

Abstract
Oroscomucoid 3 (ORMDL3), a gene localized to chromosome 17q21, has been linked in epidemiologic studies to childhood asthma and rhinoviral (RV) infections. As the single nucleotide polymorphisms linking ORMDL3 to asthma is associated with increased expression of ORMDL3, we have utilized hORMDL3zp3-Cre mice (which have universal increased expression of human ORMDL3) to determine whether infection of these transgenic mice with RV influences levels of airway inflammation or RV viral load. RV infection of hORMDL3zp3-Cre mice resulted in reduced RV viral load assessed by RT-qPCR (lung and airway epithelium), as well as reduced airway inflammation (total BAL cells, neutrophils, macrophages, and lymphocytes) compared to RV infected WT mice. Levels of the anti-viral pathways including interferons (IFNα, IFNβ, IFNλ) and RNAse L were significantly increased in the lungs of RV infected hORMDL3zp3-Cre mice. Levels of the anti-viral mouse oligo-adenylate synthetase 1g (mOas1g) pathway and RNAse L were upregulated in the lungs of unchallenged hORMDL3zp3-Cre mice. In addition, levels of mOas2, but not mOas1 (mOas1a, mOas1b, mOas1g), or mOas3 pathways were significantly more upregulated by IFNs (IFNα, IFNβ, IFNλ) in epithelial cells from hORMDL3zp3-Cre mice compared to RV infected WT mouse epithelial cells. RNAse L deficient mice infected with RV had increased RV viral load. Overall, these studies suggest that increased levels of ORMDL3 contribute to antiviral defense to RV infection in mice through pathways that may include interferons (IFNα, IFNβ, IFNλ), OAS, and RNAse L.
INTRODUCTION
Oroscomucoid 3 (ORMDL3), a gene localized to chromosome 17q21, has been linked in multiple genome wide association (GWAS) and non-GWAS epidemiologic studies to asthma (1–2), and has also been linked to the frequency of rhinoviral (RV) wheezing illness in childhood and the subsequent development of childhood asthma (3). Although there are multiple genetic epidemiologic studies linking chromosome 17q21 with asthma (2) there are limited functional studies of ORMDL3 in the lung to better understand how it may contribute to the pathogenesis of asthma precipitated by RV. We have previously reported that in wild type (WT) mice in vivo, inhalation allergen challenge induces a significant increase in levels of expression of ORMDL3 in airway epithelium and in macrophages (4), cells associated with anti-viral pathways that may modulate levels of RV infection. As the SNP linking ORMDL3 to asthma and RV infection (3) is associated with increased levels of ORMDL3 expression (5), we utilized universal ORMDL3 transgenic (TG) mice that express increased levels of human ORMDL3 (6) to determine whether increased levels of expression of ORMDL3 in the lung in these mice would influence RV viral load, and RV induced levels of airway inflammation.
From what is currently known of ORMDL3 regulated pathways, RV infection in the context of increased ORMDL3 expression could theoretically either increase airway inflammation (through upregulation of ORMDL3 regulated chemokine pathways)(4, 6), or alternatively through ORMDL3 upregulation of anti-viral pathways such as oligo-adenylate synthetase (OAS) or interferons (4, 6) reduce RV viral load, and as a consequence reduce airway inflammation. Previous studies have demonstrated that ORMDL3 regulates CXC (CXCL10, CXCL11, IL-8) and CC (CCL20) chemokine pathways in human bronchial epithelium in vitro (4) and in ORMDL3 TG mice in vivo (6). If RV infection of ORMDL3 TG mice triggered chemokine pathways (CC and CXC chemokines) in airway epithelium known to be downstream of ORMDL3 (4, 6) this could result in increased airway inflammation. In contrast, our studies of ORMDL3 transfection into normal human airway epithelial cells demonstrated that increased ORMDL3 induced high levels of anti-viral defense pathways including oligoadenylate synthetase (OAS) genes (7) which may mediate a reduction in RV viral load and airway inflammation. In humans, the OAS gene family has enzymatically active OAS1, OAS2, and OAS3 (7, 8), as well as OASL which is devoid of enzymatic activity (7, 8). Our prior studies demonstrated that OAS1, OAS2, and OAS3 were all significantly upregulated in human lung epithelial cells transfected with ORMDL3 (4). The enzymatically active Oas in mice (mOas1a, mOas2, and mOas3) are similar to human OAS (8). OAS catalyze the polymerization of ATP into 2′-5′-linked oligoadenylates which activate a constitutively expressed latent endonuclease, RNaseL, to block viral replication at the level of mRNA degradation (8, 9). OAS1 is upregulated by a wide range of viruses and has been shown to play an anti-viral role during viral infections including in studies with the asthma associated virus respiratory syncytial virus (RSV) (10). At present it is not known how RV infection of asthmatics with a SNP associated with increased ORMDL3 expression (3) would influence RV viral load or airway inflammation. In this study we have therefore used a mouse model of RV infection to determine whether increased lung expression of ORMDL3 influences RV viral load and levels of airway inflammation.
MATERIALS AND METHODS
Rhinovirus RV1B
RV1B was a generous gift of Dr. Michael Croft (La Jolla Inst for Allergy and Immunology, La Jolla, CA)(11). The RV1B was propagated in Ohio HeLa cells (ECACC, UK), at 33°C, in a humidified, 5% CO2 incubator as described (11). When the full RV1B induced cytopathic effect developed, HeLa cell lysates were harvested and cellular debris was pelleted by low speed centrifugation. RV1B in HeLa cell lysates was concentrated and partially purified by centrifugation with a 100,000 molecular weight cut-off Centricon filter (2,000 rpm at 4°C for 8 h; Millipore)(12,13). RV1B was titered by plaque assay as described (11). In brief, HeLa cell monolayers inoculated with serial 10-fold dilutions of RV1B, were incubated for 3 days at 33°C, and then fixed with 10 % formalin. Viral plaques were visualized by staining with crystal violet, counted, and viral titer was expressed as p.f.u./ml.
Mouse model of Rhinovirus RV1B infection
hORMDL3zp3-Cre mice on a C57Bl/6 background were generated as previously described (6). Six to 7-wk-old female hORMDL3zp3-Cre mice, and their WT littermate controls (n=8 mice/group/experiment; each experiment repeated 3 times) were lightly anesthetized with isofluorane and inoculated intranasally with 50ul of RV1B (2.8 X 108 p.f.u./ml), or an equal volume of sham control. Mice were euthanized 1, 2, or 4 days after infection. All the mouse experimental protocols described in the methods were approved by the UCSD Institutional Animal Care and Use Committee.
Quantitation of RV1B in Lungs and Epithelial Cells of hORMDL3zp3-Cre mice
RV1B viral RNA expression in lung and bronchial epithelial cells was compared to RV1B standard curves and expressed as copies/mg RNA using methods previously described (14). In brief, for RNA extractions, lungs were initially snap-frozen in liquid nitrogen and stored at −80°C. Total RNA was extracted from both lungs and bronchial brushing derived airway epithelial cells with RNA STAT-60 (TelTest) and reverse transcribed with oligo (dT) and SuperScript II kit (Life Technologies). To study levels of RV1B in airway epithelial cells, epithelial cells were isolated by bronchial brushing as previously described in this laboratory (15). In brief, the bronchial brushing was performed using sterile plastic feeding tubes (Solomon Scientific) modified by removal of the rubber bulb, sanding to create roughness, and autoclaving. The tube was inserted into the right main and left main bronchus. After gentle brushing of each bronchus, tubes containing the brushed epithelial cells were immediately placed in RNA STAT-60 (TelTest). We have previously demonstrated that the bronchial brushing epithelial cells are > 95% epithelial cells as assessed by ultrastructure, FACS, and RT-qPCR expression of epithelial, but not fibroblast or smooth muscle, genes (15). RT-qPCR for positive-strand viral RNA was conducted using RV1B-specific primers and probes (forward primer, 5′-GTG AAG AGC CSC RTG TGC T-3′; reverse primer, 5′-GCT SCA GGG TTA AGG TTA GCC-3′; probe. 5′-FAM-TGA GTC CTC CGG CCC CTG AAT G-TAMRA-3′ (11–13).
Quantitation of bronchoalveolar lavage (BAL) total and differential cell counts
BAL fluid was collected by lavaging the lung with 1 ml of PBS via a tracheal catheter (6). Total BAL leukocytes were counted using a hemocytometer. BAL fluid cells were cytospun onto slides which were then stained with Wright-Giemsa and differential cell counts performed under a light microscope.
Quantitation of peribronchial neutrophils, CD8+ lymphocytes, and F4/80 macrophages
Lungs were processed for immunohistology (paraffin-embedded lung sections) as previously described in this laboratory (4, 6). In brief, lungs were equivalently inflated with an intra-tracheal injection of the same volume of 4% paraformaldehyde solution (Sigma-Aldrich, St. Louis, MO) to preserve the pulmonary architecture. Lung sections were processed for immunohistochemistry to detect neutrophils (anti-mouse neutrophil elasatase or species and isotype matched control Ab; Santa Cruz Biotechnology), CD8+ lymphocytes (anti-mouse CD8 or species and isotype matched control Ab; GeneTex), or F4/80+ macrophages (anti-mouse F4/80 or species and isotype matched control Ab; Santa Cruz Biotechnology). The number of peribronchial neutrophils, CD8+ lymphocytes, or macrophages staining positive in the peribronchial space was counted using a light microscope. Results are expressed as the number of peribronchial cells staining positive per bronchiole with 150–200 mm internal diameter. At least 5 bronchioles were counted in each slide.
Quantitation of IFN-α, IFN-β, and IFN-λ in lungs of RV1B infected hORMDL3zp3-Cre mice
Total RNA was extracted from mouse lungs as described above for quantitation of RV1B mRNA. RT-qPCR to quantitate IFN-α1, IFN-β, and IFN-λ mRNA was performed with TaqMan PCR Master Mix and mouse IFN-α1, IFN-β, and IFN-λ primers (all from Applied Biosystems) as described above for quantitation of RV1B mRNA. While humans express three members of the IFN-λ family i.e. IFN-λ1 (IL-29), IFN-λ2 (IL-28A), and IFN-λ3 (IL-28B)(16, 17), mice differ from humans in expressing only two IFN-λ isoforms (IFN-λ2, IFN-λ3, but not IFN-λ1) (16,17). The mouse IFN-λ primers used in this study detected both murine IFN-λ2 (IL-28A) and IFN-λ3 (IL-28B).
Quantitation of mOas1, mOas2, and mOas3 in hORMDL3zp3-Cre mouse airway epithelial cells stimulated with IFN ex vivo
Mouse tracheal epithelial cells were obtained by dissection and culture from hORMDL3zp3-Cre mice and WT mice as previously described (18). Cultured tracheal epithelial cells were of >95% purity as assessed by E-cadherin expression on FACS and histologic detection of ciliated epithelial cells. The epithelial cells were incubated at 37°C with 100 ng/ml of either IFNα, IFNβ, or IFNλ (IFN-λ2 and IFN-λ3 in combination), and the cells were collected for RNA extraction after for 24 h. Total RNA was extracted with RNA-STAT-60 (Tel-Test) and reverse transcribed with Oligo-dT and SuperScript II kit (Life Technologies). RT-qPCR was performed with TaqMan PCR Master Mix and mOas1 (mOas1a, mOas1b, mOas1g), mOas2, and mOas3 primers (all from Life Technology). The relative amounts of transcripts were normalized to those of housekeeping gene (GAPDH) mRNA and compared between the different genes by the ΔΔ cycle threshold method as previously described in this laboratory (4, 6).
Quantitation of RNAse L in mouse lung airway epithelial cells
RNAse L is constitutively expressed in most cells (9). Total RNA was extracted from mouse lungs of RV infected WT and RV infected hORMDL3zp3-Cre mice as described above. RNAse L mRNA in lungs were quantitated by RT-qPCR using TaqMan PCR Master Mix and RNase L primers (all from Applied Biosystems) as described above for quantitation of RV1B mRNA.
Quantitation of RV1B in lungs of RNAse L deficient mice by RT-qPCR
RNAse L deficient mice were generated as previously described (19). RNAse L deficient mice or WT controls aged 6–7 weeks (n= 8 mice/group) were infected with RV1B and lungs processed to determine levels of RV1B mRNA by RT-qPCR as described above. Levels of total BAL cells and BAL differential cell counts were quantitated as described above
Statistical Analysis
All results are presented as mean + SEM. A statistical software package (Graph Pad Prism, San Diego, CA) was used for the analysis. A t test was used for analysis of two groups. ANOVA analysis was use when more than two groups were compared. P values of < 0.05 were considered statistically significant.
RESULTS
RV viral load is reduced in lung and bronchial epithelial cells of RV infected hORMDL3zp3-Cre mice
RV viral load is maximal in WT mice challenged with RV at day 1 post RV infection, and RV is not detectable on day 2 or day 4 (Figure 1A–D). Levels of RV in hORMDL3zp3-Cre mice are significantly reduced compared to WT mice at day 1 in both the lung (p<0.05) (Figure 1A), and in bronchial brush epithelial cells (p<0.005)(Figure 1B). No RV is detectable at day 2 or day 4 in hORMDL3zp3-Cre mice, results similar to that noted in WT mice (Figure 1C–D). Thus, RV challenge in hORMDL3zp3-Cre mice does not postpone peak viral load to a later time point.
Figure 1. RV viral load is reduced in lung and bronchial epithelial cells of RV infected hORMDL3zp3-Cre mice.

WT and hORMDL3zp3-Cre mice were infected with RV1B and sacrificed 1, 2, or 4 days after infection. Levels of RV1B viral load were quantitated in the lungs (Figure 1A), and bronchial brushing epithelial cells (Figure 1B) by RT-qPCR. Levels of RV viral load were significantly lower in the lungs (p<0.05), and the bronchial epithelium (p<0.005) of hORMDL3zp3-Cre mice compared to WT mice one day after infection with RV. RV was not detected in the lungs of WT or hORMDL3zp3-Cre mice at 2 days (Figure 1C), or 4 days (Figure 1D), post-RV infection.
RV induced BAL inflammation is reduced in hORMDL3zp3-Cre mice
WT mice had a significant increase in the total number of BAL cells (p<0.0001)(Figure 2A), BAL neutrophils (p< 0.0001)(Figure 2B), BAL macrophages (p<0.005) (Figure 2C), and BAL lymphocytes (p<0.05) (Figure 2D) on day 1 post-RV infection. The total number of BAL cells were significantly lower in hORMDL3zp3-Cre mice compared to WT mice one day post infection with RV (p<0.0001)(Figure 2A). In addition, the total number of BAL neutrophils (p<0.0001)(Figure 2B), and BAL macrophages (p<0.001) (Figure 2C) were significantly lower in hORMDL3zp3-Cre mice compared to WT mice one day post infection with RV.
Figure 2. RV induced BAL inflammation is reduced in hORMDL3zp3-Cre mice.

WT and hORMDL3zp3-Cre mice were infected with RV1B and sacrificed 1, 2, or 4 days after infection. BAL fluid was collected by lavaging the lung with 1 ml of PBS via a tracheal catheter. Total BAL leukocytes were counted using a hemocytometer. BAL fluid cells were cytospun onto slides which were then stained with Wright-Giemsa and differential cell counts performed under a light microscope. One day post-RV infection in WT mice there was a significant increase in the total number of BAL cells (p<0.0001)(Figure 2A), BAL neutrophils (p<0.0001)(Figure 2B), BAL macrophages (p<0.005)(Figure 2C), and BAL lymphocytes (p<0.05)(Figure 2D). The total number of BAL cells were significantly lower in hORMDL3zp3-Cre mice compared to WT mice one day after infection with RV (p<0.0001)(Figure 2A). In addition, the total number of BAL neutrophils (p<0.0001)(Figure 2B), and BAL macrophages (p<0.001)(Figure 2C) were significantly lower in hORMDL3zp3-Cre mice compared to WT mice one day after infection with RV. Levels of BAL lymphocytes were significantly lower in in hORMDL3zp3-Cre mice compared to WT mice four days after infection with RV (p<0.001)(Figure 2D).
WT mice infected with RV had a significant increase in BAL neutrophils (peaks on day 1) (Figure 2A), and a significant increase in BAL lymphocytes (peaks on day 4) (Figure 2D). At day 2 and day 4 post RV infection, total BAL cell inflammation, as well as BAL neutrophil and BAL macrophage inflammation were reduced in both hORMDL3zp3-Cre mice and WT mice (Figure 2A, 2B, 2D). There was no increased BAL inflammation (total cell, neutrophils, macrophages, or lymphocytes) in hORMDL3zp3-Cre mice compared to WT mice at day 2 and day 4 to suggest that ORMDL3 over-expression induced increased BAL inflammation at later time points following RV inoculation (Figure 2A–D). Indeed the number of BAL lymphocytes at 4 days was reduced in hORMDL3zp3-Cre mice compared to WT mice (p<0.001)(Figure 2C).
RV induced peribronchial inflammation is reduced in lungs of hORMDL3zp3-Cre mice
WT mice had a significant increase in the total number of peribronchial neutrophils (p<0.0001)(Figure 3A–E), peribronchial F4/80+ macrophages (p<0.001)(Figure 3F–J), and peribronchial CD8 positive lymphocytes (p<0.05)(Figure 3K–O), on day 1 post-RV infection as assessed by immunohistochemistry. The total number of peribronchial neutrophils (p< 0.0001)(Figure 3A–E), peribronchial F4/80+ macrophages (p<0.05)(Figure 3F–J), and peribronchial CD8 positive lymphocytes (p<0.05)(Figure 3K–O) were significantly lower in hORMDL3zp3-Cre mice compared to WT mice one day post infection with RV.
Figure 3. RV induced peribronchial inflammation is reduced in lungs of hORMDL3zp3-Cre mice.

WT and hORMDL3zp3-Cre mice were infected with RV1B and sacrificed 1, 2, or 4 days after infection. Lung sections were processed for immunohistochemistry to detect neutrophils (anti-mouse neutrophil elastase Ab), CD8 positive lymphocytes (anti-mouse CD8 Ab), or macrophages (anti-mouse F4/80 Ab). The number of neutrophils, CD8 positive lymphocytes, or macrophages staining positive in the peribronchial space was counted using a light microscope. Results are expressed as the number of peribronchial neutrophils, CD8 positive lymphocytes, or macrophages per bronchiole with 150–200 mm internal diameter. At least 5 bronchioles were counted in each slide. RV infection in WT mice induced a significant increase in the total number of peribronchial neutrophils (p<0.0001)(Figure 3A–E), peribronchial macrophages (p<0.05)(Figure 3F–J), and peribronchial CD8 positive lymphocytes (p<0.05)(Figure 3K–O) one day after infection with RV. The total number of peribronchial neutrophils (p<0.0001)(Figure 3A–E) and peribronchial macrophages (p<0.05)(Figure 3F–J) were significantly lower in hORMDL3zp3-Cre mice infected with RV compared to WT mice one day after infection with RV. Levels of CD8+ lymphocytes were significantly lower in hORMDL3zp3-Cre mice infected with RV compared to WT mice one, two, and four days after infection with RV (p<0.05)(Figure 3K–O).
We also quantitated levels of airway inflammation at later time points following RV inoculation (2 days, and 4 days)(Figure 3A–O) to compare to day 1. WT mice infected with RV had a significant increase in peribronchial neutrophils (peaks on day 1) (Figure 3E), and a significant increase in BAL lymphocytes (peaks on day 4) (Figure 3O). At day 2 and day 4 post RV infection, peribronchial neutrophil (Figure 3A–E) and peribronchial macrophage (Figure 3F–J) inflammation were reduced in both hORMDL3zp3-Cre mice and WT mice. There was no increased peribronchial inflammation (neutrophils, macrophages, or CD8+ lymphocytes) in hORMDL3zp3-Cre mice compared to WT mice at day 2 and day 4 post infection with RV to suggest that ORMDL3 over-expression induced increased peribronchial inflammation at later time points following RV inoculation. Indeed the number of peribronchial CD8+ lymphocytes at 2 and 4 days post-RV infection was reduced in hORMDL3zp3-Cre mice compared to WT mice (p<0.05) (Figure 3K–O).
Increased lung levels of IFN-α1, IFN-β, and IFN-λ in RV1B infected hORMDL3zp3-Cre mice
RV1B infection in WT mice induced an increase in lung levels of IFN-α1 (p<0.05), IFN-β (p<0.05), and IFN-λ (p<0.05) as assessed by RT-qPCR (Figure 4). However, RV1B infection in hORMDL3zp3-Cre mice induced a significantly enhanced increase compared to RV1B infected WT mice in lung levels of IFN-α1 (232 fold increase vs 7 fold increase)(hORMDL3zp3-Cre RV1B vs WT RV1B)(p<0.05), IFN-β (143 fold increase vs 16 fold increase) (hORMDL3zp3-Cre RV1B vs WT RV1B)(p<0.05), and IFN-λ (232 fold increase vs 48 fold increase) (hORMDL3zp3-Cre RV1B vs WT RV1B)(p<0.05) as assessed by RT-qPCR (Figure 4).
Figure 4. Increased lung levels of IFN-α1, IFN-β, and IFN-λ in RV1B infected hORMDL3zp3-Cre mice.

WT and hORMDL3zp3-Cre mice were infected with RV1B and sacrificed 24 hours after infection. Lungs were processed to detect levels of IFN-α1, IFN-β and IFN-λ mRNA by RT-qPCR. RV1B infection in WT mice induced an increase in lung levels of IFN-α1 (p<0.05), IFN-β (p<0.05), and IFN-λ (p<0.05). RV1B infection in hORMDL3zp3-Cre mice induced a significantly enhanced increase in lung levels of IFN-α1 (p<0.05), IFN-β (p<0.05), and IFN-λ (p<0.05)(RV1B hORMDL3zp3-Cre mice vs RV1B WT mice).
Levels of mOas2 in hORMDL3zp3-Cre airway epithelial cells are significantly increased upon stimulation with IFN-α, IFN-β, and IFN-λ
As lung levels of IFN-α, IFN-β, and IFN-λ were significantly increased in RV1B infected hORMDL3zp3-Cre mice compared to RV1B infected WT mice, we examined whether hORMDL3zp3-Cre mouse tracheal epithelial cells would have an increased ability to express the anti-viral mOas pathway when stimulated in vitro with either IFN-α, IFN-β, or IFN-λ. These studies demonstrated that levels of mOas2 increased in WT mouse tracheal epithelial cells stimulated in vitro with either IFN-α (p<0.05)(Fig 5A), IFN-β (p<0.05) (Fig 5C), or IFN-λ (p<0.05) (Fig 5E) as assessed by RT-qPCR. In WT epithelial cells the level of induction of mOas were greatest with IFN-β (749 fold), compared to IFN-α (130 fold) and IFN-λ (60 fold) (Fig 5A, C, E). However, IFN stimulation in vitro of epithelial cells derived from the hORMDL3zp3-Cre mice induced a significantly enhanced increase compared to IFN stimulated WT mouse epithelial cells in levels of mOas2 when stimulated with either IFN-α (399 fold increase vs 130 fold increase)(hORMDL3zp3-Cre vs WT)(p<0.05), IFN-β (1,351 fold increase vs 749 fold increase) (hORMDL3zp3-Cre vs WT)(p<0.05), and IFN-λ (666 fold increase vs 60 fold increase) (hORMDL3zp3-Cre vs WT)(p<0.02) as assessed by RT-qPCR (Figure 5A, C, E). In contrast to the enhanced mOas2 response to IFNs in hORMDL3zp3-Cre compared to WT mice, there was a similar increase in mOas1 (mOas1a, mOas1b, mOas1g), and mOas3 in response to IFNα (Figure 5B), and IFNβ (Figure 5D) in epithelial cells derived from hORMDL3zp3-Cre compared to WT mice. IFNλ (Figure 5F) also induced a similar increase in mOas1b, mOas1g, and mOas3 in epithelial cells derived from hORMDL3zp3-Cre compared to WT mice. However, in contrast to IFNα and IFNβ, IFNλ (Figure 5F) did not increase levels of mOas1a in epithelial cells derived from hORMDL3zp3-Cre compared to WT mice (p<0.05). Baseline expression of mOas1g (but not mOas1a, mOas1b, mOas2, or mOas3) in tracheal epithelial cells (p<0.05) (Figure 5B) and lungs (p<0.02) (Figure 5G) of hORMDL3zp3-Cre mice was statistically significantly higher than baseline expression of mOas1g in WT mice (p<0.05).
Figure 5. Levels of mOas2 in hORMDL3zp3-Cre lung airway epithelial cells are significantly increased upon stimulation with IFN-α, IFN-β, and IFN-λ.

Mouse lung epithelial cells derived from WT or hORMDL3zp3-Cre mice were incubated for 24 hrs in vitro with 100 ng/ml of either IFN-α (Figure 5A–B), IFN-β (Figure 5C–D), or IFN-λ (Figure 5E–F). Levels of mOas1 (Oas1a, Oas1b, Oas1g), mOas2, and mOas3 mRNA were quantitated by RT-qPCR. Levels of mOas2 increased in WT mouse lung epithelial cells stimulated with either IFN-α (p<0.05)(Fig 5A), IFN-β (p<0.05) (Fig 5C), or IFN-λ (p<0.05) (Fig 5E). IFN stimulation of epithelial cells derived from the lungs of hORMDL3zp3-Cre mice induced a significantly enhanced increase in levels of mOas2 compared to WT mice when stimulated with either IFN-α (p<0.05), IFN-β (p<0.05), or IFN-λ )(p<0.05)(hORMDL3zp3-Cre vs WT) (Figure 5A, C, E). There was a similar increase in mOas1 (mOas1a, mOas1b, mOas1g), and mOas3 in response to IFNα (Figure 5B), IFNβ (Figure 5D), and IFNλ (Figure 5F) in epithelial cells derived from hORMDL3zp3-Cre mice compared to WT mice. Baseline expression of mOas1g (but not mOas1a, mOas1b, mOas2, or mOas3) in epithelial cells (Figure 5B)(Figure 5G) of hORMDL3zp3-Cre mice is higher than baseline expression in WT mice (p<0.05).
Levels of RNAse L are increased in lungs of hORMDL3zp3-Cre mice
There was a statistically significant baseline up-regulation of RNAse L mRNA in the lungs of hORMDL3zp3-Cre compared to WT mice prior to RV infection (p<0.05) (Figure 6A). Levels of RNAse L quantitated by RT-qPCR were higher in the lungs of hORMDL3zp3-Cre mice infected with RV compared to WT mice infected with RV (p< 0.02) (Figure 6A).
Figure 6. RV infected RNase L deficient mice have increased RV viral load and neutrophil inflammation.

WT and hORMDL3zp3-Cre mice were infected with RV1B and sacrificed 24 hours after infection. There is a statistically significant baseline up-regulation of RNase L mRNA in the lungs of hORMDL3zp3-Cre compared to WT mice prior to RV infection (p< 0.05) (Figure 6A). Levels of RNase L quantitated by RT-qPCR were significantly higher in the lungs of hORMDL3zp3-Cre mice infected with RV compared to WT mice infected with RV (p< 0.02) (Figure 6A). WT and RNase L deficient mice were infected with RV1B and sacrificed 24 hours after infection. Levels of RV viral load as assessed by RT-qPCR were significantly higher in the lungs of RNase L deficient mice infected with RV compared to WT mice infected with RV (p<0.05)(Figure 6B). The total number of BAL cells (p<0.02)(Figure 6C) and BAL neutrophils (p<0.02)(Figure 6D) were significantly higher in RNase L deficient mice infected with RV compared to WT mice infected with RV. The levels of BAL macrophages (Figure 6E) and BAL lymphocytes (Figure 6F) were similar in RV infected RNase L deficient mice and RV infected WT mice.
RV infected RNAse L deficient mice have increased lung RV viral load
Levels of RV viral load as assessed by RT-qPCR were significantly higher in the lungs of RNAse L deficient mice infected with RV compared to WT mice infected with RV (p<0.05)(Figure 6B).
RV induced BAL inflammation is increased in RNAse L deficient mice
The total number of BAL cells were significantly higher in RNAse L deficient mice compared to WT mice infected with RV (p<0.02)(Figure 6C). In addition, the total number of BAL neutrophils were significantly higher in RNAse L deficient mice compared to WT mice infected with RV(p<0.02)(Figure 6D). The levels of BAL macrophages (Figure 6E) and BAL lymphocytes (Figure 6F) were similar in RV infected RNAse L deficient mice and RV infected WT mice
DISCUSSION
This study demonstrates that RV infection of hORMDL3zp3-Cre mice (which have increased levels of expression of human ORMDL3) results in reduced RV viral load in the lung and airway epithelium, as well as reduced airway inflammation (total BAL cells, neutrophils, macrophages, and CD8 positive lymphocytes) compared to RV infected WT mice. The potential pathway(s) by which increased levels of ORMDL3 reduce RV viral load may either be mediated by inhibition of RV viral replication and/or increase in RV viral clearance. Although the mouse model of RV infection we have used induces increases in IFNα, IFNβ, and IFNλ suggestive of RV replication, similar mouse models of RV infection have low levels of RV replication (14). As we have measured RV viral load and not RV replication, further studies are needed to determine the relative contribution of inhibition of RV viral replication and/or increase in RV viral clearance to the reduced RV viral load we have detected in hORMDL3zp3-Cre mice. ORMDL3 upregulates levels of the anti-viral pathways including interferons (IFNα, IFNβ, IFNλ), RNase L, mOas2, and mOas1g. In particular, RV challenged hORMDL3zp3-Cre mice had a significantly enhanced lung IFNα, IFNβ, and IFNλ response compared to WT mice challenged with RV. Interferons are a family of proteins with anti-viral properties that are produced and released in response to viral infections including RV, and function in establishing both an anti-viral state in host cells such as epithelial cells infected with RV, and also activate immune cells to clear the RV infection. Interferons bind to cell surface IFN receptors on viral infected cells to activate anti-viral pathways. For example IFNα and IFNβ bind to the IFNα/β receptor complex IFNAR1/2 (20), and IFN-λ to the IL-10Rβ/IL28Rα which can induce OAS, which catalyze the polymerization of ATP into 2′-5′-linked oligoadenylates, which activate a constitutively expressed latent endonuclease, RNase L, to cleave viral and cellular ssRNAs, thereby blocking viral replication (Figure 7). The 2′-5′A-OAS-RNase L pathway is typically latent but is tightly regulated by type I IFNs (including IFNα and IFNβ) and type III IFNs (including IFN-λ) (21, 22). Active OASs then synthesize 2′-5′ linked oligo-adenylates from ATP which bind to inactive RNase L and in the presence of ADP or ATP activates its enzymatic activity (21, 22). Active RNase L then recognizes and cleaves ssRNA which can result in inhibition of cellular and viral protein synthesis, autophagy, and apoptosis to prevent virus replication (21, 22). Although ORMDL3 induces RNase L, and RNase L deficient mice have increased RV viral load, further study is needed to determine whether hORMDL3zp3-Cre mice reduction in RV viral load is mediated by Oas and RNase L. Future experiments in which hORMDL3zp3-Cre mice are crossed with either RNase L deficient or OAS deficient mice and are studied using a mouse protocol with more robust RV viral replication (23) will help to define whether there is role for the ORMDL3, Oas, RNase L, pathway in reducing RV viral replication, and/or whether ORMDL3, Oas, and RNase L contribute to RV viral clearance.
Figure 7. ORMDL3, IFN, OAS and RNase L anti-viral pathway.
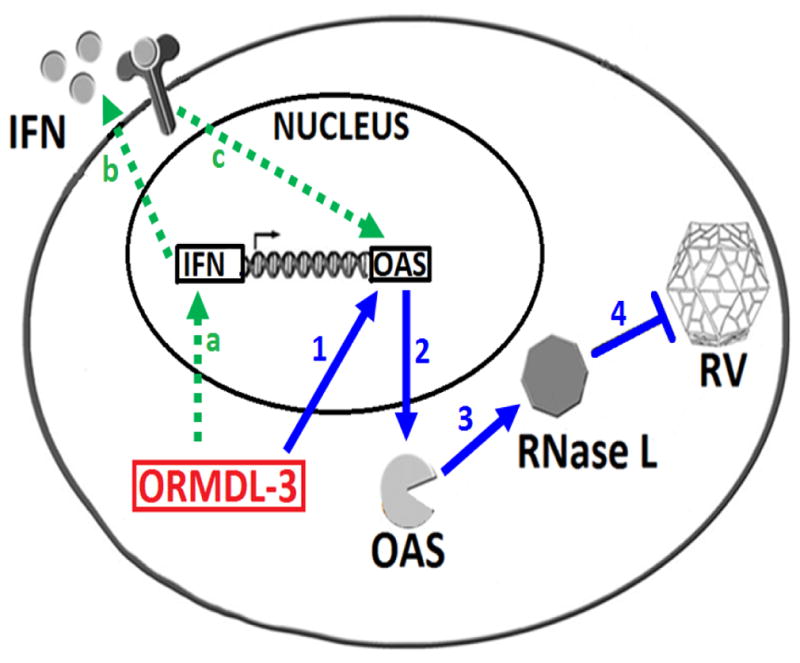
A potential antiviral pathway induced by ORMDL3. ORMDL3 induces expression of both oligoadenylate synthetase (OAS) and IFN. ORMDL3 induce OAS (blue arrow 1–2) which activates RNase L (arrow 3) to reduce RV load arrow (arrow 4). ORMDL3 also induces IFN (green arrow; a) which through autocrine pathway binds to its receptor (arrow b) and also activates OAS antiviral pathway (arrow c). (RV =rhinovirus).
In addition to IFNs activating mOas, our studies have shown that in vitro epithelial cells with increased expression of ORMDL3 exhibit enhanced activation of mOas2 in response to IFNs and may therefore contribute to the RNAse L antiviral pathway. Thus, ORMDL3 may exert anti-viral effects by both activating IFNs as well as by IFNs inducing mOas2 in cells expressing increased levels of ORMDL3 (Figure 7). Thus, the SNP on chromosome 17q21 associated with increased ORMDL3 expression may have been evolutionary conserved to function as an innate anti-viral defense pathway. At present our studies have only demonstrated that ORMDL3 plays a role in reducing RV viral load and further studies are needed to determine whether ORMDL3 also plays a role in reducing viral load of other important respiratory viruses such as RSV and influenza. Our prior studies demonstrated that human bronchial epithelial cells transfected with ORMDL3 upregulated OAS1, OAS2, and OAS3 (4, 6), while current studies demonstrate that incubation of mouse tracheal epithelial cells derived from hORMDL3zp3-Cre mice with IFNs (IFNα, IFNβ, or IFNλ) resulted in a significantly enhanced increase in mOas2 compared to WT mice. In contrast, IFNs (IFNα, IFNβ, or IFNλ) did not enhance the mOas1 (mOas1a, mOas1b, mOas1g) or mOas3 response of murine tracheal epithelial cells from hORMDL3zp3-Cre mice compared to WT mice. There was also a constitutive increase in mOas1g (but not mOas1a, mOas1b, mOas2, or mOas3) in mouse tracheal epithelial cells, and mouse lung, derived from hORMDL3zp3-Cre mice compared to WT mice. In addition, there was a statistically significant up-regulation of lung RNase L mRNA in hORMDL3zp3-Cre compared to WT mice at baseline, without RV infection. Further studies will need to investigate whether the baseline increase in expression of mOas1g and RNase L in the lungs of hORMDL3zp3-Cre mice enables a very early activation of RNase L and degradation of RV RNA. Of the eight mouse oas1 genes (oas1a-oas1h), only oas1a and oas1g are believed to be catalytically active (7). Mouse oas1b, encodes a catalytically inactive OAS but it has an alternative antiviral activity (7, 8, 24). Our studies of ORMDL3 thus demonstrate differences in the ORMDL3 regulation of OAS family members in human (OAS1, OAS2, OAS3) and mouse (mOas2, mOas1g) cells which may relate to differences in mouse and human OAS amino acid sequence identity and function (mouse Oas1c and human OAS1 share 52% identity; mouse Oas2 and human OAS2 share 61% identity; mouse Oas3 and human OAS3 share 66% identity)(7, 8, 24), the difference in methods used to increase expression of ORMDL3 in human cells (transfection) and mouse cells (hORMDL3zp3-Cre mice), or to differences in the cell types studied (bronchial epithelial cells in humans vs tracheal epithelial cells in mice).
Although our studies in mice demonstrate that RV induces innate IFNs, there is controversy in human studies as to whether asthmatics have a defective innate IFN response. Some (25, 26) but not all studies (27–29) suggest that airway epithelial cells derived from asthmatics have a reduced capacity for innate IFN synthesis in response to RV infection compared to normal airway epithelial cells. For example a deficiency in expression of IFNα, IFNβ, and IFNλ have each been described in asthma (25, 26). However these deficiencies in expression of IFNα, IFNβ, and IFNλ have not been confirmed in all studies of asthmatics (27, 28). Studies have also shown increased levels of IFNλ in the upper airway of wheezing asthmatic children compared to non-wheezing asthmatic children with a URI (29). In addition, during a viral infection in asthmatics increased levels of IFNα, IFNβ, and IFNλ have been detected which correlate with more severe asthma symptoms and the presence of an asthma exacerbation (30). In contrast, after the resolution of a viral infection in asthmatics lower levels of IFNλ are related to more severe asthma symptoms (30). At present it is not known what accounts for some but not all studies demonstrating a deficient IFN response in asthma. Whether genetics, asthma medications, the timing of measuring IFN responses (i.e. during the infection vs after resolution of the infection), or other factors influence the results in the different studies requires further investigation.
Epidemiologic studies in birth cohorts of children at high risk for the development of asthma have shown that there is a significant gene (chromosome 17q21) and environment (RV infection wheezing illness) interaction in early childhood associated with the development of asthma (3). The combined effect of RV infection in the first 3 years of life with a SNP on chromosome 17q21 was significantly associated with the development of asthma evaluated from age 6 (3). The SNP on chromosome 17q21 was associated with the number of HRV wheezing illnesses in the first 3 years of life, but not with the number of wheezing illnesses from another respiratory virus RSV (3). At present it is unknown whether the RV infection in early childhood is causal in the development of asthma, or only unmasks an underlying propensity to asthma with no direct effect on asthma risk (31). Our study in hORMDL3zp3-Cre mice infected with RV in vivo do not suggest that these mice have a deficient ability to clear RV infection. In addition, hORMDL3zp3-Cre mice infected with RV in vivo did not have more severe airway inflammation which may predispose to the development of asthma in childhood. Previous studies of RV infection in childhood have clearly linked the SNP on chromosome 17q21 with the frequency of RV wheezing illness in the first 3 years of life and the development of asthma (3). There are several potential explanations as to why hORMDL3zp3-Cre mice infected with RV in vivo may have less severe RV infection, rather than increased infection as might be predicted based on the human RV and chromosome 17q21 studies. For example, the SNP linking chromosome 17q21 to RV and the development of asthma includes four genes (ORMDL3, GSDMB, IKZF3, ZPB2) and not only ORMDL3 (2). Thus, it might be possible that GSDMB, IKZF3, or ZPB2 rather than ORMDL3 could be the gene contributing to the development of RV induced wheezing illness and asthma in early childhood. It should also be noted, that the mouse model we have used has several differences from the human study linking ORMDL3 to RV induced wheezing illness and development of asthma. The mouse model evaluated the RV viral load and airway inflammatory response to an acute RV infection. The human RV and chromosome 17q21 genetic epidemiology study examined whether RV infection influenced the development of asthma several years later in children at high risk for the development of asthma who had an RV induced wheezing illness (not investigated in the mouse study). RV infection also differs in humans and mice in that in mice we have used a model of RV1b infection which gains entry into the cell using the low density lipoprotein receptor (32), which is not the predominant receptor used by the majority of the > 160 RV serotypes (32). In contrast in humans, RV utilizes three major types of cellular membrane glycoproteins to gain entry into the host cell including intercellular adhesion molecule 1 (ICAM-1) (the majority of RV-A and RV-B), low-density lipoprotein receptor family members (12 RV-A types), and cadherin-related family member 3 (CDHR3) (RV-C)(32).
Thus, in summary in this study we demonstrated that RV infection of hORMDL3zp3-Cre mice resulted in reduced RV viral load (lung and airway epithelium), as well as reduced airway inflammation (total BAL cells, neutrophils, macrophages, and CD8+ lymphocytes) compared to RV infected WT mice. Levels of the anti-viral pathways including interferons (IFNα, IFNβ, IFNλ) and RNase L were significantly increased in the lungs of RV infected hORMDL3zp3-Cre mice. In addition, levels of the anti-viral oligo-adenylate synthetase 2 (mOas2) pathway, but not the mOas1 (mOas1a, mOas1b, mOas1g), or mOas3 pathways were significantly more upregulated by IFNs (IFNα, IFNβ, IFNλ) in epithelial cells from hORMDL3zp3-Cre mice compared to RV infected WT mouse epithelial cells. RNase L deficient mice infected with RV had increased RV viral load implying an important role for RNase L in reducing RV viral load. Overall, these studies suggest that increased levels of ORMDL3 contribute to antiviral defense to RV infection in mice through pathways that may include interferons (IFNα, IFNβ, IFNλ), OAS, and RNase L.
Acknowledgments
Grant support: NIH grants AI 107779, AI 38425, AI 07535, and AI 07469 to D.H.B. MK was supported by NIH T32 AI07469.
We acknowledge the generous provision of RNase L deficient mice by Robert Silverman PhD (Cleveland Clinic Lerner Research Institute, Cleveland).
Abbreviations
- BAL
bronchoalveolar lavage
- OAS
oligoadenylate synthetase
- ORMDL3
Oroscomucoid like 3
- RV
rhinovirus
- WT
wild-type
References
- 1.Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, Depner M, von Berg A, Bufe A, Rietschel E, Heinzmann A, Simma B, Frischer T, Willis-Owen SA, Wong KC, Illig T, Vogelberg C, Weiland SK, von Mutius E, Abecasis GR, Farrall M, Gut IG, Lathrop GM, Cookson WO. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 2.Ober C, Yao TC. The genetics of asthma and allergic disease: a 21st century perspective. Immunol Rev. 2011;242:10–30. doi: 10.1111/j.1600-065X.2011.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calışkan M, Bochkov YA, Kreiner-Møller E, Bønnelykke K, Stein MM, Du G, Bisgaard H, Jackson DJ, Gern JE, Lemanske RF, Jr, Nicolae DL, Ober C. Rhinovirus wheezing illness and genetic risk of childhood-onset asthma. N Engl J Med. 2013;368:1398–1407. doi: 10.1056/NEJMoa1211592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller M, Tam AB, Cho JY, Doherty TA, Pham A, Khorram N, Rosenthal P, Mueller JL, Hoffman HM, Suzukawa M, Niwa M, Broide DH. ORMDL3 is an inducible lung epithelial gene regulating metalloproteases, chemokines, OAS, and ATF6. Proc Natl Acad Sci U S A. 2012;109:16648–16653. doi: 10.1073/pnas.1204151109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verlaan DJ, Berlivet S, Hunninghake GM, Madore AM, Larivière M, Moussette S, Grundberg E, Kwan T, Ouimet M, Ge B, Hoberman R, Swiatek M, Dias J, Lam KC, Koka V, Harmsen E, Soto-Quiros M, Avila L, Celedón JC, Weiss ST, Dewar K, Sinnett D, Laprise C, Raby BA, Pastinen T, Naumova AK. Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am J Hum Genet. 2009;85:377–393. doi: 10.1016/j.ajhg.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller M, Rosenthal P, Beppu A, Mueller JL, Hoffman HM, Tam AB, Doherty TA, McGeough MD, Pena CA, Suzukawa M, Niwa M, Broide DH. ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. J Immunol. 2014;192:3475–3487. doi: 10.4049/jimmunol.1303047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kristiansen H, Gad HH, Eskildsen-Larsen S, Despres P, Hartmann R. The oligoadenylate synthetase family: an ancient protein family with multiple antiviral activities. J Interferon Cytokine Res. 2011;31:41–47. doi: 10.1089/jir.2010.0107. [DOI] [PubMed] [Google Scholar]
- 8.Kakuta S, Shibata S, Iwakura Y. Genomic structure of the mouse 2′,5′-oligoadenylate synthetase gene family. J Interferon Cytokine Res. 2002;22:981–993. doi: 10.1089/10799900260286696. [DOI] [PubMed] [Google Scholar]
- 9.Drappier M, Michiels T. Inhibition of the OAS/RNase L pathway by viruses. Curr Opin Virol. 2015:19–26. doi: 10.1016/j.coviro.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behera AK, Kumar M, Lockey RF, Mohapatra SS. 2′-5′ Oligoadenylate synthetase plays a critical role in interferon-gamma inhibition of respiratory syncytial virus infection of human epithelial cells. J Biol Chem. 2002;277:25601–25608. doi: 10.1074/jbc.M200211200. [DOI] [PubMed] [Google Scholar]
- 11.Mehta AK, Duan W, Doerner AM, Traves SL, Broide DH, Proud D, Zuraw BL, Croft M. Rhinovirus infection interferes with induction of tolerance to aeroantigens through OX40 ligand, thymic stromal lymphopoietin, and IL-33. J Allergy Clin Immunol. 2016;137:278–288. doi: 10.1016/j.jaci.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagarkar DR, Wang Q, Shim J, Zhao Y, Tsai WC, Lukacs NW, Sajjan U, Hershenson MB. CXCR2 is required for neutrophilic airway inflammation and hyperresponsiveness in a mouse model of human rhinovirus infection. J Immunol. 2009;183:6698–6707. doi: 10.4049/jimmunol.0900298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanders SP, Siekierski ES, Porter JD, Richards SM, Proud D. Nitric oxide inhibits rhinovirus-induced cytokine production and viral replication in a human respiratory epithelial cell line. J Virol. 1998;72:934–942. doi: 10.1128/jvi.72.2.934-942.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartlett NW, Walton RP, Edwards MR, Aniscenko J, Caramori G, Zhu J, Glanville N, Choy KJ, Jourdan P, Burnet J, Tuthill TJ, Pedrick MS, Hurle MJ, Plumpton C, Sharp NA, Bussell JN, Swallow DM, Schwarze J, Guy B, Almond JW, Jeffery PK, Lloyd CM, Papi A, Killington RA, Rowlands DJ, Blair ED, Clarke NJ, Johnston SL. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat Med. 2008;14:199–204. doi: 10.1038/nm1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doherty TA, Khorram N, Sugimoto K, Sheppard D, Rosenthal P, Cho JY, Pham A, Miller M, Croft M, Broide DH. Alternaria induces STAT6-dependent acute airway eosinophilia and epithelial FIZZ1 expression that promotes airway fibrosis and epithelial thickness. J Immunol. 2012;188:2622–2629. doi: 10.4049/jimmunol.1101632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 17.Lazear HM, Nice TJ, Diamond MS. Interferon-λ: Immune Functions at Barrier Surfaces and Beyond. Immunity. 2015;43:15–28. doi: 10.1016/j.immuni.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davidson DJ, Kilanowski FM, Randell SH, Sheppard DN, Dorin JR. A primary culture model of differentiated murine tracheal epithelium. Am J Physiol Lung Cell Mol Physiol. 2000;279:L766–L778. doi: 10.1152/ajplung.2000.279.4.L766. [DOI] [PubMed] [Google Scholar]
- 19.Zhou A, Paranjape J, Brown TL, Nie H, Naik S, Dong B, Chang A, Trapp B, Fairchild R, Colmenares C, Silverman RH. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 1997;16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kroeker AL, Coombs KM. Systems biology unravels interferon responses to respiratory virus infections. World J Biol Chem. 2014;5:12–25. doi: 10.4331/wjbc.v5.i1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chakrabarti A, Jha BK, Silverman RH. New insights into the role of RNase L in innate immunity. J Interferon Cytokine Res. 2011;31:49–57. doi: 10.1089/jir.2010.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banerjee S. RNase L and the NLRP3-inflammasome: An old merchant in a new trade. Cytokine Growth Factor Rev. 2016;29:63–70. doi: 10.1016/j.cytogfr.2016.02.008. [DOI] [PubMed] [Google Scholar]
- 23.Newcomb DC, Sajjan US, Nagarkar DR, Wang Q, Nanua S, Zhou Y, McHenry CL, Hennrick KT, Tsai WC, Bentley JK, Lukacs NW, Johnston SL, Hershenson MB. Human rhinovirus 1B exposure induces phosphatidylinositol 3-kinase-dependent airway inflammation in mice. Am J Respir Crit Care Med. 2008;177:1111–1121. doi: 10.1164/rccm.200708-1243OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elbahesh H, Jha BK, Silverman RH, Scherbik SV, Brinton MA. The Flvr-encoded murine oligoadenylate synthetase 1b (Oas1b) suppresses 2–5A synthesis in intact cells. Virology. 2011;409:262–270. doi: 10.1016/j.virol.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Contoli M, Message SD, Laza-Stanca V, Edwards MR, Wark PA, Bartlett NW, Kebadze T, Mallia P, Stanciu LA, Parker HL, Slater L, Lewis-Antes A, Kon OM, Holgate ST, Davies DE, Kotenko SV, Papi A, Johnston SL. Role of deficient type III interferon-lambda production in asthma exacerbations. Nature Medicine. 2006;12:1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- 26.Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, Holgate ST, Davies DE. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez-Souza N, Favoreto S, Wong H, Ward T, Yagi S, Schnurr D, Finkbeiner WE, Dolganov GM, Widdicombe JH, Boushey HA, Avilla PC. In vitro susceptibility to rhinovirus infection is greater for bronchial than for nasal airway epithelial cells in human subjects. J Allergy Clin Immunol. 2009;123:1384–1390. doi: 10.1016/j.jaci.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bochkov YA, Hanson KM, Keles S, Brockman-Schneider RA, Jarjour NN, Gern JE. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunology. 2010;3:69–80. doi: 10.1038/mi.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller EK, Hernandez JZ, Wimmenauer V, Shepherd BE, Hijano D, Libster R, Serra ME, Bhat N, Batalle JP, Mohamed Y, Reynaldi, Rodriguez A, Otello M, Pisapia N, Bugna J, Bellabarba M, Kraft D, Coviello S, Ferolla FM, Chen A, London SJ, Siberry GK, Williams JV, Polack FP. A mechanistic role for type III interferon-λ1 in asthma exacerbations mediated by human rhinoviruses. Am J Respir Crit Care Med. 2012;185:508–516. doi: 10.1164/rccm.201108-1462OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwantes EA, Denlinger LC, Evans MD, Gern JE, Jarjour NN, Mathur SK. Severity of virus-induced asthma symptoms is inversely related to resolution IFN-λ expression. J Allergy Clin Immunol. 2015;135:1656–1659. doi: 10.1016/j.jaci.2015.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bønnelykke K, Ober C. Leveraging gene-environment interactions and endotypes for asthma gene discovery. J Allergy Clin Immunol. 2016;137:667–79. doi: 10.1016/j.jaci.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bochkov YA, Gern JE. Rhinoviruses and Their Receptors: Implications for Allergic Disease. Curr Allergy Asthma Rep. 2016;16:30. doi: 10.1007/s11882-016-0608-7. [DOI] [PMC free article] [PubMed] [Google Scholar]