

Abstract
Autosomal recessive primary microcephaly (MCPH) is a disorder of neurodevelopment resulting in a small brain1,2. We identified WDR62 as the second most common cause of MCPH after finding homozygous missense and frame-shifting mutations in seven MCPH families. In human cell lines, we found that WDR62 is a spindle pole protein, as are ASPM and STIL, the MCPH7 and MCHP7 proteins3–5. Mutant WDR62 proteins failed to localize to the mitotic spindle pole. In human and mouse embryonic brain, we found that WDR62 expression was restricted to neural precursors undergoing mitosis. These data lend support to the hypothesis that the exquisite control of the cleavage furrow orientation in mammalian neural precursor cell mitosis, controlled in great part by the centrosomes and spindle poles, is critical both in causing MCPH when perturbed and, when modulated, generating the evolutionarily enlarged human brain6–9.
INTRODUCTION
Genetic linkage of the second most common type of MCPH to chromosome 19q12 was discovered in 1999, but despite discovery of five other MCPH genes since then, the gene responsible for MCPH2 has eluded discovery10–14. Using classical and neo-classical reverse genetics, we identified the MCPH2 gene to be WDR62 (Fig. 1a). Two previously published multi-affected consanguineous families were used to define the MCPH2 gene region on chromosome 19q12 (ref. 10). In each of these families, all affected individuals fulfilled the diagnostic criteria for MCPH and shared only one concordant homozygous region10,15. As this genomic region was sequenced and ordered, polymorphic microsatellite markers were used to iteratively define the meiotic crossovers in each family. Final refinement was achieved through analysis of SNPs in a critical affected individual from each family using the Affymetrix GeneChip Human Mapping 250K Nsp Array. This led to the delineation of a 3.2 cM and 2.6 Mb region. Candidate gene sequencing within this region, based on selection for embryonic brain expression and/or neurogenic, centrosome, spindle pole and/or mitotic function, failed to identify MCPH2.
Figure 1.
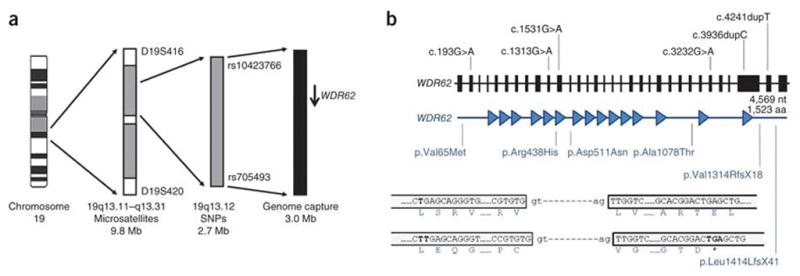
A summary of the linkage strategy used to define the MCPH2 region and mutations found in WDR62 in MCPH2 families. (a) From left to right, chromosome 19, shown as a G-banded cartoon; the initial linkage region defined by homozygous microsatellite markers; the final minimal linkage region defined by homozygous SNPs; the region subject to genome capture (slightly larger and overlapping the minimal linkage region); and WDR62 shown as an arrow pointing from 5′ _to 3′. Critical defining heterozygous markers are shown that bound each defined region. (b) WDR62 is shown from 5′ _to 3′, from left to right. Exons are shown to scale and introns are shown as an artificial fixed interval for clarity. The position of each homozygous mutation is shown. The lengths of the gene and protein are given. Below the gene, the WDR62 protein is shown with each WD repeat detected by PFAM, shown as a filled in triangle. The position that each DNA mutation affects the WDR62 protein is shown beneath, with indication of the resultant amino acid or peptide change. For the c.4241dupT mutation (resulting in the p.Leu1414LeufsX41 alteration), an explanatory cartoon is shown at the bottom of the figure. This mutation causes a frame shift in the penultimate exon of WDR62, which does not give rise to a new stop codon until the terminal exon. After the frame shift, 17 novel amino acids were found, and those amino acids at the start of the frame shift, at the splice sites and at the position of the new premature stop codon are shown.
We concluded that MCPH2 was either uncharacterized or that the mutational mechanism leading to disease was unusual. To address both of these possibilities, we used genome capture followed by massive parallel sequencing in one affected individual from each of our two MCPH2-mapping families16. We searched for a gene that contained a potentially pathogenic homozygous mutation in both families. This process identified only one gene, WDR62, with the homozygous missense mutation c.1313G>A and a homozygous one-base–pair insertion, c.4241dupT (Fig. 1, Table 1 and Supplementary Table 1). We then screened WDR62 in five further consanguineous families with an MCPH phenotype that displayed linkage to the MCPH2 locus and for which mutations in all other MCPH genes had been eliminated (Online Methods). We found four further homozygous mutations: three missense mutations (one of which occurred in two families) and one protein-truncating mutation (Fig. 1b, Table 1 and Supplementary Fig. 1). All WDR62 mutations segregated as expected for a recessive pathogenic change, were not recorded in genomic databases and were not present in ethnically matched controls (c.1531G>A was found in 1 out of 284 control chromosomes; Table 1).
Table 1.
Mutations found in WDR62 in MCPH2 families
| Mutation | Alteration | Exon | Pedigree | Affected + sibships | Control panel chromosomes and alleles screened |
|---|---|---|---|---|---|
| c.193G>A | p.Val65Met | 2 | P29436 | 3+1 | 184/184 WT Arab |
| c.1313G>A | p.Arg438His | 10 | P22011a | 6+4 | 298/298 WT Pakistani |
| c.1531G>A | p.Asp511Asn | 11 | P22009 | 3+2 | 283 WT and 1G>G/A of 284 Pakistani |
| c.1531G>A | p.Asp511Asn | 11 | P22021 | 2+1 | 283 WT and 1G>G/A of 284 Pakistani |
| c.3232G>A | p.Ala1078Thr | 27 | MCP91 | 5+2 | 190/190 WT Pakistani |
| c.3936dupC | p.Val1314ArgfsX18 | 30 | PC12 | 1 | 296/296 WT Caucasian |
| c.4241dupT | p.Leu1414LeufsX41 | 31 | P22016a | 9+6 | 298/298 WT Pakistani |
The two primary mapping families.
The mutation nomenclature for the cDNA and the protein are given, as well as the mutation type and in which exon the mutation occurs. The pedigree designation is given. For each family, the number of affected individuals seen and included in the study is given as well as the number of affected sibships in each pedigree. Finally, the details and results of control panel screen for each mutation is given. WT, wild type.
WDR62 is a gene with 32 exons and a single CpG island and polyadenylation signal (Fig. 1b). High expression has been recorded in lymphocytes and testes, but it has not previously been reported in embryonic or adult brain. There are two alternative WDR62 transcripts that have been recorded in human: exon 27 contains an in-frame, intra-exonic alternative splice acceptor site resulting in the exclusion of the first 12 nucleotides. WDR62 consists of 1,523 amino acids, contains at least 15 WD repeats, but has no ascribed functions either in organisms or in cells (Fig. 1b).
Four of the six WDR62 mutations are missense (Table 1 and Fig. 1b). The mutation c.4241dupT is expected to give rise to a stable RNA with a frame shifting insertion in the penultimate exon, resulting in a new premature stop codon in the final exon, yielding a C-terminal deletion of 109 amino acids in a region that contains neither predicted protein domains nor post-translational modification sites (Fig. 1b). Only the mutation c.3936dupC (resulting in the p.Val1314ArgfsX18 alteration) is expected to cause nonsense-mediated decay by forming a new premature stop codon in exon 30 and can be regarded as a probable null mutation.
None of the missense mutations or c.4241dupT is predicted to alter any of the WD repeat domains of WDR62. However, three of the missense mutations (resulting in p.Val65Met, p.Arg438His and p.Asp511Asn) alter evolutionarily highly conserved amino acids (including in all seven Drosophila species) (Supplementary Fig. 2). WDR62 has a mammalian homolog, MAPKBP1, in which again the three amino acids affected by the missense mutations are conserved. However, the alteration p.Ala1078Thr occurs in the C terminus of WDR62, and this region has diverged from the Drosophila ortholog of WDR62 and the human homolog MAPKBP1. This is not a highly conserved residue, as the wild-type amino acid is restricted to primates. We regard this mutation as one of unknown pathogenicity (Supplementary Fig. 2). Notably, mutations in the multiple WD repeat–containing BRWD3 cause X-linked mental retardation associated with macrocephaly17.
WDR62 was reported to be phosphorylated during mitosis in one study but not in a subsequent, more detailed study18,19. We therefore determined the subcellular distribution of WDR62 by immunocytochemistry and confocal microscopy in the following three human cell lines: HeLa (cervical cancer derived), HEK293 (embryonic renal derived) and lymphoblastoid (from B lymphocytes). All cells showed the same pattern (Fig. 2). In interphase cells, WDR62 had weak, diffuse cytoplasmic expression and was not nuclear. During mitosis, WDR62 accumulated strongly at the spindle poles but was not present at the midbody in cytokinesis. The expression pattern of WDR62 we found is identical to that of ASPM, another MCPH protein expressed at and able to focus the spindle poles in neural precursor cells (Supplementary Fig. 3 and refs. 8,20).
Figure 2.

Subcellular localization of WDR62 throughout the cell cycle. Confocal microscopy analysis of HeLa cells during each stage of the cell cycle. WDR62 staining is weak and cytoplasmic during interphase, with a concentration at a perinuclear position suggestive of the Golgi apparatus, and shows spindle pole localization during mitosis. Cells were stained with antibodies against human WDR62 (red), γ-tubulin (green) as a centrosome marker, α-tubulin (white) as a microtubule marker, and DNA (blue) stained with DAPI. Scale bar, 5 μm.
We investigated the pathogenicity of the missense mutations found in our two largest MCPH2 families, c.1313G>A and c.4241dupT. We engineered a gene construct of full length wild-type WDR62 with a C-terminal GFP fusion. We transfected cells and showed that this construct displayed diffuse interphase expression followed by spindle pole accumulation during mitosis (Fig. 3). Thus, the construct mimicked the wild-type subcellular localization pattern of WDR62. We then introduced the c.1313G>A missense mutation and the c.4241dupT frame shift mutation into a WDR62-GFP construct. These mutation constructs both led to expression of the fusion protein WDR62-GFP in the cytoplasm but not at the spindle poles during mitosis (Fig. 3 and Supplementary Fig. 4). This suggests that the c.1313G>A point mutation and the frame shift mutation c.4241dupT are both capable of independently abrogating the accumulation of WDR62 at the spindle pole and suggests that the common disease mechanism for MCPH2 microcephaly might be an absence of WDR62 at the spindle pole of dividing cells. These results do not allow us to determine if the mutant proteins have lost essential spindle targeting domains or if they are misfolded and become rapidly degraded21.
Figure 3.
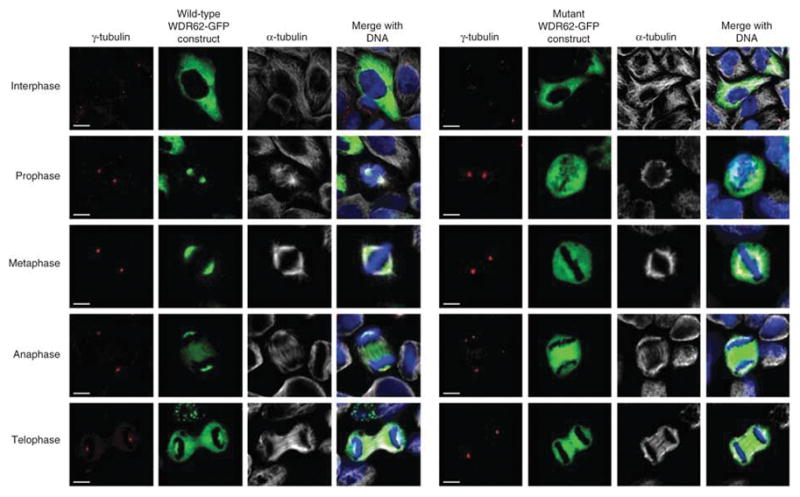
Overexpression of WDR62-GFP wild type and c.1313G>A ( p.Arg438His) mutant constructs in HeLa cells. The first 4.5 panel of images (on the left) shows the results for the wild-type WDR62-GFP construct, whereas the second 4 . 5 panel of images (on the right) shows the comparable results for the c.1313G>A (p.Arg438His) missense mutation in the WDR62-GFP construct. The wild-type WDR62-GFP protein localized to the spindle pole during mitosis (in 50 of 50 cells analyzed), paralleling the localization found for the endogenous wild type protein. Conversely, in cells expressing the mutant construct, there was no spindle pole accumulation of GFP during mitosis (in 50 of 50 cells analyzed). Cells were stained with γ-tubulin (red) as a centrosome marker and α-tubulin (white) identifying microtubules and the mitotic spindle. Both WDR62-GFP (green) proteins were directly visualized with DNA (blue) stained with DAPI. (See Supplementary Fig. 4 for the results for the c.4241dupT mutation). Scale bar, 5 μm.
It is hypothesized that MCPH is caused by a mitotic deficiency in the neural precursors in embryonic brain. Thus, we determined the tissue expression pattern of WDR62 in mouse and human embryonic brain using immunohistochemistry and confocal microscopy. We examined embryonic mouse brain at embryonic day (E) 11, E13 and E15 (ref. 22). These time points span the start of the generation of neurons destined for the cerebral cortex through to the establishment of the cortical plate23,24. We saw significant results in the neuroepithelium of the future cerebral cortex (Fig. 4). Firstly, Wdr62 expression in the neuroepithelium was found exclusively in apical precursors undergoing mitosis at the apical-ventricular surface. Apical precursors are the only cells recorded to undergo mitosis in the apical region of the neuroepithelium and can be identified by Nestin expression, the morphology of cellular DNA as a ‘metaphase plate’, and by having two centrosomes opposed to each other on either side of the ‘metaphase plate’. Secondly, intermediate neural precursors (also called basal precursors25,26) found in the sub-ventricular zone of the embryonic brain also expressed Wdr62 exclusively during mitosis.
Figure 4.
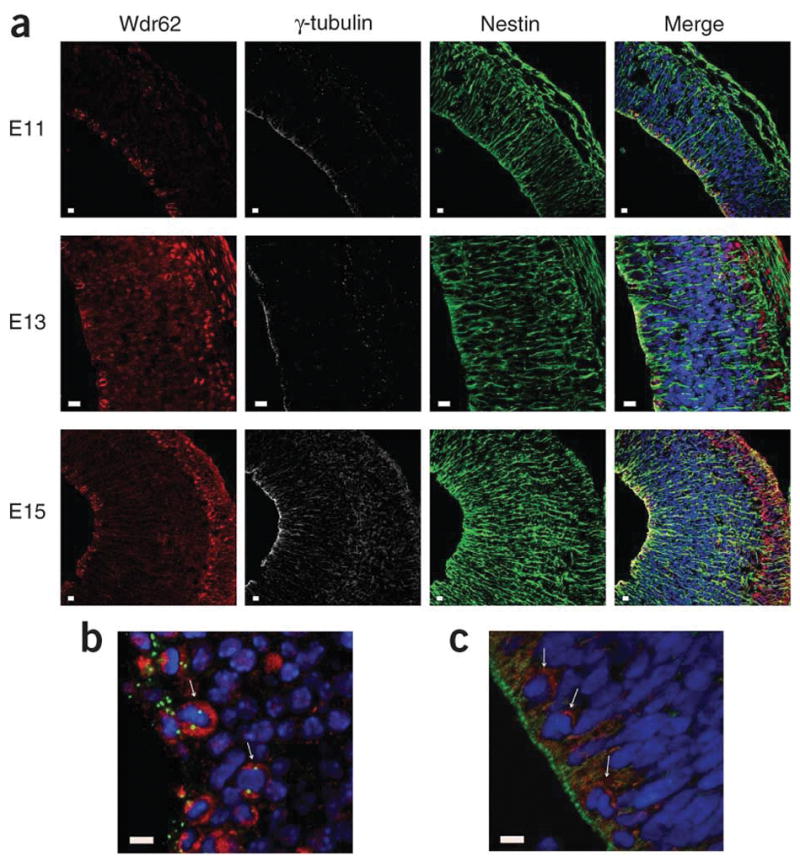
Endogenous expression pattern of WDR62 in human and mouse embryonic brain. (a) Wdr62 expression in mouse cerebral cortex neuroepithelium from early to late neurogenesis: E11, E13 and E15. Wdr62 is seen to concentrate in the cytoplasm of mitotic apical and basal neural precursor cells. In the cortical plate containing newly born neurons, seen after E13, Wdr62 is also found in the nucleus of newly born neurons. (b) A magnified image of mouse E13 neuroepithelium clearly showing Wdr62 cytoplasmic aggregation in apical mitotic precursors. The arrows indicate examples of apical neural precursor cells undergoing mitosis with a pair of centrosomes on either side of a metaphase DNA plate. In the neuroepithelium, Wdr62 expression is only clearly seen in cells undergoing mitosis. (c) A magnified image of human CS22 cerebral cortex neuroepithelium showing WDR62 cytoplasmic aggregation in apical mitotic precursors. Arrows indicate cells in which WDR62 expression can be most clearly seen, which by their position and nuclear morphology are likely to be undergoing mitosis. Due to collection, fixation and subsequent paraffin embedding, immunohistochemistry is more difficult in human embryonic brain sections; however, the apically positioned centrosomes of the apical neural precursors are clearly visible. For each set of images, the result are shown for WDR62 and Wdr62 (red), with γ-tubulin (white) as a centrosome marker, Nestin (green) marking apical neural precursors in the cytoplasm and DNA (blue) stained with DAPI. Scale bars, 10 μm.
Tissue processing does not allow the preservation of microtubules and, hence, mitotic spindle visualization; therefore we were unable to show that, as in cell lines, Wdr62 was a spindle pole protein in neural precursor cells in vivo. Magnified views of mouse apical neural progenitors highlight Wdr62 expression in mitotic cells (Fig. 4b). We then assessed the expression of WDR62 in human embryonic brain at gestation stage CS22, which is equivalent to mouse neurogenesis at stage E13 (ref. 27). By staining cells with γ-tubulin along with a WDR62 antibody, we saw the same pattern of expression as in mouse, with expression of WDR62 only in mitotic neural precursor cells (Fig. 4c and Supplementary Fig. 5).
We then investigated the developing cortical plate. We found WDR62 expression in newborn neurons and in the outermost layer of neurons that had just migrated to the cortical plate (Fig. 5a). The subcellular localization of WDR62 in these neurons was nuclear (WDR62 has no canonical basic amino acid nuclear-localization domain) and was not at spindle poles or centrosomes, as seen in neural precursors (Fig. 5a,b). A recent study reported WDR62 mutations in individuals with severe brain malformations and found interphase nuclear localization of WDR62 (ref. 28). The subcellular localization data agree for newly migrated cortical neurons but not for other cells where our and previously published data all strongly support a spindle pole localization of WDR62 in dividing cells18,19.
Figure 5.
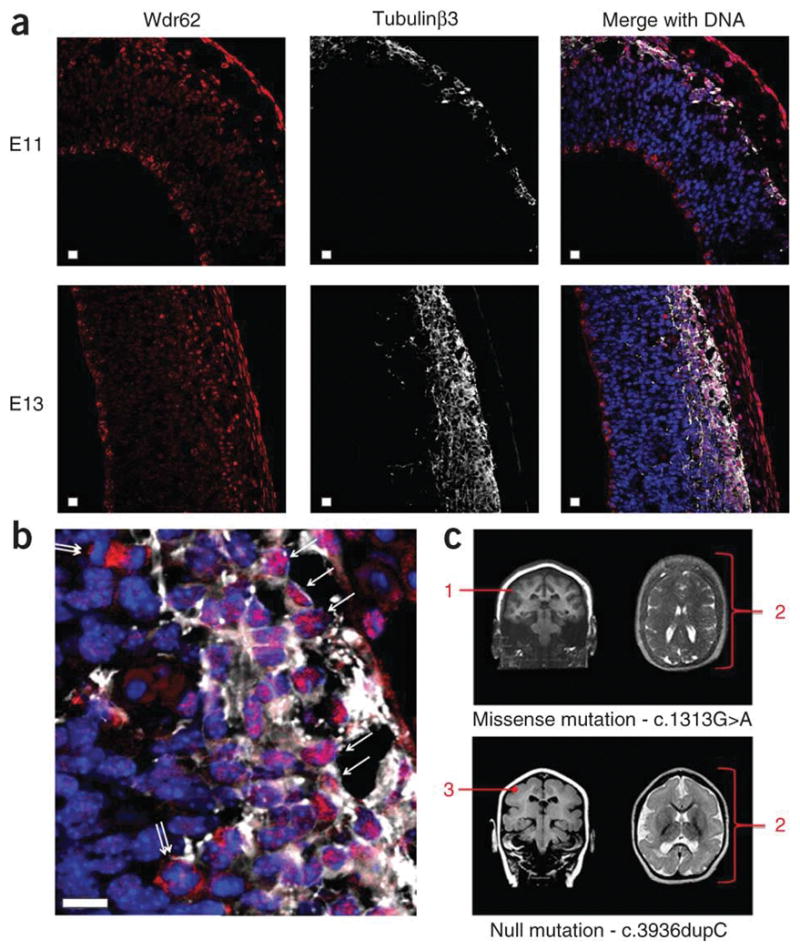
Wdr62 expression in newborn, newly arrived cortical neurons in the developing cerebral cortex and brain imaging from two individuals with WDR62 mutations. (a) Newborn neuron data. Mouse cerebral cortex neuroepithelium in early and later neurogenesis stages E11 and E13 stained with antibodies to Wdr62 (red) and costained against the neuronal marker protein Tubulinβ3 (white) with DNA (blue) stained with DAPI. The apical ventricular margin of the neuroepithelium is shown on the left of each image; on the right of each image is the outer, pial surface of the developing cerebral cortex. The developing cerebral cortex is bounded by Tubulinβ3 staining to the right of each image. Localization of Wdr62 to the developing cortical plate can be seen at the right of each image. An outermost Wdr62 staining is also seen, beyond the cerebral cortex, which represents staining of pial membrane cells. Scale bars, 10 μm. (b) An enlargement of an E13 merged image allowing easier visualization of neurons staining with Tubulinβ3 (white). The single arrows indicate neurons in which Wdr62 staining is nuclear. Also visible are cells, indicated by double arrows, undergoing mitosis in the subventricular zones with prominent Wdr62 staining in the cytoplasm, which are assumed to be basal or intermediate neural progenitors. (c) MRI images of individuals with MCPH2 from this study with homozygous WDR62 mutations; the c.1313G>A missense mutation is shown above and the protein truncating c.3936dupC mutation is shown below. The annotation in red indicates: 1, cortex normal thickness (3–4 mm) but having an indistinct border; 2, diffuse simplified gyral pattern with frontal cortex most severely affected (this feature is more marked for the null mutation); and 3, cortex appears mildly thickened (~5 mm).
We had only two brain scans available from study families (Fig. 5c); these scans were from a child with the missense mutation c.1313G>A, which showed a simplified gyral pattern, and from a child with a null mutation c.3936dupC, which also showed the simplified gyral pattern but also that the cerebral cortex was thickened. The latter individual had a substantially more severe clinical course (Online Methods). This expression and clinical data together suggest that WDR62 may also have a role in cortical lamination. It is therefore possible that missense mutations leading to WDR62 proteins that cannot be targeted to spindle poles cause a deficiency of neurogenesis resulting in primary microcephaly, but that nonsense mutations leading to a complete lack of WDR62 production cause a more severe microcephaly phenotype because of the addition of a cerebral cortex lamination defect. Future studies should determine if this conjecture is correct.
In conclusion, WDR62 is the cause of the second commonest form of MCPH and encodes a spindle pole protein that is expressed in neuronal precursor cells undergoing mitosis in the mammalian embryonic neuroepithelium. ASPM, the MCPH5 protein, is also a spindle pole protein and in Aspm-deficient mice, increased numbers of apical precursor cells undergo asymmetric (neuron generating) rather than symmetric (neural precursor generating) cell division6. In the human brain, symmetric rather than asymmetric divisions are initially needed to generate a large pool of neural precursors. This observation may explain the reduced neuron numbers seen in primary microcephaly. That WDR62, ASPM and STIL are all spindle pole proteins suggests the unique importance of focused spindle poles in neural progenitor cell division. The spindle poles are attached to the mature centrosomes in these cells and together control the position of the central spindle and hence the direction of the last stage of the cytokinesis cleavage furrow9,29,30. The furrow has to exactly bisect a small apical-membrane fate-determining domain (the cadherin hole) to ensure a symmetric division and production of two daughter cell neural precursors8,31,32. The cadherin hole diminishes in size as neurogenesis progresses from 2% to 1% of the total cell surface30. Once a cell fails to inherit part of a cadherin hole, its fate changes and it differentiates into a neuron, becomes post-mitotic and migrates out of the neuroepithelium. Our data suggest that WDR62 is a key protein in enabling spindle poles to position the cytokinetic furrow and prolong neural precursor generation, a process that is uniquely vital to human cerebral cortex growth.
URLs
Human genome browser, http://genome.ucsc.edu/; PFAM for protein domain recognition, http://pfam.sanger.ac.uk/search?tab=searchSequenceBlock; BLAST for nucleotide and protein alignment and similarity assessment, http://blast.ncbi.nlm.nih.gov/Blast.cgi; MRC-Wellcome Trust Human Developmental Biology Resource, http://www.hdbr.org/.
ONLINE METHODS
Clinical details of the seven MCPH families described in this paper
For families with WDR62 mutations, excluding c.3936dupC, microcephaly was observed in all affected children at birth or during the first months of life when no birth records were available. Height and weight were normal in all affected children, and head circumferences were between −4 and −7 standard deviations. No malformations or congenital anomalies were present in these children. Microcephalic individuals were not dysmorphic but had a sloping forehead and a disproportionate sized face and ears as compared to the skull. No neurological deficits were observed. Fundoscopy, hearing and vision were all normal. None of the affected children had seizures. Initial psychomotor development was normal, but all the children exhibited delayed speech acquisition. All had mild or moderate non-progressive mental retardation. A standard 400-bands karyotype and Affymetrix GeneChip Human Mapping 250K Nsp Array analysis was normal in a proband from each family. All parents were in good health, with normal head circumferences, were of normal intelligence and were consanguineous. Several family members had computerized axial tomography (CT) or magnetic resonance imaging (MRI) scans; all family members were reported as showing a structurally normal brain, a normal gyral pattern and a normal thickness of the cerebral cortex.
For the individual with the WDR62 mutation c.3936dupC, initial development was more delayed than the other individuals with MCPH and by the time this individual was 4 years old, it was clear that she was severely retarded. She did not speak her first words until 4 years, at 6 years her developmental quotient was found to be 60, and at 10 years she used gestures and pictures to communicate only simple needs. At 10 years, she was incontinent and socially withdrawn. She had no other neurological, dysmorphic features or a growth deficit, and she had had no seizures (electroencephalograms at birth and at an age of 6 years were normal). At birth, her head circumference was 31.5 cm, and at 5 and 10 years, her head circumference was −5 standard deviations from the expected norms. An MRI scan at 11 months showed a simplified gyral pattern with a thickened cortex (5 mm compared to the expected 3–4 mm).
All families gave informed consent to enter the study which had Cambridge Research Ethic Committee approval, Cambridge, UK.
Genome capture and parallel sequencing
A NimbleGen Sequence Capture 385K custom-designed microarray was used to capture the entire region in one affected individual from each of our two largest MCPH2 families. The captured DNA was sequenced using the Roche 454 Genome Sequencer FLX. The sequence reads were mapped to the human genome reference NCBI Build 36.1 using the GS Reference Mapper application of the Roche Genome Sequencer Data Analysis software and were annotated using UCSC Genome Browser refGene and SNP130. Supplementary Table 1 details the bioinformatics analysis of the resulting data.
WDR62 expression plasmids
The longest isoform of WDR62 was PCR amplified from HEK293 cDNA and tagged at the C terminus with enhanced gene fluorescent protein (eGFP) by cloning into the EcoRI and KpnI sites of the mammalian expression vector pEGFP-N1 (Clontech). The c.1313G>A substitution and c.4241dupT insertion were created using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer’s instructions. The inserts from the wild-type and two mutant constructs were fully sequenced. HeLa cells were transfected with the wild-type WDR62-GFP construct and checked to ensure that antibodies to WDR62 and GFP gave coincident subcellular localizations (Supplementary Fig. 6).
Transfection and immunocytochemistry
HeLa cells cultured in DMEM supplemented with 10% FCS were transiently transfected with either the wild-type or the mutant WDR62-EGFP constructs using Lipofectamine 2000 (Invitrogen). One day after transfection, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X100 and blocked with 3% BSA for 40 min. Staining was carried out using the following antibodies: mouse monoclonal anti-gamma tubulin (Abcam, ab27074) detected with the secondary Alexa Fluor 546 goat anti-mouse IgG (Invitrogen); and rat monoclonal anti-α _tubulin (AbD serotec, MCA77G) detected with Alexa Fluor 633 chicken anti-rat (Invitrogen). Slides were counterstained with Hoechst 33342 (Sigma), mounted using Fluoromount-G (Southern Biotech) and examined using an LSM 510 META Confocal laser scanning inverted microscope (Carl Zeiss) equipped with an argon-krypton laser beam. Positively transfected mitotic cells were identified by GFP fluorescence and the spindle pole localization of WDR62-GFP was assessed by colocalization with γ-tubulin (at least 50 cells were counted over 3 coverslips). The subcellular expression profile of endogenous WDR62 was analyzed in HeLa cells fixed with ice-cold methanol for 5 min and permeabilized with acetone. WDR62 protein was detected using a rabbit polyclonal antibody to WDR62 (Bethyl Laboratories, A301–560A). We further assessed the specificity of the WDR62 antibody by performing protein blots of HeLa cells with and without wild-type WDR62-GFP transfection. Using antibodies to GFP and then WDR62-only, bands of the appropriate expected sizes were seen (Supplementary Fig. 7).
Immunohistochemistry
Pregnant CD-1 mice were purchased from a UK supplier. Central nervous system embryonic tissues were harvested in accordance with the Animals Act 1986 (Scientific Procedures). E11, E13 and E15 tissues were fixed with 4% paraformaldehyde overnight at 4 °C and incubated in 30% sucrose before cryostat sectioning. Human CS22 paraffin-embedded sections fixed with 4% paraformaldehyde were sourced from the Medical Research Council-Wellcome Trust Human Developmental Biology Resource (see URLs, Newcastle). Mouse and human sections were boiled for 10 min in 10mM sodium citrate buffer (pH 6) and then treated with a blocking buffer containing 0.3% Triton X100 detergent and bovine serum albumin (3%) for 1 h. Primary antibodies were incubated overnight in the blocking buffer. Appropriate Alexa Fluor dye–conjugated secondary antibodies (Invitrogen) were incubated for 1 h along with Hoechst 33342 for nuclei staining. Primary antibodies used on tissue sections were as follows: mouse monoclonal anti-gamma tubulin (Abcam, ab11316); chicken anti-nestin (Neuromics, CH23001); rabbit anti-WDR62 (Bethyl Laboratories, A301–560A); and mouse monoclonal anti-β _tubulin III (Sigma, T8660).
Supplementary Material
Acknowledgments
The authors would like to thank the research families for their participation in this project and the Wellcome Trust, Medical Research Council, Action Research and the Higher Education Commission of Pakistan for funding (to A.K.N., M.K., O.P.C., J.J.C., G.T. and E.R.). J.D. was supported by the Belgian Kids’ Fund. M.A. was supported by grants from the Fonds Erasme and the Belgian Fonds de la Recherche Scientifique Médicale (FRSM). We thank S. Strollo for expert technical assistance. We thank the Medical Research Council (MRC)-Wellcome Trust Human Developmental Biology Resource (HDBR), Newcastle for providing the human tissue for the expression studies
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
AUTHOR CONTRIBUTIONS
The following authors contributed to the design of the study: A.K.N., M.K., J.J.C., F.G., E.R., M. Abramowicz and C.G.W. The following authors generated experimental data: A.K.N., M.K., J.D., O.P.C., G.T., R.K., M. Ansar, F.G., W.B.D., E.R. and C.G.W. Reagents were contributed by R.K., M. Abramowicz, W.A., A.L., S.P., J.-P.M., S.L., M. Abramowicz and C.G.W. The paper was written by A.K.N., M.K., O.P.C., J.J.C., W.A., S.L., F.G., W.B.D. and C.G.W.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Mochida GH, Walsh CA. Genetic basis of developmental malformations of the cerebral cortex. Arch Neurol. 2004;61:637–640. doi: 10.1001/archneur.61.5.637. [DOI] [PubMed] [Google Scholar]
- 2.Woods CG, Bond J, Enard W. Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. Am J Hum Genet. 2005;76:717–728. doi: 10.1086/429930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bond J, et al. ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002;32:316–320. doi: 10.1038/ng995. [DOI] [PubMed] [Google Scholar]
- 4.Pfaff KL, et al. The zebra fish cassiopeia mutant reveals that SIL is required for mitotic spindle organization. Mol Cell Biol. 2007;27:5887–5897. doi: 10.1128/MCB.00175-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bond J, Woods CG. Cytoskeletal genes regulating brain size. Curr Opin Cell Biol. 2006;18:95–101. doi: 10.1016/j.ceb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Fish JL, Kosodo Y, Enard W, Pääbo S, Huttner WB. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci USA. 2006;103:10438–10443. doi: 10.1073/pnas.0604066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fish JL, Dehay C, Kennedy H, Huttner WB. Making bigger brains–the evolution of neural-progenitor-cell division. J Cell Sci. 2008;121:2783–2793. doi: 10.1242/jcs.023465. [DOI] [PubMed] [Google Scholar]
- 8.Marthiens V, ffrench-Constant C. Adherens junction domains are split by asymmetric division of embryonic neural stem cells. EMBO Rep. 2009;10:515–520. doi: 10.1038/embor.2009.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thornton GK, Woods CG. Primary microcephaly: do all roads lead to Rome? Trends Genet. 2009;25:501–510. doi: 10.1016/j.tig.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts E, et al. The second locus for autosomal recessive primary microcephaly (MCPH2) maps to chromosome 19q13.1–13.2. Eur J Hum Genet. 1999;7:815–820. doi: 10.1038/sj.ejhg.5200385. [DOI] [PubMed] [Google Scholar]
- 11.Jackson AP, et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet. 2002;71:136–142. doi: 10.1086/341283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bond J, et al. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet. 2005;37:353–355. doi: 10.1038/ng1539. [DOI] [PubMed] [Google Scholar]
- 13.Kumar A, Girimaji SC, Duvvari MR, Blanton SH. Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am J Hum Genet. 2009;84:286–290. doi: 10.1016/j.ajhg.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Passemard S, et al. GeneReviews. University of Washington; Seattle, WA: 2009. Primary autosomal recessive microcephaly. < http://www.ncbi.nlm.nih.gov/pubmed/20301772>. [Google Scholar]
- 15.Roberts E, et al. Autosomal recessive primary microcephaly: an analysis of locus heterogeneity and phenotypic variation. J Med Genet. 2002;39:718–721. doi: 10.1136/jmg.39.10.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mardis ER. New strategies and emerging technologies for massively parallel sequencing: applications in medical research. Genome Med. 2009;1:40. doi: 10.1186/gm40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Field M, et al. Mutations in the BRWD3 gene cause X–linked mental retardation associated with macrocephaly. Am J Hum Genet. 2007;81:367–374. doi: 10.1086/520677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nousiainen M, Silljé HH, Sauer G, Nigg EA, Körner R. Phosphoproteome analysis of the human mitotic spindle. Proc Natl Acad Sci USA. 2006;103:5391–5396. doi: 10.1073/pnas.0507066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malik R, et al. Quantitative analysis of the human spindle phosphoproteome at distinct mitotic stages. J Proteome Res. 2009;8:4553–4563. doi: 10.1021/pr9003773. [DOI] [PubMed] [Google Scholar]
- 20.do Carmo Avides M, Tavares A, Glover DM. Polo kinase and Asp are needed to promote the mitotic organizing activity of centrosomes. Nat Cell Biol. 2001;3:421–424. doi: 10.1038/35070110. [DOI] [PubMed] [Google Scholar]
- 21.Caspi M, et al. LIS1 missense mutations: variable phenotypes result from unpredictable alterations in biochemical and cellular properties. J Biol Chem. 2003;278:38740–38748. doi: 10.1074/jbc.M301147200. [DOI] [PubMed] [Google Scholar]
- 22.Gardette R, Courtois M, Bisconte JC. Prenatal development of mouse central nervous structures: time of neuron origin and gradients of neuronal production. A radioautographic study. J Hirnforsch. 1982;23:415–431. [PubMed] [Google Scholar]
- 23.Uylings HB. Development of the cerebral cortex in rodents and man. Eur J Morphol. 2000;38:309–312. doi: 10.1076/0924-3860(200012)38:5;1-h;ft309. [DOI] [PubMed] [Google Scholar]
- 24.Bystron I, Blakemore C, Rakic P. Development of the human cerebral cortex: Boulder Committee revisited. Nat Rev Neurosci. 2008;9:110–122. doi: 10.1038/nrn2252. [DOI] [PubMed] [Google Scholar]
- 25.Attardo A, Calegari F, Haubensak W, Wilsch-Brauninger WM, Huttner WB. Live imaging at the onset of cortical neurogenesis reveals differential appearance of the neuronal phenotype in apical versus basal progenitor progeny. PLoS ONE. 2008;3:e2388. doi: 10.1371/journal.pone.0002388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noctor SC, Martinez-Cerdeno V, Kriegstein AR. Distinct behaviors of neural stem and progenitor cells underlie cortical neurogenesis. J Comp Neurol. 2008;508:28–44. doi: 10.1002/cne.21669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Rahilly R, Müller F. Developmental Stages in Human Embryos: Revised and New Measurements. Cells Tissues Organs. 2010;192:73–84. doi: 10.1159/000289817. [DOI] [PubMed] [Google Scholar]
- 28.Bilgüvar K, et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature. 2010 Aug 22; doi: 10.1038/nature09327. published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barr FA, Gruneberg U. Cytokinesis: placing and making the final cut. Cell. 2007;131:847–860. doi: 10.1016/j.cell.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Barr AR, Kilmartin JV, Gergely F. CDK5RAP2 functions in centrosome to spindle pole attachment and DNA damage response. J Cell Biol. 2010;189:23–39. doi: 10.1083/jcb.200912163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Götz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- 32.Kosodo Y, et al. Asymmetric distribution of the apical plasma membrane during neurogenic divisions of mammalian neuroepithelial cells. EMBO J. 2004;23:2314–2324. doi: 10.1038/sj.emboj.7600223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.