

Abstract
CmlI catalyzes the six-electron oxidation of an aryl-amine precursor (NH2-CAM) to the aryl-nitro group of chloramphenicol (CAM). The active site of CmlI contains a (hydr)oxo- and carboxylate-bridged dinuclear iron cluster. During catalysis, a novel diferric-peroxo intermediate P is formed and is thought to directly effect oxygenase chemistry. Peroxo intermediates can facilitate at most two-electron oxidations, so the biosynthetic pathway of CmlI must involve at least three steps. Here, kinetic techniques are used to characterize the rate and/or dissociation constants for each step by taking advantage of the remarkable stability of P in the absence of substrates (decay t1/2 = 3 h at 4 °C) and the visible chromophore of the diiron cluster. It is found that diferrous CmlI (CmlIred) can react with NH2-CAM and O2 in either order to form a P-NH2-CAM intermediate. P-NH2-CAM undergoes rapid oxygen transfer to form a diferric CmlI (CmlIox) complex with the aryl-hydroxylamine (NH(OH)-CAM) pathway intermediate. CmlIox-NH(OH)-CAM undergoes a rapid internal redox reaction to form CmlIred -nitroso-CAM (NO-CAM) complex. O2 binding results in formation of P-NO-CAM that converts to CmlIox-CAM by enzyme-mediated oxygen atom transfer. The kinetic analysis indicates that there is little dissociation of pathway intermediates as the reaction progresses. Reactions initiated by adding pathway intermediates from solution occur much more slowly than those in which the intermediate is generated in the active site as part of the catalytic process. Thus, CmlI is able to preserve efficiency and specificity while avoiding adventitious chemistry by performing the entire six-electron oxidation in one active site.
Graphical Abstract
Introduction
The final steps in the biosynthesis of chloramphenicol (CAM, Figure 1) by Streptomyces venezuelae are catalyzed by the N-oxygenase CmlI.1, 2 The substrate for CmlI is the aryl-amine analog of CAM (D-threo-1-(4-aminophenyl)-2-dichloroacetylamino-1,3-propanediol or NH2-CAM), which undergoes a six-electron oxidation, including two oxygenation steps, to yield the aryl-nitro group of the active antibiotic (Figure 1). Spectroscopic studies and the X-ray crystal structure of CmlI have shown that it has a (hydr)oxo and carboxylate-bridged dinuclear iron cluster in the active site.2, 3 Structurally similar clusters are found in many enzymes that activate O2 and then catalyze reactions such as functionalization of unactivated C-H bonds, desaturation reactions, aromatic hydroxylation, and radical formation, but notably, not aryl-amine oxygenation. 4-14 Conversely, CmlI and its structural and function homolog AurF15-19 do not catalyze the types of reactions common to other diiron oxygenases. Kinetic and spectroscopic studies of CmlI and AurF have shown that, despite the structural similarities to other diiron oxygenases, a different type of reactive oxygen intermediate is generated during catalysis. Both N-oxygenase enzymes generate long-lived peroxo intermediates that react directly with substrates,2, 18 as opposed to the high-valence oxo intermediates often formed by the other types of diiron oxygen-activating enzymes. 5, 20, 21 In CmlI, the peroxo intermediate (P) is exceptionally stable (decay t1/2 = 3 h at 4 °C and pH 9) when generated by adding O2 to the diferrous enzyme (Cmlred) in the absence of substrates.2 When P is mixed with aryl-amine substrates, it reacts in milliseconds to yield oxygenated products. The stability of P greatly facilitates transient kinetic experiments designed to investigate the kinetics of its reactivity with substrates. In recent studies, we have shown that it will react with the likely intermediates of the chloramphenicol biosynthetic pathway including aryl-nitroso-CAM (NO-CAM), which has electronic properties different from those of NH2-CAM, demonstrating the catalytic diversity of P.22 Spectroscopic studies have also shown that P has a peroxo-bridging structure significantly different from the bridging cis-μ-1,2-peroxo structure found for almost all other characterized diiron-peroxo intermediates.2, 23
Figure 1.
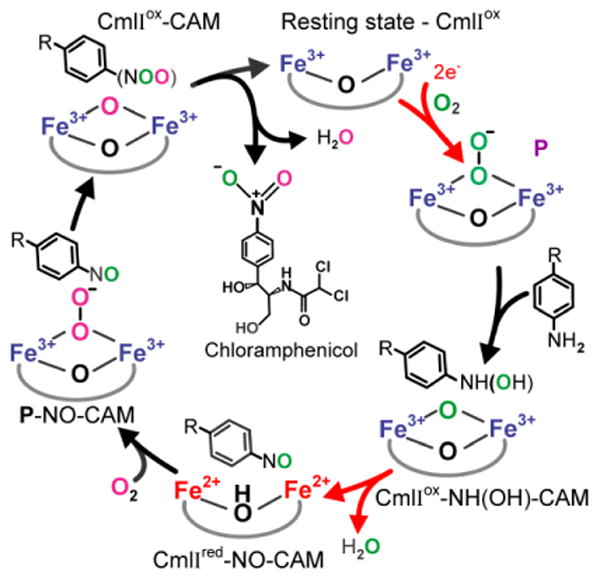
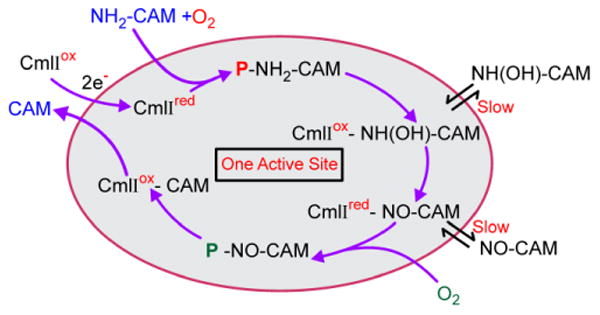
Proposed mechanistic cycle for N-oxygenation by CmlI.22, 23 Diiron cluster reduction steps are shown with red arrows. The color coding of the oxygen atoms highlights the fact that oxygen atoms from two different O2 molecules are incorporated into the final product. R = 2-(dichloroacetylamino)-1,3-propanediol.
Peroxo intermediates, like the high-valence iron-oxo intermediates of other diiron oxygenases,5, 21, 24, 25 can carry out at most a two-electron oxidation reaction, and thus the biosynthetic pathway of CmlI must have at least three steps. Studies of both CmlI and AurF 19, 22 have defined mechanistic proposals that differ substantially from the most straightforward scheme of simply repeating the oxygenation reaction of P three times with three pathway intermediates, a process that would require six external electrons.16, 26 The iteration of this mechanism that we have proposed for CmlI is shown in Figure 1. 22, 23 In this scheme, P is formed and reacted with NH2-CAM to make diferric CmlI (CmlIox) and an aryl-hydroxylamino-CAM (NH(OH)-CAM) intermediate. The resulting CmlIox-NH(OH)-CAM complex undergoes an internal redox reaction to yield Cmlred-NO-CAM that can bind O2 to re-form P as a P-NO-CAM complex. The final oxygenation reaction by this complex yields CAM and CmlIox. The internal reduction step means that this mechanism requires only two external electrons that are used to form the first P. This mechanism is supported by stepwise product formation studies showing the generation and reactivity of the predicted intermediates.22 Also, it has been observed that forming P with one isotope of O2 and then carrying out the reaction in a vessel with a different isotope of O2 in the headspace results in a mixed isotope nitro-group in CAM, thereby demonstrating re-formation of P as part of the biosynthetic pathway. 22
In the studies reported here, the dynamics of the predicted reaction pathway of CmlI are studied using transient kinetic techniques to determine rate and/or dissociation constants for each step in the process. In this way, the main tenets of the mechanism including the internal reduction step and the reaction of P with the chemically distinct substituent groups of the pathway intermediates can be independently demonstrated and quantified. In the course of this study, more subtle aspects of the process have also come to light, including the mechanism by which the enzyme catalyzes the multistep reaction without a loss of intermediates and with perfect specificity.
Experimental Procedures
Standard materials and procedures are described in the Supporting Information. The D-threo-1-(4-aminophenyl)-2-dichloroacetylamino-1,3-propanediol (NH2-CAM) substrate was obtained from Toronto Research Chemicals, Toronto, ON. The NH(OH)-CAM and NO-CAM substrates were synthesized as previously described. 22
Stopped-Flow Analysis of P Formation and Single-Turnover Reactions
Stopped-flow reactions were performed using an Applied Photophysics model SX.18MV stopped-flow device configured for single-wavelength collection at 300 nm (P formation reaction), 390 nm (reactions with NH(OH)-CAM) or 480 nm (reactions with NH2-CAM or NO-CAM). All reactions were performed in 50 mM Bicine (pH 9) at 4 °C. CmlI is fully functional between pH 6 and 9, but the stability of P is greatly enhanced at high pH. Consequently, it is possible to initiate transient kinetic experiments with CmlI in nearly a single form at pH 9, simplifying the setup of experiments and the data analysis. The stopped-flow instrument was made anaerobic by being flushed with dithionite solution followed by anaerobic buffer. CmlI was reduced as described in the Supporting Information and rapidly mixed with buffer containing varying concentrations of O2 and substrate using the stopped-flow instrument (see figure legends). In some cases, substrate was added anaerobically to the diferrous CmlI (CmlIred) solution before it was mixed with O2-containing buffer. In cases in which the P was preformed, it was generated by rapid mixture with an equivalent volume of O2-saturated buffer (or in some cases, buffer containing 0.9 equivalents of O2) in the gastight syringe. In the reactions with NH(OH)-CAM, CmlI was not reduced, but was made anaerobic before the reaction was performed.
Methods for analyzing reaction kinetics to determine rate constants and or dissociation constants are described in the Supporting Information. These methods include procedures for fitting the time course to multiexponential equations and concentration dependencies to hyperbolic expressions.
Mössbauer Analysis of the Reaction of CmlIred with O2 and NO-CAM
Mössbauer samples were prepared using an Update 715 ram syringe controller to mix solutions and dispense the mixtures onto counter-rotating aluminum wheels cooled with liquid nitrogen.27, 28 The time between mixing and freezing was controlled using a calibrated set of delay tubing between the mixer and a nozzle over the wheels. The counter rotating wheels grind the frozen solution into a powder which is deposited into the liquid nitrogen bath below the wheels. It is then collected and packed into a Mössbauer cup. In the experiments described here, one syringe held 57Fe-enriched1.25 mM CmlIred in 50 mM Bicine (pH 9), and the other held O2-saturated buffer with 6.25 mM NO-CAM. Data were collected were 20 ms, 80 ms, 250 ms, and 30 s. The Mössbauer spectra were analyzed as described in the Supporting Information.
Results
P Formation Reaction
The P diferric peroxo intermediate of CmlI forms in <1s when chemically reduced diferrous CmlI (CmlIred) is combined with O2. P is highly stable in the absence of substrate, exhibiting a t1/2 ∼ 3 h at 4 °C and pH 9,2 and thus the formation reaction is expected to be irreversible. To determine the rate constant(s) for P formation, anaerobic CmlIred was mixed with 5-36 equivalents of O2 using a stopped-flow ultraviolet-visible (UV-vis) instrument. The formation of P was followed at 300 nm, the wavelength at which optical feature changes due to P formation are maximized (see Figure S1 for rationalization of wavelength choice here and in reactions described below). The resulting traces are best fit by a three-summed-exponential expression (Figure 2). A complete description of the techniques used for the analysis of transient kinetic data presented in this study can be found in the Supporting Information. Plotting the reciprocal relaxation times (1/τ or RRT) versus O2 concentration shows that all three phases have a linear dependence on O2 concentration with apparent second-order rate constants of 58.1 ± 2.0, 19.6 ± 0.4, and 1.4 ± 0.1 s-1 mM-1 (Figure 3). The magnitude of the total amplitude change stays the same over the range of O2 concentrations tested, and the amplitudes of the three observed phases are approximately equal.
Figure 2.
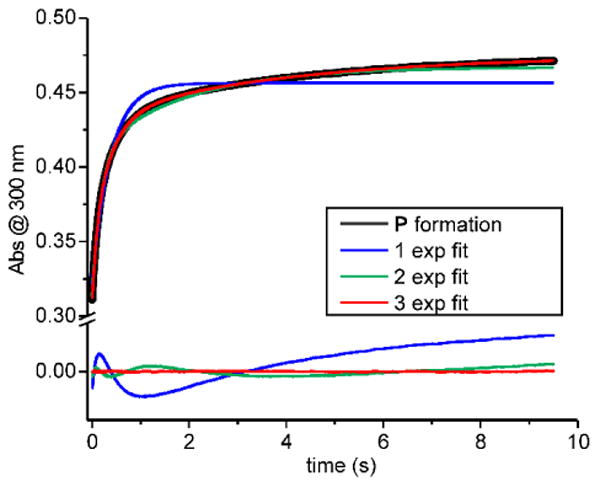
Optical changes correlated with the formation of P by combining 40 μM CmlIred with 450 μM O2 in buffer (final, postmix concentrations). Single- (blue), double- (green), and triple- (red) exponential fits of the P formation process shown by the black trace. Fit residuals are shown below the trace and highlight the need for a three-exponential fit. (50 mM Bicine at pH 9 and 4 °C).
Figure 3.
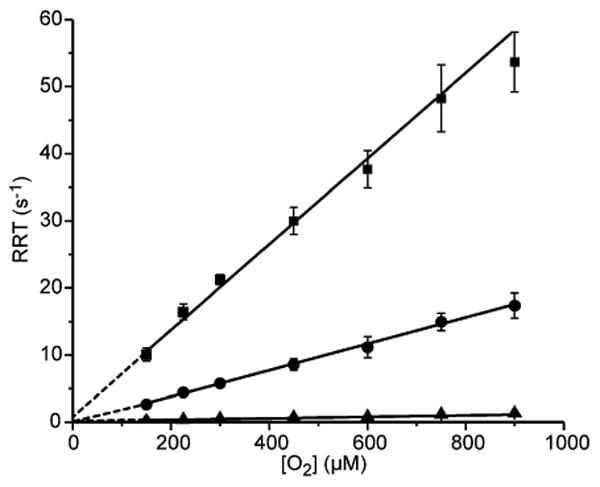
O2 concentration dependence of P formation. 40 μM CmlIred (final, postmix concentration) was mixed in a 1:1 ratio with varying concentrations of O2 in buffer. RRT-1, -2, and -3 (■,●,▲, respectively) from multiexponential fitting of stopped-flow traces are plotted vs O2 concentration, all showing linear relationships. The second-order rate constant for each process was determined from the slope of the linear fit to the data (solid lines), and they are reported in the text. The reported error of each point is one standard deviation from the mean. 50 mM Bicine at pH 9 and 4 °C.
The presence of three phases that are linearly dependent on substrate is most easily ascribed to three parallel reactions to form P, rather than a single P formation process that employs three consecutive steps, which would yield a nonlinear or no concentration dependence in two of the three phases. The Mössbauer spectrum of P shows only one type of cluster.2 If there are multiple Mössbauer-indistinguishable forms of P, they all appear to react with substrates (see below). The multiple-phase P formation behavior is likely to be the result of either or both of the following scenarios: (a) three different pathways for O2 binding that all lead to the same P, or (b) the existence of CmlIred in three different structural or protonation states that differentially affect P formation. While the Mössbauer spectra of CmlIred show only a single species, the spectra of CmlIox reveal that several forms exist in a pH dependent distribution.2 The concentration dependence plots of all phases have a y-intercept close to zero, showing that the formation of P in the absence of substrate is nearly irreversible, as expected. This finding is also in accord with our previous demonstration that P does not exchange with headspace O2 over the course of hours.22
Kinetic Parameters of the Reaction of P with NH2-CAM
The next step of the CmlI cycle that can be isolated is the reaction of P with NH2-CAM to form NH(OH)-CAM and CmlIox, followed by oxidation of NH(OH)-CAM to NO-CAM and reduction of the diferric cluster to form CmlIred. To explore this reaction, P was formed with 0.9 equivalent of O2 (to ensure no excess O2 in the reaction) and then loaded immediately on the stopped-flow instrument for reaction with 5-40 equivalents of NH2-CAM dissolved in anaerobic buffer. When monitored at 480 nm, both P decay and the subsequent reduction of the diiron cluster appear as a decrease in absorbance. Both processes are first-order or pseudo-first order, and the reaction stops after the last step, therefore summed exponential fitting is appropriate (see Supporting Information for additional details).
The time courses are fit well by a two-summed-exponential expression (Figure 4A). Both exponentials have positive amplitudes (a positive amplitude sign correlates with a decrease in absorbance), and the phase with larger RRT contributes ∼85% of the total absorbance change. Because the types of reactions involved are effectively irreversible (O-O bond cleavage and 100% diiron cluster reduction, see below), it is possible to assign the two RRTs to rate constants for two steps of the reaction. The simplest scenario for the reaction shown in Scheme 1A is to assign one RRT to the rate constant for conversion of P to CmlIox and the second to the rate constant for reduction of CmlIox to CmlIred. However, analysis of the absorbance changes makes this scenario unlikely.
Figure 4.
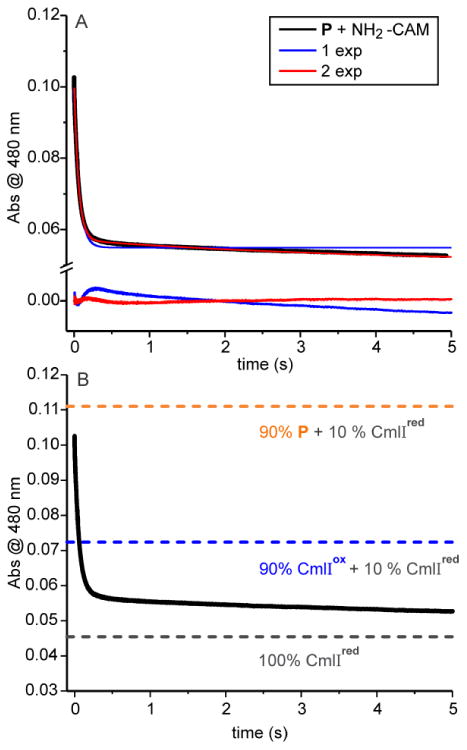
Optical changes at 480 nm correlated with the reaction of 180 μM P with 2 mM NH2-CAM. A: Single- (blue) and double- (red) exponential fits of the substrate-mediated P decay process (black). Fit residuals are shown below the trace. B: Reaction as in panel A. Dashed lines indicate the calculated absorbance of 180 μM P + 20 μM CmlIred, 180 μM CmlIox + 20 μM CmlIred, and 200 μM CmlIred. 50 mM Bicine at pH 9 and 4 °C.
Scheme 1. Proposed Schemes for the Reaction of P with NH2-CAMa.
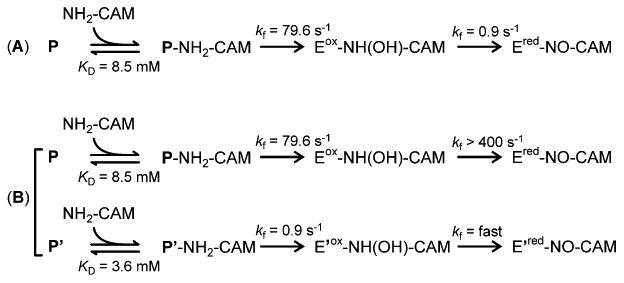
aRate constants are derived from the plots in Figure 5. For the sake of simplicity, CmlIox and CmlIred are abbreviated as Eox and Ered, respectively. P' and E' represent CmlI species that are kinetically distinct from P and E, respectively. Part A represents the case in which there is a single reaction pathway for P reacting with NH2-CAM. Part B represents the case in which two parallel pathways are possible beginning from two forms of P.
The expected absorbance due to P, CmlIox, and CmlIred at 480 nm at various stages of the reaction can be determined using the extinction coefficients of the species (Table S1) and the concentration of CmlI in the experiment (there is negligible absorption due to any of the organic CAM substrates at 480 nm). The starting sample (absorbance = 0.11, Figure 4B, orange dashed line) was prepared to have 10% CmlIred and 90% P. The first step in the proposed mechanistic model is the conversion of P and NH2-CAM to CmlIox and NH(OH)-CAM. The corresponding predicted absorbance of 90% CmlIox and the remaining 10% CmlIred would give an absorbance of 0.072 (Figure 4B, blue dashed line). After re-reduction of the CmlI by NH(OH)-CAM formed in situ, the end point pure CmlIred spectrum would give an absorbance of 0.046 (Figure 4B, black dashed line). Consequently, approximately equal changes in absorbance should result from each step. The observation that the rapid segment of the time course results in an absorbance significantly below the blue dashed line shows that more than just the P conversion to CmlIox has occurred during the fast phase.
An alternative interpretation for the overall two-phase fit of the time course with unequal amplitudes illustrated in Scheme 1B is that there are two independent processes starting from two different forms of P in an 85:15 ratio (based on the observed ratio of fast and slow phase amplitudes). If the reduction step following the reaction of the dominant form of P with NH2-CAM is much faster (> 5-fold), the reaction will appear as a single exponential time course proceeding below the absorbance demarcated by the blue dashed line. Thus, the dominant phase would be reflective of both P decay and fast reduction. The second observed phase would then describe a smaller fraction of P reacting similarly, but with a lower initial rate constant.
The selection of part B of Scheme 1 over part A is also supported by plots of the RRTs from each exponential phase versus the concentration of NH2-CAM (Figure 5). The resulting plots are hyperbolic, suggesting that an unobserved, fast, reversible second-order NH2-CAM binding process precedes P decay (see the Supplemental Experimental Procedures of Supporting Information). The dissociation constants (KD) for complex formation reactions for the faster and slower processes are 8.5 ± 1.5 mM and 3.6 ± 1 mM, respectively. Analysis of the hyperbolic curves (see Figure 5A and the Supporting Information) show that the rate constants for the step following binding are as follows: kf = 79.6 ± 5 s-1 and kr ≈ 0 s-1 for the faster process and kf = 0.9 ± 0.1 s-1 and kr ≈ 0 for the slower process. The irreversible nature of the reaction is reasonable considering that it involves the cleavage of an O-O bond and insertion of an O atom into an N-H bond as P reacts with NH2-CAM. If the reaction is irreversible, then in Scheme 1A, the concentration dependence on NH2-CAM will be lost in the second phase, which is not the case. Moreover, it is shown below that modeling the time course of the complete biosynthetic pathway requires a reduction step with a rate constant that is 2-3 orders of magnitude greater than that shown in Scheme 1A.
Figure 5.
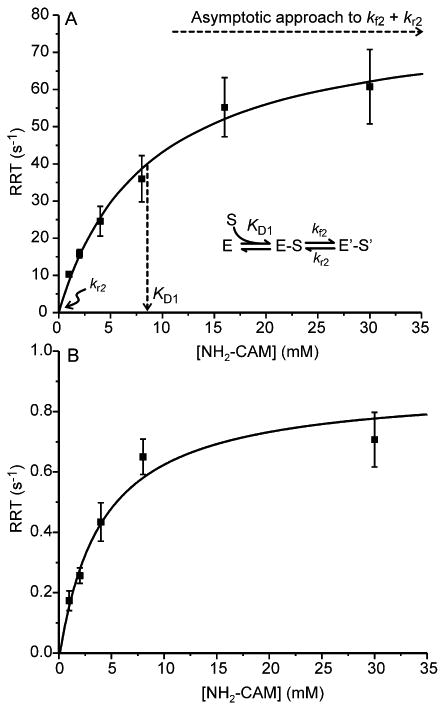
NH2-CAM concentration dependence of the RRTs for the reaction of P with NH2-CAM. Extracted RRTs plotted vs [NH2-CAM] for the reaction of P (180 μM, final postmix concentration) with varying concentrations of NH2-CAM in anaerobic buffer. The generic method for analysis is illustrated in panel A where E and E' are two forms of the enzyme, S is a substrate, and S' is a modified form of S (see the Supporting Information). (A) For RRT-1 a hyperbolic fit gives the following: KD = 8.5 ± 1.5 mM, kf = 79.6 ± 5 s-1, and kr ≈ 0 s-1. (B) For RRT-2 a hyperbolic fit gives the following: KD = 3.6 ± 1 mM, kf = 0.9 ± 0.1 s-1, and kr ≈ 0 s-1. (50 mM Bicine at pH 9 and 4 °C).
Reaction of CmlIox with NH(OH)-CAM
The reduction of CmlIox by NH(OH)-CAM can also be monitored by simply mixing these two reactants anaerobically. Careful scrubbing of O2 from the reaction buffers and the stopped-flow instrument ensures that the reaction stops after the reduction process and does not proceed to re-form P for the final oxidation step. To study this step, CmlIox was made anaerobic and then mixed in a 1:1 ratio with anaerobic buffer containing varying concentrations of NH(OH)-CAM, all of which were appropriate for establishing pseudo-first-order conditions. The resulting optical change at 390 nm was a decrease in absorbance; the time course trace fit to a double summed-exponential equation (Figure 6). The monitoring wavelength was chosen to maximize the optical change while minimizing the contribution of the NH(OH)-CAM that occurs at lower wavelengths. The net absorbance change of ∼0.03 matched the expected absorbance change for the conversion of 40 μM CmlIox to CmlIred.
Figure 6.
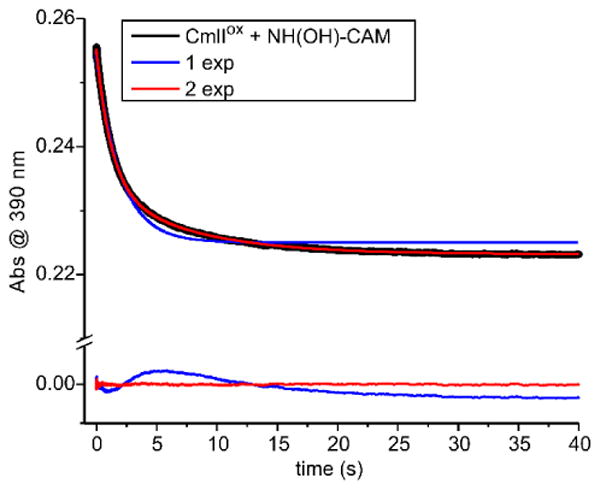
Optical change at 390 nm upon the anaerobic mix of 40 μM CmlIox and 2 mM NH(OH)-CAM (final, postmix concentrations). The trace fits best to a two exponential equation, as shown by the colored fit lines and the residuals for 1- (blue) and 2- (red) exponential fits shown below the trace. (50 mM Bicine at pH 9 and 4 °C.
The plots of RRT-1 and RRT-2 versus NH(OH)-CAM concentration are both hyperbolic (Figure 7), suggesting that a relatively fast step, presumably NH(OH)-CAM binding (KD = 1.1 mM), precedes the CmlI reduction occurring in the observable steps. The rate constants extracted from these plots (RRT-1: kf = 1.3 ± 0.1 s-1 and kr ≈ 0 and RRT-2: kf = 0.23 ± 0.03 s-1, kr ≈ 0) are very slow compared with the rate constant for reduction of the diiron cluster observed when starting the reaction with NH2-CAM as described above. The slower phase disappears as the concentration of NH(OH)-CAM increases, suggesting that the biphasic kinetics arise from some sort of allosteric effect of substrate that will not be pursued further here. A model for the reaction of CmlIox and NH(OH)-CAM is shown in Scheme 2.
Figure 7.
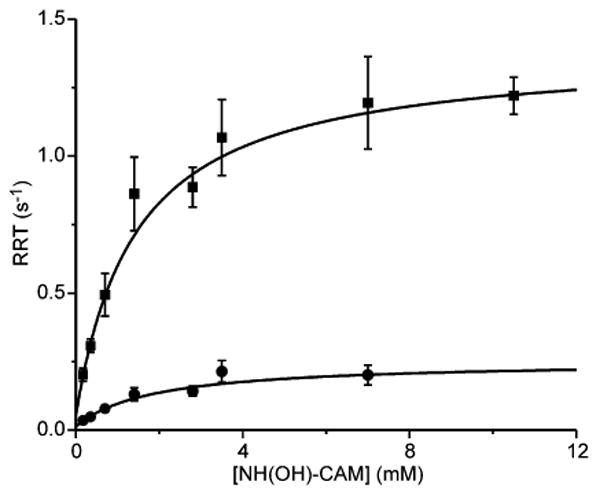
NH(OH)-CAM concentration dependence of the RRTs for the reaction of 40 μM anaerobic CmlIox (final, postmix concentration) and varying concentrations of NH(OH)-CAM in anaerobic buffer. (■) hyperbolic fits of RRT-1s extracted from the traces of the reaction, giving KD = 1.1 ± 0.05 mM, kf = 1.3 ± 0.1 s-1 and kr ≈ 0, and (●) hyperbolic fits of RRT-2s extracted from the traces of the reaction, giving KD = 1.2 ± 0.3 mM, kf = 0.23 ± 0.03 s-1, and kr ≈ 0. The amplitude of RRT-2 decreases progressively and is not observed above 8 mM NH(OH)-CAM. (50 mM Bicine at pH 9 and 4 °C).
Scheme 2. Proposed Scheme for the Reaction of CmlIox with NH(OH)-CAMa.
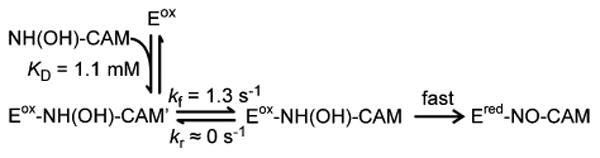
aFor the sake of simplicity CmlIox and CmlIred are abbreviated as Eox and Ered, respectively. The intermediate Eox-NH(OH)-CAM' and the assignment of the rate constant for the reduction step as “fast” are proposed on the basis modeling of the overall biosynthetic reaction pathway kinetics (see Discussion, Simulation of the Overall Reaction).
The results indicate that the reduction of the diiron cluster by NH(OH)-CAM occurs much faster when this pathway intermediate is generated in situ in the active site rather than having to bind from solution. Moreover, if NH(OH)-CAM dissociates from the active site after it is generated, but before it can reduce the diiron cluster, the subsequent reduction step would presumably occur with the slow rate constants observed for the reaction of NH(OH)-CAM in solution with CmlIox. This route for cluster reduction should thus be considered an off-pathway process.
Reaction of P with NO-CAM
Anaerobic P was formed as described above and mixed with NO-CAM dissolved in anaerobic buffer. Surprisingly, little to no reaction was observed when the reaction was monitored at 480 nm (Figure S2). Alternative ways to approach this reaction allow the kinetic time course to be monitored and the observation of product as described below.
Reaction of CmlIred with NO-CAM and O2
The reaction in which NO-CAM is converted to CAM can be examined by mixing CmlIred with a buffer solution containing a 5-150-fold excess of NO-CAM and a 5-fold excess of O2 (a saturated solution) (Figure 8A). At 480 nm, one would expect to observe an increase in absorbance as P is formed and then a decrease as the NO-CAM is converted to CAM with the formation of CmlIox. Conducting the reaction in this way gave time courses that are fit well by a three-summed exponential equation. The resulting RRTs were plotted versus NO-CAM concentration as shown in Figure 8B. The faster two phases show a hyperbolic dependence on NO-CAM concentration, while the slowest phase shows no dependence. In this case, the amplitudes of the two faster phases have opposite signs, making it unlikely that they arise from parallel reactions. The hyperbolic dependencies indicate that there is a fast, reversible step that precedes the observable reaction, which must itself consist of at least three additional steps in accord with the three-summed-exponential fit.
Figure 8.
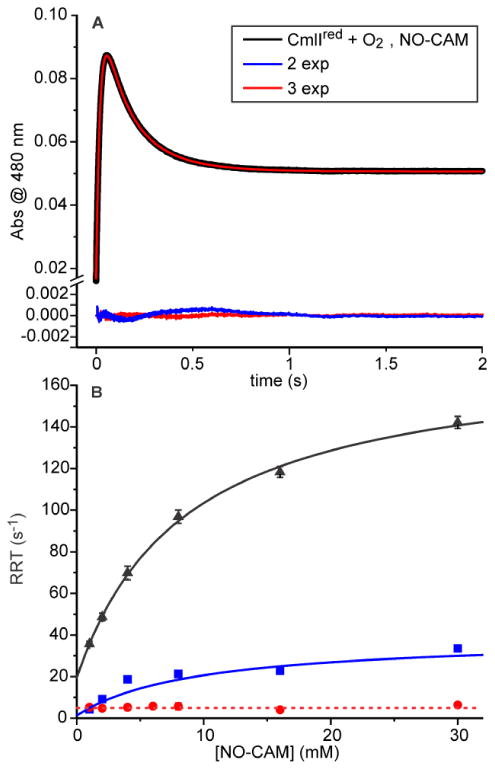
Reaction of CmlIred with NO-CAM in O2-saturated buffer. (A) Optical changes at 480 nm upon mixing 200 μM CmlIred with buffer containing 2 mM NO-CAM and 0.9 mM O2 (all postmix, final concentrations). A three-exponential fit (red) best simulates the time course and is shown superimposed on the data (black). Residuals for two- (blue line) and three- (red) exponential fits are shown below the data. (B) Fits of the RRTs versus NO-CAM concentration. For the RRT-1 hyperbolic fit (black), kf = 150.4 ± 8.8 s-1 mM-1 (at 0.9 mM O2), kr = 19.4 ± 2 s-1, KD-binding = 8.6 ± 1 mM, and KD-observed step = 0.13 mM. For the RRT-2 hyperbolic fit (blue), kf = 37.7 ± 5 s-1, and kr ∼ 0 s-1. For the RRT-3 linear fit (red), an estimate of the concentration-independent rate constant (kf + kr') is 5.2 ± 0.1 s-1. The error ranges for the blue and red points are within the size of the symbol. (50 mM Bicine at pH 9 and 4 °C.
Although the observed kinetics of the reaction of CmlIred with O2 and NO-CAM would support a variety of four-step processes that include a binding step, Scheme 3 shows one possibility that is reasonable based on the chemistry of the biosynthetic pathway. In this model, NO-CAM binding is the fast reversible step that precedes the optically observable steps with a KD of 8.6 ± 1 mM. Reversible (Figure 8B, black curve, non-zero y-intercept) O2 binding occurs in the following step to form P, giving a fast increase in absorbance. The reversible nature of this step would be expected to yield nonlinear plot for another RRT, as observed. In the model in Scheme 3, the P reacts in the subsequent step with NO-CAM irreversibly to give a decrease in absorbance. The presence of two reversible steps in the three-step series to this point (beginning with binding) should, in principle, be analyzed using the roots of a cubic equation. However, the large difference in the observed maximum RRTs allows the rate constants for the individual steps to be approximated from the maximum (kf' + kr) and intercept (kr) of the two hyperbolic plots. This approximation gives kf = 150.4 s-1 mM-1 (at 0.9 mM O2) and kr = 19.4 s-1 for O2 binding (KD = 0.13 mM) to form P and kf = 37.7 s-1 and kr = 0 for the reaction of P with bound NO-CAM. The irreversible nature of the latter step (zero intercept for the plot in Figure 8B, blue curve) uncouples the third RRTs from NO-CAM concentration, so the final concentration-independent step occurs with kf + kr' = 5.2 s-1 (Figure 8B, red curve). This step is likely to be product release and to represent the rate constant limiting substrate flux through the cycle. The small amplitude of this slow phase (< 10% of those of the faster phases) is consistent with the very small change in absorbance at 480 nm caused by the binding of CAM to CmlIox.22
Scheme 3. Proposed Scheme for the Reaction of CmlIred with NO-CAMa.
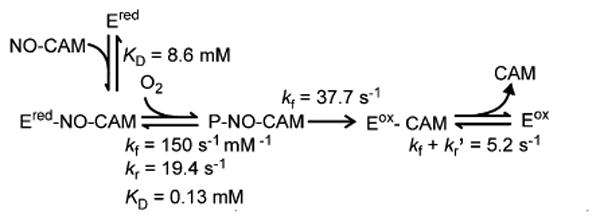
aFor the sake of simplicity, CmlIox and CmlIred are abbreviated as Eox and Ered, respectively.
At high NO-CAM concentrations, the faster RRT reaches values that far exceed the expected values based on the rate constant for P formation in the absence of NO-CAM (see Figure 3). This suggests that in the NO-CAM complex, O2 binding is facilitated, allowing much faster P formation. It also suggests that NO-CAM binding precedes O2 binding, consistent with the increasing maximum value of P formed as the concentration of NO-CAM increases (Figure 9). Any P that forms prior to NO-CAM binding would yield a dead-end complex, as the results presented above and in Figure S2 show that P does not react with NO-CAM. Because there is no substantial buildup of P at the end of the reaction, it is likely that NO-CAM binding is very fast.
Figure 9.
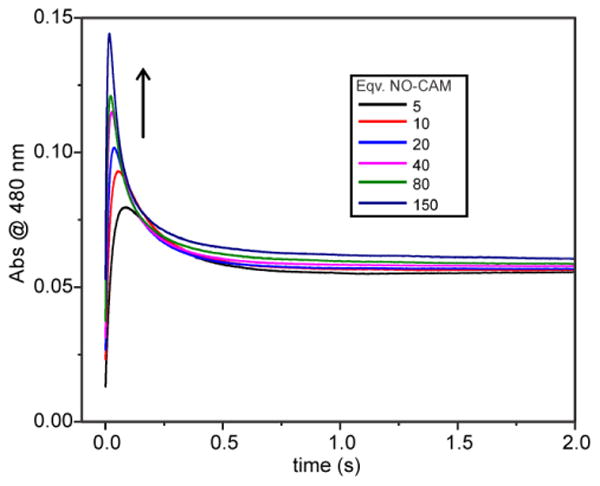
NO-CAM concentration dependence of the time course of the reaction of CmlIred with pseudo-first order concentrations of O2 and NO-CAM. The traces follow the reaction of 200 μM CmlIred with 1-30 mM NO-CAM in O2-saturated buffer. (50 mM Bicine at pH 9 and 4 °C).
Verification of P as the Active Oxidant That Acts on NO-CAM
One possible explanation for the failure of preformed P to react with NO-CAM is that a different form of P is made when NO-CAM is present in the active site. To examine this possibility, short time point samples of the reaction of CmlIred with an excess of NO-CAM and O2 were prepared by a rapid-freeze quench procedure and analyzed by Mössbauer spectroscopy (Figure 10, top).
Figure 10.
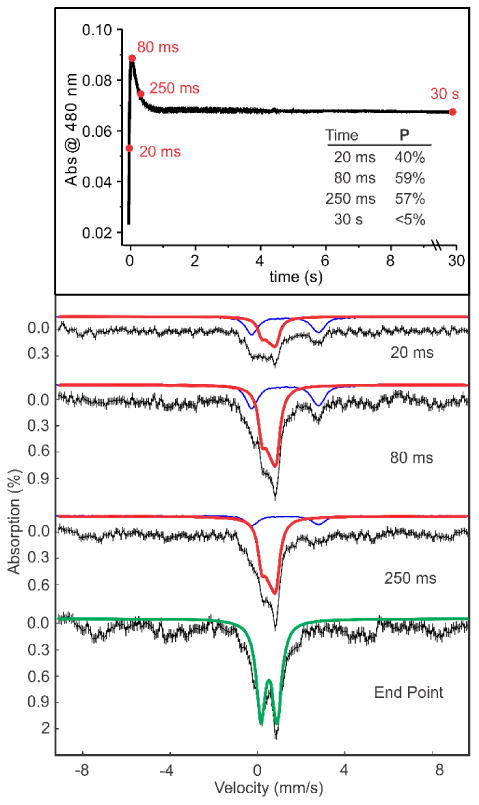
Time points (top) of RFQ samples for Mössbauer analysis of the reaction of 200 μM CmlIred with 0.9 mM O2 and 2 mM NO-CAM (final, postmix concentrations). The inset table shows the fractions of the sample present as P quantified from the Mössbauer spectra. Mössbauer spectra of the samples (bottom). The black lines are the experimental data, and the colored lines are fits to various species: diferrous (blue), intermediate P (red), and resting state diferric (green). A small fraction of mononuclear ferric ion also appears at the end point. (50 mM Bicine at pH 9 and 4 °C.
Mössbauer analysis (Figure 10, bottom) showed a decrease in the level of CmlIred over the course of the reaction, concomitant with an increase in the level of P. The spectral parameters for P formed in this way are the same as those for P formed in the absence of substrate (Table S2).2 The level of P increases to a maximum and decays following approximately the time course of the 480 nm species observed in the reaction of NO-CAM with CmlIred and O2 (Figure 9). At a long time (30 s), < 5% P is present and the sample is predominantly diferric. Previous experiments have shown that CAM is formed in this reaction.22 Thus, Mössbauer analysis suggests that intermediate P formed in the presence or absence of substrate has identical Mössbauer parameters and is the oxidant that acts on NO-CAM prebound in the active site.
Kinetic Parameters of the Complete Reaction Pathway
The complete biosynthetic pathway was examined in two ways: (a) by pre-equilibrating NH2-CAM anaerobically with CmlIred to allow the complex to reach equilibrium and then initiating the reaction by mixing with O2-saturated buffer, or (b) by mixing CmlIred with O2-saturated buffer containing various concentrations of NH2-CAM. No significant difference in trends or magnitudes of RRTs was observed between the two iterations. The data presented are for the latter combination. All optical changes were observed by stopped-flow UV-vis spectroscopy at 480 nm, 4 °C and pH 9. This wavelength was chosen because it gives the maximum optical change for the P formation and P decay processes. Using a shorter wavelength gives a greater optical change for P formation, but P decay is masked by the reappearance of the Fe-O-Fe charge transfer band of diferric CmlI, which has a shoulder at ∼375 nm. Initiating the reaction in this way allows both P formation and substrate-mediated P decay to be observed (Figure 11). After P forms and decays, the resulting CmlIox is reduced by the NH(OH)-CAM product of the first reaction to give a further decrease in absorbance. This reduction is followed by binding of O2 to re-form P, resulting in an increase in absorbance. Finally, P decays as CAM is formed to give a decrease in absorbance.
Figure 11.
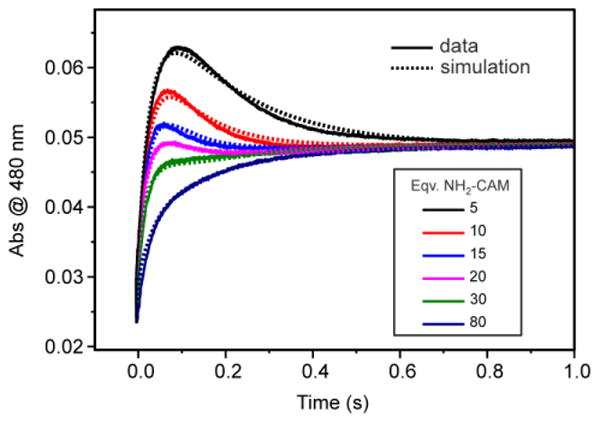
NH2-CAM concentration dependence of the overall reaction time course. The solid curves are for optical changes at 480 nm from the reaction of CmlIred (200 μM) with 1-16 mM NH2-CAM (final, postmix concentrations) in O2-saturated buffer and the dashed curves are for simulation of the same reaction using numerical integration. The rate constants and reactions used for the simulation are shown in Scheme 4 (see Discussion). The postmix O2 concentration is ∼ 0.9 mM at 4 °C. (50 mM Bicine at pH 9 and 4 °C).
In sharp contrast to the trend shown above for the reaction of CmlIred with O2 and NO-CAM, the maximum amount of P formed decreases with an increasing substrate concentration when NH2-CAM is the substrate. This suggests that the two reactions proceed differently. The traces in Figure 11 show that as the concentration of NH2-CAM increases, the pseudo-first-order rate constants for P formation and decay converge. Unfortunately, this convergence makes the resolution of the RRTs by summed-exponential fitting unreliable.
Some insight into this complex overall reaction and the differences in P reactivity at different steps can be gained by combining the measured kinetic and thermodynamic parameters for each step with numerical integration of the overall reaction. This approach to the analysis is explored in the Discussion.
Discussion
The results presented here show that the complex reaction pathway for the final steps in the biosynthesis of chloramphenicol can be studied in segments to extract rate constants and/or thermodynamic parameters for each step. The major tenets of the proposed reaction cycle shown in Figure 1 are supported by these studies. In particular, the data demonstrate the oxygenase reactivity of peroxo intermediate P with NH2-CAM and NO-CAM as well as the ability of the pathway intermediate NH(OH)-CAM to reduce the CmlI diferric cluster in preparation for the proposed midpathway re-formation of P. The results also reveal several new aspects of the pathway and its regulation, including insight into the effect of substrate binding on the rate constant for O2 binding and the dramatic difference in the rate constant for cluster reduction by NH(OH)-CAM formed in situ versus that binding from solution. Kinetic evidence is found for progress along the biosynthetic pathway at rates that are high compared with those for dissociation of intermediates from the enzyme, providing insight into how the enzyme efficiently accelerates catalysis while maintaining specificity. These new aspects of CmlI catalysis are discussed here.
Regulation of the P Formation Rate and Substrate Binding Order
The set of time courses shown in Figure 11 represents the overall biosynthetic process beginning with NH2-CAM and O2 binding to CmlIred. The time courses look similar when NH2-CAM is preincubated with CmlIred before being mixed with O2. It is evident that this process differs in two respects from that later in the pathway in which CmlIred reacts with NO-CAM and O2. First, the NO-CAM reaction has a strict binding order such that preincubation with O2 to form P stops the reaction (Figure S2). Second, the maximal amount of P formed and the apparent rate at which it is formed increase with and increase in NO-CAM concentration (Figure 9) but decrease with an increase in NH2-CAM concentration (Figure 11). This observation is readily rationalized in the case of NO-CAM because the CmlIred-NO-CAM complex must form first to allow P formation in a way that can go on to form CAM (Figure S2). In contrast, the rationale for the decreasing level of accumulation of P in the NH2-CAM reaction proposed in Scheme 4 is more complex. The lack of a preferred order of addition of substrates in this case suggests that it can proceed via two parallel routes. Experimental data are reported here for the rate constants and binding KD values for the route in which O2 can bind first (constants colored black in Scheme 4) (Figures 3 and 5). The results show that in the absence of NH2-CAM, P forms by three kinetically distinguishable routes. However, as shown by the numerical integration fit to the data shown in Figure 11 (see below), when even low levels of NH2-CAM (or NO-CAM, see Figure 8 and Scheme 3) are present, only one rate constant for P formation is required to fit the time course. Interestingly, the observed rate constant is not the same as any found in the absence of a substrate. This finding suggests that the substrate makes the CmlIred population homogeneous with respect to P formation while altering the rate of the process, by a mechanism that is currently not understood. The dissociation and rate constants for the branch of the pathway starting with NH2-CAM binding are more difficult to determine directly because of the lack of a spectral change upon NH2-CAM binding, the similarity of the rate constants for P formation and decay, and the inability to limit the reaction to a few steps. However, the presence of this alternative pathway is necessary to account for the decreasing maximal level of accumulation of P with an increasing NH2-CAM concentration. This observation is true because, at low concentrations of NH2-CAM, the initial O2 binding pathway is preferred and P decays slowly after it is formed due to the relatively low affinity of P for NH2-CAM (KD = 8.5 mM). Consequently, P maximally accumulates. As the NH2-CAM concentration increases, the rapid, high-affinity NH2-CAM binding pathway is preferred, leading efficiently to the P-NH2-CAM intermediate, which is very rapidly converted to CmlIox-NH(OH)-CAM. Consequently, neither P nor the complex of P with a substrate accumulates.
Scheme 4. Proposed Scheme for the Overall Reaction of CmlIred with NH2-CAM and O2 To Form CAMa.
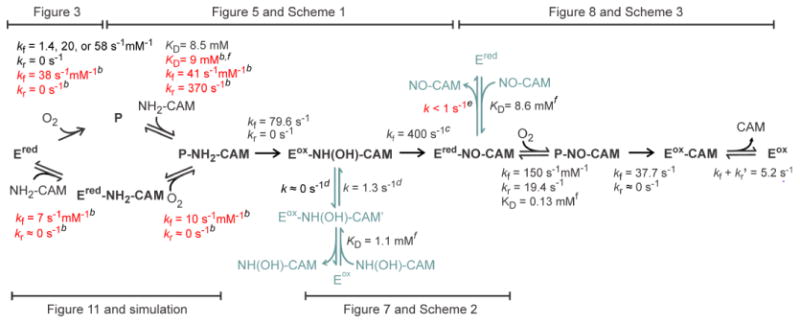
a The proposed scheme synthesizes all the kinetic data collected in this study. The figure or scheme that first introduced each rate constant or KD value is indicated. For the sake of simplicity CmlIox and CmlIred are abbreviated as Eox and Ered, respectively. Bold black intermediate names and arrows denote components of the main pathway. Blue-green intermediate names and arrows denote experimentally accessible components but are thought to be side pathways. The rate constants for intermediates leaving by the side pathways are slow and have little effect on the observed time course of the reaction. The rate constants and dissociation constants shown in black were determined experimentally in this work and are fixed in the numerical integration shown in Figure 11 (dashed curves). The rate constants and dissociation constants shown in red were determined by the numerical integration program to best fit the data over a range of substrate concentrations.
bThese constants can only be varied over only a small (> 5%) range and still give a reasonable fit to the data.
cThis constant can assume any of value >300 s-1.
dThese constants can be varied over a large range and still give a reasonable fit to the data.
eThe rate constant for dissociation of NO-CAM for this step must be <1 s-1.
fThe KD refers to the binding reaction.
In Situ versus Bimolecular Reactions
After CmlIox-NH(OH)-CAM is formed in either the reaction pathway beginning with CmlIred reacting with O2 and NH2-CAM or that beginning with P reacting with NH2-CAM, a very rapid reduction occurs to form Cmlred-NO-CAM. It is likely that both of these reaction pathways involve in situ reduction of the diiron cluster by NH(OH)-CAM before it can escape the active site. The best supporting evidence of this proposal is the very slow reduction observed when the reaction is started by mixing CmlIox with NH(OH)-CAM. This latter reaction proceeds with a fast, reversible binding reaction followed by slow reduction. It is possible that before reduction can occur, NH(OH)-CAM must reorient from the position of initial binding (CmlIox-NH(OH)-CAM') to the position adjacent to the diiron cluster that it would occupy if it were formed in situ (CmlIox-NH(OH)-CAM). We speculate that this reorientation is the slow step, so that the reduction reaction itself remains very fast as required by the overall flux through the reaction pathway (see below). The net effect of in situ reduction without intermediate dissociation is an 80-fold increase in flux through this step as well as a guarantee that most of the NH(OH)-CAM formed will not be lost from the pathway or undergo adventitious reactions.
A similar in situ versus biomolecular scenario appears to apply to the following steps in the pathway in which CmlIred-NO-CAM is converted to P-NO-CAM and then CmlIox-CAM. Our results show that the bimolecular reaction in which CmlIred binds NO-CAM and then reacts with O2 is a viable reaction. However, if NO-CAM is generated in situ by starting with the NH2-CAM reaction, the dissociation of NO-CAM before O2 binding would lead to an inefficient reaction. This observation is true because unbound NO-CAM generated in this way would be at a very low concentration compared with the concentration used to study the reaction of CmlIred with NO-CAM in O2-saturated buffer (Figure 8). Consequently, the fast O2 binding reaction to CmlIred would lead to the irreversible formation of P, which cannot react with NO-CAM not already bound to the enzyme (Figure S2). Our previously published results show that labeled NO-CAM can exchange from the diferrous site in a single turnover reaction initiated by reacting P with NH2-CAM.22 However, in this case, labeled NO-CAM is at a high concentration in solution. Even so, the exchange is inefficient and approximately half of the NO-CAM does not exchange.
Simulation of the Overall Reaction
Using the assumptions of a dual pathway for the starting NH2-CAM oxidation and limited exchange for the NH(OH)-CAM and NO-CAM intermediates, the overall mechanism shown in Scheme 4 was evaluated versus the experimental time course using numerical integration. To limit overparametrization of the simulation, all of the rate constants and most of the KD values determined experimentally in this study were fixed (black values) leaving only a few others as variable (red values). The numerical integration program adjusted these latter values to achieve an acceptable fit as shown by the dashed curves in Figure 11. In steps in which only KD values were determined or in which multiple rate constants were found experimentally, values were also allowed to float. In these cases the best fit values (red) were consistent with the experimental approximations (black). Very few of the fit values on the main biosynthetic pathway could be altered by >5 % and still give an acceptable fit (see notes for Scheme 4). However, the rate constants for the side pathways shown in blue in Scheme 4, could be varied over a much larger range without affecting the fit so long as they did not exceed specific limits. At their maximum values, these constrained rate constants were all very slow in comparison with the rate constants found in the main biosynthetic pathway.
The excellent fit to the data obtained supports the basic tenets of Scheme 4. In particular, the dual pathway for the starting NH2-CAM oxidation does accurately model the decreasing yield of P as the concentration of NH2-CAM increases, the in situ NH(OH)-CAM-mediated reduction reaction must be very fast, and slow steps, such as those found in the reaction of CmlIox reduced by direct mixing with NH(OH)-CAM, cannot be part of the main biosynthetic pathway.
It is shown by the data in Figure 3, as well as our past investigations, that P formation is effectively irreversible in the absence of substrate.22 The simulation on Scheme 4 indicates that it is also nearly irreversible in the presence of NH2-CAM but becomes reversible when NO-CAM is bound. The interaction of substrate and O2 binding in the active site is also evident from the order dependence of NO-CAM and O2 binding in the reaction with CmlIred, as noted above. It seems likely that this steric or electronic interaction affects the stability of the P intermediate. Indeed, our recently published model for the structure of P places the distal oxygen of the peroxo moiety immediately adjacent to the nitrogen substituent of the bound aromatic substrate.23 It remains unclear whether all substrates affect P stability, because the kinetics of the reaction pathway may dictate whether the reversibility is actually observed. For example, the very fast steps following P-NH2-CAM formation may mask a slow rate of release of O2 from this species.
Comparison of the Biosynthetic Pathways of CmlI and AurF
CmlI and AurF share a very high degree of sequence similarity, and they have nearly identical structures in the vicinity of the active site. The N-oxygenation reactions that they catalyze are analogous, although AurF has a smaller natural substrate, p-aminobenzoate versus NH2-CAM. Nevertheless, detailed mechanistic studies of both enzymes support a key difference in their biosynthetic pathway involving the identity of the pathway intermediate responsible for the internal reduction step.19, 22 Where we have proposed that NH(OH)-CAM fills this role in the CmlI mechanism, Bollinger, Krebs and co-workers have proposed that dihydroxylamino-benzoate serves as the reductant.19 The latter species is proposed to be formed from the preceding hydroxylamino-benzoate intermediate by reaction with the AurF peroxo species equivalent to P. One would usually assume that two biological systems with this level of similarity would use the same mechanism. However, our study suggests that, in this case, both mechanisms may be correct. The results presented here indicate that CmlI is designed to largely retain the pathway intermediates in the active site. If the retention of intermediates is not as stringent in the case of AurF, much more of the analogous hydroxylamino-benzoate may dissociate. Once released, it could then react with the AurF peroxo intermediate to yield the dihydroxylamino-benzoate or a similar intermediate. Indeed, if NH(OH)-CAM is reacted anaerobically with CmlI P, a reaction similar to that reported for AurF occurs, resulting in CAM and CmlIred.22 Thus, a delicate balance between the rate of intermediate dissociation and the rate of reduction may control the course of the reaction chemistry even in nearly identical enzymes. The CmlI mechanism is controlled by the fact that the reduction step occurs at a rate faster than the rate of NH(OH)-CAM dissociation. The enzyme system uses kinetic control to drive the reaction forward with very high efficiency and specificity.
Conclusion
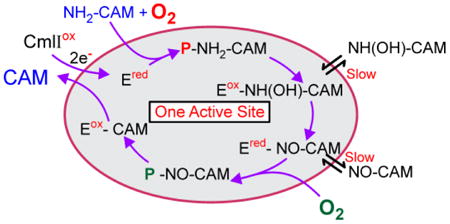
CmlI catalyzes a rare biochemical reaction that must overcome at least three mechanistic hurdles. (a) Three individual two-electron oxidation reactions must be catalyzed by the same enzyme and involve (as shown here) the same reaction cycle reactive species, P. (b) The inherent reactivity of the resulting biosynthetic pathway intermediates must be controlled to avoid adventitious reactions and loss of the eventual product. (c) The reactive intermediate of the reaction cycle must carry out oxidative chemistry with the various oxidation states of aromatic amines without adventitious oxidation of other moieties such as C-H bonds. CmlI has apparently solved some or all of these mechanistic problems by stabilizing a peroxo rather than a high-valence reactive species, thereby limiting the range of substrates that can be oxidized. This solution required the evolution of a new, more reactive, type of peroxo intermediate, P. We have described P as being ambiphilic, because a peroxo species can be either nucleophilic or electrophilic as required to match the electron donating capacity of the range of pathway intermediates.22, 23 On the other hand, the peroxo intermediate is not expected to have the extreme oxidizing potential of high-valence oxo species common to enzymes that catalyze C-H oxidation. The unusual characteristic of CmlI that emerges from this study is its ability to avoid the release of pathway intermediates from the first oxidation step through the release of CAM as illustrated in Scheme 5. This protects the cell from the reactive pathway intermediates and ensures both specificity and efficiency in antibiotic production.
Scheme 5. Model for the CmlI Biosynthetic Pathway in One Active Site.
Supplementary Material
Acknowledgments
Funding: The authors acknowledge the financial support of this work National Institutes of Health (NIH) GM118030 (to J.D.L.), NIH Grant GM38767 (to L.Q.), National Science Foundation Grant CHE-1654060 (to Y.G.), and NIH Graduate Traineeship GM08700 (to A.J.K.). Y.G. also acknowledges financial support from Carnegie Mellon University.
Footnotes
Associated Content: Supplemental Experimental Procedures, Table S1 of extinction coefficients, Figures S1-S2 showing supplemental results. This material is available free of charge via the Internet at http://pubs.acs.org.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00695.
Supplemental Experimental Procedures, a list of extinction coefficients (Table S1), and supplemental results (Figures S1 and S2) (PDF)
ORCID: Anna J. Komor: 0000-0003-4806-4233
Ruixi Fan: 0000-0002-6996-4276
Yisong Guo: 0000-0002-4132-3565
John D. Lipscomb: 0000-0002-8158-5594
Lawrence Que, Jr.: 0000-0002-0989-2813
The authors declare no competing financial interest.
References
- 1.Lu HG, Chanco E, Zhao HM. CmlI is an N-oxygenase in the biosynthesis of chloramphenicol. Tetrahedron. 2012;68:7651–7654. doi: 10.1016/j.tet.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Makris TM, Vu VV, Meier KK, Komor AJ, Rivard BS, Münck E, Que L, Jr, Lipscomb JD. An unusual peroxo intermediate of the arylamine oxygenase of the chloramphenicol biosynthetic pathway. J Am Chem Soc. 2015;137:1608–1617. doi: 10.1021/ja511649n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knoot CJ, Kovaleva EG, Lipscomb JD. Crystal structure of CmlI, the arylamine oxygenase from the chloramphenicol biosynthetic pathway. J Biol Inorg Chem. 2016;21:589–603. doi: 10.1007/s00775-016-1363-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bollinger JM, Jr, Tong WH, Ravi N, Huynh BH, Edmondson DE, Stubbe J. Mechanism of assembly of the tyrosyl radical-diiron(III) cofactor of E. coli ribonucleotide reductase. 2. Kinetics of the excess Fe2+ reaction by optical, EPR, and Mössbauer spectroscopies. J Am Chem Soc. 1994;116:8015–8023. [Google Scholar]
- 5.Wallar BJ, Lipscomb JD. Dioxygen activation by enzymes containing binuclear non-heme iron clusters. Chem Rev. 1996;96:2625–2657. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 6.Austin RN, Chang HK, Zylstra GJ, Groves JT. The non-heme diiron alkane monooxygenase of Pseudomonas oleovorans (AlkB) hydroxylates via a substrate radical intermediate. J Am Chem Soc. 2000;122:11747–11748. [Google Scholar]
- 7.Makris TM, Knoot CJ, Wilmot CM, Lipscomb JD. Structure of a dinuclear iron cluster-containing β-hydroxylase active in antibiotic biosynthesis. Biochemistry. 2013;52:6662–6671. doi: 10.1021/bi400845b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 9.Fox BG, Shanklin J, Somerville C, Münck E. Stearoyl-acyl carrier protein Δ9 desaturase from Ricinus communis is a diiron-oxo protein. Proc Natl Acad Sci U S A. 1993;90:2486–2490. doi: 10.1073/pnas.90.6.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pikus JD, Studts JM, Achim C, Kauffmann KE, Münck E, Steffan RJ, McClay K, Fox BG. Recombinant toluene-4-monooxygenase: Catalytic and Mössbauer studies of the purified diiron and Rieske components of a four-protein complex. Biochemistry. 1996;35:9106–9119. doi: 10.1021/bi960456m. [DOI] [PubMed] [Google Scholar]
- 11.Bollinger JM, Jr, Diao Y, Matthews ML, Xing G, Krebs C. myo-Inositol oxygenase: a radical new pathway for O2 and C-H activation at a nonheme diiron cluster. Dalton Trans. 2009:905–914. doi: 10.1039/b811885j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Acheson JF, Bailey LJ, Brunold TC, Fox BG. In-crystal reaction cycle of a toluene-bound diiron hydroxylase. Nature. 2017;544:191–195. doi: 10.1038/nature21681. [DOI] [PubMed] [Google Scholar]
- 13.Sazinsky MH, Bard J, Di Donato A, Lippard SJ. Crystal structure of the toluene/o-xylene monooxygenase hydroxylase from Pseudomonas stutzeri OX1. Insight into the substrate specificity, substrate channeling, and active site tuning of multicomponent monooxygenases. J Biol Chem. 2004;279:30600–30610. doi: 10.1074/jbc.M400710200. [DOI] [PubMed] [Google Scholar]
- 14.Knoot CJ, Makris TM, Lipscomb JD. Dinuclear iron cluster-containing oxygenase CmlA. In: Scott RA, editor. Encyclopedia of Inorganic and Bioinorganic Chemistry Online. John Wiley & Sons, Ltd.; Chichester, U.K.: 2015. pp. 1–10. http://dx.doi.org/10.1002/9781119951438.eibc2329. [Google Scholar]
- 15.He J, Hertweck C. Biosynthetic origin of the rare nitroaryl moiety of the polyketide antibiotic aureothin: Involvement of an unprecedented N-oxygenase. J Am Chem Soc. 2004;126:3694–3695. doi: 10.1021/ja039328t. [DOI] [PubMed] [Google Scholar]
- 16.Simurdiak M, Lee J, Zhao H. A new class of arylamine oxygenases: evidence that p-aminobenzoate N-oxygenase (AurF) is a di-iron enzyme and further mechanistic studies. ChemBioChem. 2006;7:1169–1172. doi: 10.1002/cbic.200600136. [DOI] [PubMed] [Google Scholar]
- 17.Zocher G, Winkler R, Hertweck C, Schulz GE. Structure and action of the N-oxygenase AurF from Streptomyces thioluteus. J Mol Biol. 2007;373:65–74. doi: 10.1016/j.jmb.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 18.Korboukh VK, Li N, Barr EW, Bollinger JM, Jr, Krebs C. A long-lived, substrate-hydroxylating peroxodiiron(III/III) intermediate in the amine oxygenase, AurF, from Streptomyces thioluteus. J Am Chem Soc. 2009;131:13608–13609. doi: 10.1021/ja9064969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li N, Korboukh VK, Krebs C, Bollinger JM., Jr Four-electron oxidation of p-hydroxylaminobenzoate to p-nitrobenzoate by a peroxodiferric complex in AurF from Streptomyces thioluteus. Proc Natl Acad Sci USA. 2010;107:15722–15727. doi: 10.1073/pnas.1002785107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee SK, Fox BG, Froland WA, Lipscomb JD, Münck E. A transient intermediate of the methane monooxygenase catalytic cycle containing a FeIVFeIV cluster. J Am Chem Soc. 1993;115:6450–6451. [Google Scholar]
- 21.Dassama LMK, Silakov A, Krest CM, Calixto JC, Krebs C, Bollinger JM, Jr, Green MT. A 2.8 Å Fe-Fe separation in the Fe2III/IV intermediate (X) from Escherichia coli ribonucleotide reductase. J Am Chem Soc. 2013;135:16758–16761. doi: 10.1021/ja407438p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komor AJ, Rivard BS, Fan R, Guo Y, Que L, Jr, Lipscomb JD. Mechanism for six-electron aryl-N-oxygenation by the non-heme diiron enzyme CmlI. J Am Chem Soc. 2016;138:7411–7421. doi: 10.1021/jacs.6b03341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jasniewski AJ, Komor AJ, Lipscomb JD, Que L., Jr An unprecedented (μ-1,1-peroxo)diferric structure for the ambiphilic orange peroxo intermediate of the nonheme N-oxygenase CmlI. J Am Chem Soc. 2017;139:10472–10485. doi: 10.1021/jacs.7b05389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sturgeon BE, Burdi D, Chen S, Huynh BH, Edmondson DE, Stubbe J, Hoffman BM. Reconsideration of X, the diiron intermediate formed during cofactor assembly in E. coli ribonucleotide reductase. J Am Chem Soc. 1996;118:7551–7557. [Google Scholar]
- 25.Brazeau BJ, Lipscomb JD. Kinetics and activation thermodynamics of methane monooxygenase compound Q formation and reaction with substrates. Biochemistry. 2000;39:13503–13515. doi: 10.1021/bi001473l. [DOI] [PubMed] [Google Scholar]
- 26.Winkler R, Hertweck C. Sequential enzymatic oxidation of aminoarenes to nitroarenes via hydroxylamines. Angew Chem Int Ed. 2005;44:4083–4087. doi: 10.1002/anie.200500365. [DOI] [PubMed] [Google Scholar]
- 27.Lin Y, Gerfen GJ, Rousseau DL, Yeh SR. Ultrafast microfluidic mixer and freeze-quenching device. Anal Chem. 2003;75:5381–5386. doi: 10.1021/ac0346205. [DOI] [PubMed] [Google Scholar]
- 28.Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Münck E, Lipscomb JD. Trapping and spectroscopic characterization of an FeIII-superoxo intermediate from a nonheme mononuclear iron-containing enzyme. Proc Natl Acad Sci U S A. 2010;107:16788–16793. doi: 10.1073/pnas.1010015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.