

Abstract
Gastroenteropancreatic neuroendocrine tumors (GEP-NETs) are a heterogeneous group of rare tumors whose site-specific tumor incidence and clinical behavior vary widely. Genetic alterations associated with familial inherited syndromes have been well defined; however, the genetic profile of sporadic tumors is less clear as their tumorigenesis does not appear to be controlled by classic oncogenes such as P53, RB, or KRAS. Even within GEP-NETs, there are no common oncogenic drivers; for example, DAXX/ATRX mutations are strongly implicated in the tumorigenesis of pancreatic but not small bowel NETs. Accordingly, the dysregulation of epigenetic mechanisms has been hypothesized as a potential regulator of GEP-NET tumorigenesis and has become a major focus of recent studies. Despite the heterogeneity of tumor cohorts evaluated in these studies, it is obvious that there are methylation patterns, chromatin remodeling alterations, and microRNA and long non-coding RNA (lncRNA) differential expression profiles that are distinctive of GEP-NETs, some of which are correlated with significant differences in clinical outcomes. Several translational studies have provided convincing data identifying potential prognostic biomarkers, and some of these have demonstrated preliminary success as serum biomarkers that can be used clinically. Nevertheless, there are many opportunities to further define the mechanisms by which these epigenetic modifications influence tumorigenesis, and this will provide better insight into their prognostic and therapeutic utility. Furthermore, these findings form the foundation for future studies evaluating the clinical efficacy of epigenetic modifications as prognostic biomarkers, as well as potential therapeutic targets.
Keywords: Epigenetics, Carcinoid, Neuroendocrine, MicroRNA, Methylation, Histone modifications, Chromatin remodeling, LncRNA
Core tip: Herein, we describe a review of the literature addressing known epigenetic changes which are thought to lead to the development of gastroenteropancreatic neuroendocrine tumors (GEP-NETs). Through a variety of scientific works, methylation patterns, chromatin remodeling alterations, and microRNA and long non-coding RNA differential expression profiles have been identified and in many cases correlated with GEP-NET malignancy and clinical outcomes. This overview shows the strong foundation which exists and underlines the importance of future work to evaluate the clinical efficacy of epigenetic modifications as prognostic biomarkers, as well as potential therapeutic targets.
INTRODUCTION
Overview of gastroenteropancreatic neuroendocrine tumors
Gastroenteropancreatic neuroendocrine tumors (GEP-NETs) are a heterogeneous group of rare tumors thought to arise from the malignant transformation of neuroendocrine cells located in the digestive tract. The incidence of GEP-NETs has risen from 1.0 per 100000 in 1977 to 3.7 per 100000 in 2007, with site-specific tumor incidence and behavior varying widely[1]. These differences may have roots in genomic and epigenetic differences, but the rarity of GEP-NETs has limited the progress of large-scale comprehensive analyses that could provide definitive insight into oncogenic molecular mechanisms. Genetic alterations associated with familial inherited syndromes have been well defined, such as in the multiple endocrine neoplasia syndromes, von-Hippel Lindau syndrome, neurofibromatosis, and tuberosclerosis. On the other hand, the genetic profile of sporadic tumors, which are more common, is less clear as their tumorigenesis does not appear to be controlled by classic oncogenes such as P53, RB, or KRAS[2-6].
Recent studies have identified key molecular pathways, such as activation of the PI3K/Akt/mTOR cascade secondary to PTEN and TSC2 downregulation in sporadic pancreatic NETs[7], which has led to the clinical success of the targeted mTOR inhibitor everolimus for metastatic pancreatic NETs[8]. While everolimus has been shown to extend progression free survival in phase 3 trials, the lack of a complete response for all patients implies there are other factors contributing to malignant transformation, either by alteration of other signal transduction pathways or by dysregulation at the gene expression level. Further investigation into sporadic pancreatic NETs revealed genetic alterations in MEN1 and DAXX /ATRX (death-domain-associated protein/α-thalassemia-mental retardation syndrome X-linked), which impair histone methyltransferase activity and chromatin stabilization, respectively[9]. These findings provide a link between genomic alterations and their control of gene expression, and thus have sparked further investigation into the epigenetic regulation of neuroendocrine tumors.
Epigenetics is the study of heritable modifications that control gene expression without alterations to the gene’s DNA sequence itself[10]. It provides a deeper level of understanding phenotypic changes in the absence of alterations in classical genetics. This review summarizes the different types of epigenetic mechanisms that have been implicated in GEP-NET tumorigenesis, as well as key developments in the potential for epigenetic alterations to serve as biomarkers or therapeutic targets.
Epigenetic mechanisms
Table 1 describes the different types of epigenetic mechanisms. Perhaps the most well understood epigenetic alteration is DNA methylation, which occurs at the 5’-position of the cytosine ring within dinucleotide CpG islands typically located in a gene’s regulatory region[11]. Overall, methylation has a stabilizing effect on cellular function. In cancer development, methylation patterns are tissue- and chromosome-specific - hypomethylation has been associated with chromosomal instability, reactivation of transposable elements, and loss of imprinting, whereas hypermethylation is associated with inactivation of tumor-suppressor genes[12]. Methylation is regulated by DNA methyltransferase enzymes (DNMTs); cells with defects in DNMTs are also shown to have marked nuclear abnormalities[13]. DNMT inhibitors such as 5-azacytidine (AZA) and decitabine have been shown to effectively induce demethylation in pre-clinical and clinical trials[14].
Table 1.
Overview of epigenetic mechanisms
| Mechanism | Effect | Regulation | Ref. |
| DNA Methylation | DNMTs | ||
| Hypomethylation | Chromosomal instability, reactivation of transposable elements, loss of imprinting | [12] | |
| Hypermethylation | Inactivation of tumor suppressor genes | [12] | |
| Histone modification | HDACs, histone methyltransferases | ||
| Lysine acetylation | Transcription activation | [12] | |
| Methylation | Transcription activation or suppression | [12] | |
| Noncoding RNAs | Variable | ||
| LncRNAs | Pre-transcriptional regulation | [17] | |
| MiRNAs | Post-transcriptional binding to 3’-untranslated regions to inhibit translation or promote mRNA degradation | [18] |
DNMTs: DNA methyltransferase enzymes; HDACs: Histone acetyltransferases and deacetylases; LncRNA: Long non-coding RNA.
Another epigenetic change that is frequently seen is histone modification. Histone proteins are the main component of chromatin and form the nucleosome backbone for DNA packaging. Modifications in their structure contribute to the epigenetic regulation of gene expression, including acetylation, methylation, and phosphorylation. The exact regulatory effect is dependent on the type of chemical modification; for example, lysine acetylation is generally associated with transcriptional activation, whereas methylation can either suppress or activate transcription depending on amino acid (arginine vs lysine) and histone site[12]. Several enzymes are responsible for its regulation, including histone acetyltransferases and deacetylases (HDACs) as well as histone methyltransferases. Modification of DNA packaging components is crucial to chromatin remodeling, which alters between heterochromatin (tightly packaged, transcriptionally silent) and euchromatin (less condensed, transcriptionally active) states[15].
Finally, noncoding RNAs also play a role in epigenetic changes. Noncoding RNAs account for 98% of the transcribed genome, but do not undergo subsequent translation[16]. Instead, they act as pre- and post-translational modifiers, and have gained recent attention as having regulatory roles in both normal cellular development and oncogenesis. These include large RNAs (> 200 nucleotides) such as long noncoding RNAs (lncRNA), and small RNAs (< 200 nucleotides) such as microRNAs (miRNA)[15]. lncRNAs have a pre-transcriptional function by providing molecular scaffolds for chromatin regulators, and have been implicated as biomarkers for prostate, hepatocellular, and metastatic colorectal carcinomas[17,18]. There is also evidence of their involvement of gene regulation at the transcriptional, translational, and post-translational levels[18]. On the other hand, miRNAs appear to have a mainly post-transcriptional role by binding to complementary target sites of 3’-untranslated regions (UTRs) of messenger RNAs (mRNA), which then either inhibit translation or promote mRNA degradation. The over- and under-expression of various miRNAs have been implicated in nearly all solid tumors including breast, cervical, colorectal, lung, prostate, pancreatic, and thyroid cancers[19].
Major advancements in epigenetics
As the functional mechanisms of epigenetic regulation have become better understood, the association between these modifications and malignant transformation has fueled further investigation into the role of epigenetics in tumorigenesis and patient outcomes. Traditional methods of epigenomic analysis, such as bisulfite sequencing for DNA methylation and chromatin immunoprecipitation (ChIP) for analyzing chromatin modifications and DNA-protein interactions, provided the fundamental platform necessary for the development of more efficient and accurate techniques. These methods are becoming integrated with microarray and Next-Generation Sequencing (NGS) technology to provide even more comprehensive analyses; for example, NGS can be used to elucidate a tumor’s “methylome” at single-nucleotide resolution[20].
These technologies have enabled significant epigenomic discoveries in other tumors including myelodysplastic syndrome (MDS) and cutaneous T-cell lymphoma. While it is beyond the scope of this review to detail their comprehensive history, it is important to highlight several therapeutic breakthroughs that have had an impact on clinical practice. Specifically, there are currently four FDA-approved drugs that have an epigenetic mechanism of action: 5-azacytidine and decitabine (DNMT inhibitors), and suberoylanilide hydroxamic acid and romidepsin (HDAC inhibitors). The DNMT inhibitors have demonstrated significantly higher response rates and reduced risk of leukemic transformation in the treatment for high risk myelodysplastic syndrome[21,22], and the HDAC inhibitors have been shown to induce durable responses in patients with cutaneous T-cell lymphoma[23,24]. Additionally, there have been promising preliminary in vivo data using microRNAs to improve survival in hepatocellular carcinoma[25]. While this led to a multicenter Phase I study of a liposome-based miR-34 mimic for patients with advanced HCC (ClinicalTrials.gov identifier: NCT0182997), this trial was withdrawn for immune-related serious adverse events, thus highlighting the need for further research into developing safe epigenetic therapies.
Overall, these advancements have proven the clinical value of using epigenomic modifications not only as oncologic biomarkers, but as potential therapeutic targets as well. Accordingly, current studies are investigating the epigenetic mechanisms that may contribute to GEP-NET tumorigenesis, and hopefully these advancements will serve as the foundation for future therapeutic developments.
CURRENT STATUS OF EPIGENETICS IN NETS
Hypermethylation in NETs
Methylation profiles of candidate genes in GEP-NETs has been extensively studied. There are data to suggest that these profiles are uniquely different between pancreatic tumors (PNETs) and gastrointestinal tumors (GI-NETs). Chan et al[26] detected a significantly higher degree of promoter methylation of 14 candidate genes in 14 GI-NETs compared to 11 PNETs, including MGMT (25% vs 0%), THSB1 (44% vs 9%), P14 (44% vs 9%), INK4a/P16 (31% vs 9%) and RARb (25% vs 0%). While INK4a/P16 methylation was associated with GEP-NET liver metastasis, the small sample size of this study limited a comprehensive clinical outcomes analysis. A larger, more recent analysis by How-Kit et al[27] was able to distinguish between GEP-NET subtypes by evaluating the methylation status of 807 oncogenic genes in 60 tumors using the Illumina GoldenGate technology. This study found unique DNA methylation patterns on hierarchical clustering between small bowel NETs (SB-NETs) and PNETs, as well as between functional PNET subtypes (insulinoma, gastrinoma, non-functioning). The analysis also found that gastrinomas are characterized predominantly by hypomethylated genes including metalloproteinases (MMP1, MMP3, TIMP2, TIMP3) and genes of the serpin family (SERPINA5, SERPINB5), whereas insulinomas and non-functioning PNETs had mixed hyper- and hypo-methylation profiles. Lastly, they reported hypermethylation of tumor suppressors (SMARCB1, CASP8 and NBL1) and hypomethylation of oncogenes (IL2, MCF2 and MOS); gene ontology and network analysis integrating these results detected cellular growth, apoptosis, cellular movement, and cell-cell signaling as the main molecular and cellular functions affected by the differentially methylated gene profiles. Overall, these results describe how epigenetic modifications differ between GEP-NET location and functionality, and have begun to shed light as to which genes and signaling pathways may be responsible for their differentiation.
The promoter regions of several individual genes in GEP-NETs warrant discussion, including RASSF1A, INK4a/P16, TIMP3, MGMT, IGF2, UCHL1, among others (Table 2). Although this section will describe the methylation patterns of individual genes’ promoters, it is important to note that studies have correlated hypermethylation of multiple tumor suppressor genes with more advanced disease[28,29]. For instance, in GEP-NETs, Arnold et al[30] found that a high degree of methylation across CpG sites of multiple tumor suppressors - known as CpG island methylator phenotype (CIMP) positivity - is correlated with worse clinical outcomes in a mixed cohort of 71 well-differentiated tumors of the foregut and midgut. Specifically, CIMP positivity was found in 74% of foregut and midgut tumors and was associated with higher grade tumors (ki67 > 10%), whereas CIMP negativity had a non-significant trend towards better overall survival (7 years vs 4 years)[30]. Additionally, methylation of two or more tumor suppressor genes has been shown to be associated with liver metastases in a separate GEP-NET cohort[31].
Table 2.
Methylation profiles in neuroendocrine tumors
| Modification | Gene | Gene function | Clinical effect | Tumor | Ref. |
| Hypermethylation (Inactivation) | |||||
| RASSF1 | Induces cell cycle arrest | Correlated with malignancy, levels highest in metastases | PNET > GI-NET | [32-34] | |
| INK4a/p16 | Induces cell cycle arrest and apoptosis | Decreased 5-yr survival, liver metastases | PNET, gastrinoma | [38,39] | |
| MGMT | DNA repair | Improved response to temozolomide | PNET | [32] | |
| TIMP-3 | Inhibits metalloproteinases | Correlated with metastases | PNET | [44] | |
| UCHL-1 | Post-translational modifier, de-ubiquitinates proteins marked for lysosomal degradation | Correlated with metastases | GEP-NET | [45-47] | |
| IGF2 | Chromatin packaging | Specific for insulinomas, increased stage | Insulinoma | [51] | |
| MLH1 | DNA repair | Correlated with malignancy | PNET, insulinoma | [52] | |
| Global hypomethylation | |||||
| LINE-1 | Repeating long interspersed nucleotide elements | Correlated with malignancy and lymph node metastases | Ileal NET > GEP-NET | [53-55] | |
| Alu | Repeating long interspersed nucleotide elements | Correlated with malignancy | GEP-NET | [54] |
GEP-NETs: Gastroenteropancreatic neuroendocrine tumors; PNETs: Pancreatic tumors; GI-NETs: Gastrointestinal tumors.
Multiple groups have shown clinically relevant methylation patterns in single gene promoter regions in GEP-NETs specifically. One of the initial large cohorts evaluating the methylation patterns of GEP-NETs analyzed 11 selected tumor suppressors in 48 PNETs, 75% of which were well-differentiated[32]. RASSF1 was hypermethylated in 75% of cases, followed by INK4a/p16 (40%), O6-MGMT (40%), RAR-β (25%), and hMLH1 (23%). Interestingly, the methylation patterns were largely preserved when comparing metastatic tumor deposits to the matched primary tumor.
Ras-association domain gene family 1 (RASSF1) is a tumor suppressor that induces cell cycle arrest whose promoter region has been well-studied and is hypermethylated in 57% of GEP-NETs[31]. It appears that there are differences in RASSF1 hypermethylation depending on the site of origin of the GEP-NET as 75%-100% of PNETs[32-34], 33% of gastric NETs[34], and 0%-61% of SB-NETs[34,35] were found to have RASSF1 hypermethylation. The degree of methylation in PNETs has been found to be higher than adjacent normal pancreas and is expectedly inversely correlated with its level of gene expression[36]. Slightly counterintuitive to the statistically similar methylation profiles found between matched metastatic vs primary PNET cohorts described above, the degree of RASSF1 methylation has been reported to be higher in lymphatic metastases in a GEP-NET cohort[31] and distant metastatic deposits in a SB-NET cohort[35,37] when compared to primary tumors. Nonetheless, while RASSF1 promoter methylation itself has not been correlated with changes in survival, its gene expression on an mRNA level is correlated with survival in one cohort of SB-NETs[37].
The methylation status of INK4a/p16 (also known as cyclin-dependent kinase inhibitor 2A or CDKN2A) is another well-studied area in GEP-NET epigenetics. Methylation at three or more tumor suppressor genes, specifically at the INK4a/p16 locus, is predictive of decreased 5-year survival as well as tumor recurrence within 2 years of operation on multivariate analysis[32]. Additional studies have documented that methylation of the INK4a/p16 locus occurs in up to 58% of PNETs and gastrinomas[38,39] but less than 15% of benign insulinomas[40], and is associated with liver metastasis in GEP-NETs[26,31] and poorer overall survival rates in poorly differentiated colorectal NETs[41]. The gene is located on chromosome 9p21 and is known to induce cell cycle arrest and apoptosis. Its inactivation by homozygous deletion is well-documented in many cancers; however, hypermethylation of its promoter has also been associated with colorectal, lung, breast, renal, and prostate cancers[42]. Despite its prevalence and association with cell cycle regulation, further clinical association is unclear as some studies have found the degree of INK4a/p16 promoter methylation to be independent of disease stage in gastrinomas[39]. Nevertheless, since INK4a/p16 promoter hypermethylation is a prevalent finding in PNETs, it still is thought be a driver for early tumorigenesis and should have continued focus as a major epigenetic modification in GEP-NETs.
The silencing of several other individual loci by promoter hypermethylation may have important implications in GEP-NET tumorigenesis. MGMT is a DNA methyltransferase that serves an important role in DNA repair. The MGMT promoter is methylated in up to 40% of PNETs[32]. Several studies have correlated hypermethylation of the MGMT promoter (and subsequent protein loss) with improved response to temozolomide, an oral chemotherapeutic agent, specifically longer progression-free and overall survival[43]. TIMP-3 is a tumor suppressor involved in the inhibition of proteolytic activity of the matrix metalloproteinases; its decreased expression allows MMPs to contribute to tumor growth, angiogenesis and invasion. Hypermethylation of its promoter occurs in 44% of sporadic PNETs, and appears to be more prevalent in metastatic tumors[44]. UCHL-1 is a post-translational modifier that de-ubiquitinates proteins otherwise destined for lysosomal degradation; it is a known tumor suppressor in multiple tumor types and has been shown to stabilize p53 levels and induce cell cycle arrest[45,46]. In a cohort of well-differentiated GEP-NETs, loss of UCHL1 expression by CpG promoter hypermethylation has been shown to be associated with metastatic GEP-NETs in well-differentiated tumors[47]. Additionally, hypermethylation of CTNNB1 (beta-catenin), WIF1 (wnt inhibitory factor), TCEB3C (elongin A3), and SEMA3F (semaphorin 3F) have been identified in SB-NETs[35,37,48,49], with CTNNB1 and SEMA3F hypermethylation also having been identified in metastatic tumors[35,49]. Most recently, hypermethylation of the tumor suppressor homeobox-only protein has been associated with worse recurrence free survival in a cohort of 36 PNETs[50].
To further illustrate the complexity of NETs, insulinomas have a unique methylation profile compared to other sporadic NETs. In a cohort of insulinomas, gastrinomas, non-functioning pancreatic NETs, and SB-NETs, hypermethylation of the CpG-rich differentially methylated region 2 (DMR2) region of the imprinted gene IGF2 was specific for insulinomas, which results in inhibition of chromatin packaging and thus allows continued expression of IGF2. Furthermore, there was a correlation between decreasing degree of IGF2 methylation with increasing stage of malignancy as defined by the WHO classification across the entire cohort[51]. In a separate study, insulinomas had a higher rate of MLH1 promoter hypermethylation, which has a regulatory role in DNA repair; decreased gene expression of MLH1 was further correlated with tumor malignancy[52].
Contrary to many reports of hypermethylated genes described above, global hypomethylation of GEP-NETs has been reported as well. Global hypomethylation is assessed by analyzing the methylation status of long interspersed nucleotide elements (LINE)-1 and Alu, which are heavily methylated repeating elements that comprise 15% and 10% of the human genome, respectively. A recent meta-analysis concluded that global hypomethylation is associated with a worse prognosis in colorectal tumors, melanoma, gastric cancer, hepatocellular carcinoma, amongst others[53]. Within GEP-NETs, the significance and incidence of hypomethylation seems to vary by tumor type. Choi et al[54] compared a cohort of 35 well-differentiated GEP-NETs to normal tissue, finding that global hypomethylation was found more frequently in tumor samples than in normal tissue. This same study showed that relative tumor hypomethylation of LINE-1 was more prevalent in ileal carcinoid tumors than in non-ileal carcinoid tumors and PNETs, and was also more prevalent in tumors with lymph node metastasis, chromosome 18 loss, and RASSF1 methylation. Interestingly, Alu methylation was inversely correlated with methylation of MGMT[54]. There were no survival differences between degrees of LINE-1 and Alu methylation, and more importantly, methylation of LINE-1 and Alu did not appear to be a sensitive marker for generalized CpG methylation of multiple genes of interest (e.g., MGMT).
In contrast to the Choi study[54] cited above, a study of 58 GEP-NETs of variable grades showed that LINE-1 hypomethylation was highest in PNETs compared to SB-NETs, and is correlated with worse tumor staging in PNETs[55]. The most convincing data regarding global hypomethylation in PNETs was described by Stefanoli et al[56] who performed quantitative bisulfite pyrosequencing to determine methylation status on 56 PNETs and 8 normal pancreas samples. LINE-1 methylation was significantly lower in PNETs compared to normal tissue. Additionally, in tumor samples, LINE-1 methylation was significantly lower in stage III and IV disease as compared to stage I and II disease (57.4% vs 61.7%, respectively, P = 0.002). Furthermore, PNETs with less than 58% LINE-1 methylation were correlated with worse overall survival (P < 0.0001). This study also analyzed the methylation status of 33 tumor suppressor genes, and identified three PNET clusters each with increasing frequency of gene-specific hypermethylation. The PNET cluster with the highest degree of methylation was associated with stage IV disease (P = 0.04) and poor overall survival (P = 0.004), and implicated ten tumor suppressors: DAPK1, TIMP3, PAX5, HIC1, CADM1, PYCARD, ESR1, VHL, RARB and WT1. Interestingly, most of the LINE-1 hypomethylated PNETs were distributed within the clusters containing a higher frequency of hypermethylated tumor suppressors, thus highlighting that global hypomethylation and gene-specific hypermethylation may be found concurrently in aggressive PNETs.
Chromatin remodeling in NETs
One of the initial investigations into chromatin remodeling in GEP-NETs was by Jiao et al[9]. The authors performed exome sequencing of 10 sporadic PNETs and identified mutations in several genes involved in chromatin remodeling that were subsequently confirmed in a 58-sample validation cohort. The top three mutated genes, MEN1 (44%), DAXX (25%), and ATRX (18%) are all recognized to have a prominent role in chromatin remodeling, and may even provide a link between DNA methylation and histone modifications. Interestingly, in this study, patients with mutations in MEN1, DAXX/ATRX, or a combination of both had improved survival when compared to those without any mutation, particularly in those with metastatic disease[9]. While the clinical implications of MEN1, DAXX, and ATRX mutations vary in subsequent studies (discussed below), these findings prompted further investigation into the genes’ downstream mechanistic effects. MEN1 encodes menin, a nuclear scaffold protein that serves as a transcriptional regulator by remodeling chromatin and is also an essential component of a histone methyltransferase complex containing MLL2 and Ash2L[57]. ATRX is a chromatin remodeling protein that interacts with DNA methyltransferase 3A and 3L - this promotes DNA methylation of histone H3K4 when it is unmodified, which can result in telomere/chromatin changes as well as transcriptional activation[58]. DAXX is an H3.3-specific histone chaperone, which interacts with ATRX for H3.3 incorporation and heterochromatin assembly at telomeres[59,60]. Ultimately, these complex interactions between DNA methylation and chromatin remodeling are essential for maintaining histone methylation patterns in newly replicated chromatin and preserving the stability of gene expression[58].
Further work has defined the role of these mutations in the cellular behavior of GEP-NETs. The defining feature of telomerase-independent telomere maintenance associated with DAXX/ATRX gene mutations is alternative lengthening of telomeres (ALT). This phenomenon can be detected by telomere-specific fluorescence in situ hybridization (FISH) and was seen in 25/41 (61%) of PNET tumors in one cohort[61]. All PNETs with DAXX/ATRX mutations (n = 19) had positive ALT during FISH analysis in this study, and tumors without mutations that were ALT positive (n = 6) had lost nuclear expression of DAXX/ATRX. Other studies in GEP-NETs have reported similar findings - specifically that the ALT positive phenotype is associated with loss of ATRX or DAXX expression, particularly in PNETs[62]. Comparable findings have been reported in other tumors as well, including pediatric glioblastomas, where 44% of patients harbor a mutation in the DAXX/ATRX pathway resulting in ALT positivity[63].
Several subsequent studies have corroborated the above DAXX/ATRX mutation findings in GEP-NETs. de Wilde et al[64] evaluated 109 well-differentiated PNETs from 28 patients with MEN-1 syndrome and found that expression of DAXX/ATRX was normal and there was no ALT positivity in tumors < 0.5 cm. However, 25% of tumors > 3 cm (6% of tumors > 0.5 cm) had lost expression; in each of these tumors, there was ALT positivity. Additionally, available lymph node metastases (2/3) had the same phenotype, and tumor grading was more likely to be WHO grade 2. The authors concluded that since loss of DAXX/ATRX expression was found only in larger, more aggressive tumors, DAXX/ATRX defects might be a later event in MEN-1 syndrome PNET tumorigenesis. Unfortunately, the cohort was too small (n = 12 for size > 3 cm) to perform other translational analyses, such as correlating DAXX/ATRX expression with survival or other clinicopathologic features.
In addition to these findings, Marinoni et al[65] evaluated DAXX/ATRX expression in a derivation cohort of 61 well-differentiated PNET tumors and validation cohort of 70 tumors. Their analysis showed that loss of DAXX or ATRX protein occurred in 42% of the cohort and was correlated with ALT positivity. Furthermore, DAXX/ATRX loss of expression was also correlated with chromosome instability, which has previously been associated with poor outcomes in PNETs. Both cohorts in this study also demonstrated that a loss of DAXX or ATRX correlated significantly with a decreased relapse-free survival; only the derivation cohort detected a correlation with decreased tumor-specific survival. These findings are contrary to the initial report of DAXX/ATRX mutations described above, which appeared to be correlated with improved prognosis[9]. These discrepancies are likely secondary to the initial report’s cohort consisting of all metastatic PNETs, whereas only 18% of the Marinoni et al[65] tumors were metastatic. Therefore, it is more important to appreciate that DAXX/ATRX loss of expression identifies only a subset of PNETs, and might have a different role in localized and distant tumor stages.
Notably, another study by Pipinikas et al[66] found similar clinical outcomes as Marinoni et al[65] - PNETs that were DAXX-negative (i.e., DAXX-deficient) had the worst progression-free survival at five years (DAXX-negative: 16%, P < 0.001; ATRX-negative: 52%, P = 0.15; and DAXX/ATRX-positive: 85%). There was a significantly higher proportion of DAXX/ATRX loss in intermediate grade tumors compared to low-grade tumors (68% vs 27%, P = 0.008)[66]. This study also bridged the relationship between DAXX/ATRX, DNA methylation, and histone modifications. Using genome-wide methylation analysis, they found a significantly higher number of methylation-variable positions in DAXX-negative vs ATRX/DAXX-positive tumors compared to ATRX-negative vs ATRX/DAXX-positive tumors. This highlights the functional importance of DAXX (a H3.3-specific histone chaperone) as a driver for genome-wide methylation changes. Lastly, they found a higher incidence of copy number variations in DAXX/ATRX-negative tumors compared to DAXX/ATRX-positive tumors.
These data were definitively confirmed in the largest series evaluating ALT positivity and DAXX/ATRX loss in PNETs. Singhi et al[67] performed telomere-specific FISH and DAXX/ATRX immunohistochemistry on 321 patients with resected PNETs, as well as 191 paired distant metastases from 52 patients. ALT-positivity and DAXX/ATRX-loss were present in 31% and 26% of resected PNETs, respectively, and associated with greater tumor size, worse WHO grade, lymph node metastasis, and distant metastasis. The rates of ALT-positivity and DAXX/ATRX-loss were higher in distant metastases (67% and 52%, respectively), and in fact, distant metastases only occurred in the setting of ALT-positive and DAXX/ATRX-negative primary tumors. Comparing long-term outcomes to wild-type PNETs, ALT-positive patients had worse 5-year disease-free survival (40% vs 96%, P < 0.001) and 10-year disease-specific survival (50% vs 89%, P < 0.001). Similar to the Marioni and Pipinikis studies, ALT was an independent prognostic factor for disease free survival (HR = 7.1, P < 0.001), but not for disease-specific survival (similar results were found when substituting DAXX/ATRX-loss for ALT positivity). These studies provide strong evidence suggesting that ALT-positivity and DAXX/ATRX-loss are associated with aggressive clinicopathologic features and worse disease-free survival, but not necessarily disease-specific survival.
Another gene of interest in chromatin remodeling of GEP-NETs is NAP1L1 (nucleosome assembly protein 1-like 1), whose function is thought to be in nucleosome assembly and exchange of histone dimers, which may contribute to the regulation of cell proliferation as a transcriptional modifier[68,69]. In a cohort of 43 PNETs, NAP1L1 was significantly overexpressed in metastasis and inversely correlated with expression of p57Kip2[70]. This inverse correlation was confirmed in vitro by silencing NAP1L1 in the BON cell line, which correlated with a subsequent increase in p57Kip2 mRNA and protein levels. Furthermore, there was decreased signaling of the mammalian target of rapamycin (mTOR) pathway, which lead to less aggressive phenotypes in vitro and in vivo. Most importantly, though, the p57Kip2 promoter in NAP1L1-silenced BON cells was significantly less methylated, and using ChIP analysis, NAP1L1-bound DNA fragments were found to include the p57Kip2 promoter. The authors concluded that although NAP1-like proteins typically control gene expression via histone H3 acetylation[71], NAP1L1 may have the ability to regulate p57Kip2 through promoter methylation in GEP-NETs. This finding highlights the overlapping complexity of epigenetic modifications, and while these recent studies have provided a major step in advancing our understanding of chromatin remodelers as PNET prognostic indicators, the mechanisms by which these mutations affect survival on a cellular level remain to be fully elucidated.
MicroRNAs in NETs
Recent studies have begun to focus on global miRNA profiling of GEP-NETs, and several of these are reviewed here. miRNAs primarily play a post-transcriptional role by either inhibiting translation of mRNA or promoting mRNA degradation. Many different specific miRNAs have been identified with a breadth of clinical implications in GEP-NETs (Figure 1, Supplementary Table 1). While the prognostic and therapeutic implications of these changes are still controversial, these findings provide a strong foundation for future investigations.
Figure 1.
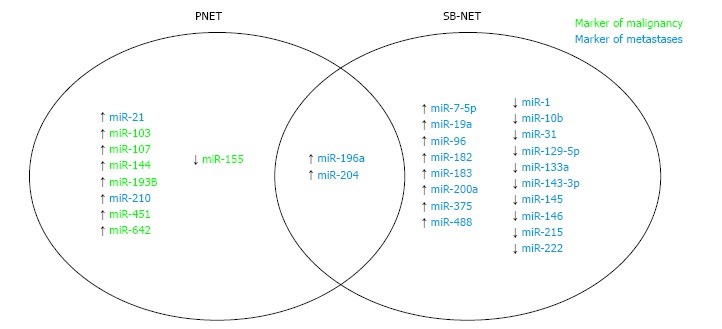
MicroRNA in neuroendocrine tumors. PNET: Pancreatic tumors; SB-NETs: Small bowe neuroendocrine tumors.
Roldo et al[72] in one of the first GEP-NET miRNA studies, evaluated 12 insulinomas, 28 nonfunctioning PNETs, and four acinar carcinomas in comparison to normal pancreatic tissue. Their analysis demonstrated that overexpression of miR-103 and miR-107 and underexpression of miR-155 could discriminate the whole tumor cohort from normal tissue. Interestingly, overexpression of miR-204 was specific to insulinomas and correlated with immunohistochemical staining of insulin better than insulin mRNA expression. Lastly, miR-21 was correlated with Ki67 > 2% as well as liver metastasis. While these data do not necessarily explain the downstream mechanistic effects of these expression differences, this initial report gave insight into the miRNA expression differences of PNETs - in addition to those mentioned above, ten microRNAs (miR-125a, miR-99a, miR-99b, miR-125b-1, miR-342, miR-130a, miR-132, miR-129-2, miR-125b-2) were able to differentiate PNETs from normal tissue and acinar tumors independently, and the miRNA profiles between nonfunctioning tumors and insulinomas were noted to be indistinguishable.
miRNA profiling of PNETs has been further analyzed by Thorns et al[73] in a cohort including PNET tumor samples of various grades, normal tissue, as well as serum. This study found that expression of miR-642 correlated with Ki-67 score, and miR-210 correlated with metastatic disease. Interestingly, 13 miRNAs were more abundant in the serum of patients with PNETs compared to normal subjects; specifically, miR-193b was a marker for PNETs in both tumor and serum samples[73].
Investigation into miRNA profiling in SB-NETs first gained traction in a study by Ruebel et al[74] evaluating the differential expression of 95 miRNAs in eight matched primary and metastatic ileal NETs. There was downregulation of miR-133a, miR-145, miR-146, miR-222, and miR-10b in all metastatic samples compared to primary tumors, and up-regulation of miR-183, miR-488 and miR-19a in six of eight metastatic tumors compared to the primary. miR-133a was significantly downregulated in a validation cohort of six additional cases. This study presented an initial understanding of miRNA expression differences in SB-NETs. However, perhaps one of the best designed miRNA profiling studies in GEP-NETs was performed in a cohort of five primary SB-NETs, five lymph node metastases, and five liver metastases[75]. In this cohort, the Affymetrix Genechip miRNA array detected 15 upregulated and 18 downregulated miRNAs when comparing mesenteric metastases to primary tumors. These 33 miRNAs were further differentially expressed when comparing liver metastases to lymph node metastases; in fact, 14 of 15 upregulated miRNAs had a further increase in expression in the liver metastases. Furthermore, nine miRNAs of interest were validated in a cohort of three primary tumors, three lymph node metastases, and three liver metastases - miR-96, miR-182, miR-183, miR-196a, and miR-200a were confirmed to be upregulated in metastases, and miR-31, miR-129-5p, miR-133a, and miR-215 were confirmed to be downregulated in metastases. These results provide insight into the differential expression of miRNAs that may be implicated in disease progression.
A subsequent comprehensive analysis of 90 patients by Miller et al[76] profiled the global miRNAome of SB-NETs and their metastases. The authors identified 39 differentially expressed miRNAs between primary tumors and normal tissue. Interestingly, they found significant overlap between upregulated miRNAs in both primary SB-NETs and their metastases compared to their respective normal tissues. While the most differentially expressed microRNAs were upregulation of miR-204, miR-7-5p, and miR-375, the profile included microRNAs identified in the above studies, specifically miR-31, miR-96, miR-129-5p, miR-182, miR-196a, miR-200a and miR-215. When comparing lymph node and liver metastases to their matched primary tumors, several significantly downregulated microRNAs were identified, including miR-1, miR-133a, miR-143-3p, and miR-145-5p - again, several of these were also identified in the above studies. The authors then analyzed the target genes of miR-1 and miR-143 in the existing GSE27162 dataset. They identified both miRNAs are predicted to target the NUAK2 and FOSB oncogenes, which are significantly upregulated in SB-NET lymph node metastasis. The inverse correlation identified in this bioinformatics analysis was confirmed in vitro, indicating that this miRNA/mRNA interaction may be a crucial step in metastatic progression of SB-NETs.
miR-196a has been identified in several studies, and has been further investigated by Li et al[77]. The authors used miRNA target prediction software, which identified HOXA9, HOXB7, LRP4, and RSPO2 as potential downstream targets of miR-196a. The transcripts of these targets were found to be downregulated in tumor and serum samples of patients with SB-NETs compared to healthy donors. More interestingly, when miR-196a was silenced in the SB-NET cell line CNDT2.5, all four targets’ gene and protein expression significantly increased. Further downstream targets of these genes, including those in the WNT signaling pathway, were also expectedly upregulated. This did not result in any phenotypic changes in cell growth; however, the only in vitro assay performed was an MTT (cell viability) assay.
miR-196a has also been implicated in PNET clinicopathologic outcomes in a study by Lee et al[78]. The authors analyzed 37 resected PNETs and found that increased expression of miR-196a was significantly associated with advanced pathologic T stage (50% vs 7%), N stage (50% vs 4%), higher mitotic counts (60% vs 4%), and greater ki-67 index (60% vs 22%). Furthermore, patients with increased expression had a higher risk for recurrence (hazard ratio: 16.3, 95% confidence interval: 1.7 to 154, P = 0.015), worse disease-free survival (P < 0.001), and worse overall survival (P = 0.046). These findings demonstrate that miR-196a may have prognostic utility in both pancreatic and small bowel NETs.
miRNAs may also play a role in the development of insulinomas. Jiang et al identified 114 differentially expressed microRNAs when comparing four insulinomas to 4 normal pancreatic islet cells - 28 of these belonged to three miRNA families that localized to the epigenetically-regulated imprinted chromosome 14q32[79]. The most significant differentially expressed cluster, miR-144/451, was validated in 25 insulinomas and 8 normal pancreatic islets. The authors further demonstrated that in mouse pancreatic beta-cells, overexpression of miR-144/451 increased cell viability and proliferation - miR-144 was found to inhibit the tumor suppressor PTEN leading to increased AKT pathway activation, while miR-451 directly downregulated p19, a cell-cycle regulator. Further investigation into the downstream effects of the other differentially expressed microRNAs in this study could provide even more insight into the tumorigenesis of insulinomas.
Lastly, there is increasing evidence that serum miRNA expression can serve as a biomarker for patients with SB-NETs. The panel of nine miRNAs described above (miR-96, miR-182, miR-183, miR-196a, miR-200a, miR-31, miR-129-5p, miR-133a, and miR-215) was evaluated in the serum of patients with SB-NETs and compared to healthy volunteers by Li et al[80]. The study found that miR-96, miR-182, miR-196a, and miR-200a were upregulated and miR-31, miR-129-5p, miR-133a, and miR-215 were downregulated when comparing patients with lymph node metastases compared to healthy volunteers - these findings are largely congruent with the authors’ original study in tumor tissue[75], although serum expression of miR-183 did not differ between groups. The study further shows that patients treated with somatostatin analogues (SSA) have even further upregulation of miR-96, miR-182, miR-183, miR-196a, and miR-200a at all stages of disease (i.e., in primary tumors, lymph node and liver metastases), with the exception that miR-200a is not significantly upregulated in patients with liver metastases. Therefore, these results suggest that there are certain serum miRNA profiles that are not only detectable in patients with SB-NETs, but also have a specific response to somatostatin analogues.
lncRNA in NETs
The current knowledge regarding the pathogenesis and prognostic implications of lncRNAs is still in its preliminary stages. However, one study thus far has described a potentially relevant lncRNA in GEP-NETs[81]. Modali et al[82] specifically investigated the relationship between menin and MEG3, a monoallelic, maternally-expressed lncRNA whose loss of expression has been described in several tumors. The authors demonstrated that in a mouse insulinoma cell line, overexpression of menin significantly increased MEG3 expression by histone-H3 lysine-4 trimethylation (a marker of transcriptional activation) and CpG hypomethylation at the Meg3 promoter CRE site, which allows binding of the transcription factor cAMP response element-binding protein. Furthermore, MEG3 overexpression was shown to reduce cell proliferation rates, induce cell cycle arrest, and downregulate c-Met proto-oncogene expression. This inverse relationship of menin-Meg3 to c-Met was confirmed in an in vivo PNET mouse model, as well as in human MEN1 PNET and sporadic insulinoma samples. In all of these, MEG3 expression was decreased, c-Met expression was elevated, and the MEG3 promoter was hypermethylated in tumors compared to normal islets. This initial study of lncRNA in PNETs has highlighted the potential importance of lncRNA epigenetic regulation in neuroendocrine tumor development.
CLINICAL APPLICATIONS AND FUTURE DIRECTIONS
Several studies discussed above have provided data identifying potential prognostic biomarkers, and some have even hypothesized clinical translation by evaluating their utility as serum biomarkers. Continued investigation is required to identify the mechanisms by which these epigenetic modifications influence tumorigenesis, providing better insight into their prognostic and therapeutic utility.
There has been limited success in developing effective epigenetic therapies, as most of these studies have only evaluated drug treatments in GEP-NET cell lines such as BON (pancreas) and CNDT2.5 (midgut). However, they have shown initial promising effects by demonstrating attenuation of aggressive GEP-NET tumor phenotypes with drug treatments. For instance, overexpression of DNA methyltransferases 1, 3a, and 3b have been implicated in GEP-NET tumorigenesis[83]. Accordingly, AZA therapy (i.e., DNMT inhibition) in BON and CDNT2.5 cells has been shown to reduce cell proliferation, induce cell cycle arrest at the G2 to M transition, and decrease expression of the neuroendocrine markers chromogranin A (CgA) and neuron-specific enolase[84]. Similarly, HDAC inhibition with trichostatin A (TSA), sodium butyrate (NaB), and MS-275 in BON cells inhibited cell proliferation, induced apoptosis via caspase-3 activation and Bcl-2 downregulation, and promoted cell cycle arrest at the G1-S transition[85]. Furthermore, combination treatment of BON cells with both valproic acid (an HDAC inhibitor) and lithium has been shown to suppress CgA expression, upregulate Notch1 signaling, and inhibit glycogen synthase kinase-3β activity, thereby increasing cellular proliferation[86]. Currently, no targeted miRNA therapies have been attempted.
Despite these in vitro findings with DNMT and HDAC inhibitors, clinical trials have been unsuccessful. Only one published phase 2 clinical trial of any epigenetic therapy for neuroendocrine tumors has been attempted[87]. The HDAC inhibitor depsipeptide was administered to 15 patients with metastatic GEP-NET and lung neuroendocrine tumors, but the study was terminated prematurely due to a high rate of adverse cardiac events (ventricular tachycardia, prolonged QTc, sudden death) precluding observation of an objective response rate. More studies evaluating the anti-tumor pharmacology and adverse events in in vitro and in vivo experiments are necessary before future attempts at clinical trials are indicated.
CONCLUSION
The field of epigenetics is constantly evolving and major strides have been made to help define its relevance in GEP-NET tumorigenesis. The current body of literature suggests there are methylation patterns, chromatin remodeling alterations, and microRNA and lncRNA differential expression profiles that are distinctive of GEP-NETs, some of which are correlated with poorer clinical outcomes. However, given the variety of GEP-NETs, many studies’ results are confounded by heterogeneity of tumor cohorts. Thus, larger studies with more stringent inclusion criteria are required to evaluate the utility of epigenetic modifications as prognostic biomarkers as well as potential therapeutic targets.
Footnotes
Conflict-of-interest statement: There are no conflicts of interest to report.
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: February 27, 2017
First decision: June 12, 2017
Article in press: August 4, 2017
P- Reviewer: Cerwenka H, Chetty R S- Editor: Kong JX L- Editor: A E- Editor: Lu YJ
Contributor Information
Brendan M Finnerty, Department of Surgery, New York Presbyterian Hospital, Weill Cornell Medicine, New York, NY 10065, United States. bmf9002@nyp.org.
Katherine D Gray, Department of Surgery, New York Presbyterian Hospital, Weill Cornell Medicine, New York, NY 10065, United States.
Maureen D Moore, Department of Surgery, New York Presbyterian Hospital, Weill Cornell Medicine, New York, NY 10065, United States.
Rasa Zarnegar, Department of Surgery, New York Presbyterian Hospital, Weill Cornell Medicine, New York, NY 10065, United States.
Thomas J Fahey III, Department of Surgery, New York Presbyterian Hospital, Weill Cornell Medicine, New York, NY 10065, United States.
References
- 1.Lawrence B, Gustafsson BI, Chan A, Svejda B, Kidd M, Modlin IM. The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011;40:1–18, vii. doi: 10.1016/j.ecl.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 2.Öberg K. The genetics of neuroendocrine tumors. Semin Oncol. 2013;40:37–44. doi: 10.1053/j.seminoncol.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Williams GT. Endocrine tumours of the gastrointestinal tract-selected topics. Histopathology. 2007;50:30–41. doi: 10.1111/j.1365-2559.2006.02570.x. [DOI] [PubMed] [Google Scholar]
- 4.Yoshimoto K, Iwahana H, Fukuda A, Sano T, Saito S, Itakura M. Role of p53 mutations in endocrine tumorigenesis: mutation detection by polymerase chain reaction-single strand conformation polymorphism. Cancer Res. 1992;52:5061–5064. [PubMed] [Google Scholar]
- 5.Chung DC, Smith AP, Louis DN, Graeme-Cook F, Warshaw AL, Arnold A. Analysis of the retinoblastoma tumour suppressor gene in pancreatic endocrine tumours. Clin Endocrinol (Oxf) 1997;47:523–528. doi: 10.1046/j.1365-2265.1997.2861110.x. [DOI] [PubMed] [Google Scholar]
- 6.Yashiro T, Fulton N, Hara H, Yasuda K, Montag A, Yashiro N, Straus F 2nd, Ito K, Aiyoshi Y, Kaplan EL. Comparison of mutations of ras oncogene in human pancreatic exocrine and endocrine tumors. Surgery. 1993;114:758–763; discussion 763-764. [PubMed] [Google Scholar]
- 7.Missiaglia E, Dalai I, Barbi S, Beghelli S, Falconi M, della Peruta M, Piemonti L, Capurso G, Di Florio A, delle Fave G, et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28:245–255. doi: 10.1200/JCO.2008.21.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, Tomassetti P, Pavel ME, Hoosen S, Haas T, Lincy J, Lebwohl D, Öberg K; RAD001 in Advanced Neuroendocrine Tumors, Third Trial (RADIANT-3) Study Group. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–523. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holliday R. The inheritance of epigenetic defects. Science. 1987;238:163–170. doi: 10.1126/science.3310230. [DOI] [PubMed] [Google Scholar]
- 11.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 12.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 13.Espada J, Ballestar E, Santoro R, Fraga MF, Villar-Garea A, Németh A, Lopez-Serra L, Ropero S, Aranda A, Orozco H, et al. Epigenetic disruption of ribosomal RNA genes and nucleolar architecture in DNA methyltransferase 1 (Dnmt1) deficient cells. Nucleic Acids Res. 2007;35:2191–2198. doi: 10.1093/nar/gkm118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rius M, Lyko F. Epigenetic cancer therapy: rationales, targets and drugs. Oncogene. 2012;31:4257–4265. doi: 10.1038/onc.2011.601. [DOI] [PubMed] [Google Scholar]
- 15.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 16.Amaral PP, Dinger ME, Mercer TR, Mattick JS. The eukaryotic genome as an RNA machine. Science. 2008;319:1787–1789. doi: 10.1126/science.1155472. [DOI] [PubMed] [Google Scholar]
- 17.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yarmishyn AA, Kurochkin IV. Long noncoding RNAs: a potential novel class of cancer biomarkers. Front Genet. 2015;6:145. doi: 10.3389/fgene.2015.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee YS, Dutta A. MicroRNAs in cancer. Annu Rev Pathol. 2009;4:199–227. doi: 10.1146/annurev.pathol.4.110807.092222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tollefsbol TO. Advances in epigenetic technology. Methods Mol Biol. 2011;791:1–10. doi: 10.1007/978-1-61779-316-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, Stone RM, Nelson D, Powell BL, DeCastro CM, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429–2440. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- 22.Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, Klimek V, Slack J, de Castro C, Ravandi F, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 23.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 24.Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, Zain J, Prince HM, Leonard JP, Geskin LJ, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27:5410–5417. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov. 2013;12:847–865. doi: 10.1038/nrd4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan AO, Kim SG, Bedeir A, Issa JP, Hamilton SR, Rashid A. CpG island methylation in carcinoid and pancreatic endocrine tumors. Oncogene. 2003;22:924–934. doi: 10.1038/sj.onc.1206123. [DOI] [PubMed] [Google Scholar]
- 27.How-Kit A, Dejeux E, Dousset B, Renault V, Baudry M, Terris B, Tost J. DNA methylation profiles distinguish different subtypes of gastroenteropancreatic neuroendocrine tumors. Epigenomics. 2015;7:1245–1258. doi: 10.2217/epi.15.85. [DOI] [PubMed] [Google Scholar]
- 28.Ueki T, Toyota M, Skinner H, Walter KM, Yeo CJ, Issa JP, Hruban RH, Goggins M. Identification and characterization of differentially methylated CpG islands in pancreatic carcinoma. Cancer Res. 2001;61:8540–8546. [PubMed] [Google Scholar]
- 29.House MG, Guo M, Iacobuzio-Donahue C, Herman JG. Molecular progression of promoter methylation in intraductal papillary mucinous neoplasms (IPMN) of the pancreas. Carcinogenesis. 2003;24:193–198. doi: 10.1093/carcin/24.2.193. [DOI] [PubMed] [Google Scholar]
- 30.Arnold CN, Sosnowski A, Schmitt-Gräff A, Arnold R, Blum HE. Analysis of molecular pathways in sporadic neuroendocrine tumors of the gastro-entero-pancreatic system. Int J Cancer. 2007;120:2157–2164. doi: 10.1002/ijc.22569. [DOI] [PubMed] [Google Scholar]
- 31.Liu L, Broaddus RR, Yao JC, Xie S, White JA, Wu TT, Hamilton SR, Rashid A. Epigenetic alterations in neuroendocrine tumors: methylation of RAS-association domain family 1, isoform A and p16 genes are associated with metastasis. Mod Pathol. 2005;18:1632–1640. doi: 10.1038/modpathol.3800490. [DOI] [PubMed] [Google Scholar]
- 32.House MG, Herman JG, Guo MZ, Hooker CM, Schulick RD, Lillemoe KD, Cameron JL, Hruban RH, Maitra A, Yeo CJ. Aberrant hypermethylation of tumor suppressor genes in pancreatic endocrine neoplasms. Ann Surg. 2003;238:423–431; discussion 431-432. doi: 10.1097/01.sla.0000086659.49569.9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dammann R, Schagdarsurengin U, Liu L, Otto N, Gimm O, Dralle H, Boehm BO, Pfeifer GP, Hoang-Vu C. Frequent RASSF1A promoter hypermethylation and K-ras mutations in pancreatic carcinoma. Oncogene. 2003;22:3806–3812. doi: 10.1038/sj.onc.1206582. [DOI] [PubMed] [Google Scholar]
- 34.Pizzi S, Azzoni C, Bottarelli L, Campanini N, D’Adda T, Pasquali C, Rossi G, Rindi G, Bordi C. RASSF1A promoter methylation and 3p21.3 loss of heterozygosity are features of foregut, but not midgut and hindgut, malignant endocrine tumours. J Pathol. 2005;206:409–416. doi: 10.1002/path.1784. [DOI] [PubMed] [Google Scholar]
- 35.Zhang HY, Rumilla KM, Jin L, Nakamura N, Stilling GA, Ruebel KH, Hobday TJ, Erlichman C, Erickson LA, Lloyd RV. Association of DNA methylation and epigenetic inactivation of RASSF1A and beta-catenin with metastasis in small bowel carcinoid tumors. Endocrine. 2006;30:299–306. doi: 10.1007/s12020-006-0008-1. [DOI] [PubMed] [Google Scholar]
- 36.Malpeli G, Amato E, Dandrea M, Fumagalli C, Debattisti V, Boninsegna L, Pelosi G, Falconi M, Scarpa A. Methylation-associated down-regulation of RASSF1A and up-regulation of RASSF1C in pancreatic endocrine tumors. BMC Cancer. 2011;11:351. doi: 10.1186/1471-2407-11-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fotouhi O, Adel Fahmideh M, Kjellman M, Sulaiman L, Höög A, Zedenius J, Hashemi J, Larsson C. Global hypomethylation and promoter methylation in small intestinal neuroendocrine tumors: an in vivo and in vitro study. Epigenetics. 2014;9:987–997. doi: 10.4161/epi.28936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muscarella P, Melvin WS, Fisher WE, Foor J, Ellison EC, Herman JG, Schirmer WJ, Hitchcock CL, DeYoung BR, Weghorst CM. Genetic alterations in gastrinomas and nonfunctioning pancreatic neuroendocrine tumors: an analysis of p16/MTS1 tumor suppressor gene inactivation. Cancer Res. 1998;58:237–240. [PubMed] [Google Scholar]
- 39.Serrano J, Goebel SU, Peghini PL, Lubensky IA, Gibril F, Jensen RT. Alterations in the p16INK4a/CDKN2A tumor suppressor gene in gastrinomas. J Clin Endocrinol Metab. 2000;85:4146–4156. doi: 10.1210/jcem.85.11.6970. [DOI] [PubMed] [Google Scholar]
- 40.Bartsch DK, Kersting M, Wild A, Ramaswamy A, Gerdes B, Schuermann M, Simon B, Rothmund M. Low frequency of p16(INK4a) alterations in insulinomas. Digestion. 2000;62:171–177. doi: 10.1159/000007810. [DOI] [PubMed] [Google Scholar]
- 41.Arnold CN, Nagasaka T, Goel A, Scharf I, Grabowski P, Sosnowski A, Schmitt-Gräff A, Boland CR, Arnold R, Blum HE. Molecular characteristics and predictors of survival in patients with malignant neuroendocrine tumors. Int J Cancer. 2008;123:1556–1564. doi: 10.1002/ijc.23690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, Sidransky D, Baylin SB. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55:4525–4530. [PubMed] [Google Scholar]
- 43.Walter T, van Brakel B, Vercherat C, Hervieu V, Forestier J, Chayvialle JA, Molin Y, Lombard-Bohas C, Joly MO, Scoazec JY. O6-Methylguanine-DNA methyltransferase status in neuroendocrine tumours: prognostic relevance and association with response to alkylating agents. Br J Cancer. 2015;112:523–531. doi: 10.1038/bjc.2014.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wild A, Ramaswamy A, Langer P, Celik I, Fendrich V, Chaloupka B, Simon B, Bartsch DK. Frequent methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene in pancreatic endocrine tumors. J Clin Endocrinol Metab. 2003;88:1367–1373. doi: 10.1210/jc.2002-021027. [DOI] [PubMed] [Google Scholar]
- 45.Xiang T, Li L, Yin X, Yuan C, Tan C, Su X, Xiong L, Putti TC, Oberst M, Kelly K, et al. The ubiquitin peptidase UCHL1 induces G0/G1 cell cycle arrest and apoptosis through stabilizing p53 and is frequently silenced in breast cancer. PLoS One. 2012;7:e29783. doi: 10.1371/journal.pone.0029783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li L, Tao Q, Jin H, van Hasselt A, Poon FF, Wang X, Zeng MS, Jia WH, Zeng YX, Chan AT, et al. The tumor suppressor UCHL1 forms a complex with p53/MDM2/ARF to promote p53 signaling and is frequently silenced in nasopharyngeal carcinoma. Clin Cancer Res. 2010;16:2949–2958. doi: 10.1158/1078-0432.CCR-09-3178. [DOI] [PubMed] [Google Scholar]
- 47.Kleiman DA, Beninato T, Sultan S, Crowley MJ, Finnerty B, Kumar R, Panarelli NC, Liu YF, Lieberman MD, Seandel M, et al. Silencing of UCHL1 by CpG promoter hyper-methylation is associated with metastatic gastroenteropancreatic well-differentiated neuroendocrine (carcinoid) tumors. Ann Surg Oncol. 2014;21 Suppl 4:S672–S679. doi: 10.1245/s10434-014-3787-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edfeldt K, Ahmad T, Åkerström G, Janson ET, Hellman P, Stålberg P, Björklund P, Westin G. TCEB3C a putative tumor suppressor gene of small intestinal neuroendocrine tumors. Endocr Relat Cancer. 2014;21:275–284. doi: 10.1530/ERC-13-0419. [DOI] [PubMed] [Google Scholar]
- 49.Bollard J, Massoma P, Vercherat C, Blanc M, Lepinasse F, Gadot N, Couderc C, Poncet G, Walter T, Joly MO, et al. The axon guidance molecule semaphorin 3F is a negative regulator of tumor progression and proliferation in ileal neuroendocrine tumors. Oncotarget. 2015;6:36731–36745. doi: 10.18632/oncotarget.5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ushiku H, Yamashita K, Kawamata H, Waraya M, Katoh H, Yokoi K, Tanaka T, Ishii S, Nishizawa N, Kikuchi M, et al. Homeobox-Only Protein Expression Is a Critical Prognostic Indicator of Pancreatic Neuroendocrine Tumor and Is Regulated by Promoter DNA Hypermethylation. Pancreas. 2016;45:1255–1262. doi: 10.1097/MPA.0000000000000646. [DOI] [PubMed] [Google Scholar]
- 51.Dejeux E, Olaso R, Dousset B, Audebourg A, Gut IG, Terris B, Tost J. Hypermethylation of the IGF2 differentially methylated region 2 is a specific event in insulinomas leading to loss-of-imprinting and overexpression. Endocr Relat Cancer. 2009;16:939–952. doi: 10.1677/ERC-08-0331. [DOI] [PubMed] [Google Scholar]
- 52.Mei M, Deng D, Liu TH, Sang XT, Lu X, Xiang HD, Zhou J, Wu H, Yang Y, Chen J, et al. Clinical implications of microsatellite instability and MLH1 gene inactivation in sporadic insulinomas. J Clin Endocrinol Metab. 2009;94:3448–3457. doi: 10.1210/jc.2009-0173. [DOI] [PubMed] [Google Scholar]
- 53.Li J, Huang Q, Zeng F, Li W, He Z, Chen W, Zhu W, Zhang B. The prognostic value of global DNA hypomethylation in cancer: a meta-analysis. PLoS One. 2014;9:e106290. doi: 10.1371/journal.pone.0106290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi IS, Estecio MR, Nagano Y, Kim DH, White JA, Yao JC, Issa JP, Rashid A. Hypomethylation of LINE-1 and Alu in well-differentiated neuroendocrine tumors (pancreatic endocrine tumors and carcinoid tumors) Mod Pathol. 2007;20:802–810. doi: 10.1038/modpathol.3800825. [DOI] [PubMed] [Google Scholar]
- 55.Stricker I, Tzivras D, Nambiar S, Wulf J, Liffers ST, Vogt M, Verdoodt B, Tannapfel A, Mirmohammadsadegh A. Site- and grade-specific diversity of LINE1 methylation pattern in gastroenteropancreatic neuroendocrine tumours. Anticancer Res. 2012;32:3699–3706. [PubMed] [Google Scholar]
- 56.Stefanoli M, La Rosa S, Sahnane N, Romualdi C, Pastorino R, Marando A, Capella C, Sessa F, Furlan D. Prognostic relevance of aberrant DNA methylation in g1 and g2 pancreatic neuroendocrine tumors. Neuroendocrinology. 2014;100:26–34. doi: 10.1159/000365449. [DOI] [PubMed] [Google Scholar]
- 57.Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 58.Hashimoto H, Vertino PM, Cheng X. Molecular coupling of DNA methylation and histone methylation. Epigenomics. 2010;2:657–669. doi: 10.2217/epi.10.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Drané P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24:1253–1265. doi: 10.1101/gad.566910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim HS, Lee HS, Nam KH, Choi J, Kim WH. Telomere length abnormalities and telomerase RNA component expression in gastroenteropancreatic neuroendocrine tumors. Anticancer Res. 2015;35:3501–3510. [PubMed] [Google Scholar]
- 63.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tönjes M, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 64.de Wilde RF, Heaphy CM, Maitra A, Meeker AK, Edil BH, Wolfgang CL, Ellison TA, Schulick RD, Molenaar IQ, Valk GD, et al. Loss of ATRX or DAXX expression and concomitant acquisition of the alternative lengthening of telomeres phenotype are late events in a small subset of MEN-1 syndrome pancreatic neuroendocrine tumors. Mod Pathol. 2012;25:1033–1039. doi: 10.1038/modpathol.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marinoni I, Kurrer AS, Vassella E, Dettmer M, Rudolph T, Banz V, Hunger F, Pasquinelli S, Speel EJ, Perren A. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology. 2014;146:453–60.e5. doi: 10.1053/j.gastro.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 66.Pipinikas CP, Dibra H, Karpathakis A, Feber A, Novelli M, Oukrif D, Fusai G, Valente R, Caplin M, Meyer T, et al. Epigenetic dysregulation and poorer prognosis in DAXX-deficient pancreatic neuroendocrine tumours. Endocr Relat Cancer. 2015;22:L13–L18. doi: 10.1530/ERC-15-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Singhi AD, Liu TC, Roncaioli JL, Cao D, Zeh HJ, Zureikat AH, Tsung A, Marsh JW, Lee KK, Hogg ME, et al. Alternative Lengthening of Telomeres and Loss of DAXX/ATRX Expression Predicts Metastatic Disease and Poor Survival in Patients with Pancreatic Neuroendocrine Tumors. Clin Cancer Res. 2017;23:600–609. doi: 10.1158/1078-0432.CCR-16-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park YJ, Chodaparambil JV, Bao Y, McBryant SJ, Luger K. Nucleosome assembly protein 1 exchanges histone H2A-H2B dimers and assists nucleosome sliding. J Biol Chem. 2005;280:1817–1825. doi: 10.1074/jbc.M411347200. [DOI] [PubMed] [Google Scholar]
- 69.Zlatanova J, Seebart C, Tomschik M. Nap1: taking a closer look at a juggler protein of extraordinary skills. FASEB J. 2007;21:1294–1310. doi: 10.1096/fj.06-7199rev. [DOI] [PubMed] [Google Scholar]
- 70.Schimmack S, Taylor A, Lawrence B, Alaimo D, Schmitz-Winnenthal H, Büchler MW, Modlin IM, Kidd M. A mechanistic role for the chromatin modulator, NAP1L1, in pancreatic neuroendocrine neoplasm proliferation and metastases. Epigenetics Chromatin. 2014;7:15. doi: 10.1186/1756-8935-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Attia M, Rachez C, De Pauw A, Avner P, Rogner UC. Nap1l2 promotes histone acetylation activity during neuronal differentiation. Mol Cell Biol. 2007;27:6093–6102. doi: 10.1128/MCB.00789-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roldo C, Missiaglia E, Hagan JP, Falconi M, Capelli P, Bersani S, Calin GA, Volinia S, Liu CG, Scarpa A, et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J Clin Oncol. 2006;24:4677–4684. doi: 10.1200/JCO.2005.05.5194. [DOI] [PubMed] [Google Scholar]
- 73.Thorns C, Schurmann C, Gebauer N, Wallaschofski H, Kümpers C, Bernard V, Feller AC, Keck T, Habermann JK, Begum N, et al. Global microRNA profiling of pancreatic neuroendocrine neoplasias. Anticancer Res. 2014;34:2249–2254. [PubMed] [Google Scholar]
- 74.Ruebel K, Leontovich AA, Stilling GA, Zhang S, Righi A, Jin L, Lloyd RV. MicroRNA expression in ileal carcinoid tumors: downregulation of microRNA-133a with tumor progression. Mod Pathol. 2010;23:367–375. doi: 10.1038/modpathol.2009.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li SC, Essaghir A, Martijn C, Lloyd RV, Demoulin JB, Oberg K, Giandomenico V. Global microRNA profiling of well-differentiated small intestinal neuroendocrine tumors. Mod Pathol. 2013;26:685–696. doi: 10.1038/modpathol.2012.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miller HC, Frampton AE, Malczewska A, Ottaviani S, Stronach EA, Flora R, Kaemmerer D, Schwach G, Pfragner R, Faiz O, et al. MicroRNAs associated with small bowel neuroendocrine tumours and their metastases. Endocr Relat Cancer. 2016;23:711–726. doi: 10.1530/ERC-16-0044. [DOI] [PubMed] [Google Scholar]
- 77.Li SC, Shi H, Khan M, Caplin M, Meyer T, Öberg K, Giandomenico V. Roles of miR-196a on gene regulation of neuroendocrine tumor cells. Mol Cell Endocrinol. 2015;412:131–139. doi: 10.1016/j.mce.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 78.Lee YS, Kim H, Kim HW, Lee JC, Paik KH, Kang J, Kim J, Yoon YS, Han HS, Sohn I, et al. High Expression of MicroRNA-196a Indicates Poor Prognosis in Resected Pancreatic Neuroendocrine Tumor. Medicine (Baltimore) 2015;94:e2224. doi: 10.1097/MD.0000000000002224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jiang X, Shan A, Su Y, Cheng Y, Gu W, Wang W, Ning G, Cao Y. miR-144/451 Promote Cell Proliferation via Targeting PTEN/AKT Pathway in Insulinomas. Endocrinology. 2015;156:2429–2439. doi: 10.1210/en.2014-1966. [DOI] [PubMed] [Google Scholar]
- 80.Li SC, Khan M, Caplin M, Meyer T, Öberg K, Giandomenico V. Somatostatin Analogs Treated Small Intestinal Neuroendocrine Tumor Patients Circulating MicroRNAs. PLoS One. 2015;10:e0125553. doi: 10.1371/journal.pone.0125553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Modali SD, Parekh VI, Kebebew E, Agarwal SK. Epigenetic regulation of the lncRNA MEG3 and its target c-MET in pancreatic neuroendocrine tumors. Mol Endocrinol. 2015;29:224–237. doi: 10.1210/me.2014-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou Y, Zhang X, Klibanski A. MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol. 2012;48:R45–R53. doi: 10.1530/JME-12-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rahman MM, Qian ZR, Wang EL, Yoshimoto K, Nakasono M, Sultana R, Yoshida T, Hayashi T, Haba R, Ishida M, et al. DNA methyltransferases 1, 3a, and 3b overexpression and clinical significance in gastroenteropancreatic neuroendocrine tumors. Hum Pathol. 2010;41:1069–1078. doi: 10.1016/j.humpath.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 84.Alexander VM, Roy M, Steffens KA, Kunnimalaiyaan M, Chen H. Azacytidine induces cell cycle arrest and suppression of neuroendocrine markers in carcinoids. Int J Clin Exp Med. 2010;3:95–102. [PMC free article] [PubMed] [Google Scholar]
- 85.Baradari V, Huether A, Höpfner M, Schuppan D, Scherübl H. Antiproliferative and proapoptotic effects of histone deacetylase inhibitors on gastrointestinal neuroendocrine tumor cells. Endocr Relat Cancer. 2006;13:1237–1250. doi: 10.1677/erc.1.01249. [DOI] [PubMed] [Google Scholar]
- 86.Adler JT, Hottinger DG, Kunnimalaiyaan M, Chen H. Combination therapy with histone deacetylase inhibitors and lithium chloride: a novel treatment for carcinoid tumors. Ann Surg Oncol. 2009;16:481–486. doi: 10.1245/s10434-008-0194-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shah MH, Binkley P, Chan K, Xiao J, Arbogast D, Collamore M, Farra Y, Young D, Grever M. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res. 2006;12:3997–4003. doi: 10.1158/1078-0432.CCR-05-2689. [DOI] [PubMed] [Google Scholar]