

Editorial
Cholestasis is a pathological condition where normal bile flow out of the liver is reduced or disrupted, leading to intrahepatic accumulation of bile acids. High levels of toxic bile acids damage the bile duct epithelium and elevate biliary pressure to rupture the bile duct and expose hepatocytes to high concentrations of bile acids, macrophage infiltration, inflammation and leads to hepatocyte cell death. Cholestasis could result from genetic defects in canalicular transporters, mechanical obstruction of the bile duct by gallstones or tumors, pregnancy (intrahepatic cholestasis of pregnancy), autoimmune destruction of the bile ducts, or drug-induced liver toxicity (1, 2). Primary sclerosing cholangitis (PSC) and primary biliary cirrhosis (PBC) are two common types of progressive chronic cholestasis in humans. PSC is small bile duct inflammation and blockage causing accumulation of bile acids in hepatocytes, portal hypertension and biliary fibrosis and cirrhosis. PBC is a chronic autoimmune liver disease with progressive destruction of the bile ducts. The majority of PSC patients are male whereas most PBC patients are female.
Maintaining bile acid homeostasis is essential for protection against inflammatory liver diseases in the gastrointestinal tract, such as liver fibrosis and cirrhosis, non-alcoholic and alcoholic liver disease, liver cancer, diabetes, and inflammatory bowel diseases. Enterohepatic circulation of bile acids from the liver to the intestine and back to the liver occurs 6–8 times a day (Fig. 1). This physiological pathway not only regulates bile acid synthesis and maintains a constant bile acid pool, but also controls gut microbial overgrowth and intestinal barrier function. Bile acids are physiological detergents required for nutrient and drug absorption and are signaling molecules that activate farnesoid X receptor (FXR) and the membrane G protein-coupled bile acid receptor-1 (Gpbar-1 or TGR5) in the gastrointestinal tract. Activation of FXR and TGR5 by agonists is known to protect against inflammatory liver diseases. Activation of FXR induces small heterodimer partner (SHP), a negative transcription factor known to inhibit transcription of Cyp7a1, a key regulatory enzyme in the synthesis of cholic acid and chenodeoxycholic acid, two primary bile acids in human bile (Fig. 1) (3).
Fig. 1. Bile acid and LncRNA H19 signaling in hepatic fibrosis.
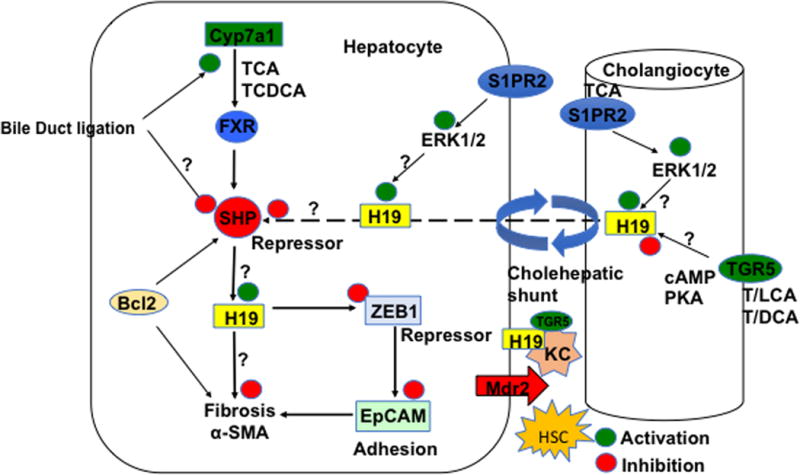
In hepatocytes, Cyp7a1 initiates bile acid synthesis from cholesterol to form cholic acid (CA) and chenodeoxycholic acid (CDCA) in human liver. Bile acids activate FXR to induce the repressor SHP, which inhibits H19 expression. In cholestasis, bile acids induce H19, which represses the repressor zinc finger E box-binding homeobox 1 (ZEB1). ZEB1 binds to and represses epithelial cell adhesion molecule (EpCAM) involved in fibrosis. This negative regulatory loop mediates bile acid-induced liver fibrosis. In this study, bile duct ligation induced Cyp7a1 but inhibited Shp mRNA expression. AAV8-H19 restored Cyp7a1 and Shp involved in bile acid synthesis. The anti-apoptotic factor Bcl1 suppresses SHP by promoting SHP decay and reduces H19 expression to protect against liver fibrosis. In hepatocytes and cholangiocytes, taurocholic acid activates the sphingosine-1-phosphate receptor 2 (S1PR2)/ERK1/2 pathway to induce H19. TGR5 is activated by secondary bile acid lithocholic acid and deoxycholic acid and is expressed in cholangiocytes and Kupffer cells, but not in hepatocytes. Differential regulation of these bile acid receptors may modulate H19 levels in hepatocytes. H19 may be shuffling between hepatocytes and cholangiocytes via the cholehepatic shunt to cause inflammation and cholestatic liver disease. Hepatic macrophages, Kupffer cells, and stellate cells (HSC) maybe also be involved in H19-mediated fibrosis. In bile duct-ligated mice and Mdr2−/− mice, bile acids induced H19 levels. Bile acid activation of TGR5 signaling may protect against cholestasis. The “?” indicates that the underlying mechanism is not clear and needs to be further studied.
Non-coding RNAs including microRNAs (miRNAs) and long non-coding RNAs (LncRNAs) play key roles in liver diseases and hepatocellular carcinoma (4). LncRNAs are about 200 nucleotides in length. Recent studies have linked LncRNA H19 (H19) to cholestatic liver injury and provide new insight into the mechanism and pathogenesis of cholestasis (5, 6). H19 is imprinted and maternally expressed during embryo development (7). H19 expression is low in adult human liver and is highly induced in livers with cholestatic fibrosis and cirrhosis, indicating H19 may be involved in the pathogenesis of liver fibrosis (5). H19 is induced by c-Myc oncogene in tumors (8) and anti-apoptotic protein Bcl2 activates caspase 8 to degrade SHP and results in increased H19 expression and bile acid pool size, and promotes hepatic fibrosis (5). Thus, bile acids may induce SHP to inhibit H19 expression as protection against cholestatic liver injury (Fig. 1).
In this issue of Hepatology, Song et al (9) further studied the mechanism of H19 in promoting cholestasis and liver fibrosis using bile duct ligation (BDL) -induced cholestasis in mice. They found that adenovirus associated virus 8 (AAV8)-mediated overexpression of H19 augmented BDL-induced liver fibrosis, increased bile acid levels and induced H19 expression. However, BDL markedly increased liver mRNA levels of Cyp7a1 but reduced Cyp8b1, Cyp7b1 and SHP involved in bile acid synthesis, and AAV8-H19 restored Cyp7a1 and Shp, but not Cyp8b1 and Cyp7b1 levels. Alteration of bile acid synthesis genes would alter bile acid composition and hepatic metabolic homeostasis. These data are in contradictory to their previous report that bile acids induced SHP to repress H19 (6). On the other hand, H19 produced in hepatocytes or cholangiocytes may inhibit SHP in hepatocytes (Fig. 1). This BDL+AAV8-H19 model may complicate the results in this study. Other mouse models, such as multi-drug resistant 2 deficient (Mdr−/−) cholestatic mouse model may be used to verify their finding. It is possible that sphingosine-1-phosphate receptor 2 (S1PR2) may induce H19 expression in hepatocytes. Further study is need to confirm whether selective activation of FXRSHP and S1PR2 by primary bile acids in hepatocytes, or TGR5 by secondary bile acids in Kupffer cells and cholangiocytes regulates H19 expression (Fig. 1). H19 decreased hepatic zinc finger E box-binding homeobox 1 (ZEB1) but increased epithelial cell adhesion molecule (EpCAM) expression in BDL mice. Either overexpression of ZEB1 or knockdown EpCAM ameliorates H19-induced fibrosis. Hepatic stellate cells (HSC) are activated to increase α-smooth muscle actin and fibrogenesis. α-smooth muscle actin is increased in H19-BDL mice and decreased in H19−/−-BDL mice, suggesting that activation of HSCs also contributed to H19-mediated fibrosis. Further mechanistic study revealed that ZEB1 repressed EpCAM promoter activity and gene transcription by binding to a ZEB1 binding site on the EpCAM gene promoter, and H19 interacted with ZEB1 protein to block its binding to the EpCAM promoter (Fig 1). Additional in vitro experiments show that bile acids induced H19 in mouse small cholangiocytes, but much less so in mouse large cholangiocytes, characteristic of inflammation in small bile ducts in PBC. In addition, they found that H19 expression levels were increased in human PSC and PBC livers. Consistently, EpCAM proteins are significantly induced in PSC, and even higher in PBC samples, while ZEB1 proteins were lower in PSC and PBC in livers with high H19. Thus, human patient liver specimens support the role of H19 in the pathogenesis of human cholestatic diseases. Overall, this study demonstrated that activation of hepatic H19 promoted cholestatic liver fibrosis in mice through the ZEB1/EpCAM signaling pathway. The dysregulation of bile acid homeostasis induces H19 in cholangiocytes and hepatocytes, and results in induction of EpCAM leading to biliary hyperplasia. This study provides a mechanistic insight into the role of LncRNA in cholestatic liver diseases. Further study is needed to show how H19 specifically interacts with the zinc finger on the ZEB1 protein and how H19 represses expression of ZEB1 and induces or represses of SHP, two repressors involved in the negative regulatory loop in the regulation of H19 expression (Fig. 1).
Another study published in Hepatology reports that H19 is markedly induced in female Mdr2−/− mice but not in aged-matched male Mdr2−/− mice at advanced stages of cholestasis, and estrogen might play a role in enhancing H19 expression in intrahepatic cholestasis (6). The taurocholate-activated S1PR2 may activate ERK1/2 to induce H19 by a mechanism that is not yet known. This study did not exam H19 expression in small and large cholangiocytes. The differential expression and induction of H19 by bile acids in different population of cholangiocytes is not known. It should be noted that Mdr2−/− mice is a model of PSC, a male- dominate cholestatic liver disease. The cause of gender disparity in PSC and PBC is not clear and requires further studies.
Currently there is no effective treatment for liver fibrosis, PSC, or PBC. Ursodeoxycholic acid (Urso) exhibits anti-inflammatory effects but is not effective for treating PSC. Nor-Urso promotes biliary HCO3− secretion to alleviate liver injury in Mdr2−/− mice. Fibrates, which activate peroxisomal proliferator-activated receptor α, have been used to treat PBC patients who do not respond to Urso. Obeticholic acid (OCA) has been approved recently to treat PBC. H19 LncRNA may be used as a biomarker for diagnosis of cholestasis and drugs targeting to reduce H19 expression in cholestasis may alleviate chronic cholestatic liver diseases.
Acknowledgments
This study is supported by NIH grants DK58379 and DK44442.
Abbreviations
- FXR
farnesoid X receptor
- LncRNA
long non-coding RNA
- Mdr2
multi-drug resistant 2
- PBC
primary biliary cirrhosis
- PSC
primary sclerosing cholangitis
- SHP
small heterodimer partner
- Urso
Ursodeoxycholic acid
Footnotes
Financial conflict of interest: Nothing to declare.
References
- 1.Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. 2014;4:25–36. doi: 10.1016/j.jceh.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zollner G, Trauner M. Mechanisms of cholestasis. Clin Liver Dis. 2008;12:1–26. vii. doi: 10.1016/j.cld.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 3.Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takahashi K, Yan IK, Kogure T, Haga H, Patel T. Extracellular vesicle-mediated transfer of long non-coding RNA ROR modulates chemosensitivity in human hepatocellular cancer. FEBS Open Bio. 2014;4:458–467. doi: 10.1016/j.fob.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Liu C, Barbier O, Smalling R, Tsuchiya H, Lee S, Delker D, et al. Bcl2 is a critical regulator of bile acid homeostasis by dictating Shp and lncRNA H19 function. Sci Rep. 2016;6:20559. doi: 10.1038/srep20559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Liu R, Yang J, Sun L, Zhang L, Jiang Z, Puri P, et al. The role of LncRNA H19 in gender disparity of cholestatic liver injury in Mdr2−/− mice. Hepatology. 2017 doi: 10.1002/hep.29145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poirier F, Chan CT, Timmons PM, Robertson EJ, Evans MJ, Rigby PW. The murine H19 gene is activated during embryonic stem cell differentiation in vitro and at the time of implantation in the developing embryo. Development. 1991;113:1105–1114. doi: 10.1242/dev.113.4.1105. [DOI] [PubMed] [Google Scholar]
- 8.Barsyte-Lovejoy D, Lau SK, Boutros PC, Khosravi F, Jurisica I, Andrulis IL, Tsao MS, et al. The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res. 2006;66:5330–5337. doi: 10.1158/0008-5472.CAN-06-0037. [DOI] [PubMed] [Google Scholar]
- 9.Song Y, Liu C, Liu X, Trottier J, Beaudoin M, Zhang L, Pope C, et al. H19 promotes cholestatic liver fibrosis by preventing ZEB1-mediated inhibition of EpCAM. Hepatology. 2017 doi: 10.1002/hep.29209. [DOI] [PMC free article] [PubMed] [Google Scholar]