

Abstract
Lower glycolysis involves a series of reversible reactions, which interconvert intermediates that also feed anabolic pathways. 3-phosphoglycerate (3-PG) is an abundant lower glycolytic intermediate that feeds serine biosynthesis via the enzyme phosphoglycerate dehydrogenase, which is genomically amplified in several cancers. Phosphoglycerate mutase (PGAM1) catalyzes the isomerization of 3-PG into the downstream glycolytic intermediate 2-phosphoglycerate (2-PG). Catalytic activity of PGAM1 requires its histidine phosphorylation. We show that the primary PGAM1 histidine phosphate donor is 2,3-bisphosphoglycerate (2,3-BPG), which is made from the glycolytic intermediate 1,3-bisphosphoglycerate (1,3-BPG) by bisphosphoglycerate mutase (BPGM). When BPGM is knocked out, 1,3-BPG can directly phosphorylate PGAM1. In this case, PGAM1 phosphorylation and activity are decreased, but nevertheless sufficient to maintain normal glycolytic flux and cellular growth rate. 3-PG, however, accumulates, leading to increased serine synthesis. Thus, one biological function of BPGM is to control glycolytic intermediate levels and thereby serine biosynthetic flux.
Graphical Abstract
INTRODUCTION
Glycolysis fulfills two fundamental tasks: anaerobic ATP generation and production of biomass precursors to support cell growth1. These two functions of glycolysis are balanced at multiple branching points along the glycolytic pathway. For example, glucose-6-phosphate can either pass through glycolysis for energy production, undergo isomerization to glucose-1-phosphate to support glycogen synthesis, or feed into pentose phosphate pathway (PPP) for the production of NADPH and ribose-phosphate, which are precursors for lipids and nucleotides, respectively. Extensive regulation of branching points is expected to be involved in balancing cellular needs. Such regulation can involve either local feedbacks or more distal interactions. For example, 3-PG was recently proposed to inhibit the PPP enzyme 6-phosphogluconate dehydrogenase (6PGD)2. Interestingly, 3-PG itself also sits at a branching point, which can either go to serine biosynthesis through phosphoglycerate dehydrogenase (PHGDH), or continue into glycolysis via conversion to 2-PG by the enzyme phosphoglycerate mutase 1 (PGAM1). Because of the genomic amplification of PHGDH and importance of serine in cancer2–6, there is particular interest in understanding how the partitioning of 3-PG between glycolysis and serine synthesis is controlled.
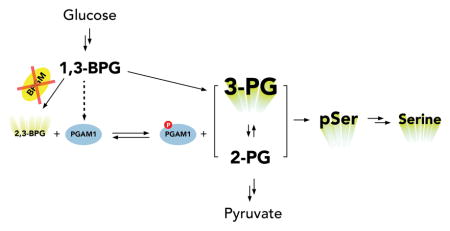
Even though most 3-PG is directed into lower glycolysis through PGAM1, very little is known about how this enzyme is activated in cells to carry out its glycolytic function. For PGAM1 to participate in glycolysis, it must first be primed through phosphorylation on His-11 within its active site (Fig. 1a)7,8. The textbook mechanism of this priming event is the donation of a phosphoryl group from the metabolite 2,3-bisphosphoglycerate (2,3-BPG)9,10. The production of 2,3-BPG can occur through the activity of bisphosphoglycerate mutase (BPGM), which catalyzes the rearrangement of the glycolytic intermediate 1,3-BPG11. BPGM has a well-known role in red blood cells where it is highly expressed12,13 and participates in the Luebering-Rapoport pathway that functions to generate high levels of 2,3-BPG for regulation of oxygen transport via direct binding to deoxyhemoglobin14,15. However, little is known about the importance of BPGM for maintaining PGAM1 activity and glycolytic flux. Yeast achieve high glycolytic flux in the absence of a dedicated BPGM enzyme, likely via direct phosphorylation of glycerate phosphomutase (GPM) by 1,3-BPG16,17. Moreover, BPGM expression is extremely low or undetectable in most mammalian cell types13, which challenges the notion that BPGM activity is the source for 2,3-BPG to activate PGAM1.
Figure 1. BPGM deletion diminishes cellular 2,3-BPG and PGAM1 phosphorylation.

a) Schematic depicting phosphoryl-transfer step between 3-PG, 2-PG and PGAM1. b) Western blot analysis of different mammalian cell lysates using an α-pHis antibody. Top panel shows untreated lysates and bottom panel shows lysates treated with hydroxylamine prior to Western blot analysis (see Supplementary Fig. 6 for Coomassie stain loading control). c) LC-MS analysis of 2,3-BPG levels in wt and BPGM knockout HEK 293T cells (n = 3, mean ± s.d., * = p<0.05). d) Western blot analysis of wt and BPGM knockout HEK 293T cells using an α–pHis or α–PGAM1 antibody (α–actin antibody was used as a loading control). e) Western blot analysis of wt and BPGM knockout HCT116 or MDA-MB-231 cells using an α–pHis or α–PGAM1 antibody (α–actin antibody was used as a loading control). f) LC-MS analysis of 2,3-BPG levels in wt and BPGM knockout HCT116 cells (n = 3, mean ± s.d., *** = p<0.001). See Supplementary Figure 23 for full Western blot images.
Given the well-defined role of 2,3-BPG in PGAM1 activation18, we sought to address the extent to which BPGM-mediated 2,3-BPG production serves as a regulator of PGAM1 activity and how this ultimately effects glycolysis. Specifically, we investigated 2,3-BPG generation and PGAM1 activation in transformed cultured cells, which like most tissues have minimal BPGM expression. By combining α-phosphohistidine (α-pHis) immunoassays, LC-MS-based metabolomic analysis, and CRISPR-mediated gene disruption of BPGM, we show that BPGM is responsible for generating the vast majority of 2,3-BPG. When BPGM is knocked out, both PGAM1 phosphorylation and protein levels drop but, surprisingly, glycolysis and cell growth continue unabated. While this demonstrates a primary role for 2,3-BPG in PGAM1 histidine phosphorylation, it also reflects the ability of an alternate phosphorylation source, 1,3-BPG, to phosphorylate and thereby activate PGAM1. Thus, cells have redundant capabilities to activate the key glycolytic enzyme, with either 2,3-BPG made by trace BPGM or 1,3-BPG made by glycolysis. We further observed that BPGM knockout cells display increased production of both phosphoserine and serine due to increased glucose to serine flux. Collectively, these results demonstrate that, at least in transformed cultured cells, BPGM primarily impacts 3-PG concentration and thereby serine pathway flux.
RESULTS
PGAM1 is phosphorylated by 2,3-BPG produced by BPGM
Using a recently developed α–pHis antibody19–22, we observed an abundant pHis-containing protein in lysates from a number of mammalian cell lines (Fig. 1b). The Western blot signal was lost upon treatment with hydroxylamine (HA), a reagent known to cause selective hydrolysis of phosphohistidine (Fig. 1b), confirming the identity of this modification20. The apparent molecular weight of this pHis-containing protein was consistent with PGAM1. To verify this, we subjected the pHis-protein band from HEK 293T cells to tryptic digestion followed by LC-MS/MS analysis. This confirmed our initial assignment and showed that phosphorylation was on the active site histidine, i.e. His-11 (Supplementary Results, Supplementary Fig. 1). These results demonstrate that PGAM1 is phosphorylated on histidine at detectable levels in mammalian cell lysates.
Next we turned to the question of how PGAM1 becomes histidine phosphorylated. The most plausible phosphoryl donor is 2,3-BPG, which is made from 1,3-BPG by BPGM. However, BPGM protein expression has only been detected in erythrocytes12,13 and placenta tissue23, which led us to investigate its levels in the HEK 293T cell line. We failed to detect BPGM protein in HEK 293T cells and the mRNA levels were found to be extremely low (Supplementary Fig. 2). The low expression prevented us from reliably targeting BPGM at the protein or RNA level for further study. As a result, we turned to CRISPR-Cas924 mediated gene disruption in order to study the physiological function of BPGM in glycolysis. Accordingly, we generated three unique clones of HEK 293T cells, all containing frameshift deletions in the BPGM coding sequence (Supplementary Fig. 3). We found that, despite the presumably infinitesimal amount of BPGM in HEK 293T cells, 2,3-BPG is dramatically depleted upon BPGM deletion (Fig. 1c). The depletion of 2,3-BPG results in complete loss of PGAM1 phosphorylation as detected by the α–pHis antibody (Fig. 1d). This demonstrates that BPGM activity is responsible for the vast majority of cellular 2,3-BPG production and PGAM1 phosphorylation. Furthermore, we found that depleting 2,3-BPG in the BPGM deletion cells lowered the PGAM1 protein levels, but not the PGAM1 mRNA levels (Supplementary Fig. 4). This observation is consistent with previous in vitro data that 2,3-BPG promotes the stability of the PGAM1 protein25. Importantly, re-expression of BPGM in BPGM deletion cells restored 2,3-BPG levels and increased both the PGAM1 protein level and phosphorylation level (Supplementary Fig. 5), showing that the production of 2,3-BPG and phosphorylation of PGAM1 is BPGM dependent.
To investigate whether this BPGM deletion effect extends to other mammalian cell lines, we generated three individual BPGM knockout clonal cell lines from human colon carcinoma HCT116 cells (HCT-ΔBPGM-1, HCT-ΔBPGM-2, HCT-ΔBPGM-3) as well as two polyclonal BPGM knockout cell lines from human breast adenocarcinoma MDA-MB-231 cells (MDA-ΔBPGM-1 and MDA-ΔBPGM-2). Disruption of BPGM in these cell lines resulted in a similar loss of PGAM1 histidine phosphorylation (Fig. 1e). We further analyzed each of the HCT116 BPGM knockout clonal cell lines and found that both 2,3-BPG and PGAM1 protein levels decreased compared to wt (Fig. 1f and Supplementary Fig. 4).
BPGM deletion does not affect the glycolysis rate
Having confirmed BPGM as the major source of cellular 2,3-BPG and PGAM1 phosphorylation, we next investigated the central question of whether the BPGM-induced perturbations on PGAM1 would impact glycolytic flux. To our surprise, although we observed dramatic loss of phosphorylated PGAM1, BPGM disruption did not change the rate of HEK 293T cell growth, nor the rates of glucose uptake or lactate production (Fig. 2a,b). A similar trend was observed in HCT116 cells (Supplementary Fig. 7). We also measured the growth rates of wt and BPGM disrupted HEK 293T and HCT116 cells under hypoxic conditions to determine whether the combination of an increased glycolytic demand and deficient BPGM impacts the growth rate, but we did not observe any changes (Supplementary Fig. 8).
Figure 2. BPGM deletion does not affect cell growth nor glycolytic flux.

a) Cell growth rates for wt and BPGM deficient HEK 293T cells (ΔBPGM-1, ΔBPGM-2, ΔBPGM-3) were determined by measuring packed cell volumes (PCV) over the indicated time course (n = 3, mean ± s.d.). b) Media glucose and lactate were measured at t = 24 hours for wt and BPGM deficient HEK 293T cells (ΔBPGM-1, ΔBPGM-2, ΔBPGM-3) and the consumption and production fluxes were calculated and plotted (n = 3, mean ± s.d.). c) 3-PG and 2-PG metabolite levels were quantified in wt and BPGM deficient HEK 293T cells (ΔBPGM-1, ΔBPGM-2, ΔBPGM-3) based on differences in MS/MS-based fragmentation patterns. (n = 3, mean ± s.d., * = p<0.05, ** = p<0.01, *** = p<0.001, n.s. = no significance).
Despite this result, we found evidence that BPGM deletion does indeed disrupt PGAM1 activity. Specifically, we observed higher mono-PG levels in ΔBPGM HEK 293T cells that could be rescued through re-expression of BPGM (Supplementary Fig. 9). Using the differential MS/MS fragmentation patterns of 3-PG and 2-PG (Supplementary Fig. 10), we further analyzed the total mono-PG pool and found that the increase in mono-PG is entirely due to increased 3-PG levels (Fig. 2c). This increase in 3-PG over 2-PG implies that PGAM1 has reduced turnover when BPGM is deleted. The reduced PGAM1 activity is likely compensated by the buildup of 3-PG, which drives the PGAM1 reaction forward into lower glycolysis.
PGAM1 can be directly phosphorylated by 1,3-BPG
When BPGM is disrupted, PGAM1 activity is dramatically decreased, but not completely eliminated as evidenced by the lack of change to the overall glycolytic flux. Also, while phosphorylated PGAM1 in ΔBPGM cells is below the detection limit, the overexpression of FLAG-PGAM1 in ΔBPGM cells does show the presence of PGAM1 histidine phosphorylation (Fig. 3a). This observation led us to consider alternate sources of PGAM1 phosphorylation that can occur in the absence of BPGM activity. It has recently been reported that PEP is able to indirectly phosphorylate PGAM1 on His-11 in HEK 293T hypotonic cell lysates26. Using our α-pHis antibody we were unable to detect PEP-induced phosphorylation of PGAM1 in aqueous buffer (Supplementary Fig. 11). We further investigated the ability of PEP to phosphorylate PGAM1 in hypotonic lysates using either Western blot analysis or 32P-PEP labeling and found that addition of PEP decreases, rather than increases, PGAM1 phosphorylation (Supplementary Fig. 11). This decrease in phosphorylation occurs because the addition of PEP in the lysates causes, through enolase activity, a temporary increase in mono-PG levels. Mono-PG + phosphorylated PGAM1 are in quasi-equilibrium with 2,3-BPG + dephosphorylated PGAM1 and, through its conversion to 2,3-BPG, mono-PG consumes PGAM1 phosphorylation (Supplementary Fig. 11). Collectively, these results show that PEP does not play a direct or indirect role in promoting PGAM1 histidine phosphorylation.
Figure 3. 1,3-BPG is an alternative source of PGAM1 phosphorylation.

a) Western blot analysis of PGAM1 overexpression lysates from wt and BPGM knockout HEK 293T cell lysates using an α–pHis and α–PGAM1 antibody (α–actin antibody was used as a loading control). b) Western blot analysis of a PGAM1 phosphorylation time course in the presence of 2,3-BPG or 1,3-BPG (membranes were stained with Coomassie to serve as a loading control). c) 1,3-BPG-treated PGAM1 was subjected to trypsinization and LC-MS analysis. Shown is the MS/MS spectrum of the tryptic peptide of PGAM1 containing the pHis site (His-11) and in mirror image is shown the MS/MS spectrum of 2,3-BPG induced phosphorylation of PGAM1 to demonstrate that both 1,3-BPG and 2,3-BPG result in His-11 phosphorylation. d) LC-MS analysis of PGAM1 phosphorylation with 1,3-BPG in the absence of 2-PG, low 2-PG (50 μM), or high 2-PG (1000 μM). Extracted ion chromatograms for 1,3-BPG and 2,3-BPG (m/z=264.952) are shown for each of the described reaction conditions. See Supplementary Figure 24 for full Western blot images.
Another potential phospho-donor is 1,3-BPG which is a highly reactive glycolytic intermediate involved in ATP production through phosphoglycerate kinase to generate 3-PG27. 1,3-BPG is not commercially available due to the instability of the acyl phosphate group. Accordingly, we prepared fresh 1,3-BPG using coupled enzymatic reactions (Supplementary Fig. 12): glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to convert glyceraldehyde-3-phosphate (GAP) and NAD+ into 1,3-BPG and NADH, which was recycled to NAD+ by lactate dehydrogenase (LADH) catalyzed reduction of pyruvate28. HPLC-purified 1,3-BPG was analyzed by LC-MS and shown to be free of 2,3-BPG and to serve as an effective substrate to phosphoglycerate kinase (PGK) (Supplementary Fig. 12), confirming that we prepared the desired bisphospho-metabolite. When incubated with purified 1,3-BPG, PGAM1 was histidine phosphorylated within seconds (Fig. 3b); as expected, the modification was hydroxylamine sensitive (Supplementary Fig. 13). LC-MS/MS analysis of the 1,3-BPG-treated PGAM1 confirmed that phosphorylation occurs on the canonical pHis site (His-11)29 in similar fashion to 2,3-BPG-treated PGAM1 (Fig. 3c).
1,3-BPG rapidly phosphorylates PGAM1 on the active site histidine, suggesting that 1,3-BPG can serve as an effective backup to 2,3-BPG for PGAM1 phosphorylation. After phosphorylation by 1,3-BPG, PGAM1 can donate its phosphate to mono-PG to produce 2,3-BPG (Fig. 3d and Supplementary Fig. 14). In similar fashion, BPGM’s catalytic cycle involves histidine phosphorylation by 1,3-BPG and donation to mono-PG to make 2,3-BPG8,30. Despite similarity in the mechanism, 1,3-BPG to 2,3-BPG conversion by PGAM1 is much less efficient, as reflected in loss of most cellular 2,3-BPG upon BPGM knockout. We were curious how direct phosphorylation of PGAM1 by 1,3-BPG could maintain adequate PGAM1 phosphorylation for glycolytic flux despite very low cellular 2,3-BPG in BPGM knockout cells. Accordingly, we measured the equilibrium constant between 2,3-BPG/free PGAM1 and mono-PG/phosphorylated-PGAM1 by titrating mono-PG in the presence of 1 μM 2,3-BPG and PGAM1 and monitored the PGAM1 phosphorylation levels by Western blot (Supplementary Fig. 14). The calculated Keq value of 70 (Supplementary Fig. 14) indicates that the in vitro equilibrium for 2,3-BPG reacting with PGAM1 favors the formation of phosphorylated PGAM1.
BPGM disruption causes increase in glucose-derived serine
Even though 1,3-BPG is capable of phosphorylating PGAM1, the enzyme remains hypophosphorylated when BPGM is deleted and the deficiency of cellular PGAM1 activity causes a buildup of 3-PG. 3-PG has been suggested as an inhibitor of the oxidative pentose phosphate pathway at the phosphogluconate dehydrogenase step2. We therefore set out to test the metabolic consequences of 3-PG accumulation by first addressing its impact on the oxidative pentose phosphate pathway. LC-MS-based metabolome profiling of HEK 293T cells showed no increase of phosphogluconate in BPGM deletion cells (Fig. 4a and Supplementary Fig. 15). We further investigated the impact of 3-PG accumulation on flux through the oxidative pentose phosphate pathway by monitoring the labeling pattern of ribose-5-phosphate (R5P) from cells fed with 1,2-13C glucose. However, we observed no difference in the R5P labeling pattern between wt and ΔBPGM cells (Supplementary Fig. 15). Therefore, 3-PG accumulation caused by BPGM deletion is not sufficient to inhibit the pentose phosphate pathway.
Figure 4. BPGM deletion results in higher serine de novo synthesis flux.

a) Heat map of metabolite levels measured by LC-MS in wt and BPGM deficient HEK 293T cells. Yellow and blue color scale indicates the changes to metabolite levels. b) Cellular serine production pathways. 13C6-glucose makes M+3 serine through fGlc (pathway 1). M+3 serine makes M+2 glycine and M+1 Me-THF through fSer→Gly (pathway 2). This same flux makes M+0 glycine and M+0 Me-THF from M+0 serine in DMEM. The M+0 and M+2 glycine plus M+0 and M+1 Me-THF makes Serine M+0−M+3 through fGly→Ser. c) A bar graph measuring the media serine concentration at t = 24 hours of each labeled fraction is plotted for wt and BPGM deficient HEK 293T cells (n = 3, mean ± s.d.). d) M+3 serine concentrations measured at different time points over 10 hours are plotted for wt (orange dots) and BPGM deficient (blue dots) HEK 293T cells. Based on the fluxes calculated from the 10 h time points only, the model predictions of M+3 serine concentration are plotted as single lines for wt (orange) and BPGM deficient (blue) HEK 293T cells. Error bars represent 95% CI (n=3). e) Serine and glycine biosynthetic fluxes in wt and BPGM deficient HEK 293T cells. Error bars represent 95% CI (n=3). f) Bar graph indicating the serine de novo synthesis flux from glucose (fGlc) for wt and BPGM deficient HEK 293T cells overexpressing active BPGM or catalytically inactive BPGM (H11A). Error bars represent 95% CI (n=3).
Interestingly, metabolomics analysis did reveal that phosphoserine and serine levels increased in all three of the BPGM deletion cell lines compared to the wild-type HEK 293T cells (Fig. 4a). These metabolic changes were BPGM-deletion-dependent, since the overexpression of active BPGM fully rescued this metabolic phenotype while overexpression of an inactive BPGM mutant (BPGM H11A) did not (Supplementary Fig. 16). In contrast, the overexpression of active PGAM1 could not decrease the mono-PG and phosphoserine levels back to wt levels (Supplementary Fig. 17) showing that elevating PGAM1 cannot completely compensate for BPGM disruption. Interestingly, the opposite perturbation of reducing PGAM1 protein levels in the presence of fully active BPGM in wild-type HEK 293T cells via shRNA knockdown caused a significant decrease to the amount of total PGAM1 protein but not the pool of activated PGAM1 (Supplementary Fig. 18). This is because the 2,3-BPG level remains stable in PGAM1 knockdown cells when active BPGM is present. As a result, no dramatic changes are observed to mono-PG, serine flux, or the oxidative pentose phosphate pathway in HEK 293T cells when only the PGAM1 levels are attenuated (Supplementary Fig. 18).
To further study the impact of BPGM deletion on de novo serine biosynthesis, we fed both wt and BPGM deletion HEK 293T cells with 13C6-Glucose, which generates M+3 serine (Fig. 4b). It was observed that BPGM deficient cells produce more M+3 serine compared to wt (Fig. 4c). This is consistent with our hypothesis that BPGM deletion leads to a higher rate of de novo serine biosynthesis from glucose (fGlc→Ser). In order to fully address this question, however, we considered all the sources of serine production. Upon addition of 13C6-Glucose, the generated M+3 serine can be made either from M+6 glucose through de novo serine synthesis (pathway 1) or from M+2 glycine + M+1 methylene-THF through serine hydroxymethyltransferase (SHMT) (pathway 2) (Fig. 4b). In addition, it is possible that the increased M+3 serine levels that we observe in the BPGM deletion cells could result from a decrease in the total net serine consumption. Therefore, in order to accurately determine fGlc→Ser in wt and BPGM deletion cells, we developed a series of differential equations describing the labeling kinetics with fluxes as the model parameters. Because glucose-derived serine de novo synthesis (pathway 1) only provides M+3 serine and SHMT flux (pathway 2) provides M+0, M+1, M+2, and M+3 serine, the flux parameters can be uniquely determined. Fluxes based on a single end point labeling measurement were sufficient to predict experimental measurements over earlier time points (Fig. 4d). The non-linearity of the M+3 serine fraction is predicted by the model, and underscores the value of using differential equations to model this process. Using this method, we detect a 60% higher fGlc→Ser in BPGM disruption cells (6.4 vs. 4.0 nmol/μl PCV/h), and we find that the SHMT forward and reverse fluxes (fGly→Ser and fSer→Gly) do not change with BPGM disruption (Fig. 4e). Importantly, the increased fGlc→Ser could be rescued to wt levels upon re-expression of catalytically active BPGM (Fig. 4f), indicating that BPGM activity controls de novo serine biosynthesis. This increase in serine biosynthesis flux in BPGM deletion cells led to a small but significant increase in purine de novo synthesis (Supplementary Fig. 19) demonstrating a downstream effect of BPGM activity on serine biosynthesis and ultimately purine production. Furthermore, the knockout of BPGM in HCT116 cells also resulted in increased serine precursor levels and de novo serine biosynthesis demonstrating that this effect is not unique to HEK 293T cells (Supplementary Fig. 20). Given the ability of BPGM disrupted cells to generate increased levels of pSer and serine over wt cells, we tested whether depleting both serine and glycine from the media would impact cell growth, but found that removing these two amino acids had no impact (Supplementary Fig. 21).
We were intrigued by the unaltered growth of BPGM deletion cells in normal, hypoxic or serine/glycine limited media conditions, especially since the disruption of this gene dramatically impacts 2,3-BPG and PGAM1 phosphorylation levels. Thus, we moved to a xenograft tumor model using BPGM disrupted HCT116 cells to see whether the lack of BPGM activity impacts the ability of tumor cells to grow in an in vivo setting. Compared to wt HCT116 cells, we observed minor but significant growth impairment in BPGM deletion mouse tumors (Supplementary Fig. 22); the small magnitude of the effect demonstrates that BPGM, although it may subtly facilitate glycolysis or metabolic balance, does not play an essential role in in vivo tumor growth. Collectively, these results demonstrate an underappreciated robustness in lower glycolysis.
DISCUSSION
Histidine phosphorylation is a required step for activating PGAM1 catalytic activity and has been known for several decades10. How PGAM1 is phosphorylated in cells, however, has remained less clear. Here we take advantage of recent developments in phosphohistidine antibody research20,21,31, metabolomics analysis32, and targeted gene disruption strategies24 to revisit this fundamental question. Our initial analysis of PGAM1 phosphorylation in different mammalian cell lines indicated that PGAM1 is phosphorylated on histidine at high enough levels for detection by Western blot (Fig. 1b). The fact that all tested cell lines showed strong PGAM1 histidine phosphorylation suggested that abundant phospho-PGAM1 is required to sustain glycolysis for normal cell function. Using the techniques described above, we sought to fully characterize the basis for PGAM1 phosphorylation in a non red-blood cell system by specifically addressing the ability of the known PGAM1 phosphodonor, 2,3-BPG, to activate PGAM1 through histidine phosphorylation. We therefore focused our attention to modulating cellular levels of 2,3-BPG by targeting BPGM, the enzyme responsible for its production, by gene disruption. Previous reports13, as well as our own analysis of HEK 293T cells, indicate extremely low levels of BPGM expression in non-red blood cells, leading to doubts on the role of BPGM in 2,3-BPG production. However, to our surprise, most PGAM1 histidine phosphorylation was lost, and PGAM1 protein levels were reduced, when the BPGM gene was disrupted. This correlated with a dramatic loss of 2,3-BPG indicating that, despite extremely low expression levels, BPGM is playing the primary role in generation of 2,3-BPG for PGAM1 activation. Furthermore, we observed similar effects in HCT116 cells as well as loss of PGAM1 phosphorylation in BPGM-disrupted MDA-MB-231 cells demonstrating that this effect may be general across mammalian cells.
Remarkably, in HEK 293T and HCT116 cell lines we observed no differences in the glycolysis or cell growth rates between wt and BPGM deletion cells, despite the profound changes to 2,3-BPG levels and PGAM1 phosphorylation. Given that the PGAM1 mutase reaction to convert 3-PG to 2-PG is reported to be extremely fast (>1000 s−1)33, it is likely that only a small amount of activated PGAM1 is required to sustain the glycolytic flux. Thus, the fact that, in HEK 293T and HCT116 cells, greatly reduced levels of PGAM1 phosphorylation in the BPGM deletion cells is sufficient to sustain normal levels of glycolysis raises an obvious and bigger question of what, if anything, is BPGM doing? Conceivably, its metabolic activity may be necessary for growth in a different cellular type or context, or play a distinct physiological role, such as in enabling rapid glycolysis to support burst anaerobic exercise. A number of cancer cell lines have been shown to be more dependent on PGAM1 protein levels and/or activty2,18, highlighting the possibility that BPGM-mediated 2,3-BPG production may play a more prominent role in sustaining PGAM1 activity, glycolysis, and/or cell growth in other cellular types or environments outside of the HEK 293T and HCT116 cell models that we tested.
The observation that glycolysis proceeds normally (Fig. 2b) and that PGAM1 can still be phosphorylated on histidine (Fig. 3a) in the absence of BPGM led us to consider other routes that activate PGAM1 through histidine phosphorylation. We tested both PEP and 1,3-BPG based on previous reports that these small molecules can phosphorylate PGAM126,33. Our results show that PEP is not involved in direct phosphorylation and, rather, can promote the hydrolysis of phosphorylated PGAM1 (Supplementary Fig. 11). In the case of 1,3-BPG, we directly show that this phosphorylation can rapidly occur on the active site histidine. While it has been previously suspected that 1,3-BPG may serve to activate PGAM1 in the absence of 2,3-BPG34, we now provide both biochemical and cellular evidence that this indeed can be the case. Moreover, our results indicate that 1,3-BPG acts as an alternate source of PGAM1 phosphorylation that is sufficient to support normal glycolytic flux in the presence or absence of BPGM activity.
BPGM deletion also leads to the upregulation of serine de novo synthesis. Serine is not only used for protein synthesis, but also provides one-carbon units to sustain nucleotide biosynthesis. Phosphoglycerate dehydrogenase (PHGDH) is the rate-limiting enzyme in the serine de novo synthesis pathway and is frequently amplified in tumors3,35. Loss of BPGM appears to mimic the effect of PHGDH amplification, leading to an increase of incorporation into nucleotides of one-carbon units from glucose-derived serine (Supplementary Fig. 19). Furthermore, though 2-PG has been proposed to activate PHGDH in different cancer cell lines2, our results in HEK 293T BPGM deletion cells showed that both phosphoserine and serine levels increased even though 2-PG remained unchanged. The increased levels of 3-PG that we observed in HEK 293T BPGM deletion cells suggested a simple mechanism in which accumulation and overflow of 3-PG causes serine de novo synthesis upregulation (Fig. 5). Therefore, BPGM is dispensable in maintaining glycolytic flux, but is indispensable for 3-PG and serine homeostasis. Though it appears that 1,3-BPG can phosphorylate a smaller fraction of PGAM1 to support normal glycolytic rate, it is not sufficient to maintain normal 3-PG levels and thereby properly tune glycolysis-driven anabolism.
Figure 5. Effect of BPGM disruption on lower glycolysis.

a) BPGM activity converts 1,3-BPG to 2,3-BPG for activation of PGAM1 via histidine phosphorylation. b) Upon BPGM disruption, 2,3-BPG levels and PGAM1 protein and phosphorylation levels decrease without impacting overall glycolysis likely because of 1,3-BPG mediated phosphorylation of PGAM1. Furthermore, as a result of decreased PGAM1 protein and phosphorylation levels, 3-PG, phosphoserine, and serine metabolite levels increase.
Online Methods
General materials
All buffering salts, sodium dodecyl sulfate, and sodium deoxycholate were purchased from Fisher Scientific. HPLC-grade MeOH (646377), Coomassie brilliant blue, bovine serum albumin (BSA), hydroxylamine (HA), dithiothreitol (DTT), iodoacetamide, bromoethylamine, 2,3-BPG (D5764), 2-PG (79470), 3-PG (P8877), PEP (860077), 2-Phosphoglycolic acid, ADP, ATP, NAD, NADH, rabbit pyruvate kinase, yeast enolase, rabbit lactate dehydrogenase, formic acid, A431 cells, Fetal Bovine Serum (F4135), Anti-FLAG M2 antibody (catalog number: F3165-.2MG), and Dialyzed Fetal Bovine Serum (F0392) were purchased from Sigma-Aldrich. D-Glucose (U-C13, 99%), and D-Glucose (1,2-13C2, 99%) were from Cambridge Isotope Laboratories. D-[5-2H]-Glucose was from Omicron Biochemicals. NP-40, Pierce BCA Protein Assay kit, and anti-BPGM antibody (PA5-21821) were purchased from Thermo Scientific. 12% Criterion TGX gels, 4–20% Criterion Tris-HCl gels, 0.2 μm PVDF membrane, and Micro Bio-spin P-6 gel desalting columns were purchased from BioRad. Goat anti-rabbit 800CW and goat anti-mouse 680RD secondary antibodies were from LI-Cor Biosciences. Trypsin (Sequencing grade modified trypsin) was from Promega. Anti-β-actin mouse monoclonal antibody (ab8226) and anti-PGAM1 rabbit monoclonal antibody (ab129191) were from Abcam. Packed cell volume tubes (87005) were from TPP. HEK 293T, HeLa, HCT116, MDA-MB-231, and 4T1 cells were purchased from ATCC. U2OS cells were a kind gift from the Cristea Lab (Princeton University).
Statistics
Unless noted otherwise, the data are presented as the mean ± standard deviation (s.d.) of n number of independent experiments, and the P values were calculated using a two-tailed Student’s t-test. The 95% confidence interval (95% CI) was calculated using the bootstrap method. Statistics were generated using R or Microsoft Excel.
General cell culturing methods
Unless otherwise stated, all cell lines were cultured in DMEM (4.5g/L Glucose, 110 mg/L Pyruvate, Gibco product number 11995-065) supplemented with 10% FBS (Sigma) and penicillin/streptomycin (Gibco). Cells were grown in an incubator at 37 °C with 5% CO2 in 10-cm dishes (Falcon, product number 353003), 6cm dishes (Falcon, product number 353004), or 6-well plates (Falcon, 353046) as described below. For passaging, all cells were detached from the plate using 0.05% Trypsin-EDTA (Gibco). All transfections were performed using Opti-MEM (31985-088) from Gibco and Lipofectamine 2000 from Invitrogen as described below.
General Western blotting method
Samples were diluted into 4× basic loading buffer (160 mM Tris, pH 8.5, 40% (v/v) glycerol, 4% (w/v) SDS, 0.08% (w/v) bromophenol blue, and 8% (v/v) BME) and resolved by SDS-PAGE. Unless otherwise noted, samples were loaded onto a 12% TGX gel in Tris-Glycine running buffer (25 mM Tris base, 192 mM glycine, pH 8.3) and run for 10 min at 110 V and then for 50 min at 175 V. Proteins from the gel were then electroblotted onto a PVDF membrane in Towbin buffer (25 mM Tris base, 192 mM glycine, pH 8.3, 10 % (v/v) methanol) at 0.75 A for 1 hour. The membrane was blocked with 3% BSA in wash buffer (25 mM Tris, 137 mM NaCl, 2.7 mM KCl, 0.1% (v/v) Tween-20, pH 8.5) for 1 hour at room temperature. The membrane was then incubated overnight at 4 °C with primary antibody at the following dilutions: affinity-purified rabbit anti-pHis antibody (prepared as described previously21) at 1:100 dilution, rabbit anti-PGAM1 antibody at 1:1,000 dilution, mouse anti-B-actin at 1:1,000 dilution. Primary antibodies were diluted in wash buffer with 3% BSA. The membrane was then washed with wash buffer (3 × 5 min) and incubated with goat anti-rabbit IRDye 800CW or goat anti-mouse IRDye 680RD antibody conjugate (diluted 1:15,000 in wash buffer with 3% BSA) for 1 hour at RT. The membrane was washed with wash buffer (3 × 5 min), drained, and washed three times with MQ-H2O. The membrane was then imaged using a Li-COR Odyssey Infrared Imager. In cases where Coomassie staining was used to assess protein loading, the membrane was then washed with water, stained with Coomassie blue, and imaged on an Epson scanner.
General LC-MS/MS of pHis proteins
Peptides were subjected to a LC-MS data acquisition and analysis methodology specifically designed to aid in the identification and characterization of pHis-containing peptides and other labile phosphospecies, as we have previously described21. Briefly, concentrated peptides were resuspended in 1 mM ammonium bicarbonate, pH 8. Samples were subjected to high-resolution nano-UPLC-MS on an Orbitrap Elite platform (ThermoFisher Scientific) outfitted with a nanoACQUITY nano-flow UPLC system (Waters) and a customized PicoView ion source (New Objective). In-line sample trapping and desalting was performed on a capillary trap column (150 μm × 5 cm) packed with ReproSil-Pur C18-AQ 5 μm/120 Å resin (Dr. Maisch, ESI Source Solutions, LLC.), while separations were performed on a capillary column (75 μm X approx. 25 cm) packed with ReproSil-Pur C18-AQ 3 μm/120 Å resin (Dr. Maisch) over 2 hour elution gradients (from 5–35% acetonitrile in water with 0.1% formic acid) at a flow rate of 200 nL/min. Nanospray ionization/desolvation was achieved at the Elite using a voltage of 2.0 kV and a heated capillary temperature of 250 °C. Samples were subject to initial survey runs in which CID MS/MS was conducted in the LTQ linear ion trap on the top 20 most abundant multiply charged positive-ion species, interspersed with full-scan (m/z 360–1800) high resolution mass spectra acquired in the Orbitrap at a resolution setting of 60,000. Analysis of the CID MS/MS spectra for the triplet neutral loss pattern (−80 Da, −98 Da, and −116 Da off the parent ion) that we have shown to be associated with phosphohistidine species was conducted using the software TRIPLET21. Lists of parent ion species that produced triplet-positive spectra were used to direct subsequent targeted nano-UPLC-MS/MS runs in which multistage activation CID was used to target the parent ions and their −98 Da and −80 Da neutral loss species. Analysis was restricted to 2+ and 3+ ions only. LC-MS data acquisition was carried out at the Harvard University Faculty of Arts and Sciences (FAS) Mass Spectrometry and Proteomics Resource Laboratory (proteomics.fas.harvard.edu).
Raw data files were subject to database search using the Mascot search engine (v. 2.4, Matrix Science) in the context of ProteomeDiscoverer (v. 1.4, ThermoFisher). Searches were conducted against the SwissProt Human database, allowing for a parent ion mass window of ±6 ppm. For the 25 kDa protein band sample (PGAM1 from 293 cells), the following parameters were used: ≤ 3 missed trypsin cleavages, histidine phosphorylation, methionine oxidation and N-terminal protein acetylation as variable modifications, and carbamidomethylation of cysteines as a fixed modification. For purified PGAM1 samples from in vitro experiments, fixed modification of cysteine was omitted. Mass spectra were visualized using Xcalibur (v. 2.2, ThermoFisher), ProteomeDiscoverer and Scaffold (v. 4.4.8, Proteome Software). All phosphohistidine-bearing peptide assignments were further validated by manual inspection.
General metabolomic LC-MS analysis
Metabolites were extracted with cold 80% MeOH containing 0.5% formic acid preincubated on dry-ice. Typically, 400 μL extract was taken and dried down under N2 gas. The extracted metabolites were then reconstituted in 100 μL of water for LC-MS analysis. The LC–MS method involved reversed-phase ion-pairing chromatography coupled with negative mode electrospray ionization to a stand-alone orbitrap mass spectrometer (Thermo Scientific) scanning from m/z 85–1,000 at 1 Hz at 100,000 resolution with LC separation on a Synergy Hydro-RP column (100 mm × 2 mm, 2.5 μm particle size, Phenomenex) using a gradient of solvent A (97%:3% H2O:MeOH with 10 mM tributylamine and 15 mM acetic acid), and solvent B (100% MeOH). The gradient was 0 min, 0% B; 5 min, 0% B; 7 min, 20% B; 17 min, 80% B; 20 min, 100% B; 23.5 min, 100% B; 24 min, 0% B; 30 min, 0% B. The flow rate was 0 min, 200 μl min−1; 20 min, 200 μl min−1; 20.5 min, 300 μl min−1; 29.5 min, 300 μl min−1; 30 min, 200 μl min−1. Injection volume was 10 μL and column temperature 25 °C. Data were analyzed using the MAVEN software suite.
Western blot analysis of mammalian cell lysates
HEK 293T, A431, 4T1, HeLa, U2OS, and FLAG-PGAM1 overexpressing HEK 293T cells were grown in 10-cm dishes to a confluency of 80%. Just prior to lysis, the DMEM was removed and replaced with fresh DMEM for 30 minutes. The media was then removed followed by addition of 1 mL of RIPA buffer (25 mM Tris pH 8.0, 150 mM NaCl, 1%NP40, 1% deoxycholate, 1% SDS) to each plate. The cells were dissolved into the RIPA buffer using a cell scraper and then transferred to an Eppendorf tube and sonicated on ice for 5 seconds and 45% power (repeated for a total of three times). The samples were then mixed with 4× loading buffer (160 mM Tris, pH 8.5, 40% (v/v) glycerol, 4% (w/v) SDS, 0.08% (w/v) bromophenol blue, and 8% (v/v) BME) and analyzed by Western blot according to the General Western blotting method described above. 25 μg of each lysate was used for the analysis. Protein concentration was measured for each lysate using the BCA protein assay kit according to manufacturer’s instructions.
Mass spectrometric analysis of PGAM1 histidine phosphorylation in HEK 293T cells
SDS-PAGE gel bands corresponding to the 110 kDa and 25 kDa MW regions from HEK 293T cell lysate were excised and subjected to in-gel reduction and alkylation and trypsin digestion following the procedure of Shevchenko, et al.36 with the following alterations. All steps were performed at room temperature. For analysis of the 25 kDa MW protein band, the lysate was resolved on a 12% TGX gel, and a gel piece containing the MW region of interest was excised and then reduced by treating with DTT for 30 min (15 mM dissolved in 20 mM Tris, pH 8.5). Thiol alkylation consisted of a 30-min treatment with iodoacetamide (20 mM in 20 mM Tris, pH 8.5). The alkylation reaction was quenched by treatment with DTT (75 mM dissolved in 20 mM Tris, pH 8.5). Trypsin digestion was accomplished over 4 hours, using 2 μg enzyme in 20 mM Tris, pH 8.5. Extraction of peptides from the gel was achieved using acetonitrile and 20 mM Tris pH 8.5, and samples were concentrated and subject to desalting using STAGE-tips,37 as previously described21, then stored at −80 °C until LC-MS/MS analysis. LC-MS/MS was performed at the Harvard University Faculty of Arts and Sciences (FAS) Mass Spectrometry and Proteomics Resource Laboratory, as described above in the General LC-MS/MS analysis of pHis proteins.
Analysis of BPGM in wt 293T cells
HEK 293T cells were grown in a 10-cm dish to 80% confluency and then lysed in 1 mL of RIPA buffer (25 mM Tris pH 8.0, 150 mM NaCl, 1% NP40, 1% deoxycholate, 1% SDS) and sonicated (1×5s at 45% power). 25 μg of the lysate was the analyzed by Western blot using an anti-BPGM antibody as described above in the General Western blotting method with a 1:1000 dilution in wash buffer with 3% BSA and a 1 hour primary antibody incubation step. RNA levels of BPGM were obtained by global RNA sequencing as described below.
RNA-Seq analysis of wt and BPGM deficient 293T cells
Wt or BPGM knockout HEK 293T cells were grown to 80% confluency in 10-cm dishes and harvested in cold PBS. RNA was obtained using RNeasy (Qiagen) according to manufacturer’s protocol. The purified RNA was quantified and analyzed at the microarray facility (Princeton University). Library formation, sequencing, and data analysis were performed as described previously.38 The average gene expression for BPGM, PGAM1, GAPDH, PGK1, and ENO1 were obtained and plotted on a log2 scale.
Generation of BPGM deficient HEK 293T cells
BPGM knockout cells were generated in HEK 293T cells following the protocol described by Ran et. al.24 Briefly, sgRNA sequences that target the BPGM gene were obtained using online software (www.dna20.com/eCommerce/cas9/input) with the following input parameters: species: homo sapiens, PAM sequence: NGG, Nickase was highlighted, gRNA offset: 4–8 base pairs. The following gRNA sites were obtained from this search:
Guide Site 1a: 5′-GTGACGCTCATTTAGACGCC-3′ (targets bottom strand)
Guide Site 1b: 5′-GGGCCTTGATCGGTCTCAAC-3′ (targets top strand)
Guide Site 2a: 5′-TCCTTTAAGCTTTCCGACCG -3′ (targets bottom strand)
Guide Site 2b: 5′-TGGAGAGACTCCTTCCCTAT -3′ (targets top strand)
The guide Oligos were synthesized by IDT. The oligos were phosphorylated, annealed, and inserted into pSpCas9(BB)-Puro plasmid following the published protocol24 to give the following four plasmids (1a, 1b, 2a, 2b).
HEK 293T cells were plated into a 24-well plate containing 1 mL of DMEM and grown to a confluency of 30–50%. 40 μL of Opti-MEM was mixed with 0.5 μg of pSpCas9(BB)-Puro plasmid from 1a and 1b combined OR plasmid from 2a and 2b combined and then each was further combined with 40 μL of Opti-MEM containing 2 μL of lipofectamine and incubated for 30 min at room temperature. The media in the 24-well plate was removed and the cells were washed with 250 μL of Opti-MEM. Fresh 250 μL of Opti-MEM was then added and the lipofectamine and plasmid solution was added dropwise to the cells and they were incubated for 6 hours at 37 °C. The media was then removed and replaced with DMEM and incubated for 12 hours. The media was then removed and replaced with complete DMEM plus puromycin (Gibco, product number A11138-03, 1:5000 dilution) and grown for 48 hours.
After growing 48 hours in DMEM containing puromycin, the cells were washed 1× with 500 μL DMEM. The cells were then resuspended in 2 mL of DMEM and diluted to 10 cells/mL and added to a 96-well plate (Corning, product number 3603) in 100 μL aliquots. The plates were incubated at 37 °C until wells containing single cell colonies could be identified and transferred to 24-well plates for further cell growth and subsequent DNA analysis. DNA samples from individual colonies were generated using DNA Quick Extract solution (QE09050) from Epicentre according to manufacturer’s instructions.
The clones were screened by PCR and Sanger sequencing. The BPGM locus was amplified using primers XSp228 (5′-GCCCATTTGAAAACGCCTGG-3′) and XSp230 (5′-GTTTTAGGAGTGCCCTACTGC-3′). All PCR was performed using AccuPrime™ Pfx DNA Polymerase (ThermoFisher Scientific). 20 ng genomic DNA was used as the template and 1 μM of each primer was added to 50 μL PCR reaction. The annealing temperature was 58 °C and the extension time was 1 min. The PCR products were cleaned up with Wizard® SV Gel and PCR Clean-Up System (Promega). The purified DNA was submitted for sequencing at GENEWIZ. The PCR primers were used as sequencing primers. Due to the presence of two alleles, the sequencing traces must be deconvoluted. The online tool TIDE (Tracking of Indels by Decomposition) was used for this purpose39. The clones having a wt allele or having alleles with deletion sizes that are multiple of 3 were discarded at this point. The remaining clones were subcloned to confirm the deletion sequence.
The BPGM locus was amplified from selected clones using the primers
XSp233 (CAGTGAATTCGAGCTCGGTACCCGGGGATCCGCCCATTTGAAAACGCCTGG)
XSp235 (TTACGCCAAGCTTGCATGCCTGCAGGTCGACGTTTTAGGAGTGCCCTACTGC).
The pUC19 plasmid was linearized using BamHI and SalI. The amplified BPGM was ligated into the vector using Gibson Assembly® Master Mix (New England Biolabs) following standard protocols. Typically, for each HEK 293T clone, 36 colonies were picked and the plasmids were extracted and sequenced. Three HEK 293T clonal cell lines that each contained BPGM disruption sites resulting in a frameshift were identified by sequencing and labeled as ΔBPGM-1, ΔBPGM-2, and ΔBPGM-3. Two unique cut sites were identified for ΔBPGM-1 and ΔBPGM-3 and three cut sites were identified for ΔBPGM-2 (Supplementary Fig. 3).
Genotyping of BPGM-deficient HEK 293T cell lines
For each of the clones, the genomic DNA was extracted using DNA Quick Extract solution (QE09050) from Epicentre according to manufacturer’s instructions. 20 ng genomic DNA was used as the template and 1 μM of each primer was added to 50 μL PCR reaction. All PCR was performed using AccuPrime™ Pfx DNA Polymerase (ThermoFisher Scientific). The annealing temperature was 58 °C and the extension time was 1 min. The primers used for each clones are:
| ΔBPGM-1: | XSp248 (5′-GCTCCTGGCGTCTAAATGAGC-3′) |
| XSp230 (5′-GTTTTAGGAGTGCCCTACTGC-3′) | |
| ΔBPGM-2: | XSp257 (5′-GTCTAAATGAGCGTCACTATGG -3′) |
| XSp230 (5′-GTTTTAGGAGTGCCCTACTGC-3′) | |
| ΔBPGM-3: | XSp228 (5′-GCCCATTTGAAAACGCCTGG -3′) |
| XSp249 (5′-AGAACATCCTTTAAGCTTTCCG -3′) |
The PCR products were resolved on 1% agarose gel in 1× TAE buffer (40 mM Tris, 20 mM acetic acid, and 1 mM EDTA). The DNA bands were stained with ethidium bromide and visualized on a UV transilluminator (Supplementary Fig. 3).
Generation of BPGM deficient HCT116 cells
BPGM knockout cells were generated in HCT116 cells and screened by PCR and Sanger sequencing exactly as described above for HEK 293T cells.
The BPGM locus was amplified from selected clones using the following primers:
XSp233 (CAGTGAATTCGAGCTCGGTACCCGGGGATCCGCCCATTTGAAAACGCCTGG)
XSP235 (TTACGCCAAGCTTGCATGCCTGCAGGTCGACGTTTTAGGAGTGCCCTACTGC)
The amplified BPGM locus was inserted into the pUC19 plasmid as described above for HEK 293T cells. Three HCT116 clonal cell lines that each contained BPGM disruption sites resulting in a frameshift were identified by sequencing and labeled as ΔBPGM-1, ΔBPGM-2, and ΔBPGM-3. For each of the cell lines, only one unique cut site was identified.
ΔBPGM-1
The BPGM gene was identified to have an 11 base pair deletion (273–283). A portion of the BPGM is shown below with the missing sequence containing a double strikethrough.
GTGCCTGTGGAAAGCTCCTGGCGTCTAAATGAGCGTCA
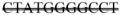 TGATCGGTCTCAACAGGGAGCAGATGGCTTTGAATCATGGTGA
TGATCGGTCTCAACAGGGAGCAGATGGCTTTGAATCATGGTGA
ΔBPGM-2
The BPGM gene was identified to have a missing base pair at position 273 which is replaced with a 105 base pair insertion. A portion of the BPGM is shown below with the inserted sequence shown in underline.
GTGCCTGTGGAAAGCTCCTGGCGTCTAAATGAGCGTCATATTCCTGAAACAACGAATTGCTGTGTGTAGTCTTTGCCCATCTCCCACGATGGGCTTTTTTTTAACATTTTTCCGCTTCGCTACCTCGCCCCCATATGGGGCCTTATGGGGCCTTGATCGGTCTCAACAGGGAGCAGATGGCTTTGAATCATGGTGA
ΔBPGM-3
The BPGM gene was identified to have an 11 base pair deletion (267–277). A portion of the BPGM is shown below with the missing sequence containing a double strikethrough.
GTGCCTGTGGAAAGCTCCTGGCGTCTAAATGA
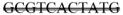 GGGCCTTGATCGGTCTCAACAGGGAGCAGATGGCTTTGAATCATGGTGA
GGGCCTTGATCGGTCTCAACAGGGAGCAGATGGCTTTGAATCATGGTGA
Generation of BPGM deficient MDA-MB-231 cells
MDA-MB-231 cells containing BPGM gene disruption were generated by lentiviral delivery of pLentiCRISPR v2 plasmid (Addgene, 52961) expressing Cas9 and BPGM targeting gRNA (guide site 2a or 2b sequences were used, see Generation of BPGM deficient HEK 293T cells above). The guide oligos were synthesized by IDT. The oligos were phosphorylated, annealed, and inserted into pLentiCRISPR v2 plasmid following the published protocol24 to give the desired plasmids (2a, 2b). Scrambled gRNA sites inserted into the pLentiCRISPR v2 plasmid were used as controls with the following sequences (taken from the Human Gecko library40):
Sequence D19- ACGGAGGCTAAGCGTCGCAA
Sequence D20 – CGCTTCCGCGGCCCGTTCAA
Lentivirus containing BPGM targeting plasmid was generated in HEK 293T cells by PEI transfection with shRNA vectors and packaging vectors (VSVG and psPAX2). MDA-MB-231 cells were infected with 0.45 um-filtered virus supplemented with 8 μg/mL polybrene for 12 hours. After lentiviral delivery, cells were treated with DMEM containing 1 μg/μL puromycin for 7 days and then transferred to DMEM with no puromycin for Western blot analysis.
Metabolomic analysis of cell lysates for 2,3-BPG levels
Wt and BPGM knockout cells (HEK 293T and HCT116 cells) were plated in a 10-cm dish at 2.0×106 cells/plate and grown for 24 hours in 10 mL of DMEM media. After 24 hours, the media was removed and replaced with fresh DMEM and grown for an additional 24 hours. The media was removed and 1.2 mL of 80% MeOH containing 0.5% formic acid pre-chilled on dry ice was added to each 10-cm dish. The cells were suspended into the solution using a cell scraper and then transferred to an Eppendorf tube, and neutralized with 60 μL of 15% NH4HCO3. The samples were then centrifuged at 17,000 xg for 5 minutes and then stored at −80 °C until analysis. Samples were analyzed according the General metabolomic LC-MS analysis method described above.
Western blot analysis of BPGM deficient cells
Wt and BPGM knockout cells (from HEK 293T, HCT116, and MDA-MB-231 cells) were plated in a 6-well plate at 400,000 cells per well and grown for 48 hours to 70% confluency. The cells were lysed in 400 μL of RIPA buffer (25 mM Tris pH 8.0, 150 mM NaCl, 1% NP40, 1% deoxycholate, 1% SDS) and sonicated (1×5s at 45% power). 25 μg of each sample was then analyzed by Western blot using anti-pHis and anti-PGAM1 antibodies as described above. Protein concentrations from each lysate were measured by the BCA protein assay kit according to the manufacturer’s instructions. For HEK 293T and HCT116 cells, the PGAM1 signal intensity was measured by densitometry using ImageStudioLite software from LICOR and graphed as a histogram.
Rescue of BPGM in BPGM deficient cells
Wt HEK 293T and ΔBPGM-1 HEK 293T cells were plated in a 6-well plate at 700,000 cells/well and grown for 24 hours at 37 °C. After 24 hours, the media was removed and 1.5 mL of Opti-MEM was added to each well followed by the addition of 1 μg of pEGFP-N1 DNA plasmid expressing FLAG-BPGM, FLAG-BPGM H11A mutant, or GFP that had been pre-incubated with 4 μL of lipofectamine in 300 μL of Opti-MEM. The cells were then incubated for 6 hours at 37 °C. The media was removed and replaced with DMEM and then incubated for 18 hours at 37 °C. The cells were then lysed in 400 μL of RIPA and transferred to an Eppendorf tube. The samples were then sonicated (1 × 5s at 45% power) and the protein concentration was measured for each sample using the BCA assay kit according to manufacturer’s instructions. 20 μg of each sample was then analyzed by Western blot using the General Western blotting method described above.
For metabolomics analysis, wt HEK 293T and ΔBPGM-1 HEK 293T cells were plated in 10-cm dishes at 2.0×106 cells/well and grown for 24 hours at 37 °C. After 24 hours, the media was removed and 5 mL of Opti-MEM was added to each well followed by the addition of 3 μg of pEGFP-N1 DNA plasmid expressing FLAG-BPGM or FLAG-BPGM H11A mutant that had been pre-incubated with 8 μL of lipofectamine in 500 μL of Opti-MEM. The cells were then incubated for 6 hours at 37 °C. The media was removed and replaced with DMEM and then incubated overnight at 37 °C. After incubation, the media was removed and the cells were lysed in 1.2 mL of 80% MeOH containing 0.5% formic acid and transferred to an Eppendorf tube, and neutralized with 60 μL of 15% NH4HCO3. The samples were then centrifuged for 5 min at 17,000 xg. The samples were stored at −80 °C until LC-MS analysis for 2,3-BPG levels according the General metabolomic LC-MS analysis method described above.
Cell proliferation measurement
Wt or BPGM knockout HEK 293T cells were plated onto 5 × 6 cm dishes at the equivalent of 1 μL of packed cell volume (pcv) per plate and grown in 2 mL of DMEM at 37 °C for 24 hours (thus at t = 0, the pcv was set to 1 μL). After 24 hours, the media from one of the plates was removed and 0.5 mL of 0.5% Trypsin-EDTA was added to the plate and incubated for 5 min at 37 °C. 1 mL of DMEM was then added and the cells were resuspended and 1 mL was transferred to a packed cell volume tube and centrifuged at 5,000 xg for 5 min to obtain the packed cell volume number (t=24-hour time point). In the remaining plates, the media was removed and replaced with fresh DMEM for continued growth at 37 °C and the packed cell volume number was measured every 24 hours as described above over a 120-hour period.
Cellular glucose consumption and lactate production measurement
Wt or BPGM knockout HEK 293T cells were plated in a 6-well plate at 600,000 cells/well in DMEM media and grown for 24 hours. The media was removed and replaced with DMEM containing dialyzed FBS and grown for an additional 24 hours. 1 mL of the media was then removed from each well and analyzed using the YSI 7100 instrument, which was set to recalibrate after every 5 samples.
The packed cell volume (pcv) was measured for each well by removing the media followed by addition of 1 mL of 0.05% Trypsin-EDTA and incubation for 5 min at 37 °C. An additional 1 mL of media was added to each well and the cells were resuspended. 1 mL of the suspension was transferred to a packed cell volume tube and centrifuged at 5,000 xg for 5 min to obtain the packed cell volume number.
LC-MS/MS analysis of 3-PG and 2-PG ratios from HEK 293T lysates
Wt and BPGM knockout cells were plated in 6-cm dishes at 0.85×106 cells/plate in 2.5 mL DMEM and grown for 24 hours. After 24 hours, the media was removed and replaced with DMEM containing dialyzed FBS and grown for an additional 24 hours. The media was then removed and 0.5 mL of 80% MeOH in MQ-H2O, pre-chilled on dry ice was added to each 6-cm dish. The cells were suspended into the 80% MeOH containing 0.5% formic acid using a cell scraper and then transferred to an Eppendorf tube, and neutralized with 60 μL of 15% NH4HCO3. The samples were then centrifuged at 17,000 xg for 5 min and stored at −80 °C until analysis.
The LC–MS method involved hydrophilic interaction chromatography coupled by negative mode electrospray ionization to the Q Exactive hybrid quadrupole-Orbitrap mass spectrometer (Thermo Scientific). The LC separation was performed on a XBridge BEH Amide column (150 mm × 2.1 mm, 2.5 μm particle size, Waters) using a gradient of solvent A (95%:5% H2O:Acetonitrile with 20 mM Ammonium Bicarbonate), and solvent B (100% Acetonitrile). The gradient was 0 min, 85% B; 2 min, 85% B; 3 min, 80% B; 5 min, 80% B; 6 min, 75% B; 7 min, 75% B; 8 min, 70% B; 9 min, 70% B; 10 min, 50% B; 12 min, 50% B; 13 min, 25% B; 16 min, 25% B; 18 min, 0% B; 23 min, 0% B; 24 min, 85% B; 30 min, 85% B. The flow rate was 150 μL min−1. Injection volume was 5 μL and column temperature 25 °C. The tandem MS scans were under negative mode with 70,000 resolution. The AGC target was 5e5 and the maximum IT was 30 ms. The parent ion m/z was set to 184.986, with an isolation window of 1.5 m/z. The collision energy was set to 30 NCE. The data were analyzed using the Xcalibur Quan Browser software suite. 3-PG and 2-PG both generate fragments with m/z 79 and 97, but at different ratios. The standard mixtures containing 3-PG and 2-PG (in the ratio of 100:0, 80:20, 60:40, 40:60, 20:80 and 0:100) were used to generate a standard curve of 97/(79+97). The 97/(79+97) value from experimental samples was calculated and converted into the 3-PG/Total Mono-PG ratio using the standard curve. The absolute concentration of mono-PG in WT cells was measured as described.41 The ion counts of 3-PG and 2-PG were converted to absolute concentrations by normalizing to the wt total mono-PG concentration.
PEP-mediated phosphorylation of PGAM1
His6-PGAM1 (prepared in the unphosphorylated form as described previously22) was diluted 5-fold in 50 mM Tris pH 7.5 and 50 mM NaCl to a final concentration of 0.3 μg/μL. 1 mM PEP or 2,3-BPG was added to the sample and incubated for 15 min at rt. The sample was then diluted 1:1 with 4× loading buffer (160 mM Tris, pH 8.5, 40% (v/v) glycerol, 4% (w/v) SDS, 0.08% (w/v) bromophenol blue, and 8% (v/v) BME). 1 μg of each sample was analyzed by Western blot according to the General western blotting method described above.
Phosphatase activity measurement of PGAM1
50 μL of 1.5 μg/μL His6-PGAM1 (prepared in the unphosphorylated form as described previously22) was treated with 500 μM 2,3-BPG for 5 minutes at room temperature in assay buffer (50 mM Tris pH 7.5, 50 mM NaCl). The sample was then desalted 2× on a Micro Bio-spin P-6 gel desalting column according to manufacturer’s instructions into assay buffer. The desalted, phosphorylated PGAM1 was added to 1 mL of reaction buffer (50 mM Tris pH 7.5, 50 mM NaCl) to a final concentration of 0.5 μM. The sample was aliquoted into 4 × 1.5 mL Eppendorf tubes at 200 μL each. 20 μM 2-Phosphoglycollate, PEP, or buffer only was then added to each of the reaction mixtures and the reactions were incubated for 0, 5, 15, 30 min at 30 °C. At each time point, 20 μL was removed and added to 20 μL of 4× loading buffer (160 mM Tris, pH 8.5, 40% (v/v) glycerol, 4% (w/v) SDS, 0.08% (w/v) bromophenol blue, and 8% (v/v) BME) and stored at −80 °C until Western blot analysis. For Western blotting, 20 μL of each sample was analyzed according to the General Western blotting method described above.
Preparation of FLAG-PGAM1 overexpression cells
HEK 293T cells were plated in a 10-cm plate and grown in complete DMEM until the cells reached 50% confluency. The cells were then transfected with a pEGFP-N1 plasmid expressing FLAG-PGAM1. The transfection was done by replacing the media with 5 mL of complete DMEM followed by addition of 3 ng of Flag-PGAM1 pEGFP-N1 plasmid that was pre-mixed for 30 min with 15 μL of lipofectamine in 800 μL Opti-mem. The cells were then grown for 12 hours at 37 °C and the media was replaced with 10 mL complete DMEM and grown for 24 hours at 37 °C. After 24 hours, the cells were then diluted into a 6-well plate and grown in DMEM containing G418 (Gibco) at 750 ng/mL final concentration for one week with media replaced every 2 days to select for stable transfecting cells. Individual colonies on the 6-well plate were picked using a pipette tip and transferred to a new 6-well plate containing 2 mL complete DMEM with 750 ng/mL G418. The cells were grown to confluency and then transferred to a 10-cm dish and grown in DMEM without G418 for further passaging. FLAG-PGAM1 overexpression was verified by Western blotting using an anti-PGAM1 antibody as described above in the General Western blotting method.
Analysis of PEP induced phosphorylation of PGAM1 overexpressed in hypotonic lysate
HEK 293T cells overexpressing FLAG-PGAM1 were grown to 70% confluency in a 10-cm dish with DMEM. Prior to cell lysis, the media was replaced with fresh DMEM and incubated for 30 min. The cells where then resuspended in the media, transferred to a falcon tube, and centrifuged for 3 min at 1000 xg to pellet the cells. The cells were then resuspended in 3 mL of PBS and pelleted by centrifugation (3 min at 1000 xg) for a total of two repeat washes. The cells were then resuspended in 3 mL hypotonic lysis buffer (20 mM HEPES pH 7.0, 5 mM KCl, 1 mM MgCl2, 5 mM DTT) and incubated for 10 min on ice. After incubation, the lysates were passed 3× through a 27 ½ gauge needle and the lysates were then centrifuged to pellet the cell debris. The supernatant was transferred to a separate Eppendorf tube and the total protein concentration was determined using the Bradford assay.
The hypotonic lysate was then diluted 5-fold in assay buffer (50 mM Tris pH 7.5, 50 mM KCl, 15 mM MgCl2, 2 mM MnCl2, 1 mM DTT). PEP (final concentration of 100 μM) or buffer was then added to the reaction and sample was removed at 1, 3, 5, 10 min (a t = 0-min time point was also taken prior to addition of PEP). For Western blotting, 30 μL of sample was removed and added to 10 μL of 4× loading buffer (160 mM Tris, pH 8.5, 40% (v/v) glycerol, 4% (w/v) SDS, 0.08% (w/v) bromophenol blue, and 8% (v/v) BME) and immediately frozen in liquid nitrogen and then stored at −80 °C until analysis according to the General Western blotting method described above (25 μL of sample was used for the analysis). For metabolomics samples, 200 μL was removed and added to 200 μL of MeOH and then flash frozen in liquid nitrogen and stored at −80 °C until LC-MS analysis according to the General LC-MS metabolomic analysis described above.
Analysis of 32P-PEP-induced phosphorylation of overexpressed PGAM1 in hypotonic lysate
Hypotonic lysate (prepared as described above) was diluted 5-fold in Assay buffer (50 mM Tris pH 7.5, 50 mM KCl, 15 mM MgCl2, 2 mM MnCl2, 1 mM DTT) followed by addition of 10 μM or 100 μM PEP containing 5.4 μCi 32P-PEP (prepared as described below). Time points were taken at 1, 3, 5, 10 min (a t = 0-min time point was also taken prior to addition of PEP). At each time point, 40 μL of the reaction was removed and mixed with 10 μL of 4× loading buffer (160 mM Tris, pH 8.5, 40% (v/v) glycerol, 4% (w/v) SDS, 0.08% (w/v) bromophenol blue, and 8% (v/v) BME) followed by immediate freezing in liquid nitrogen and storage at −80 °C until analysis. For autoradiography, 25 μL of each sample was resolved by SDS-PAGE. The gel was subsequently vacuum-dried on a filter paper and incubated with a Kodax Bio-Max film for 48 hours followed by development employing standard procedures.
Preparation of 32P-PEP
0.2 nmol (600 μCi) of [γ-32P]-ATP (60 μL) (3000 Ci/mmol, 10 mCi/mL, Perkin Elmer BLU002A001MC) was added to 1.2 mL of PEP buffer (10 mM Tris (pH 7.5), 3 mM MgCl2, 1 mM DTT) containing 800 mM pyruvate. The final concentration of ATP was raised to 12.5 μM through the addition of 15 nmol of cold ATP (this dilutes the specific activity of [γ-32P]-ATP to 40 Ci/mmol). 5 units of rabbit muscle pyruvate kinase was then added and the sample was incubated for 1 hour at 37 °C. The sample was then purified by anion exchange chromatography using AG-1-x8 bicarbonate resin that was prepared from AG-1-x8 chloride resin (Bio-Rad 731-6212) by washing the resin with 20 column volumes of 1 M NaOH, 20 column volumes of 1 M Sodium Bicarbonate, and then 10 column volumes of MQ-H2O. The sample was then added over 1 mL of AG-1-x8 bicarbonate resin and then washed with 2.5 mL of MQ-H2O. PEP was then eluted from the column by adding 2.5 mL of triethylammonium bicarbonate (TeAB) in the following sequential concentrations (0.1, 0.2, 0.4, 0.6, 0.75 M). The 0.6 M TeAB fraction was collected and assumed to contain 3 μM PEP (with a specific activity of 40 Ci/mmol, 120 μCi/mL) based on the analysis of an identical PEP preparation and purification experiment using non-radioactive materials that was quantified by LC-MS employing PEP standards of known concentrations.
Western blot analysis of PGAM1 overexpression in BPGM deficient cells
Wt HEK 293T and ΔBPGM-1 HEK 293T cells were plated in a 6-well plate at 800,000 cells/well and grown for 24 hours at 37 °C. After 24 hours, the media was removed and 1.5 mL of Opti-MEM was added to each well followed by the addition of 1.25 μg of pEGFP-N1 DNA plasmid expressing FLAG-PGAM1, or empty vector that had been pre-incubated with 4 μL of lipofectamine in 200 μL of Opti-MEM. The cells were then incubated for 6 hours at 37 °C. The media was removed and replaced with DMEM and then incubated for 24 hours at 37 °C. The cells were then lysed in 800 μL of RIPA and transferred to an Eppendorf tube. The samples were then sonicated (1×5s at 45% power) and the protein concentration was measured for each sample using the BCA assay kit according to the manufacturer’s instructions. 30 μg of each sample was then analyzed by Western blot according to the General Western blotting method described above.
In vitro production of 1,3-BPG
1,3-BPG was made enzymatically using a GAPDH and LDH coupled reaction. The reaction mixture contains 25 mM Tris pH 8.0, 50 mM NaCl, 1 mM MgCl2, 10 mM KH2PO4, 400 μM NAD, 1 mM GAP, 3.3 mM pyruvate, 1 unit/mL GAPDH, 0.1 mg/mL BSA and 16.7 units/mL LDH. The reaction mixture was incubated at room temperature for 30 min, and then quenched with 50% methanol. The purification of 1,3-BPG was performed on a Synergy Hydro-RP column (150 mm × 2 mm, 4 μm particle size, Phenomenex) using a gradient of solvent A (97%:3% H2O:MeOH with 10 mM tributylamine and 15 mM acetic acid), and solvent B (100% MeOH). The gradient was 0 min, 0% B; 5 min, 0% B; 10 min, 20% B; 20 min, 20% B; 35 min, 65% B; 38 min, 95% B; 42 min, 95% B; 43 min, 0% B; 50 min, 0% B. The flow rate was 200 μL min−1. Injection volume was 30 μL and column temperature 25 °C. The retention time of major compounds in the mixture are: 1,3-BPG 31.5 min, 2,3-BPG 33.3 min, 3-PG 25.9 min, GAP 13.3 min and pyruvate 16.7 min. The fractions containing 1,3-BPG from these multiple injections were then combined and diluted 2-fold with MQ-H2O. The sample was flash frozen in liquid nitrogen, and then lyophilized to ~200 μL total volume. The sample was then aliquoted into separate tubes, flash frozen in liquid nitrogen, and stored at −80 °C until use. The identity of purified 1,3-BPG was confirmed using a phosphoglycerate kinase (PGK) assay. 20 μM 1,3-BPG, 1 mM ADP and 3.7 units of PGK (Sigma P7634) were added to 25 mM Tris pH 7.5, 50 mM NaCl and 5 mM MgCl2. The reactions were carried out at room temperature for 30 min and quenched with equal volume of 50% MeOH/water. LC/MS analysis was performed using the General metabolomic LC-MS analysis method described above.
Preparation of 1,3-BPG and 2,3-BPG treated PGAM1 for mass spectrometric analysis
1 μM His6-PGAM1 (prepared in the unphosphorylated form as described previously22) in 50 mM Tris pH 7.5, 50 mM NaCl was treated with 1 μM 1,3-BPG or 2,3-BPG at room temperature for 5 min. The sample was then desalted using a Micro Bio-spin P-6 gel desalting column according to manufacturer’s instructions followed by treatment with 0.2 μg trypsin for 4 hours at room temperature. The samples were then frozen in liquid nitrogen, lyophilized, and stored at −80 °C until LC-MS/MS analysis. LC-MS/MS was performed the Harvard University Faculty of Arts and Sciences (FAS) Mass Spectrometry and Proteomics Resource Laboratory, using conditions described above in General LC-MS/MS of pHis proteins.
Kinetic analysis of PGAM1 phosphorylation
His6-PGAM1 (prepared in the unphosphorylated form as described previously22) was diluted in 50 mM Tris pH 7.5, 50 mM NaCl to a final concentration of 0.15 μM and then aliquoted into separate Eppendorf tubes in 500 μL reaction volumes. 0.25 μM 2,3-BPG or 1,3-BPG was then added to each sample followed by incubation at 30 °C for 5, 20, 40, 60, 120, 180 seconds (for the 0-second time point, enzyme sample was taken prior to the addition of 2,3-BPG or 1,3-BPG). (Note that the 0.25 μM concentration is below the reported cellular metabolite levels for both metabolites)41. At each time point, 25 μL of each reaction was removed and mixed with 25 μL of 4× loading buffer (160 mM Tris, pH 8.5, 40% (v/v) glycerol, 4% (w/v) SDS, 0.08% (w/v) bromophenol blue, and 8% (v/v) BME) and then placed immediately in liquid nitrogen and stored at −80 °C until Western blot analysis. 25 μL of each sample was analyzed by Western blot as described above. Densitometry was used to quantitate the signal intensity for each of the time points using ImageStudioLite software from LI-Cor and plotted as a function of time.
Metabolomic analysis of PGAM1-mediated production of 2,3-BPG from 1,3-BPG
1,3-BPG was added to reaction buffer (50 mM Tris pH 7.5, 50 mM NaCl) at a final concentration of 10 μM followed by the addition of 2-PG at 0, 50, or 1000 μM final concentration. His6-PGAM1 (prepared in the unphosphorylated form as described previously22) was then added at a final concentration of 1 μM and the samples were incubated for 30 min at 30 °C (as controls, samples were prepared with 1,3-BPG only or PGAM1 only). The samples were quenched by diluting the samples 1-fold with 100% MeOH and then immediately flash freezing in liquid nitrogen. The samples were then transferred to −80 °C until LC-MS analysis according the General metabolomic LC-MS Analysis method described above.
Keq determination of 2,3-BPG induced phosphorylation of PGAM1
His6-PGAM1 (prepared in the unphosphorylated form as described previously22) was diluted to 0.2 μM final concentration in assay buffer (50 mM Tris pH 8.0, 50 mM NaCl) to a final volume of 500 μL. The sample was then aliquoted into Eppendorf tubes in 50 μL aliquots. 0, 1, 2, 5, 10, 25, 50, 100, 250, 500, 1000 μM 2-PG was then added to each sample. 1 μM of 2,3BPG was then added to each sample and the sample was then incubated for 5 min at room temperature. 50 μL of 4× loading buffer (160 mM Tris, pH 8.5, 40% (v/v) glycerol, 4% (w/v) SDS, 0.08% (w/v) bromophenol blue, and 8% (v/v) BME) was then added to each sample and immediately stored at −80 °C until Western blotting analysis. 25 μL of each sample was analyzed by Western blot according to the General Western blotting method described above.
After Western blotting, the signal intensities were measured by densitometry using ImageStudioLite software from LI-Cor. The absolute p-PGAM1 concentration was obtained by comparing to the 0 μM 2-PG control. The equilibrium of 2,3-BPG and PGAM1 follows the equation below.
The Keq is then:
where x can be solved as:
The Keq is then chosen to minimize the deviation between the calculated p-PGAM1 concentration and experimental observations and plotted as the fitted line in Supplementary Fig. 14.
Global metabolite analysis of wt and BPGM knockout cells
Wt and BPGM knockout cells (HEK 293T and HCT116 cells) were plated in a 10-cm dish at 2.0×106 cells/plate and grown for 24 hours in 10 mL of DMEM media. After 24 hours, the media was removed and replaced with fresh DMEM and grown for an additional 24 hours. The media was removed and 1.2 mL of 80% MeOH containing 0.5% formic acid pre-chilled on dry ice was added to each 10-cm dish. The cells were suspended into the solution using a cell scraper and then transferred to an Eppendorf tube, and neutralized with 60 μL of 15% NH4HCO3. The samples were then centrifuged at 17,000 xg for 5 min and then stored at −80 °C until analysis. Samples were analyzed according the General metabolomic LC-MS analysis described above. The metabolite levels were normalized to the packed cell volume and wt cells. The heatmap is generated using Java TreeView.
Oxidative pentose phosphate pathway flux measurements
Wt HEK 293T and ΔBPGM-1 HEK 293T cells were plated in a 6-well plate at 800,000 cells/well in 2.5 mL DMEM and grown for 24 hours at 37 °C. After incubating 24 hours, the cells were washed with 2 mL Glucose/Pyruvate free DMEM (Gibco Product Number: 11966-025) containing dialyzed FBS. After washing, 2.5 m mL of Glucose/Pyruvate free DMEM supplemented with 10% dialyzed FBS, 25 mM D-Glucose (1,2-13C2, 99%), and 110 mg/mL Pyruvate. The cells were then incubated for 24 hours. The media was removed and the cells were then resuspended in 500 μL of 80% MeOH containing 0.5% formic acid pre-incubated on dry ice using a cell scraper and then transferred to an Eppendorf tube, and neutralized with 60 μL of 15% NH4HCO3. The samples were then centrifuged at 17,000 xg for 5 min and then stored at −80 °C until analysis. Samples were analyzed according the General metabolomic LC-MS analysis described above. The LC-MS data were processed with MAVEN to extract the labeling percentage of ribose-5-phosphate. The labeling percentage was corrected for isotope natural abundance.
Serine production measurements in wt and BPGM knockout cells
Wt and BPGM knockout cells were plated in a 6-cm dish at 0.85×106 cells/plate in 2.5 mL DMEM and grown for 24 hours. After 24 hours, the media was removed and 2.5 mL of DMEM (containing dialyzed FBS, pen/strep, 110 mg/mL pyruvate, 25 mM uniformly labeled C13-Glucose) was added to each plate followed by an additional 24 hour incubation period. 1 mL of media was then collected from each plate and transferred to an Eppendorf tube and stored at −80 °C until analysis. Samples were analyzed according the LC-MS analysis of media samples for serine and glycine concentration described below.
Serine biosynthesis time course and flux analysis of wt and BPGM knockout cells
Wt and BPGM knockout cells were plated in a 6-cm dish at 0.85×106 cells/plate in 2.5 mL DMEM and grown for 24 hours. After 24 hours, the media was removed and 2.5 mL of DMEM (containing dialyzed FBS, pen/strep, 110 mg/mL pyruvate, 25 mM uniformly labeled C13-Glucose) was added to each plate and incubated for 0, 1, 3, 6, and 10 hours. 1 mL of media was then collected from each plate and transferred to an Eppendorf tube and stored at −80 °C until analysis. Three plates were grown for each time point and an additional plate was grown for each time point to calculate the packed cell volume. This was done by resuspending the cells in the 2.5 mL of media and transferring 1 mL to a packed cell volume tube. The tube was then spun at 5000 xg for 5 minutes and the packed cell volume was measured. Samples were analyzed according the LC-MS analysis of media samples for serine and glycine concentration described below. Serine biosynthesis flux was calculated as described below in Calculating serine biosynthesis fluxes.
Purine nucleotide time course and flux analysis of wt and BPGM knockout cells
Wt and BPGM knockout cells were plated in a 6-cm dish at 0.85×106 cells/plate in 2.5 mL DMEM and grown for 24 hours. After 24 hours, the media was removed and 2.5 mL of DMEM (containing dialyzed FBS, pen/strep, 110 mg/mL pyruvate, 25 mM uniformly labeled C13-Glucose) was added to each plate and incubated for 0, 3, 6, 10, 20 and 24 hours. The media was removed and 1.2 mL of 80% MeOH containing 0.5% formic acid pre-chilled on dry-ice was added to each 10-cm dish. The cells were suspended into the solution using a cell scraper and then transferred to an Eppendorf tube, and neutralized with 60 μL of 15% NH4HCO3. The samples were then centrifuged at 17,000 xg for 5 minutes and then stored at −80 °C until analysis. Samples were analyzed according the General metabolomic LC-MS analysis described above. The 13C enrichment of purine was calculated using the M+6−M+10 fractions of the nucleotide labeling.
BPGM-mediated rescue of serine production in Wt and BPGM knockout cells
Wt and BPGM knockout HEK 293T cells were plated in a 6-cm dish at 0.85×106 cells/plate in 2.5 mL DMEM and grown for 24 hours. After 24 hours, the media was removed and 1.5 mL of Opti-MEM was added to each well followed by the addition of 1.25 μg of pEGFP-N1 DNA plasmid expressing FLAG-BPGM or FLAG-BPGM H11A mutant that had been pre-incubated with 4 μL of lipofectamine in 200 μL of Opti-MEM. The cells were then incubated for 6 hours at 37 °C. The media was removed and replaced with 2.5 mL of DMEM (containing dialyzed FBS, pen/strep, 110 mg/mL pyruvate, 25 mM uniformly labeled C13-Glucose) and then incubated for 10 hours at 37 °C. 1 mL of media was then collected from each plate and transferred to an Eppendorf tube and stored at −80 °C until analysis. Three plates were grown for each time point and an additional plate was grown for each time point to calculate the packed cell volume. This was done by resuspending the cells in the 2.5 mL of media and transferring 1 mL to a packed cell volume tube. The tube was then spun at 5000 xg for 5 min and the packed cell volume was measured. Samples were analyzed according the LC-MS analysis of media samples for serine and glycine concentration described below. Serine biosynthesis flux was calculated as described below in Calculating serine biosynthesis fluxes.
PGAM1-mediated rescue of mono-PG and phosphoserine production in BPGM knockout cells
For metabolomics analysis, wt 293T, ΔBPGM-1, ΔBPGM-2, and ΔBPGM-3 HEK 293T cells were plated in 10-cm dishes and plated at 2.0×106 cells/well and grown for 24 hours at 37 °C. After 24 hours, the media was removed and 5 mL of Opti-MEM was added to each well followed by the addition of 3 μg of pReceiver M11 DNA plasmid expressing FLAG-PGAM1 or FLAG-PGAM1 H11A mutant that had been pre-incubated with 8 μL of lipofectamine 2000 in 500 μL of Opti-MEM. The cells were then incubated for 6 hours at 37 °C. The media was removed and replaced with DMEM and then incubated overnight at 37 °C. After incubation, the media was removed and the cells were lysed in 1.2 mL of 80% MeOH containing 0.5% formic acid and transferred to an Eppendorf tube, and neutralized with 60 μL of 15% NH4HCO3. The samples were then centrifuged for 5 min at 17,000 xg. The samples were stored at −80 °C until LC-MS analysis according the General metabolomic LC-MS Analysis method described above.
LC-MS/MS analysis of media samples for serine and glycine concentration
The media samples were diluted 20-fold and mixed with 20 μM 13C2,15N1-Glycine and 10 μM 13C3,15N1-Serine. The mixture was analyzed on the Q Exactive hybrid quadrupole-Orbitrap mass spectrometer (Thermo Scientific). The LC separation was performed on a Synergy Hydro-RP column (100 mm×2 mm, 2.5 μm particle size, Phenomenex) using a gradient of solvent A (97%:3% H2O:MeOH), and solvent B (100% MeOH). The gradient was 0 min, 0% B; 6 min, 0% B; 12 min, 70% B; 14 min, 100% B; 18 min, 100% B; 19 min, 0% B; 24 min, 0% B and 25 min, 0% B. The flow rate was 0 min, 50 μL min−1; 6 min, 50 μL min−1; 12 min, 200 μL min−1; 24 min, 200 μL min−1 and 25 min, 50 μL min−1. Injection volume was 5 μL and column temperature 25 °C. The MS scans were under positive mode with 140,000 resolution. The AGC target was 5e6 and the maximum IT was 500 ms. The scan mass window was 75–1000. The data were analyzed using the MAVEN software suite and the absolute concentrations of serine and glycine were calculated.
Calculating serine biosynthesis fluxes
The serine and glycine synthesis and consumption involve five fluxes in the model. fGlc represents serine de novo synthesis flux from glucose. fGly→Ser represents glycine to serine flux via SHMT. fSer→Gly represents serine to glycine flux also via SHMT. Finally, fSerCon represents total serine consumption flux and fGlyCon represents total glycine consumption flux. Using these fluxes, the serine and glycine concentrations in the media can be described using the following equations.
In the equations, V represents the packed cell volume at any given time point. V0 and VT represent the packed cell volume at the beginning and the end of the growth period respectively. Seri and Glyi represent the absolute concentration of serine and glycine M+i, respectively. Me-THFi represents the absolute concentration of Me-THF M+i. Me-THF1/Me-THF0+1 is difficult to measure directly, but can be approximated by the fraction of 3-13C-Serine. Therefore, the equations can be rewritten as the following.
Given the initial conditions, which is the serine and glycine concentration in DMEM, these equations give media serine and glycine concentrations at any time point, if the fluxes are known. The flux estimation is therefore an optimization problem, in which the set of fluxes was found to minimize the deviation between the predicted serine/glycine concentrations at the end point and the measured ones. The numerical simulation of the differential equations was performed in R with the deSolve package and the optimization was performed with minqa package.
Metabolomic analysis of shRNA-mediated PGAM1 knockdown cells
shRNA constructs targeting human PGAM1 were purchased from Sigma in the pLKO.1 vector backbone from the TRC shRNA library. The following plasmids were purchased:
PGAM1 KD-1: TRCN0000029469 (TTTCTGCTTTATTGAGACCGG)
PGAM1 KD-2: TRCN0000029470 (TATTTCTTCATTCCAGAAGGG)
PGAM1 KD-3: TRCN0000029471 (ATGTTGCTGTAGAAAGGATGG)
Wt HEK 293T cells were transfected with each of the above shRNA plasmids via lentiviral delivery. As controls, pLKO.1-puro empty vector (empty control) (Sigma, product number SHC001) or pLKO.1-puro non-mammalian shRNA (scrambled control) (Sigma, product number SHC002) were also transfected into wt HEK 293T cells. Lentiviral particles were generated by PEI transfection of HEK 293T cells with shRNA vectors and packaging vectors (VSVG and psPAX2). Wt HEK 293T cells were infected with isolated virus and put under puromycin selection at 2 μg/mL. Cells were maintained in DMEM containing 2 μg/mL puromycin.
HEK 293T cells transfected with the different shRNA constructs were each plated in 6-cm dish at 0.8×106 cells/plate in 2.5 mL DMEM and grown for 24 hours. After 24 hours, the media was removed and replaced with 2.5 mL of DMEM (containing dialyzed FBS, pen/strep, 110 mg/mL pyruvate, 25 mM uniformly labeled C13-Glucose) and grown for an additional 24 hours. 1 mL of media was then collected from plate and transferred to an Eppendorf tube and stored at −80 °C until LC-MS analysis. The remaining media was removed and the cells were then resuspended in 500 μL of 80% MeOH containing 0.5% formic acid pre-incubated on dry ice using a cell scraper and then transferred to an Eppendorf tube, and neutralized with 60 μL of 15% NH4HCO3. The samples were then centrifuged at 17,000 xg for 5 min and then stored at −80 °C until analysis. Samples were analyzed according the General metabolomic LC-MS analysis and serine biosynthesis flux analysis method described above. For Western blot analysis of pHis levels on PGAM1, the cells were lysed in 400 μL of RIPA buffer (25 mM Tris pH 8.0, 150 mM NaCl, 1% NP40, 1% deoxycholate, 1% SDS) and sonicated (1×5s at 45% power). 25 μg of each sample was then analyzed by Western blot using anti-pHis and anti-PGAM1 antibodies as described above.
Cell growth measurements under hypoxic conditions
Wt or BPGM knockout HEK 293T and HCT116 cells were plated onto 10-cm dishes and grown for 48 hours to 80–90% confluency at 37 °C. The plates were harvested and diluted to a concentration of 60,000 cells/mL in DMEM. 100 μL of this dilution (6,000 cells) was added to a 96-well plate in triplicate. One set of plates was incubated in normoxic conditions (5% CO2, 37 °C) and one set was incubated in hypoxic conditions (0.5% O2, 5% CO2, 37 °C) in a Coy Laboratory incubator that was pre-flushed with N2 overnight. At t=0 hours, 20 μL of diluted EZQuant reagent (3-fold dilution in PBS) was added to each well to be analyzed. The plates were incubated for 2 hours at 37 °C and the absorbance at 450nm was recorded. This was repeated every 24 hours as described above over a 120-hour period to obtain the cell growth numbers at t = 0, 24, 48, 72, 96, and 120 hours.
Cell growth measurements in serine and glycine depleted media
Wt or BPGM knockout HEK 293T and HCT116 cells were plated onto 10-cm dishes and grown for 48 hours to 80–90% confluency at 37 °C. The plates were harvested and diluted to a concentration of 60,000 cells/mL in DMEM (25 mM glucose, 1 mM pyruvate, with or without serine and glycine). 100 μL of this dilution (6,000 cells) was added to a 96-well plate in triplicate. At t=0 hours, 20 μL of diluted EZQuant (Alstem Bio, Product# #CQ01) reagent (3-fold dilution in PBS) was added to each well to be analyzed. The plates were incubated for 2 hours at 37 °C and the absorbance at 450nm was recorded. This was repeated every 24 hours as described above over a 120-hour period to obtain the cell growth numbers at t = 0, 24, 48, 72, 96, and 120 hours.
Xenograft of HCT116 BPGM knockout cells
All procedures involving mice and experimental protocols were approved by the University Institutional Animal Care and Use Committee (IACUC). NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were ordered from Jackson Laboratory and bred in-house. Stock mice for xenograft experiments were randomized, with each group receiving an equal number of mice from each litter. Subcutaneous tumors were generated by injection of HCT116 wild-type and HCT116 ΔBPGM knockout cells into the hind flank of male 6-week-old NSG mice (one million cells/mouse were used for wt or BPGM-deficient cells). Tumor diameter was followed by weekly caliper measurements and mice were sacrificed after four weeks. Tumors were excised and weighed. Note: no statistical method was used to predetermine sample size. We used 6 mice per experimental group in all animal experiments. These experiments were repeated in two independent tests. This is sufficient (90% probability) to detect a difference of 2.0 times of standard deviation in tumor size between groups (2-sided t-test, P = 0.05).
Supplementary Material
Acknowledgments
We thank current and former members of the Muir and Rabinowitz laboratories for helpful discussions during the preparation of this manuscript and Vipin Suri of Raze Therapeutics for technical help. This work was funded by the U. S. National Institutes of Health for T.W.M. (5R01GM095880) and J.D.R. (R01 CA163591 and P30DK019525), the U. S. Department of Energy for J.D.R. (DE-SC0012461), Stand Up to Cancer for J.D.R. (SU2C-AACR-DT0509), and the Brewster Foundation and Breast Cancer Research Foundation for Y.K. R.C.O. and J-M.K. were supported by postdoctoral fellowships from the National Institutes of Health Research Service Award (1F32CA167901) and Damon Runyon Cancer Research Foundation (DRG-2005-09), respectively.
Footnotes
Author Contributions
R.C.O. designed experiments, prepared materials, analyzed data, interpreted results, generated HEK 293T and HCT116 knockout cell lines, conducted experiments for metabolomic data analysis, Western blotting, cell growth measurements, and in vitro phosphorylation analysis. X.S. designed experiments, prepared materials, analyzed data, interpreted results, designed primers and performed all DNA sequencing analysis for BPGM knockout experiments, conducted all metabolomic experiments and data analysis, prepared 1,3-BPG, and derived and conducted all serine flux calculations. M.H. designed experiments, analyzed data, interpreted results, performed cell assays for metabolomic data analysis, performed Western blots for BPGM rescue and PGAM1 knockdown experiments, and performed cell growth measurements of knockout cells with Serine/Glycine or oxygen depletion. J-M.K. designed experiments, analyzed data, interpreted results, and performed Western blot analysis of BPGM protein levels. M.E. generated MDA-MB-231 knockout cells, prepared PGAM1 knockdown cells, and designed, performed, and analyzed mouse xenograft experiments. Y.D. designed and conducted RNA-seq analysis of wt and knockout HEK 293T cells. B.W. designed and conducted 32P labeling experiments, analyzed data, and interpreted results of metabolomic experiments. E.G. performed phosphorylated PGAM1 Western blot analysis from multiple cell lines. D.H.P. performed LC-MS-based proteomic analysis of phosphorylated PGAM1, analyzed data, and interpreted results. T.W.M. designed experiments, analyzed data, and interpreted results. J.D.R. designed experiments, analyzed data, and interpreted results. R.C.O., X.S., M.H., T.W.M., and J.D.R. wrote the manuscript with contributions from all authors.
Competing financial interests
The authors declare no competing financial interests.
Additional information
Any supplementary information, chemical compound information and source data are available in the online version of the paper. Correspondence and requests for materials should be addressed to T.W. M. or J. D. R.
References
- 1.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hitosugi T, et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell. 2012;22:585–600. doi: 10.1016/j.ccr.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Possemato R, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–50. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH, Maddocks OD. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014;7:1248–58. doi: 10.1016/j.celrep.2014.04.045. [DOI] [PubMed] [Google Scholar]
- 5.Mattaini KR, et al. An epitope tag alters phosphoglycerate dehydrogenase structure and impairs ability to support cell proliferation. Cancer Metab. 2015;3:5. doi: 10.1186/s40170-015-0131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samanta D, et al. PHGDH Expression Is Required for Mitochondrial Redox Homeostasis, Breast Cancer Stem Cell Maintenance, and Lung Metastasis. Cancer Res. 2016;76:4430–42. doi: 10.1158/0008-5472.CAN-16-0530. [DOI] [PubMed] [Google Scholar]
- 7.Fothergill-Gilmore LA, Watson HC. The phosphoglycerate mutases. Adv Enzymol Relat Areas Mol Biol. 1989;62:227–313. doi: 10.1002/9780470123089.ch6. [DOI] [PubMed] [Google Scholar]
- 8.Rose ZB. The enzymology of 2,3-bisphosphoglycerate. Adv Enzymol Relat Areas Mol Biol. 1980;51:211–53. doi: 10.1002/9780470122969.ch5. [DOI] [PubMed] [Google Scholar]
- 9.Nelson DL, Lehninger AL, Cox MM. Lehninger principles of biochemistry. W.H. Freeman; New York: 2008. [Google Scholar]
- 10.Sutherland EW, Posternak TZ, Cori CF. The mechanism of action of phosphoglucomutase and phosphoglyceric acid mutase. J Biol Chem. 1949;179:501. [PubMed] [Google Scholar]
- 11.Rose ZB. The purification and properties of diphosphoglycerate mutase from human erythrocytes. J Biol Chem. 1968;243:4810–20. [PubMed] [Google Scholar]
- 12.Narita H, Yanagawa S, Sasaki R, Chiba H. Induction of 2,3-bisphosphoglycerate synthase in Friend leukemia cells. Biochem Biophys Res Commun. 1981;103:90–6. doi: 10.1016/0006-291x(81)91664-8. [DOI] [PubMed] [Google Scholar]
- 13.Joulin V, et al. Isolation and characterization of the human 2,3-bisphosphoglycerate mutase gene. J Biol Chem. 1988;263:15785–90. [PubMed] [Google Scholar]
- 14.Chiba H, Sasaki R. Functions, of 2,3-bisphosphoglycerate and its metabolism. Curr Top Cell Regul. 1978;14:75–116. doi: 10.1016/b978-0-12-152814-0.50007-1. [DOI] [PubMed] [Google Scholar]
- 15.Sasaki R, Ikura K, Narita H, Yanagawa S-i, Chiba H. 2,3-bisphosphoglycerate in erythroid cells. Trends in Biochemical Sciences. 1982;7:140–142. [Google Scholar]
- 16.Rigden DJ, Walter RA, Phillips SE, Fothergill-Gilmore LA. Sulphate ions observed in the 2.12 A structure of a new crystal form of S. cerevisiae phosphoglycerate mutase provide insights into understanding the catalytic mechanism. J Mol Biol. 1999;286:1507–17. doi: 10.1006/jmbi.1999.2566. [DOI] [PubMed] [Google Scholar]
- 17.White MF, Fothergill-Gilmore LA. Mutase versus synthase: the phosphoglycerate mutase family studied by protein engineering. Biochem Soc Trans. 1990;18:257. doi: 10.1042/bst0180257. [DOI] [PubMed] [Google Scholar]
- 18.Hitosugi T, et al. Tyr26 phosphorylation of PGAM1 provides a metabolic advantage to tumours by stabilizing the active conformation. Nat Commun. 2013;4:1790. doi: 10.1038/ncomms2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kee JM, Villani B, Carpenter LR, Muir TW. Development of stable phosphohistidine analogues. J Am Chem Soc. 2010;132:14327–9. doi: 10.1021/ja104393t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kee JM, Oslund RC, Perlman DH, Muir TW. A pan-specific antibody for direct detection of protein histidine phosphorylation. Nat Chem Biol. 2013;9:416–21. doi: 10.1038/nchembio.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oslund RC, et al. A phosphohistidine proteomics strategy based on elucidation of a unique gas-phase phosphopeptide fragmentation mechanism. J Am Chem Soc. 2014;136:12899–911. doi: 10.1021/ja507614f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kee JM, Oslund RC, Couvillon AD, Muir TW. A second-generation phosphohistidine analog for production of phosphohistidine antibodies. Org Lett. 2015;17:187–9. doi: 10.1021/ol503320p. [DOI] [PubMed] [Google Scholar]
- 23.Pritlove DC, Gu M, Boyd CA, Randeva HS, Vatish M. Novel placental expression of 2,3-bisphosphoglycerate mutase. Placenta. 2006;27:924–7. doi: 10.1016/j.placenta.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson CM, Price NC. Denaturation and renaturation of the monomeric phosphoglycerate mutase from Schizosaccharomyces pombe. Biochem J. 1987;245:525–30. doi: 10.1042/bj2450525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vander Heiden MG, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–9. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moellering RE, Cravatt BF. Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science. 2013;341:549–53. doi: 10.1126/science.1238327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Negelein E. Synthesis, determination, analysis, and properties of 1,3-diphosphoglyceric acid. Methods in Enzymology. 1957;3:216–220. [Google Scholar]
- 29.Han CH, Rose ZB. Active site phosphohistidine peptides from red cell bisphosphoglycerate synthase and yeast phosphoglycerate mutase. J Biol Chem. 1979;254:8836–40. [PubMed] [Google Scholar]
- 30.Wang Y, et al. Seeing the process of histidine phosphorylation in human bisphosphoglycerate mutase. J Biol Chem. 2006;281:39642–8. doi: 10.1074/jbc.M606421200. [DOI] [PubMed] [Google Scholar]
- 31.Fuhs SR, et al. Monoclonal 1- and 3-Phosphohistidine Antibodies: New Tools to Study Histidine Phosphorylation. Cell. 2015;162:198–210. doi: 10.1016/j.cell.2015.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu W, Kimball E, Rabinowitz JD. A high-performance liquid chromatography-tandem mass spectrometry method for quantitation of nitrogen-containing intracellular metabolites. J Am Soc Mass Spectrom. 2006;17:37–50. doi: 10.1016/j.jasms.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 33.Rose ZB, Dube S. Rates of phosphorylation and dephosphorylation of phosphoglycerate mutase and bisphosphoglycerate synthase. J Biol Chem. 1976;251:4817–22. [PubMed] [Google Scholar]
- 34.Rose ZB, Dube S. Phosphoglycerate mutase. Kinetics and effects of salts on the mutase and bisphosphoglycerate phosphatase activities of the enzyme from chicken breast muscle. J Biol Chem. 1978;253:8583–92. [PubMed] [Google Scholar]
- 35.Samanta D, Semenza GL. Serine Synthesis Helps Hypoxic Cancer Stem Cells Regulate Redox. Cancer Res. 2016;76:6458–6462. doi: 10.1158/0008-5472.CAN-16-1730. [DOI] [PubMed] [Google Scholar]
- 36.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–60. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 37.Rappsilber J, Ishihama Y, Mann M. Stop and Go Extraction Tips for Matrix-Assisted Laser Desorption/Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal Chem. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 38.David Y, Vila-Perello M, Verma S, Muir TW. Chemical tagging and customizing of cellular chromatin states using ultrafast trans-splicing inteins. Nat Chem. 2015;7:394–402. doi: 10.1038/nchem.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–4. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park JO, et al. Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat Chem Biol. 2016;12:482–489. doi: 10.1038/nchembio.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.