

Abstract
A sequence variation (I148M) in patatin‐like phospholipase domain‐containing protein 3 (PNPLA3) is strongly associated with fatty liver disease, but the underlying mechanism remains obscure. In this study, we used knock‐in (KI) mice (Pnpla3148M/M) to examine the mechanism responsible for accumulation of triglyceride (TG) and PNPLA3 in hepatic lipid droplets (LDs). No differences were found between Pnpla3148M/M and Pnpla3+/+ mice in hepatic TG synthesis, utilization, or secretion. These results are consistent with TG accumulation in the Pnpla3148M/M mice being caused by impaired TG mobilization from LDs. Sucrose feeding, which is required to elicit fatty liver in KI mice, led to a much larger and more persistent increase in PNPLA3 protein in the KI mice than in wild‐type (WT) mice. Inhibition of the proteasome (bortezomib), but not macroautophagy (3‐methyladenine), markedly increased PNPLA3 levels in WT mice, coincident with the appearance of ubiquitylated forms of the protein. Bortezomib did not increase PNPLA3 levels in Pnpla3148M/M mice, and only trace amounts of ubiquitylated PNPLA3 were seen in these animals. Conclusion: These results are consistent with the notion that the 148M variant disrupts ubiquitylation and proteasomal degradation of PNPLA3, resulting in accumulation of PNPLA3‐148M and impaired mobilization of TG from LDs. (Hepatology 2017;66:1111‐1124).
Abbreviations
- 3‐MA
3‐methyladenine
- ATGL
adipose triglyceride lipase
- CGI‐58
comparative gene identification 58
- FLD
fatty liver disease
- HSD
high‐sucrose diet
- IgG
immunoglobulin G
- KI
knock‐in
- LD
lipid droplet
- LPAAT
lysophosphatidic acid acyltransferase
- mAb
monoclonal antibody
- mRNA
messenger RNA
- PLIN2
perilipin 2
- PNPLA3
patatin‐like phospholipase‐domain containing protein 3
- SEM
standard error of the mean
- TG
triglycerides
- Tg
transgene
- VLDL
very low density lipoprotein
- WT
wild‐type
Fatty liver disease (FLD) is a burgeoning health problem due to the increased prevalence of both obesity and insulin resistance. The most significant and frequent genetic variation associated with hepatic steatosis is a nonsynonymous variant in the gene encoding patatin‐like phospholipase‐domain containing protein 3 (PNPLA3).1 Substitution of methionine for isoleucine at residue 148 is strongly associated with not only with hepatic accumulation of triglyceride (TG), but also the full spectrum of both nonalcoholic FLD and alcoholic FLD,2 including hepatic inflammation and fibrosis3 and hepatocellular carcinoma.4
Despite the robust association between PNPLA3‐I148M and FLD, the mechanistic basis of this relationship has not been fully defined. Molecular modeling of PNPLA3 predicts that the I148M substitution disrupts enzymatic activity.5 In vitro assays using purified PNPLA3 are consistent with this prediction; the I148M substitution markedly decreases TG hydrolase activity.5 These findings suggested that PNPLA3 functions as a hepatic TG hydrolase, and that the I148M substitution is a loss‐of‐function allele that causes fatty liver by impairing PNPLA3‐mediated TG hydrolysis.5 This model was brought into question by the finding that PNPLA3 knockout mice do not develop hepatic steatosis.6, 7 Thus, the hepatic TG accumulation associated with PNPLA3‐148M cannot be attributed to a simple loss of TG hydrolase activity in the liver.
In contrast to PNPLA3 knockout mice, transgenic mice overexpressing human PNPLA3‐148M (PNPLA3Tg148M/+ mice) have significantly increased liver TG content, suggesting that the variant may confer a gain‐of‐function.8 Further support for this notion came from the finding that PNPLA3 has lysophosphatidic acid acyltransferase (LPAAT) activity and that the PNPLA3‐I148M is associated with an increase in TG synthesis.9 However, we failed to observe LPAAT activity associated with PNPLA3 expression either in vitro or in vivo in PNPLA3Tg148M/+ mice.5, 8 Therefore, the hepatic steatosis associated with the 148M variant cannot be explained by simple loss‐ or gain‐of‐function models. The available data suggest that hepatic steatosis requires the presence of a catalytically dead (or significantly impaired) protein that interferes with mobilization of TG from lipid droplets (LDs). Thus, the variant causes fatty liver due to a combination of conferring both a loss‐of‐function and a gain‐of‐function.
To test this hypothesis, we introduced the susceptibility variant (148M) and a catalytically inactive variant (S47A) into mouse Pnpla3.10 Both lines developed hepatic steatosis when fed a high‐sucrose diet (HSD) (no.901683; 74% kilocalories from sucrose; MP Biomedicals, Santa Ana, CA). We also found that the PNPLA3 protein accumulates on LDs in sucrose‐fed Pnpla3148M/M and in Pnpla347A/A mice, but not in Pnpla3+/+ mice. We performed metabolic studies to further define the effects of PNPLA3‐148M on TG synthesis, oxidation, and secretion and to pinpoint the pathway responsible for the accumulation of the 148M protein on LDs.
Materials and Methods
ANIMALS
PNPLA3 knock‐in (KI) mice in which methionine was substituted for isoleucine at codon 148 [Pnpla3148M/M, also referred to as KI mice in this article], or in which alanine was substituted for the catalytic serine at residue 47 (Pnpla347A/A), and mice expressing a human PNPLA3‐WT or PNPLA3‐148M transgene (Tg) (PNPLA3TgWT/+ and PNPLA3Tg148M/+) were generated as described previously.8, 10 The transgenic mice used in the experiments were offspring of heterozygous X wild‐type (WT) matings, and the non‐Tg littermates were used as controls. The KI mice were backcrossed into C57Bl/6J mice for six generations. Mice were housed in microisolator and shoebox cages with 7090 sani‐chip bedding (Harlan Teklad, Houston, TX). All animal experiments were performed with the approval of the Institutional Animal Care and Research Advisory Committee at the University of Texas Southwestern Medical Center in Dallas, Texas.
Mice were maintained on a 12/12‐hour light (8 am to 8 pm)/dark cycle (8 pm to 8 am) and fed Teklad Mouse/Rat Diet 7001 chow diet (Harlan Teklad, Houston, TX) ad libitum. For the dietary challenge studies, mice were fed an HSD for 4 weeks unless stated otherwise. Before each experiment, the mice were metabolically synchronized using one of two protocols: protocol 1 comprised 12 hours of fasting (8 am to 8 pm, light) and 12 hours of refeeding (8 pm to 8 am, dark) and protocol 2 comprised 18 hours of fasting (2 pm to 8 am) and 6 hours of refeeding (8 am to 2 pm).
ANTIBODIES
Mouse anti‐human (h) PNPLA3 monoclonal antibodies (mAb) (clones 4C12 and 11C5) were raised against a recombinant fusion protein of hPNPLA3 and trigger factor.11 A rabbit anti‐mouse (m) PNPLA3 mAb was developed using a peptide corresponding to residues 454‐927 (clone 19A6). The following commercial antibodies were used for immunoblotting: mouse anti‐ubiquitin mAb (P4D1; Santa Cruz Biotech, Inc., 1:500), rabbit anti‐mATGL (Cell Signaling Technology, MA, 1:1,000), guinea pig anti‐mPLIN2 (Fitzgerald Industries International, Acton, MA, 1:5000), goat anti‐mPNPLA3 polyclonal antibody (R&D Systems, Minneapolis, MN, 1:1000), mouse anti‐hCGI‐58 mAb (Abnova, Walnut CA, 1:5,000), and rabbit anti‐phospho‐S6 ribosomal protein polyclonal antibody (Cell Signal Technology, Danvers, MA). Secondary antibodies used for immunoblotting included goat anti‐rabbit immunoglobulin G (IgG), goat anti‐mouse IgG, or Fab fragment‐specific horseradish peroxidase conjugated antibodies (Jackson Laboratories, West Grove, PA, 1:3,000).
LIVER CHEMISTRIES
Lipids were extracted from livers (∼100 mg). Cholesterol and TGs were measured using enzymatic assays (Infinity, Thermo Electron, Madison, WI, and Wako, Richmond, VA, respectively).12 Values were normalized to sample weight. Lipids were separated into cholesterol esters, TGs, and phospholipids using thin‐layer chromatography.13 Bands were excised and lipids were measured as described.8
IN VIVO LIPID SYNTHESIS STUDIES AND VERY LOW DENSITY LIPOPROTEIN SYNTHESIS STUDIES
Incorporation of tritum from tritiated water (3H‐H2O) into fatty acids, phosphatidic acid (PA), and TG was measured in sucrose‐fed mice. Feeding was synchronized for 3 days (protocol 1). On the third day, mice were fasted for 24 hours and stagger‐fed for 4 hours before intraperitoneal administration of 3H‐H2O (50 mCi in 250 μL 0.9% NaCl).14, 15
Sodium 14C‐palmitate‐albumin (94.5 nmol/mouse; 118 × 103 dpm/nmol) was injected intravenously (IV). Thirty minutes after the injection, mice were anesthetized and killed. Venous blood (500 μL) and livers were collected. Radiolabeled lipids were measured in liver samples (200 mg) as described previously.15
Very low density lipoprotein (VLDL) secretion and TG mobilization were measured as described previously.16
FATTY ACID OXIDATION IN PRIMARY HEPATOCYTES
Feeding was synchronized in sucrose‐fed mice (protocol 1). Mice were anesthetized using isoflurane in a glass chamber and primary hepatocytes were seeded at 6 × 104 cells per well on XF‐24 cell culture plates (Seahorse Bioscience) and oxygen (O2) consumption rates were measured exactly as described previously.17
MESSENGER RNA EXPRESSION
Total RNA was prepared from mouse tissues as described.18 RNA was extracted from the liver and levels of selected transcripts were measured using real‐time polymerase chain reaction as described.18 All reactions were done in triplicate. The relative amount of messenger RNAs (mRNAs) was calculated using the comparative threshold cycle method with mouse 36B4 as an internal control.
BORTEZOMIB AND 3‐METHYLADENINE TREATMENT OF MICE
Bortezomib (LC Laboratories, Woburn, MA) (10 mg) was dissolved in ethanol (1 mL) to prepare a stock solution. The solution was diluted in 0.9% saline to a final concentration of 5% (vol/vol). Mice were fed an HSD for 3 days (protocol 1). On the third day, either bortezomib (1 mg/kg) or vehicle control (0.9% NaCl, 5% ethanol) was injected via the tail veins. Mice were then fed an HSD ad libitum for 5 hours before being killed.
Mice were also treated with 3‐methyladenine (3‐MA) as described previously19, 20 with minor modifications. Mice were fed an HSD for 1 week and then injected intravenously with 3‐MA (15 mg/kg) in PBS or with vehicle alone.
IMMUNOBLOTTING
Immunoblot analysis of liver lysates and LD proteins was performed as described previously.21 Bands were visualized using West Pico Super Signal Chemiluminescent Substrate (Thomson Fisher Scientific, Waltham, MA) and quantified using the LICOR Odyssey Imaging System (LI‐COR, Inc., Lincoln, NE).
IMMUNOPRECIPITATION OF PNPLA3
LDs were isolated 10 and the proteins were solubilized in buffer A [0.1% Fos‐choline‐13, 50 mM Tris‐HCl, pH 7.5, 150 mM NaCl plus protease inhibitor (Roche)]. A total of 30 μg protein was incubated with rabbit anti‐mouse PNPLA3 mAb (19A6) overnight at 4°C. After 12 hours, 50 μL protein A/G agarose (Santa Cruz Biotechnology, Inc.) was added to the protein/antibody mixture and allowed to incubate at 4°C for 3 hours. Beads were washed 3 times in buffer A without the detergent. Beads were boiled and immunoblotting was performed as described.21 To avoid interference with the immunoglobulin heavy chain, goat anti‐rabbit light chain IgG and goat anti‐mouse light chain IgG and Fab fragment‐specific horseradish peroxidase–conjugated secondary antibodies were used for detection.
Results
FATTY ACID AND TG SYNTHESIS IN Pnpla3148M/M MICE
Previously, we showed that the I148M substitution in PNPLA3 is associated with hepatic steatosis in humans and in sucrose‐fed mice.1, 10 To determine whether the fatty liver is caused by an increase in TG synthesis, as has been suggested,9 we compared rates of incorporation of 3H from 3H2O into fatty acids and TG in WT and KI mice. Tissues were collected 1 hour after injection of tritiated water into the peritoneal cavity, and lipids were extracted from the indicated tissues and saponified to release free fatty acids. Tritium incorporation into 3H‐fatty acids (Fig. 1A) and TG (Fig. 1B) did not differ between the two lines in any of the tissues collected (Fig. 1B).
Figure 1.
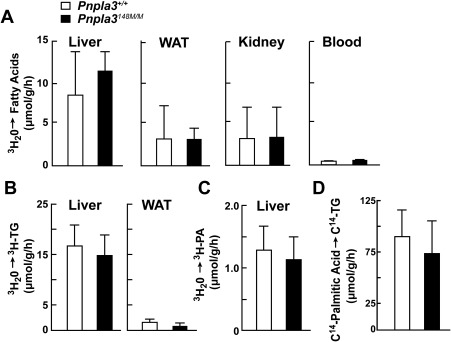
3H‐H2O and 14C‐palmitate incorporation in Pnpla3+/+(WT) and Pnpla3148/M/M (KI) mice. (A‐C) Incorporation of 3H‐H2O into (A) fatty acids, (B) TG, and (C) PA in liver, white adipose tissue (WAT), kidney, and blood of 11‐ to 13‐week‐old female PNPLA3‐WT and PNPLA3‐KI mice (n = 10 per group). Mice were fed an HSD for 4 weeks and then feeding was synchronized for 3 days (protocol 1). During the last fasting cycle, the fast was extended to 24 hours. Mice were then stagger‐fed at 4‐hour intervals before being injected intraperitoneally with 3H‐H2O (50 mCi in 250 μL 0.9% NaCl). After 1 hour, the mice were killed and tissues were processed as described in Materials and Methods. Bars represent the mean ± standard error of the mean (SEM). (D) Incorporation of 14C‐palmitate into hepatic TG in 12‐week old female WT and KI mice (10/group). Mice were handled as described in panel A but were injected through the tail vein with 14C‐palmitate‐BSA (200 μCi) and killed after 1 hour. Each bar represents the mean ± SEM. Levels were compared among lines using a Student t test. The experiment was repeated once, and the results were similar.
Kumari et al.9 reported that PNPLA3 has LPAAT activity and that the I148M variant was associated with an increase in that activity. Those authors suggested that the steatosis associated with PNPLA3‐148M is caused by increased formation of PA from lysophosphatidic acid, leading to increased TG synthesis. We failed to detect a difference between the KI and WT mice in incorporation of tritium into PA (Fig. 1C).
To compare the rates of TG synthesis from circulating fatty acids, we infused sodium 14C‐palmitate‐albumin (94.5 nmol) into sucrose‐fed WT and KI mice. The mice were killed after 30 minutes and the amount of 14C‐TG in the livers was quantified. No significant difference in the rate of appearance of 14C‐TG was found between the lines (Fig. 1D).
Thus, we found no evidence that PNPLA3‐148M causes fatty liver by promoting synthesis of TG from either endogenously synthesized or circulating fatty acids.
VLDL‐TG SECRETION AND HEPATIC FATTY ACID OXIDATION IN Pnpla3148M/M MICE
To determine whether VLDL secretion is compromised in Pnpla3148M/M mice, as occurs in mice with a sequence variant in TM6SF2 that confers susceptibility to FLD,21 we inhibited lipolysis of circulating TG by infusing Triton WR‐1339 (500 mg/kg) after a 4‐hour fast, and then monitored the appearance of TG in the circulation.16 The rates of TG secretion did not differ significantly between sucrose‐fed WT and KI mice (Fig. 2A). Thus, these data do not support the notion that the accumulation of TG associated with PNPLA3‐148M expression is caused by a defect in the incorporation of TG into VLDL or in the secretion of VLDL‐TG from the liver.
Figure 2.
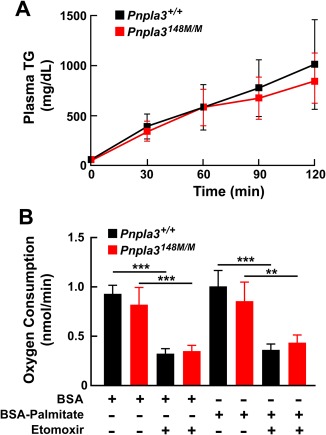
VLDL‐TG secretion (A) and fatty acid oxidation (B) in Pnpla3+/+ and Pnpla3148M/M mice. (A) Male WT and KI mice (n = 5 per group, age 14‐15 weeks) were fed an HSD for 7 days and then feedings were synchronized (protocol 2). After a 4‐hour fast, Triton WR‐1339 (500 μg/g) was injected into the tail veins. Blood was collected at the indicated time points, and plasma TG levels were measured. Mean plasma TG levels (± SEM) at each point are shown. Rates of VLDL‐TG secretion were determined by least square regression of plasma TG levels plotted against time. Slope estimates were determined from the linear portion of each graph and compared using a Student t test (P = 0.52). The experiment was repeated once, and the results were similar. (B) Male mice (n = 3 per group, age 11‐12 weeks) were fed an HSD for 4 weeks and the feedings were synchronized for 3 days (protocol 1). The mice were killed at the end of the last refeeding cycle and primary hepatocytes were isolated. Oxygen consumption was measured in the primary hepatocytes using Seahorse XF‐24 analyzer as described in Materials and Methods. Cells were treated with either etomoxir (300 μM), an inhibitor of carnitine palmitoyl transferase‐1 or with BSA‐palmitate (200 μM). Values represent the mean ± SEM. **P < 0.01, ***P < 0.001.
We also measured and compared rates of fatty acid oxidation in primary hepatocytes from KI and WT mice using the Seahorse XF‐24 Analyzer.17 Rates of O2 consumption were similar in hepatocytes from the two lines, both at baseline and after addition of palmitate to the medium (Fig. 2B). As expected, addition of etomoxir, which blocks entry of fatty acids into mitochondria, reduced O2 consumption in both mouse lines.
The data in Figures 1 and 2 suggest that the fatty liver in sucrose‐fed Pnpla3148M/M mice was caused by reduced mobilization of TG from LDs, rather than by increased TG synthesis, decreased TG oxidation or decreased VLDL‐TG secretion. Therefore, we examined the relationship between PNPLA3 mRNA and protein expression and TG mobilization after a prolonged fast in the two lines of mice.
RATE OF HEPATIC TG MOBILIZATION AFTER FASTING IS SIMILAR IN Pnpla3148M/M AND Pnpla3+/+ MICE
WT and KI mice were fasted for 24 hours and then refed a chow diet. Fasting increased the level of liver TG 10‐fold in both lines of mice (Fig. 3), which is similar to what has been seen previously in C57Bl/6 mice.14 With refeeding, levels of hepatic TG fell at similar rates in the WT and KI mice, returning to baseline within 9 hours.
Figure 3.
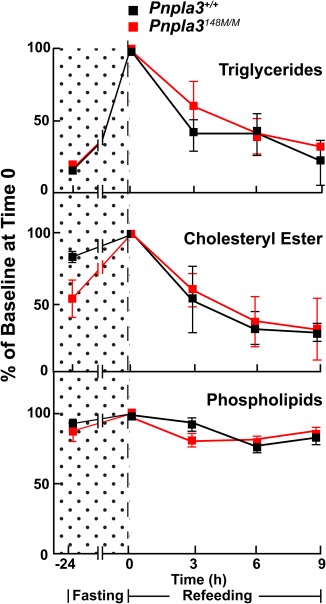
Triglyceride mobilization from livers of fasting Pnpla3+/+ and Pnpla3148M/M mice. Male mice (n = 3 per group, age 11‐12 weeks) were fed a chow diet for 3 days (12 hours of fasting [8 am to 8 pm] and 12 hours of feeding [8 pm to 8 am]). The mice were fasted 24 hours in the last cycle and then refed with a chow diet. Mice were sacrificed at the indicated time points and livers were collected. Hepatic lipids were measured as described in Materials and Methods. The experiment was repeated once, and the results were similar.
Thus, we found no evidence that expression of PNPLA3‐148M impairs mobilization of fatty acids from hepatic TG with refeeding after a prolonged fast. It could be argued that this result was predictable given that PNPLA3 is not expressed in the fasting state.22 To address this issue, we monitored hepatic PNPLA3 levels during the transition from a fed state, when PNPLA3 is expressed, to a fasted state.
DELAYED CLEARANCE OF PNPLA3 DURING THE FED TO FASTING TRANSITION IN Pnpla3148M/M (KI) MICE
Mice were fed an HSD for 3 days (protocol 1) before the food was removed. Livers were collected at the end of the last feeding cycle (time zero) and then at the indicated times. At the 2‐hour time point, PNPLA3 levels were 25‐fold higher in the KI mice than in the WT mice, despite only a 1.6‐fold difference in mRNA levels (Fig. 4A). As anticipated, PNPLA3 mRNA levels fell progressively over the 12‐hour fast. In the WT mice, PNPLA3 protein fell to undetectable levels within 6 hours. In contrast, in the KI mice, PNPLA3 levels were maintained over the same time period. The PNPLA3 protein to mRNA ratio continued to increase over the first 9 hours of the experiment in the KI mice, thus revealing a significant dissociation of PNPLA3 mRNA and protein levels in the mice expressing the mutant protein.
Figure 4.
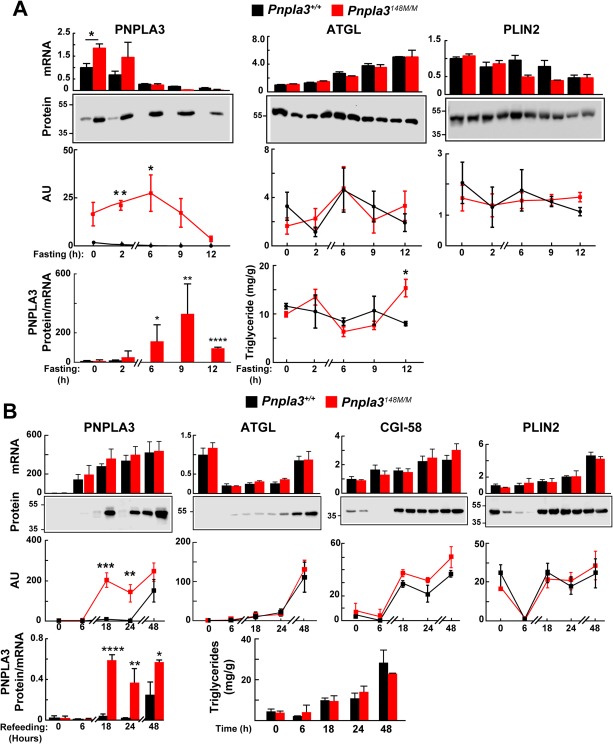
Dysregulation of PNPLA3‐148M expression in LDs of Pnpla3+/+ and PNPLA3148M/M mice upon fasting (A) and refeeding (B). (A) Male WT and KI mice (n = 3 per group, age 10‐12 weeks) were fed an HSD for 3 days (protocol 1). At the end of the last feeding cycle, mice were fasted and killed at the indicated time points. Lipid droplets and mRNA were isolated from the liver as described in Materials and Methods. Real‐time polymerase chain reaction was used to quantify selected mRNAs using 36B4 mRNA as a normalization standard. The level of PNPNLA3 mRNA in WT mice at time 0 was set at 1. LD proteins were precipitated using acetone, and immunoblotting was performed. Proteins (3 μg) were size‐fractionated by way of 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and incubated with the following antibodies: anti‐mPNPLA3 (19A6), anti‐mATGL, and anti‐mPLIN2 (see Materials and Methods for types of antibodies and dilutions). Signals were quantified using a LICOR system. (B) PNPLA3 accumulation on LDs after sucrose feeding. Male WT and KI mice (n = 3 per group, age 12‐14 weeks) were synchronized on a chow diet for 3 days (protocol 2). At the end of the last fasting cycle, the mice were fed an HSD for the indicated times and then killed. Livers were processed as described in Materials and Methods. The experiment was repeated once, and the results were similar. Values represent the mean ± SEM. Levels were compared among lines using a Student t test. *P < 0.05. **P < 0.01. ***P < 0.001. ****P < 0.0001.
In the same experiment, we examined the effects of fasting on mRNA and protein levels of two other LD proteins: adipocyte TG lipase (ATGL), the major lipase in LDs,23 and perilipin 2 (PLIN2), an LD protein that increases with TG accumulation in hepatocytes.24 ATGL mRNA levels increased with fasting, as expected.25 Despite the increase in ATGL mRNA, protein levels did not show a concomitant increase over the same time period. No correlation was found between the amount of PNPLA3 and ATGL on LDs. We also examined the levels of comparative gene identification 58 (CGI‐58), a cofactor of ATGL; no significant differences were seen in the levels of CGI‐58 between the two lines of mice until the last time point, when levels were marginally higher in the 148M mice (P = 0.03) (Supporting Fig. S1). Thus, the FLD associated with PNPLA3‐148M does not reflect a reduction in the amount of ATGL on LDs.
As a control for the experiment, we examined the effect of fasting on levels of PLIN2 mRNA and protein. The level of the PLIN2 transcript fell by ∼40% in the fasted animals. No consistent differences in PLIN2 protein were seen between the WT and KI mice over the course of the experiment.
Under the conditions of this experiment, no differences in hepatic TG levels were seen between the two lines of mice, except at the last time point, when the TG levels were modestly higher in the KI mice (Fig. 4A, bottom panel). Thus, the changes in abundance of PNPLA3 that occurred acutely at the fed‐to‐fasting transition were associated with marginal differences in hepatic TG levels.
ACCUMULATION OF PNPLA3‐148M ON LDs PRECEDES ACCUMUATION OF HEPATIC TG
Next, we examined PNPLA3 mRNA and protein levels in livers of mice transitioning from an ad libitum chow diet (protocol 2) to an HSD. Here, livers were collected at the end of the fasting cycle (time zero), when neither PNPLA3 mRNA nor protein were detectable, and then at the indicated times after mice were fed an HSD (Fig. 4B). Levels of PNPLA3 mRNA increased dramatically within 6 hours of initiating sucrose feeding and continued to increase over the course of the experiment. Levels of PNPLA3 protein were ∼200‐fold higher in KI mice than in WT mice at the 18‐hour time point and remained significantly higher until the end of the experiment, despite comparable levels of PNPLA3 mRNA. PNPLA3 levels in total liver lysates were similar to those in the isolated LDs (Supporting Fig. S2), consistent with our prior observation that >90% of PNPLA3 expressed in hepatocytes is associated with LDs.26 Despite the major differences in PNPLA3 content of LDs, the rates of accumulation of TG were similar in the two groups of mice. Thus, the effects of PNPLA3‐148M on TG accumulation are not immediate and precede the development of hepatic steatosis.
To determine whether the increase in PNPLA3‐148M displaces either ATGL or CGI‐58 from LDs, we examined the levels of the two proteins. Both proteins accumulated on the droplet over the course of the experiment, but the accumulation was similar in WT and KI mice. Therefore, the higher TG content associated with chronic expression on PNPLA3‐148M is not caused by displacement of ATGL or CGI‐58 from the droplet. We cannot rule out the possibility that the presence of PNPLA3‐148M interferes with the activity of ATGL or competes for binding of CGI‐58.
REDUCED UBIQUITYLATION OF PNPLA3 IN LIVERS OF Pnpla3148M/M MICE
We tested the hypothesis that PNPLA3‐148M accumulates in the livers of Pnpla3148M/M mice due to defective ubiquitylation of the protein. PNPLA3TgWT/+ and PNPLA3Tg148M/+ mice were fed an HSD for 3 days and given bortezomib (1 mg/kg) or vehicle control at the end of the feeding cycle. After 8 hours, the mice were killed and livers were collected. PNPLA3 mRNA levels did not change significantly with bortezomib treatment in either group (Supporting Fig. S3A). Bortezomib treatment was associated with a 15‐fold increase in PNPLA3 protein levels in the mice expressing the human WT transgene. In contrast, no significant increase in PNPLA3‐148M was seen in the bortezomib‐treated PNPLA3Tg148M/+ animals (Fig. 5A). Levels of PLIN2 increased to a similar extent in both groups of transgenic mice as has been reported previously.27
Figure 5.
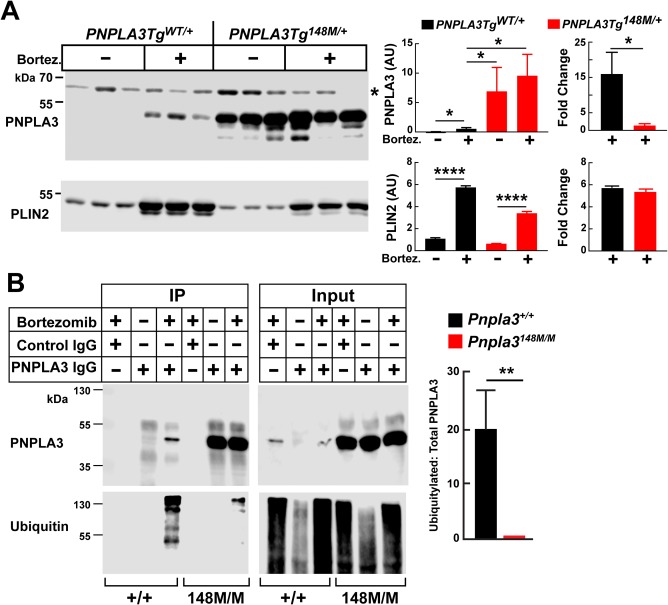
Ubiquitylation of PNPLA3 in vivo in PNPLA3TgWT/+ and PNPLA3Tg148M/+ mice. (A) Male PNPLA3 Tg mice (n = 3 per group, age 11‐13 weeks) were fed an HSD for 3 days (protocol 1). At 8 am the mice were injected with bortezomib (1 mg/kg) and vehicle control (0.9% NaCl plus 5% ethanol) through the tail vein. After 8 hours, the mice were killed and livers were collected and processed as described in Materials and Methods. Each bar represents the mean ± SEM. (B) Pnpla3+/+ and Pnpla3148M/M male mice (n = 3 per group, age 12‐14 weeks) were treated as described in panel A, except that the mice were killed after 5 hours. Liver samples were pooled and LDs were isolated as described in Materials and Methods. PNPLA3 was immunoprecipitated from the LD fraction, and immunoblot analysis was performed after the proteins (10% of input) were size‐fractionated by way of 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis using an anti‐mPNPLA3 and anti‐ubiquitin mAb (see Materials and Methods for antibody type and dilution). Signals were quantified using a LICOR system. Each bar represents the mean ± SEM. Levels were compared among lines using a Student t test. *P < 0.05. **P < 0.01. ****P < 0.0001. *Nonspecific band. The experiment was repeated twice, and the results were similar.
Bortezomib treatment was associated with TG accumulation in livers from both lines of transgenic mice (Supporting Fig. S3B, left). A similar increase in hepatic TG was seen in Pnpla3+/+ and in Pnpla3‐/‐ mice (Supporting Fig. S3B, right). Thus, the increase in TG associated with bortezomib treatment cannot be attributed to differences in PNPLA3 (or PLIN2) levels on hepatic LD in the two lines of mice. It remains possible that the increase in both PNPLA3 and PLIN2 levels associated with bortezomib treatment is secondary to its effects on TG accumulation.
REDUCED UBIQUITYLATION OF PNPLA3‐148M
Another possibility is that the accumulation of PNPLA3‐148M was caused by a reduction in ubiquitylation of the protein. To test this hypothesis, we treated WT and KI mice with vehicle alone or with bortezomib and compared PNPLA3 levels after immunoprecipitation from LDs (Fig. 5B). In WT mice, PNPLA3 levels were 20‐fold higher in the bortezomib‐treated mice than in animals treated with vehicle alone. As was seen previously in Tg mice (Fig. 5A), levels of PNPLA3 in mice treated with vehicle alone were much higher (50×) in the Pnpla3148M/M mice than in their WT counterparts. Levels of PNPLA3 in KI mice did not increase after treatment with the proteasomal inhibitor (Fig. 5B).
Next, we compared the levels of ubiquitylation of PNPLA3 in the two groups of mice. The PNPLA3 that was immunoprecipitated from LDs was subjected to immunoblot analysis with an anti‐ubiquitin antibody (Fig. 5B). A ladder of bands was detected in the WT animals, but only after bortezomib treatment. This result is consistent with the notion that bortezomib treatment causes the accumulation of poly‐ubiquitylated forms of the WT protein. Only a modest increase in the intensity of the higher molecular weight bands was seen in the bortezomib‐treated Pnpla3148M/M animals (Fig. 5B). The ratio of ubiquitylated PNPLA3 to total PNPLA3 was 20‐fold greater in WT mice than in KI mice in the group of bortezomib‐treated animals (Fig. 5B, right panel).
REDUCED UBIQUITYLATION OF PNPLA3‐47A
Previously, we showed that the TG hydrolase activity of PNPLA3 was inactivated by substitution of alanine for the catalytic serine at residue 47.5 Mice homozygous for this variant (Pnpla347A/A) phenocopy the 148M variant; the Pnpla347A/A mice accumulated PNPLA3 on LDs and developed hepatic steatosis on an HSD.10 In our initial studies, sucrose feeding induced hepatic steatosis in mice heterozygous for the 148M (Pnpla3148M/+) but not in mice heterozygous for the 47A variant (Pnpla347A/+).10 To determine whether the 47A variant causes steatosis by a different mechanism, we repeated this experiment multiple times with greater numbers of sixth‐generation animals. Sucrose‐fed heterozygous and homozygous mice with the S47A substitution accumulated PNPLA3 on LD and developed hepatic steatosis (Fig. 6A).
Figure 6.
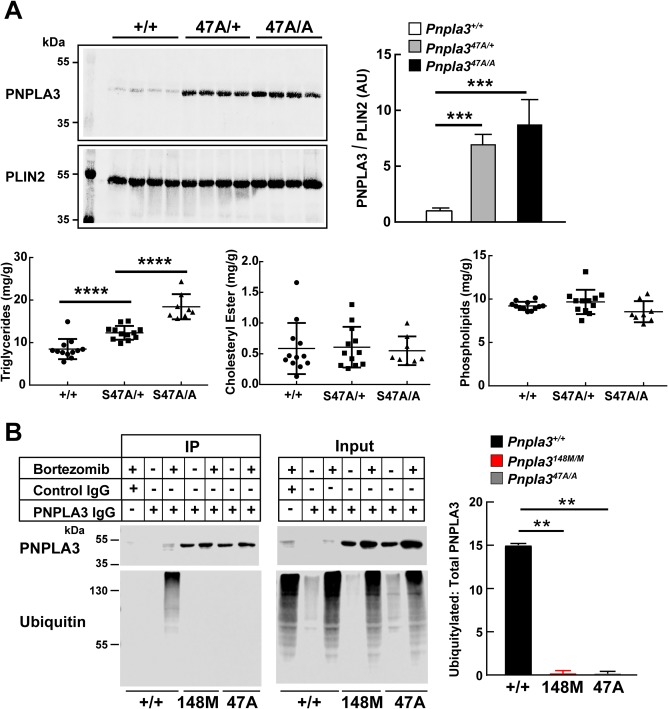
Ubiquitylated PNPLA3 in LDs of Pnpla3+/+, Pnpla347A/A, and Pnpla3148M/M mice. (A) Pnpla3+/+, Pnpla3S47A/+, and Pnpla3S47A/A female mice (n = 8 per group, age 13‐16 weeks) were fed an HSD for 4 weeks and then feedings were synchronized for 3 days (protocol 1). Mice were killed after the last refeeding cycle, and the livers were collected for lipid analyses and LD isolation. (B) Pnpla3+/+, Pnpla3148M/M, and Pnpla347A/A female mice (n = 3 per group per time point, age 11‐13 weeks) were fed an HSD for 3 days, and the feedings were then synchronized (protocol 1). After the last refeeding cycle, mice were injected with bortezomib as described in Figure 5. Mice were killed after 5 hours, and the livers were collected and pooled. LDs were isolated and samples were processed exactly as described in Figure 5B. Each bar represents the mean ± SEM. Levels were compared among lines using a Student t test. **P < 0.01. ***P < 0.001. ****P < 0.0001. The data are representative of three independent experiments.
If the accumulation of PNPLA3 on LDs in Pnpla3148M/M mice is due to defective ubiquitylation of the protein, then a similar defect in ubiquitylation should be seen in Pnpla347A/A mice. To determine whether this was the case, we performed the experiment shown in Fig. 6B in the Pnpla3148M/M and Pnpla347A/A mice. Bortezomib treatment was associated with a 9‐fold increase in PNPLA3 protein levels in the WT animals. Immunoblotting with an anti‐ubiquitin antibody revealed a ladder of higher molecular weight bands in the bortezomib‐treated animals. A more modest increase in PNPLA3 was seen with bortezomib treatment in the Pnpla3148M/M and Pnpla347A/A mice (1.6‐fold and 1.5‐fold, respectively). No higher molecular weight bands were detected when the immunoprecipitates from these mice were blotted with an anti‐ubiquitin antibody.
NO CHANGE IN PNPLA3 LEVELS IN MICE TREATED WITH 3‐MA
We also treated the mice with 3‐MA to inhibit macroautophagy.28 3‐MA treatment was associated with a reduction in the levels of phosphorylated ribosomal protein S6, as expected,29 but it had no effect on hepatic levels of PNPLA3 or TG in either group of transgenic animals (Fig. 7 and Supporting Fig. S4).
Figure 7.
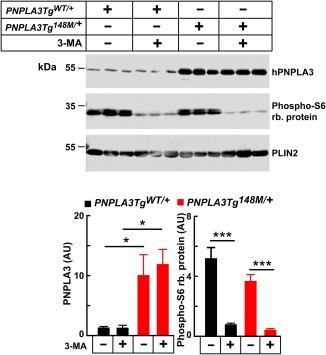
Hepatic PNPLA3 levels in 3‐MA–treated mice expressing human PNPLA3‐WT and ‐148M. Male PNPLA3TgWT/+ and PNPLA3Tg148M/+ mice (n = 3 per group, age 11‐13 weeks) were fed an HSD for 1 week. Feeding was synchronized (protocol 1), and at the end of the third feeding cycle, the mice were treated with 3‐MA (15 mg/kg) and fasted for 3 hours before being killed. LDs were harvested from the liver and immunoblotting was performed as described in Materials and Methods. Phospho‐S6 ribosomal protein was used as a positive control. The data are representative of two independent experiments. Each bar represents the mean ± SEM. Levels were compared among lines using a Student t test. *P < 0.05. ***P < 0.001.
Discussion
A major finding of this paper is that the I148M substitution in PNPLA3 does not cause hepatic steatosis either by increasing TG synthesis or by reducing TG disposal through fatty acid oxidation or VLDL‐TG secretion. We also show that the 148M variant alters posttranslational modification of PNPLA3. In mice, treatment with the proteasome inhibitor bortezomib increased hepatic PNPLA3‐WT levels ∼15‐fold (Fig. 6A), coincident with the accumulation of poly‐ubiquitylated forms of the protein. This finding indicates that ubiquitylation and proteasomal degradation is the major catabolic pathway for PNPLA3‐WT. Bortezomib treatment had little effect on the levels of PNPLA3‐148M, and a much smaller fraction (∼1/20) of the variant protein was poly‐ubiquitylated (Figs. 5B and 6B). Taken together, our data are consistent with a model in which the WT form of PNPLA3 is efficiently ubiquitylated and rapidly turned over by proteasomal degradation. In contrast, PNPLA3‐148M evades ubiquitylation and proteasomal degradation, resulting in accumulation of PNPLA3‐148M and impaired TG mobilization from LDs.
Like other genes involved in TG metabolism, PNPLA3 is a target of the transcription factor sterol regulatory element binding protein 1c and is strongly up‐regulated by carbohydrate feeding.22 Despite the coordinate regulation of these genes, our data are not consistent with the notion that the fatty liver associated with PNPLA3 is due to an increase in endogenous synthesis of fatty acids or TGs. The incorporation of 3H‐H20 into fatty acids, PA, and TG was not increased in sucrose‐fed Pnpla3148M/M mice (Fig. 1).
An alternative possibility is that the I148M variant causes a decrease in VLDL‐TG secretion. Mutations in genes required for secretion of TG‐rich lipoproteins (VLDL) from the liver lead to decreased plasma TG levels and fatty liver.30, 31 Pirazzi et al.32 reported that PNPLA3‐148M is associated with reduced plasma TG levels in very obese individuals, suggesting the possibility that the steatosis is caused by a reduction in VLDL‐TG secretion. Rates of VLDL‐TG appearance into the circulation were essentially identical in WT and PNPLA3‐148M KI mice (Fig. 2). Whereas we cannot exclude the possibility that the PNPLA3‐148M is associated with a decrease in VLDL‐TG secretion under some conditions, our data are not consistent with an essential role for PNPLA3 in VLDL secretion.
In previous studies, we showed that the TG lipase activity of PNPLA3 is impaired by I148M substitution.5 In primary hepatocytes prepared from mice expressing WT and mutant human PNPLA3 transgenes, overexpression of the 148M protein was associated with a reduction in glycerol release.8 These findings are consistent with the hypothesis that I148M substitution impairs mobilization of TG from hepatic LDs. To further evaluate this hypothesis, we took advantage of the observation that mice subjected to prolonged fasting (24 hours) develop hepatic steatosis, which resolves rapidly upon refeeding.14 This response provides a bioassay for the mobilization of liver TG. The changes in liver TG associated with refeeding after a prolonged fast did not differ between KI and WT animals (Fig. 3). The result suggests that the 148M mutation does not impair TG mobilization in fasted animals. However, hepatic expression of PNPLA3 is very low in fasted mice. This finding, taken together with the observation that inactivation of Pnpla3 does not cause hepatic steatosis, is consistent with our prior observation that the PNPLA3‐I148M is not purely a loss‐of‐function mutation. PNPLA3‐148M expression is required for PNPLA3‐associated hepatic steatosis.
The model we have developed based on our finding that accumulation of PNPLA3‐148M on LDs impairs mobilization of TG from the droplets is congruent with the results of studies of PNPLA3 in cultured cells. We have shown previously that PNPLA3 is predominantly located on LDs and that expression of PNPLA3‐148M is associated with droplets of larger size and with impaired cellular TG hydrolysis.26 Chamoun et al.33 also found that PNPLA3 is targeted to LDs and that cells expressing PNPLA3‐148M have larger droplets. More recently, Ruhanen et al.34 found that PNPLA3‐148M is preferentially located on LDs compared with PNPLA3‐WT when expressed in cultured cells and that expression of the mutant protein is associated with reduced TG turnover. We found no evidence that PNPLA3 interferes with ATGL activity by displacing it from the droplet.
Previously, we showed that PNPLA3 is tightly regulated by dietary carbohydrates.22 PNPLA3 mRNA, which is expressed at very low levels during fasting, is robustly up‐regulated by food intake.22 In the present study, we performed the converse experiment and examined the effect on fasting on PNPLA3 levels in mice fed an HSD to compare the turnover of the protein in KI and WT mice (Fig. 4A). At the start of the experiment, the levels of PNPLA3 were >15‐fold higher in the KI mice than in the WT animals. After 6 hours of fasting, no PNPLA3 was detectable in the livers of the WT animals; in contrast, PNPLA3 levels remained high in the livers of KI mice. Thus, significant posttranslational regulation of PNPLA3 occurs in WT mice in association with fasting and refeeding; the corresponding changes in PNPLA3 protein levels are greatly blunted by the 148M variant.
Our present data indicate that the 148M variant enables PNPLA3 to evade proteasomal degradation. The 148M protein is under‐ubiquitylated relative to the WT protein (Fig. 5B and 6B). A similar accumulation of non‐ubiquitylated PNPLA3 was seen in the mice expressing PNPLA3‐47A (Fig. 6B). It is possible that these variants alter the conformation of PNPLA3 so that it is no longer recognized by the E3 ligase that ubiquitylates the WT protein. Both variants impair catalytic activity by decreasing the Vmax of the enzyme and neither is expected to impair substrate binding. 5 It is possible that the uncleaved substrate remains bound to the enzyme and traps it in a conformation that is not recognized by the E3 ligase. An alternative possibility is that these variants are rapidly de‐ubiquitinylated, thus resulting in accumulation of the protein.
It remains possible that the loss of catalytic activity of PNPLA3‐148M and –47A may trap the mutant proteins on LDs and prevent their translocation to the proteasome. Binding to LDs decreases proteasomal degradation of several LD proteins.35, 36 Accumulation of the 148M and S47A isoforms may reflect changes in the composition of the droplet surface that increase the affinity of PNPLA3‐148M for LDs. However, trapping due to changes in the droplet surface seems unlikely given that levels of other LD‐associated proteins, such as perilipin 2 and ATGL, were not increased in the KI mice (Fig. 4).
Protein degradation in eukaryotic cells is mediated by two mechanistically distinct systems: the 26S proteasome, which catalyzes the targeted degradation of tightly regulated, short‐lived proteins, and autophagy, which degrades long‐lived proteins and organelles.37 The observation that PNPLA3‐WT is rapidly turned over by proteasomal degradation is consistent with this model. Treatment with bortezomib increased PNPLA3 levels in WT mice, but even after 8 hours the levels of PNPLA3 in the treated animals failed to equal those seen in untreated Pnpla3Tg148M/+ mice. It is possible that the inhibition of proteasome activity by bortezomib is incomplete at the dose used in our study, but we cannot exclude the possibility that other degradation pathways contribute to PNPLA3 turnover. Levels of PNPLA3‐WT protein were not increased by administration of 3‐MA, arguing against a role for macroautophagy. However, we cannot rule out that under other conditions, autophagy may contribute to PNPLA3 degradation.38 Further studies to identify the specific residues on PNPLA3 that undergo poly‐ubiquitylation will be required to further elucidate the specific mechanisms by which the 148M variant evades degradation.
The present findings have therapeutic implications for FLD. If the mechanistic basis for PNPLA3‐148M‐associated steatosis is similar in mice and humans, then agents that decrease PNPLA3 protein levels are likely to be efficacious in carriers, but not in individuals with two WT alleles. Conversely, agents that inhibit PNPLA3 activity are likely to exacerbate steatosis in individuals with PNPLA3‐WT alleles. We have shown that human PNPLA3‐WT is ubiquitylated when expressed in mice (Fig. 5) or human liver cells22 and that the human PNPLA3‐148M isoform accumulates on LDs when expressed in the livers of mice.10 Therefore, it appears likely that the 148M substitution promotes steatosis by similar mechanisms in mice and humans.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29273/suppinfo.
Supporting Information
Acknowledgment
We thank Regeneron for generating the Pnpla3148M/M and Pnpla3 47A/A mice; Stephanie Spaeth for excellent care of the mice; Guosheng Liang and Luke Engelking for assistance with the tritium water incorporation studies; and Liangcai Nie, Fang Xu, Christina Zhao, Maritza Thomas, Linda Donnelly, and Angela Carroll for excellent technical support.
Potential conflict of interest: none
Soumik BasuRay was supported by a postdoctoral research fellowship award from the American Liver Foundation. Jonathan C. Cohen and Helen H. Hobbs were supported by grants RO1 DK090066 and PO1 HL20948 from the National Institutes of Health.
Contributor Information
Jonathan C. Cohen, Email: jonathan.cohen@utsouthwestern.edu
Helen H. Hobbs, Email: helen.hobbs@utsouthwestern.edu
REFERENCES
- 1. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science 2011;332:1519‐1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sookoian S, Castano GO, Burgueno AL, Gianotti TF, Rosselli MS, Pirola CJ. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Res 2009;50:2111‐2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Valenti L, Dongiovanni P, Ginanni Corradini S, Burza MA, Romeo S. PNPLA3 I148M variant and hepatocellular carcinoma: a common genetic variant for a rare disease. Dig Liver Dis 2013;45:619‐624. [DOI] [PubMed] [Google Scholar]
- 5. Huang Y, Cohen JC, Hobbs HH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J Biol Chem 2011;286:37085‐37093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Basantani MK, Sitnick MT, Cai L, Brenner DS, Gardner NP, Li JZ, et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res 2011;52:318‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen W, Chang B, Li L, Chan L. Patatin‐like phospholipase domain‐containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 2010;52:1134‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest 2012;122:4130‐4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab 2012;15:691‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015;61:108‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ferbitz L, Maier T, Patzelt H, Bukau B, Deuerling E, Ban N. Trigger factor in complex with the ribosome forms a molecular cradle for nascent proteins. Nature 2004;431:590‐596. [DOI] [PubMed] [Google Scholar]
- 12. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957;226:497‐509. [PubMed] [Google Scholar]
- 13. Moon YA, Hammer RE, Horton JD. Deletion of ELOVL5 leads to fatty liver through activation of SREBP‐1c in mice. J Lipid Res 2009;50:412‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guan HP, Goldstein JL, Brown MS, Liang G. Accelerated fatty acid oxidation in muscle averts fasting‐induced hepatic steatosis in SJL/J mice. J Biol Chem 2009;284:24644‐24652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP‐1a. J Clin Invest 1996;98:1575‐1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Millar JS, Cromley DA, McCoy MG, Rader DJ, Billheimer JT. Determining hepatic triglyceride production in mice: comparison of poloxamer 407 with Triton WR‐1339. J Lipid Res 2005;46:2023‐2028. [DOI] [PubMed] [Google Scholar]
- 17. Nasrin N, Wu X, Fortier E, Feng Y, Bare OC, Chen S, et al. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J Biol Chem 2010;285:31995‐32002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Engelking LJ, Kuriyama H, Hammer RE, Horton JD, Brown MS, Goldstein JL, et al. Overexpression of Insig‐1 in the livers of transgenic mice inhibits SREBP processing and reduces insulin‐stimulated lipogenesis. J Clin Invest 2004;113:1168‐1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Palma C, Morisi F, Cheli S, Pambianco S, Cappello V, Vezzoli M, et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis 2012;3:e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carmignac V, Svensson M, Korner Z, Elowsson L, Matsumura C, Gawlik KI, et al. Autophagy is increased in laminin alpha2 chain‐deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum Mol Genet 2011;20:4891‐4902. [DOI] [PubMed] [Google Scholar]
- 21. Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. J Biol Chem 2016;291:10659‐10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang Y, He S, Li JZ, Seo YK, Osborne TF, Cohen JC, et al. A feed‐forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci U S A 2010;107:7892‐7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zimmerman R, Strauss JG, Haemmerle G, Schoiswohl G, Birner‐Gruenberger R, Riederer M, et al. Fat mobolization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004;306:1383‐1386. [DOI] [PubMed] [Google Scholar]
- 24. Kaushik S, Cuervo AM. Degradation of lipid droplet‐associated proteins by chaperone‐mediated autophagy facilitates lipolysis. Nat Cell Biol 2015;17:759‐770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marvyn PM, Bradley RM, Button EB, Mardian EB, Duncan RE. Fasting upregulates adipose triglyceride lipase and hormone‐sensitive lipase levels and phosphorylation in mouse kidney. Biochem Cell Biol 2015;93:262‐267. [DOI] [PubMed] [Google Scholar]
- 26. He S, McPhaul C, Li JZ, Garuti R, Kinch L, Grishin NV, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem 2010;285:6706‐6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu G, Sztalryd C, Lu X, Tansey JT, Gan J, Dorward H, et al. Post‐translational regulation of adipose differentiation‐related protein by the ubiquitin/proteasome pathway. J Biol Chem 2005;280:42841‐42847. [DOI] [PubMed] [Google Scholar]
- 28. Seglen PO, Gordon PB. 3‐Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A 1982;79:1889‐1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blommaart EF, Krause U, Schellens JP, Vreeling‐Sindelarova H, Meijer AJ. The phosphatidylinositol 3‐kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 1997;243:240‐246. [DOI] [PubMed] [Google Scholar]
- 30. Schonfeld G, Patterson BW, Yablonskiy DA, Tanoli TS, Averna M, Elias N, et al. Fatty liver in familial hypobetalipoproteinemia: triglyceride assembly into VLDL particles is affected by the extent of hepatic steatosis. J Lipid Res 2003;44:470‐478. [DOI] [PubMed] [Google Scholar]
- 31. Wetterau JR, Aggerbeck LP, Bouma ME, Eisenberg C, Munck A, Hermier M, et al. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science 1992;258:999‐1001. [DOI] [PubMed] [Google Scholar]
- 32. Pirazzi C, Adiels M, Burza MA, Mancina RM, Levin M, Stahlman M, et al. Patatin‐like phospholipase domain‐containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol 2012;57:1276‐1282. [DOI] [PubMed] [Google Scholar]
- 33. Chamoun Z, Vacca F, Parton RG, Gruenberg J. PNPLA3/adiponutrin functions in lipid droplet formation. Biol Cell 2013;105:219‐233. [DOI] [PubMed] [Google Scholar]
- 34. Ruhanen H, Perttila J, Holtta‐Vuori M, Zhou Y, Yki‐Jarvinen H, Ikonen E, et al. PNPLA3 mediates hepatocyte triacylglycerol remodeling. J Lipid Res 2014;55:739‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nian Z, Sun Z, Yu L, Toh SY, Sang J, Li P. Fat‐specific protein 27 undergoes ubiquitin‐dependent degradation regulated by triacylglycerol synthesis and lipid droplet formation. J Biol Chem 2010;285:9604‐9615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takahashi Y, Shinoda A, Kamada H, Shimizu M, Inoue J, Sato R. Perilipin2 plays a positive role in adipocytes during lipolysis by escaping proteasomal degradation. Sci Rep 2016;6:20975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schreiber A, Peter M. Substrate recognition in selective autophagy and the ubiquitin‐proteasome system. Biochim Biophys Acta 2014;1843:163‐181. [DOI] [PubMed] [Google Scholar]
- 38. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008;4:151‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29273/suppinfo.
Supporting Information