011). Furthermore, the enzyme sphingomyelinase (SMase) is activated by TNFα and has been shown to be elevated in acute systemic inflammation (e.g. sepsis) (Okazaki et al., 2014, Wong et al., 2000). Skeletal muscle exposed to SMase exhibits muscle weakness, which is thought to be the result of SMase-induced NOX2 activation (Bost et al., 2015, Loehr et al., 2014). Thus, SMase appears to be a mediator in the ROS/RNS stress in skeletal muscle. Its role in RA-associated muscle weakness is unknown, but worth investigating further. Nevertheless, ROS/RNS are thought to be unspecific in their reactivity, reacting with whatever is close (Kumar et al., 2012); i.e. to gain further detailed knowledge of the involvement of the free radical sources associated with RA and other inflammatory diseases, we believe that it will be important to localize and quantify intracellular hotspots of ROS/RNS production in skeletal muscle afflicted with RA. Advanced knowledge in this area of redox-induced muscle dysfunction could identify targets and lay the groundwork for future therapeutic interventions to counteract muscle dysfunction associated with inflammatory conditions.
7. Therapeutic Interventions to Counteract Arthritis-associated Muscle Weakness
According to recommendations by European Leage Against Rheumatism (EULAR), RA treatment should be initiated with conventional synthetic disease-modifying anti-rheumatic drugs (DMARDs, most commonly methotrexate) and low-dose glucocortiods (Smolen et al., 2017). If the first line of treatment fails, patients can receive conventional synthetic DMARDs in combination with targeted synthetic DMARDs or biological DMARDs (Smolen et al., 2017). TNFα inhibitors (e.g. adalimumab, certolizumab pegol, etanercept, golimumab, infliximab, biosimilars) and anti-IL6 receptor antibodies (e.g. tocilizumab) are biological DMARDs that are considered to be efficient and safe (Smolen et al., 2017). By definition DMARDs must reduce structural damage progression, whereas anti-inflammatory drugs (e.g glucocortiods) reduce pain and stiffness and improve physical function, i.e. do not interfere with joint damage and hence are not disease modifying. RA cannot be cured, but the current available treatment options allow for good therapeutic successes and tight control of the disease activity, by retaining the disease in a low-inflammatory state. Nevertheless, Lemmey et al., recently showed that well-managed RA patients with a low disease activity still performed ~ 25–35% poorer than age- and weight-matched healthy control subjects on all functional muscle measurements tested, including knee extensor and handgrip strength (Lemmey et al., 2016). This strongly suggests that muscle weakness and muscle dysfunction are not directly influenced by the patient's inflammatory status and/or disease activity. Instead, separate and unique therapy strategies appear necessary to counteract muscle dysfunction and muscle weakness present in patients with RA, and probably also translates to other chronic inflammatory disorders associated with muscle complications.
7.1. Exercise as Therapy
Regular physical exercise, both aerobic and strength exercise, are recognized as an important component of the management of RA. In patients with RA, exercise-induced beneficial effects include increased force production and muscle mass, increased aerobic capacity, lower amount of fat mass, decreased inflammation and pain, and an overall sense of well-being (Häkkinen, 2004, Häkkinen et al., 2001, Lemmey et al., 2009, Sharif et al., 2011, Sokka et al., 2008, Stenström and Minor, 2003). Thus, exercise per se appears as an effective overall therapy for patients with RA. However, this requires that the patients are active several days per week, which is not the case for many patients with RA. In fact, Sokka et al., reported that out of 5235 patients from 58 sites in 21 countries, only ~ 14% were physically active ≥ 3 times per week (Sokka et al., 2008). Furthermore, Lemmey et al., 2012 reported that in their 3-year follow-up study of patients with RA that had performed a 24 week high-intensity strength training program, no one in the exercise group was still exercising. Thus, a challenge with physical exercise as therapy is to achieve sustainability and to engage the patients in regular physical activity for the rest of their life.
7.2. Antioxidant Treatment to Counteract RA-associated Muscle Weakness
Several antioxidants (e.g. Vitamin E, Vitamin C, SS31, CoQ10) have been tested in clinical trials for a range of diseases (e.g. cardiovascular and diabetes), but the outcome has often been inconclusive (Ajith and Jayakumar, 2014, Lonn et al., 2005). However, the outcome probably reflects our limited knowledge of the temporal and spatial distribution of ROS/RNS, rather than antioxidants as such being ineffective as a therapeutic tool. Nevertheless, the involvement of ROS/RNS in muscle dysfunction associated with RA, indicates that antioxidant treatment could potentially be beneficial in improving muscle function in patients with RA. Thus far, to our knowledge, antioxidant treatments have not been tested in patients to counteract RA-associated muscle weakness. Mn-Salen compounds (e.g. EUK-134) have been proposed to possess distinct advantages, e.g. exhibit combined SOD/catalase mimetic functions, over nonspecific antioxidant scavenger effect which is how e.g. N-acetyl cysteine function. The EUK-series also exhibit high translational value as it is developed for oral administration (Baker et al., 1998, Rosenthal et al., 2009). In addition to scavenging mitochondrial O2•−/H2O2 formation, Mn-Salen catalyzes the removal of ONOO•− and ameliorates nitrosative stress (Sharpe et al., 2002). Interestingly, EUK-134 has been shown to lower amount ONOO•−-induced 3-NT modifications on actin and to prevent the loss of muscle force production in rats with RA (Yamada et al., 2015b). EUK-134 treatment has also been shown to prevent ROS/RNS-associated muscle wasting and weakness in other pathological conditions, including the mouse-models of Duchenne muscle dystrophy and pulmonary hypertension (Himori et al., 2017, Kim and Lawler, 2012, Lawler et al., 2014). For that reason, despite antioxidants reported history of not being successful in clinical trials, it would be interesting to test the effects of EUK-134 on muscle function in patients with RA.
7.3. Novel Actions to Improve Muscle Function
Based on the scientific results discussed in this review, Fig. 3 illustrates a tentative vicious cycle that may contribute to the arthritis-induced muscle weakness. This model show that nitrosative modifications of the RyR1 protein complex results in facilitated and increased Ca2 + release during muscle contractions, which further activates the Ca2 +-sensitive NOS1 that by itself can cause amplification of O2•−, NO and ONOO•−. This results in ONOO•− attacks of myofibrillar proteins and causes contractile dysfunction and muscle weakness. Thus, a novel action to counteract RA-associated muscle weakness could be to inhibit this viscous cycle by pharmacological intervention targeting RyR1 to stabilize SR Ca2 + release, hence counteract the facilitated Ca2 + release observed in arthritis (Yamada et al., 2015b, Yamada et al., 2009). AICAR (5-aminoimidazole-4carboxamideribonucleotide) and S107 are known to stabilize RyR1 activity, normalize Ca2 + release and shown to reduce the ROS/RNS burden and improve muscle function in muscle dystrophy and cancer-related muscle weakness, respectively (Lanner et al., 2012, Waning et al., 2015). Thus, AICAR and S107 could be potentially useful compounds to counteract RA-induced muscle weakness.
8. Conclusion
Muscle weakness is strongly linked with declined physical function, reduced quality of life, impaired work capacity, and increased mortality. Patients with RA are commonly afflicted by muscle weakness and also experience all listed associated risks and complications. RA is a chronic disease, but today there are effective pharmacological treatment strategies (e.g. methotrexate combined with low-dose glucocortiods) that lower inflammation, joint damage and overall disease activity. Yet, the patients' strength or physical function does not fully recover. Thus, it appears as the RA-induced muscle weakness cannot be counteracted with a treatment strategy that only attacks the disease itself. Instead, we suggest that future RA therapies should provide improvement in muscle strength as well as reducing the disease activity (i.e. inflammation and joint damage). For instance, EUK-134 or RyR1-stabilizing compounds and muscle strength exercises could be a potentially useful combination therapy together with methotrexate to improve intrinsic muscle function and reduce the disease activity, respectively. Ultimately, this combinational intervention (pharmacological and exercise) can significantly improve the quality of life for the afflicted patients and will also lower the burden on society since the ability to work will improve among these patients.
9. Outstanding Questions
-
•
Patients with RA suffer from physical impairments including muscle weakness, which type of muscle weakness affects patients with RA?
-
•
What is the underlying signaling that results in RA-induced muscle weakness?
-
•
How can muscle function be improved for patients with RA?
Search Strategy and Selection Criteria
Data from this review were identified by searches of PubMed and references from relevant articles using the following keywords, alone or in combination: “Rheumatoid arthritis”, “Muscle weakness”, “Pro-inflammatory cytokines”, “Reactive nitrogen species”, “Calcium signaling”, “Antioxidant”. Only articles published in English were included. Abstracts and reports from meetings were excluded.
Author Contribution
TY and JTL performed the literature search and drafted the manuscript. TY, MMS, EK, JTL edited and revised the manuscript. All authors approved the final version of the manuscript.
Funding/Acknowledgements
This work was supported by grants from the Japan Society for the Promotion of Science, Jeansson Foundation (2597/12), Åke Wiberg Foundation (M14-0210), KI Rheumatology fund, the Swedish Rheumatism Association and the Swedish Research Council (2016-02457).
References
- Ajith T.A., Jayakumar T.G. Mitochondria-targeted agents: future perspectives of mitochondrial pharmaceutics in cardiovascular diseases. World J. Cardiol. 2014;6:1091–1099. doi: 10.4330/wjc.v6.i10.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alamanos Y., Voulgari P.V., Drosos A.A. Incidence and prevalence of rheumatoid arthritis, based on the 1987 American College of Rheumatology criteria: a systematic review. Semin. Arthritis Rheum. 2006;36:182–188. doi: 10.1016/j.semarthrit.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Allen D.G., Westerblad H. The effects of caffeine on intracellular calcium, force and the rate of relaxation of mouse skeletal muscle. J. Physiol. 1995;487:331–342. doi: 10.1113/jphysiol.1995.sp020883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloatti G., Penna C., Mariano F., Camussi G. Role of NO and PAF in the impairment of skeletal muscle contractility induced by TNF-alpha. Am. J. Phys. Regul. Integr. Comp. Phys. 2000;279:R2156–2163. doi: 10.1152/ajpregu.2000.279.6.R2156. [DOI] [PubMed] [Google Scholar]
- Andersson D.C., Betzenhauser M.J., Reiken S., Meli A.C., Umanskaya A., Xie W., Shiomi T., Zalk R., Lacampagne A., Marks A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011;14:196–207. doi: 10.1016/j.cmet.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson D.C., Betzenhauser M.J., Reiken S., Umanskaya A., Shiomi T., Marks A.R. Stress-induced increase in skeletal muscle force requires protein kinase A phosphorylation of the ryanodine receptor. J. Physiol. 2012;590:6381–6387. doi: 10.1113/jphysiol.2012.237925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade F.H., Reid M.B., Allen D.G., Westerblad H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 1998;509:565–575. doi: 10.1111/j.1469-7793.1998.565bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronache Z., Hamilton S.L., Dirksen R.T., Melzer W. A retrograde signal from RyR1 alters DHP receptor inactivation and limits window Ca2 + release in muscle fibers of Y522S RyR1 knock-in mice. PNAS. 2009;106:4531–4536. doi: 10.1073/pnas.0812661106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydin J., Shabalina I.G., Place N., Reiken S., Zhang S.J., Bellinger A.M., Nedergaard J., Cannon B., Marks A.R., Bruton J.D. Nonshivering thermogenesis protects against defective calcium handling in muscle. FASEB J. 2008;22:3919–3924. doi: 10.1096/fj.08-113712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K., Marcus C.B., Huffman K., Kruk H., Malfroy B., Doctrow S.R. Synthetic combined superoxide dismutase/catalase mimetics are protective as a delayed treatment in a rat stroke model: a key role for reactive oxygen species in ischemic brain injury. J. Pharmacol. Exp. Ther. 1998;284:215–221. [PubMed] [Google Scholar]
- Bellinger A.M., Reiken S., Carlson C., Mongillo M., Liu X., Rothman L., Matecki S., Lacampagne A., Marks A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009;15:325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser H., Dostert C., Mak T.W., Brenner D. TNF and ROS crosstalk in inflammation. Trends Cell Biol. 2016;26:249–261. doi: 10.1016/j.tcb.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Bost E.R., Frye G.S., Ahn B., Ferreira L.F. Diaphragm dysfunction caused by sphingomyelinase requires the p47(phox) subunit of NADPH oxidase. Respir. Physiol. Neurobiol. 2015;0:47–52. doi: 10.1016/j.resp.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botti H., Moller M.N., Steinmann D., Nauser T., Koppenol W.H., Denicola A., Radi R. Distance-dependent diffusion-controlled reaction of NO and O2•− at chemical equilibrium with ONOO•−. J. Phys. Chem. B. 2010;114:16584–16593. doi: 10.1021/jp105606b. [DOI] [PubMed] [Google Scholar]
- Bredt D.S., Hwang P.M., Snyder S.H. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:768–770. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- Brennan F.M., McInnes I.B. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Invest. 2008;118:3537–3545. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwalow I.B., Minin E.A., Samoilova V.E., Boecker W., Wellner M., Schmitz W., Neumann J., Punkt K. Compartmentalization of NO signaling cascade in skeletal muscles. Biochem. Biophys. Res. Commun. 2005;330:615–621. doi: 10.1016/j.bbrc.2005.02.182. [DOI] [PubMed] [Google Scholar]
- Cangemi R., Celestini A., Del Ben M., Pignatelli P., Carnevale R., Proietti M., Calabrese C.M., Basili S., Violi F. Role of platelets in NOX2 activation mediated by TNFα in heart failure. Intern. Emerg. Med. 2014;9:179–185. doi: 10.1007/s11739-012-0837-2. [DOI] [PubMed] [Google Scholar]
- Carballal S., Bartesaghi S., Radi R. Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochim. Biophys. Acta. 2014;1840:768–780. doi: 10.1016/j.bbagen.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A.J., Bruton J.D., Lanner J.T., Westerblad H. Antioxidant treatments do not improve force recovery after fatiguing stimulation of mouse skeletal muscle fibres. J. Physiol. 2015;593:457–472. doi: 10.1113/jphysiol.2014.279398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A.J., Yamada T., Rassier D.E., Andersson D.C., Westerblad H., Lanner J.T. Reactive oxygen/nitrogen species and contractile function in skeletal muscle during fatigue and recovery. J. Physiol. 2016;594:5149–5160. doi: 10.1113/JP270650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements M.K., Siemsen D.W., Swain S.D., Hanson A.J., Nelson-Overton L.K., Rohn T.T., Quinn M.T. Inhibition of actin polymerization by peroxynitrite modulates neutrophil functional responses. J. Leukoc. Biol. 2003;73:344–355. doi: 10.1189/jlb.0802401. [DOI] [PubMed] [Google Scholar]
- Croft M., Siegel R.M. Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat. Rev. Rheumatol. 2017;13:217–233. doi: 10.1038/nrrheum.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S., Kundu S., Ghosh P., De S., Ghosh A., Chatterjee M. Correlation of oxidant status with oxidative tissue damage in patients with rheumatoid arthritis. Clin. Rheumatol. 2014;33:1557–1564. doi: 10.1007/s10067-014-2597-z. [DOI] [PubMed] [Google Scholar]
- Durham W.J., Aracena-Parks P., Long C., Rossi A.E., Goonasekera S.A., Boncompagni S., Galvan D.L., Gilman C.P., Baker M.R., Shirokova N. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:53–65. doi: 10.1016/j.cell.2008.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutka T.L., Mollica J.P., Lamb G.D. Differential effects of peroxynitrite on contractile protein properties in fast- and slow-twitch skeletal muscle fibers of rat. J. Appl. Physiol. 2011;110:705–716. doi: 10.1152/japplphysiol.00739.2010. [DOI] [PubMed] [Google Scholar]
- Ekdahl C., Broman G. Muscle strength, endurance, and aerobic capacity in rheumatoid arthritis: a comparative study with healthy subjects. Ann. Rheum. Dis. 1992;51:35–40. doi: 10.1136/ard.51.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorova M., Kuleva N., Hoffmann R. Reversible and irreversible modifications of skeletal muscle proteins in a rat model of acute oxidative stress. Biochim. Biophys. Acta. 2009;1792:1185–1193. doi: 10.1016/j.bbadis.2009.09.011. [DOI] [PubMed] [Google Scholar]
- Fedorova M., Bollineni R.C., Hoffmann R. Protein carbonylation as a major hallmark of oxidative damage: update of analytical strategies. Mass Spectrom. Rev. 2014;33:79–97. doi: 10.1002/mas.21381. [DOI] [PubMed] [Google Scholar]
- Förstermann U., Closs E.I., Pollock J.S., Nakane M., Schwarz P., Gath I., Kleinert H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension. 1994;23:1121–1131. doi: 10.1161/01.hyp.23.6.1121. [DOI] [PubMed] [Google Scholar]
- Fraser A., Vallow J., Preston A., Cooper R.G. Predicting ‘normal’ grip strength for rheumatoid arthritis patients. Rheumatology. 1999;38:521–528. doi: 10.1093/rheumatology/38.6.521. [DOI] [PubMed] [Google Scholar]
- Gehlert S., Bungartz G., Willkomm L., Korkmaz Y., Pfannkuche K., Schiffer T., Bloch W., Suhr F. Intense resistance exercise induces early and transient increases in ryanodine receptor 1 phosphorylation in human skeletal muscle. PLoS One. 2012;7:e49326. doi: 10.1371/journal.pone.0049326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon A.M., Homsher E., Regnier M. Regulation of contraction in striated muscle. Physiol. Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- Grönwall C., Amara K., Hardt U., Krishnamurthy A., Steen J., Engstrom M., Sun M., Ytterberg A.J., Zubarev R.A., Scheel-Toellner D. Autoreactivity to malondialdehyde-modifications in rheumatoid arthritis is linked to disease activity and synovial pathogenesis. J. Autoimmun. 2017 doi: 10.1016/j.jaut.2017.06.004. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Häkkinen A. Effectiveness and safety of strength training in rheumatoid arthritis. Curr. Opin. Rheumatol. 2004;16:132–137. doi: 10.1097/00002281-200403000-00011. [DOI] [PubMed] [Google Scholar]
- Häkkinen A., Sokka T., Kotaniemi A., Hannonen P. A randomized two-year study of the effects of dynamic strength training on muscle strength, disease activity, functional capacity, and bone mineral density in early rheumatoid arthritis. Arthritis Rheum. 2001;44:515–522. doi: 10.1002/1529-0131(200103)44:3<515::AID-ANR98>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Hartog A., Hulsman J., Garssen J. Locomotion and muscle mass measures in a murine model of collagen-induced arthritis. BMC Musculoskelet. Disord. 2009;10:59. doi: 10.1186/1471-2474-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helliwell P.S., Jackson S. Relationship between weakness and muscle wasting in rheumatoid arthritis. Ann. Rheum. Dis. 1994;53:726–728. doi: 10.1136/ard.53.11.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Ochoa E.O., Pratt S.J.P., Lovering R.M., Schneider M.F. Critical role of intracellular RyR1 calcium release channels in skeletal muscle function and disease. Front. Physiol. 2015;6:420. doi: 10.3389/fphys.2015.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliquin P., Borderie D., Hernvann A., Menkes C.J., Ekindjian O.G. Nitric oxide as s-nitrosoproteins in rheumatoid arthritis. Arthritis Rheum. 1997;40:1512–1517. doi: 10.1002/art.1780400820. [DOI] [PubMed] [Google Scholar]
- Himori K., Abe M., Tatebayashi D., Lee J., Westerblad H., Lanner J.T., Yamada T. Superoxide dismutase/catalase mimetic EUK-134 prevents diaphragm muscle weakness in monocrotalin-induced pulmonary hypertension. PLoS One. 2017;12:e0169146. doi: 10.1371/journal.pone.0169146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J.L., Hsieh Y., Tu C., O'Connor D., Nick H.S., Silverman D.N. Catalytic properties of human manganese superoxide dismutase. J. Biol. Chem. 1996;271:17687–17691. doi: 10.1074/jbc.271.30.17687. [DOI] [PubMed] [Google Scholar]
- Huffman K.M., Jessee R., Andonian B., Davis B.N., Narowski R., Huebner J.L., Kraus V.B., McCracken J., Gilmore B.F., Tune K.N. Molecular alterations in skeletal muscle in rheumatoid arthritis are related to disease activity, physical inactivity, and disability. Arthritis Res. Ther. 2017;19:12. doi: 10.1186/s13075-016-1215-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur H., Halliwell B. Evidence for nitric oxide-mediated oxidative damage in chronic inflammation. Nitrotyrosine in serum and synovial fluid from rheumatoid patients. FEBS Lett. 1994;350:9–12. doi: 10.1016/0014-5793(94)00722-5. [DOI] [PubMed] [Google Scholar]
- Khojah H.M., Ahmed S., Abdel-Rahman M.S., Hamza A.B. Reactive oxygen and nitrogen species in patients with rheumatoid arthritis as potential biomarkers for disease activity and the role of antioxidants. Free Radic. Biol. Med. 2016;97:285–291. doi: 10.1016/j.freeradbiomed.2016.06.020. [DOI] [PubMed] [Google Scholar]
- Kim J.H., Lawler J.M. Amplification of proinflammatory phenotype, damage, and weakness by oxidative stress in the diaphragm muscle of mdx mice. Free Radic. Biol. Med. 2012;52:1597–1606. doi: 10.1016/j.freeradbiomed.2012.01.015. [DOI] [PubMed] [Google Scholar]
- Knowles R.G., Moncada S. Nitric oxide synthases in mammals. Biochem. J. 1994;298:249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H., Lim H.-W., More S.V., Kim B.-W., Koppula S., Kim I.S., Choi D.-K. The role of free radicals in the aging brain and Parkinson's disease: convergence and parallelism. Int. J. Mol. Sci. 2012;13:10478–10504. doi: 10.3390/ijms130810478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurien B.T., Hensley K., Bachmann M., Scofield R.H. Oxidatively modified autoantigens in autoimmune diseases. Free Radic. Biol. Med. 2006;41:549–556. doi: 10.1016/j.freeradbiomed.2006.05.020. [DOI] [PubMed] [Google Scholar]
- Lambeth J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Landskron G., De la Fuente M., Thuwajit P., Thuwajit C., Hermoso M.A. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185. doi: 10.1155/2014/149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanner J.T., Georgiou D.K., Joshi A.D., Hamilton S.L. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010;2:a003996. doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanner J.T., Georgiou D.K., Dagnino-Acosta A., Ainbinder A., Cheng Q., Joshi A.D., Chen Z., Yarotskyy V., Oakes J.M., Lee C.S. AICAR prevents heat-induced sudden death in RyR1 mutant mice independent of AMPK activation. Nat. Med. 2012;18:244–251. doi: 10.1038/nm.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler J.M., Kunst M., Hord J.M., Lee Y., Joshi K., Botchlett R.E., Ramirez A., Martinez D.A. EUK-134 ameliorates nNOSmu translocation and skeletal muscle fiber atrophy during short-term mechanical unloading. Am. J. Phys. Regul. Integr. Comp. Phys. 2014;306:R470–482. doi: 10.1152/ajpregu.00371.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.S., Georgiou D.K., Dagnino-Acosta A., Xu J., Ismailov I.I., Knoblauch M., Monroe T.O., Ji R., Hanna A.D., Joshi A.D. Ligands for FKBP12 increase Ca2 + influx and protein synthesis to improve skeletal muscle function. J. Biol. Chem. 2014;289:25556–25570. doi: 10.1074/jbc.M114.586289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmey A.B., Marcora S.M., Chester K., Wilson S., Casanova F., Maddison P.J. Effects of high-intensity resistance training in patients with rheumatoid arthritis: a randomized controlled trial. Arthritis Rheum. 2009;61:1726–1734. doi: 10.1002/art.24891. [DOI] [PubMed] [Google Scholar]
- Lemmey A.B., Williams S.L., Marcora S.M., Jones J., Maddison P.J. Are the benefits of a high-intensity progressive resistance training program sustained in rheumatoid arthritis patients? A 3-year followup study. Arthritis Care Res. 2012;64:71–75. doi: 10.1002/acr.20523. [DOI] [PubMed] [Google Scholar]
- Lemmey A.B., Wilkinson T.J., Clayton R.J., Sheikh F., Whale J., Jones H.S., Ahmad Y.A., Chitale S., Jones J.G., Maddison P.J. Tight control of disease activity fails to improve body composition or physical function in rheumatoid arthritis patients. Rheumatology. 2016;55:1736–1745. doi: 10.1093/rheumatology/kew243. [DOI] [PubMed] [Google Scholar]
- Li J., Hsu H.-C., Mountz J.D. Managing macrophages in rheumatoid arthritis by reform or removal. Curr. Rheumatol. Rep. 2012;14:445–454. doi: 10.1007/s11926-012-0272-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loehr J.A., Abo-Zahrah R., Pal R., Rodney G.G. Sphingomyelinase promotes oxidant production and skeletal muscle contractile dysfunction through activation of NADPH oxidase. Front. Physiol. 2014;5:530. doi: 10.3389/fphys.2014.00530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londhe P., Guttridge D.C. Inflammation induced loss of skeletal muscle. Bone. 2015;80:131–142. doi: 10.1016/j.bone.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonn E., Bosch J., Yusuf S., Sheridan P., Pogue J., Arnold J.M., Ross C., Arnold A., Sleight P., Probstfield J. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA. 2005;293:1338–1347. doi: 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- Lu Q., Xia N., Xu H., Guo L., Wenzel P., Daiber A., Munzel T., Förstermann U., Li H. Betulinic acid protects against cerebral ischemia-reperfusion injury in mice by reducing oxidative and nitrosative stress. Nitric Oxide. 2011;24:132–138. doi: 10.1016/j.niox.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Luo S., Lei H., Qin H., Xia Y. Molecular mechanisms of endothelial NO synthase uncoupling. Curr. Pharm. Des. 2014;20:3548–3553. doi: 10.2174/13816128113196660746. [DOI] [PubMed] [Google Scholar]
- Marx S.O., Reiken S., Hisamatsu Y., Jayaraman T., Burkhoff D., Rosemblit N., Marks A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Mateen S., Moin S., Khan A.Q., Zafar A., Fatima N. Increased reactive oxygen species formation and oxidative stress in rheumatoid arthritis. PLoS One. 2016;11:e0152925. doi: 10.1371/journal.pone.0152925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen U.R., Dideriksen K., Andersen M.B., Boesen A., Malmgaard-Clausen N.M., Sorensen I.J., Schjerling P., Kjaer M., Holm L. Preserved skeletal muscle protein anabolic response to acute exercise and protein intake in well-treated rheumatoid arthritis patients. Arthritis Res. Ther. 2015;17:271. doi: 10.1186/s13075-015-0758-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moe K.T., Yin N.O., Naylynn T.M., Khairunnisa K., Wutyi M.A., Gu Y., Atan M.S., Wong M.C., Koh T.H., Wong P. Nox2 and Nox4 mediate tumour necrosis factor-alpha-induced ventricular remodelling in mice. J. Cell. Mol. Med. 2011;15:2601–2613. doi: 10.1111/j.1582-4934.2011.01261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molza A.E., Mangat K., Le Rumeur E., Hubert J.F., Menhart N., Delalande O. Structural basis of neuronal nitric-oxide nynthase interaction with dystrophin repeats 16 and 17. J. Biol. Chem. 2015;290:29531–29541. doi: 10.1074/jbc.M115.680660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy R.M., Dutka T.L., Lamb G.D. Hydroxyl radical and glutathione interactions alter calcium sensitivity and maximum force of the contractile apparatus in rat skeletal muscle fibres. J. Physiol. 2008;586:2203–2216. doi: 10.1113/jphysiol.2007.150516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noack M., Miossec P. Selected cytokine pathways in rheumatoid arthritis. Semin. Immunopathol. 2017;39:365–383. doi: 10.1007/s00281-017-0619-z. [DOI] [PubMed] [Google Scholar]
- Ochala J., Lehtokari V.L., Iwamoto H., Li M., Feng H.Z., Jin J.P., Yagi N., Wallgren-Pettersson C., Penisson-Besnier I., Larsson L. Disrupted myosin cross-bridge cycling kinetics triggers muscle weakness in nebulin-related myopathy. FASEB J. 2011;25:1903–1913. doi: 10.1096/fj.10-176727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki T., Liang F., Li T., Lemaire C., Danialou G., Shoelson S.E., Petrof B.J. Muscle-specific inhibition of the classical nuclear factor-kappaB pathway is protective against diaphragmatic weakness in murine endotoxemia. Crit. Care Med. 2014;42:e501–509. doi: 10.1097/CCM.0000000000000407. [DOI] [PubMed] [Google Scholar]
- de Oliveira Nunes Teixeira V., Filippin L.I., Viacava P.R., de Oliveira P.G., Xavier R.M. Muscle wasting in collagen-induced arthritis and disuse atrophy. Exp. Biol. Med. 2013;238:1421–1430. doi: 10.1177/1535370213505961. [DOI] [PubMed] [Google Scholar]
- Ozkan Y., Yardym-Akaydyn S., Sepici A., Keskin E., Sepici V., Simsek B. Oxidative status in rheumatoid arthritis. Clin. Rheumatol. 2007;26:64–68. doi: 10.1007/s10067-006-0244-z. [DOI] [PubMed] [Google Scholar]
- Pacher P., Beckman J.S., Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Palma L., Chillemi C., Albanelli S., Rapali S., Bertoni-Freddari C. Muscle involvement in rheumatoid arthritis: an ultrastructural study. Ultrastruct. Pathol. 2000;24:151–156. doi: 10.1080/01913120050132886. [DOI] [PubMed] [Google Scholar]
- Pearson T., McArdle A., Jackson M.J. Nitric oxide availability is increased in contracting skeletal muscle from aged mice, but does not differentially decrease muscle superoxide. Free Radic. Biol. Med. 2015;78:82–88. doi: 10.1016/j.freeradbiomed.2014.10.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinniger G.J., Lavin T., Bakker A.J. Skeletal muscle weakness caused by carrageenan-induced inflammation. Muscle Nerve. 2012;46:413–420. doi: 10.1002/mus.23318. [DOI] [PubMed] [Google Scholar]
- Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. PNAS U S A. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid M.B., Moylan J.S. Beyond atrophy: redox mechanisms of muscle dysfunction in chronic inflammatory disease. J. Physiol. 2011;589:2171–2179. doi: 10.1113/jphysiol.2010.203356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiken S., Lacampagne A., Zhou H., Kherani A., Lehnart S.E., Ward C., Huang F., Gaburjakova M., Gaburjakova J., Rosemblit N. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle. J. Cell Biol. 2003;160:919–928. doi: 10.1083/jcb.200211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat. Med. 2014;20:709–711. doi: 10.1038/nm.3624. [DOI] [PubMed] [Google Scholar]
- Rosenthal R.A., Huffman K.D., Fisette L.W., Damphousse C.A., Callaway W.B., Malfroy B., Doctrow S.R. Orally available Mn porphyrins with superoxide dismutase and catalase activities. JBIC. 2009;14:979–991. doi: 10.1007/s00775-009-0550-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rullman E., Andersson D.C., Melin M., Reiken S., Mancini D.M., Marks A.R., Lund L.H., Gustafsson T. Modifications of skeletal muscle ryanodine receptor type 1 and exercise intolerance in heart failure. J Heart Lung Transplant. 2013;32:925–929. doi: 10.1016/j.healun.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell M.L., Hanna W.M. Ultrastructural pathology of skeletal muscle in various rheumatic diseases. J. Rheumatol. 1988;15:445–453. [PubMed] [Google Scholar]
- Sakellariou G.K., Jackson M.J., Vasilaki A. Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic. Res. 2014;48:12–29. doi: 10.3109/10715762.2013.830718. [DOI] [PubMed] [Google Scholar]
- Salanova M., Schiffl G., Rittweger J., Felsenberg D., Blottner D. Ryanodine receptor type-1 (RyR1) expression and protein S-nitrosylation pattern in human soleus myofibres following bed rest and exercise countermeasure. Histochem. Cell Biol. 2008;130:105–118. doi: 10.1007/s00418-008-0399-6. [DOI] [PubMed] [Google Scholar]
- Santulli G., Nakashima R., Yuan Q., Marks A.R. Intracellular calcium release channels: an update. J. Physiol. 2017;595:3041–3053. doi: 10.1113/JP272781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif S., Thomas J.M., Donley D.A., Gilleland D.L., Bonner D.E., McCrory J.L., Hornsby W.G., Zhao H., Lively M.W., Hornsby J.A. Resistance exercise reduces skeletal muscle cachexia and improves muscle function in rheumatoid arthritis. Case Rep. Med. 2011;2011:205691. doi: 10.1155/2011/205691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe M.A., Ollosson R., Stewart V.C., Clark J.B. Oxidation of nitric oxide by oxomanganese-salen complexes: a new mechanism for cellular protection by superoxide dismutase/catalase mimetics. Biochem. J. 2002;366:97–107. doi: 10.1042/BJ20020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokova N., Rios E. Activation of Ca2 + release by caffeine and voltage in frog skeletal muscle. J. Physiol. 1996;493:317–339. doi: 10.1113/jphysiol.1996.sp021386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolen J.S., Landewe R., Bijlsma J., Burmester G., Chatzidionysiou K., Dougados M., Nam J., Ramiro S., Voshaar M., van Vollenhoven R. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017;76:960–977. doi: 10.1136/annrheumdis-2016-210715. [DOI] [PubMed] [Google Scholar]
- Sokka T., Häkkinen A., Kautiainen H., Maillefert J.F., Toloza S., Mork H.T., Calvo-Alen J., Oding R., Liveborn M., Huisman M. Physical inactivity in patients with rheumatoid arthritis: data from twenty-one countries in a cross-sectional, international study. Arthritis Rheum. 2008;59:42–50. doi: 10.1002/art.23255. [DOI] [PubMed] [Google Scholar]
- Stasko S.A., Hardin B.J., Smith J.D., Moylan J.S., Reid M.B. TNF signals via neuronal-type nitric oxide synthase and reactive oxygen species to depress specific force of skeletal muscle. J. Appl. Physiol. 2013;114:1629–1636. doi: 10.1152/japplphysiol.00871.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenström C.H., Minor M.A. Evidence for the benefit of aerobic and strengthening exercise in rheumatoid arthritis. Arthritis Rheum. 2003;49:428–434. doi: 10.1002/art.11051. [DOI] [PubMed] [Google Scholar]
- Stuehr D., Pou S., Rosen G.M. Oxygen reduction by nitric-oxide synthases. J. Biol. Chem. 2001;276:14533–14536. doi: 10.1074/jbc.R100011200. [DOI] [PubMed] [Google Scholar]
- Sun J., Xin C., Eu J.P., Stamler J.S., Meissner G. Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. PNAS. 2001;98:11158–11162. doi: 10.1073/pnas.201289098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supinski G.S., Callahan L.A. Free radical-mediated skeletal muscle dysfunction in inflammatory conditions. J. Appl. Physiol. 2007;102:2056–2063. doi: 10.1152/japplphysiol.01138.2006. [DOI] [PubMed] [Google Scholar]
- Supinski G., Stofan D., Callahan L.A., Nethery D., Nosek T.M., DiMarco A. Peroxynitrite induces contractile dysfunction and lipid peroxidation in the diaphragm. J. Appl. Physiol. 1999;87:783–791. doi: 10.1152/jappl.1999.87.2.783. [DOI] [PubMed] [Google Scholar]
- Szabó C., Ischiropoulos H., Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- Tiago T., Simao S., Aureliano M., Martin-Romero F.J., Gutierrez-Merino C. Inhibition of skeletal muscle S1-myosin ATPase by peroxynitrite. Biochemistry. 2006;45:3794–3804. doi: 10.1021/bi0518500. [DOI] [PubMed] [Google Scholar]
- Tong J., McCarthy T.V., MacLennan D.H. Measurement of resting cytosolic Ca2 + concentrations and Ca2 + store size in HEK-293 cells transfected with malignant hyperthermia or central core disease mutant Ca2 + release channels. J. Biol. Chem. 1999;274:693–702. doi: 10.1074/jbc.274.2.693. [DOI] [PubMed] [Google Scholar]
- van Vilsteren M., Boot C.R., Knol D.L., van Schaardenburg D., Voskuyl A.E., Steenbeek R., Anema J.R. Productivity at work and quality of life in patients with rheumatoid arthritis. BMC Musculoskelet. Disord. 2015;16:107. doi: 10.1186/s12891-015-0562-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsmith J., Roubenoff R. Cachexia in rheumatoid arthritis. Int. J. Cardiol. 2002;85:89–99. doi: 10.1016/s0167-5273(02)00237-1. [DOI] [PubMed] [Google Scholar]
- Waning D.L., Mohammad K.S., Reiken S., Xie W., Andersson D.C., John S., Chiechi A., Wright L.E., Umanskaya A., Niewolna M. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat. Med. 2015;21:1262–1271. doi: 10.1038/nm.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M.L., Xie B., Beatini N., Phu P., Marathe S., Johns A., Gold P.W., Hirsch E., Williams K.J., Licinio J. Acute systemic inflammation up-regulates secretory sphingomyelinase in vivo: a possible link between inflammatory cytokines and atherogenesis. PNAS. 2000;97:8681–8686. doi: 10.1073/pnas.150098097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Bers D.M. Free and bound intracellular calmodulin measurements in cardiac myocytes. Cell Calcium. 2007;41:353–364. doi: 10.1016/j.ceca.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T., Place N., Kosterina N., Östberg T., Zhang S.J., Grundtman C., Erlandsson-Harris H., Lundberg I.E., Glenmark B., Bruton J.D. Impaired myofibrillar function in the soleus muscle of mice with collagen-induced arthritis. Arthritis Rheum. 2009;60:3280–3289. doi: 10.1002/art.24907. [DOI] [PubMed] [Google Scholar]
- Yamada T., Abe M., Lee J., Tatebayashi D., Himori K., Kanzaki K., Wada M., Bruton J.D., Westerblad H., Lanner J.T. Muscle dysfunction associated with adjuvant-induced arthritis is prevented by antioxidant treatment. Skelet. Muscle. 2015;5:20. doi: 10.1186/s13395-015-0045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T., Fedotovskaya O., Cheng A.J., Cornachione A.S., Minozzo F.C., Aulin C., Friden C., Turesson C., Andersson D.C., Glenmark B. Nitrosative modifications of the Ca2 + release complex and actin underlie arthritis-induced muscle weakness. Ann. Rheum. Dis. 2015;74:1907–1914. doi: 10.1136/annrheumdis-2013-205007. [DOI] [PMC free article] [PubMed] [Google Scholar]