

Conspectus
The unique role that stereochemistry plays in molecular recognition events continues to provide a driving force for synthesizing organic compounds in enantioenriched form. The tendency of enantioenriched organic compounds to revert to an entropically favored racemic state in the presence of viable racemization pathways (e.g., the enolization of stereogenic carbonyl derivatives) can sometimes interfere with this objective; however, beginning with Noyori's foundational disclosure of a dynamic kinetic transfer hydrogenation, the ability to channel racemic, configurationally labile starting materials through stereoconvergent reaction pathways has been recognized as a powerful strategy in asymmetric synthesis. Proton transfer, retro-aldol, retro-Michael, reversible redox events, and other processes that can be deleterious to asymmetric synthesis are exploitable in enantioconvergent reactions using chiral small molecules and enzymes as asymmetric catalysts. Enantioselective reduction of configurationally labile carbonyl derivatives bearing a C–H acidic chiral center are particularly common. Because facile racemization is vital to stereocontrol in these transformations, hydrogenations of β-dicarbonyls are commonplace, while less activated substrates have been used less commonly. Our entry into enantioconvergent catalysis evolved from a long-standing interest in the synthesis of complex glycolates and began with the development of a general Noyori-type transfer hydrogenation of α-keto esters. Key innovations in this work include the identification of a new terphenylsulfonamide–Ru(II) complex, which displays unusual preference toward reduction of α-keto esters, and the observation that α-keto esters racemize under mildly basic conditions. This work was extended to the dynamic kinetic hydrogenation of racemic acyl phosphonates. Moreover, the recent recognition that the mechanistic paradigm underlying enantioconvergent hydrogenation chemistry can be extended to diverse carbon-centered nucleophiles has led to advances in the art. Our lab has developed a number of enantioconvergent tertiary alcohol syntheses. In the context of carbon-centered nucleophiles, we have focused on the use of α-keto esters; however, in the latter part of this Account, we will briefly describe our nascent efforts to develop dynamic kinetic additions of carbon-centered nucleophiles to β-oxo acid derivatives. While the enantioconvergent hydrogenation of β-keto acid derivatives is carried out on 100-ton scale annually, non-hydrogenative transformations of these compounds constitute an underexplored subclass of enantioconvergent reactions.
With regard to future prospects, a trend toward transformations that afford increasing levels of molecular complexity is apparent. It can be expected that the burgeoning field of asymmetric 1,2-addition chemistry will further drive this chemistry to encompass a wider array of enantioconvergent additions. Additionally, the continued exploration of these chemistries in the context of less conventional electrophiles, as well as identifying novel or overlooked modes of racemization, holds considerable potential.
Graphical abstract
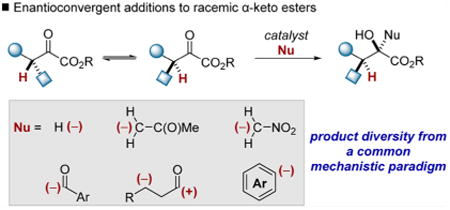
1. Introduction
Nucleophilic addition to α-stereogenic carbonyl derivatives is a robust strategy for the synthesis of complex alcohols.1 The requisite chiral electrophiles are readily prepared by the functionalization of enolate derivatives. The addition of acyl anion equivalents to prochiral electrophiles constitutes an alternative approach that can be deployed in the synthesis of α-stereogenic carbonyls.2,3 Both approaches require basic reaction conditions, and the inclination of optically active carbonyls bearing acidic protons to racemize via enolization can pose a significant challenge.4 Enantioconvergent catalysis represents a powerful solution to this problem.5 The ability to channel configurationally labile starting materials through stereoconvergent reaction pathways, disclosed by Noyori and co-workers in their foundational report of a dynamic kinetic hydrogenation (Scheme 1A),6 represents a significant advance in organic synthesis. Racemization, typically viewed as a nuisance, can be harnessed to achieve streamlined syntheses of complex molecular frameworks.7 Numerous transformations that operate under this mechanistic paradigm have been developed.5 The complexity found in the products ascends according to the number of stereocenters centers formed and the reagents coupled in the enantioselective step (Scheme 1B). Beginning with enantioconvergent reactions that furnish a single stereo-center,8,9 the next stratum of complexity includes Noyori-type hydrogenations that establish two chiral centers.6,10,11 In many enantioconvergent reactions, facile substrate racemization is a prerequisite for obtaining high stereoselectivity;5,12 therefore, β-oxo ester derivatives have emerged as the factotum substrate class for dynamic kinetic hydrogenations. Non-hydrogenative transformations that establish two chiral centers constitute the third echelon of complexity.13 The final echelon of complexity comprises a handful of reactions that involve the addition of prochiral nucleophiles and create three chiral centers.14
Scheme 1. Enantioconvergent Reactions of Carbonyl Compounds.
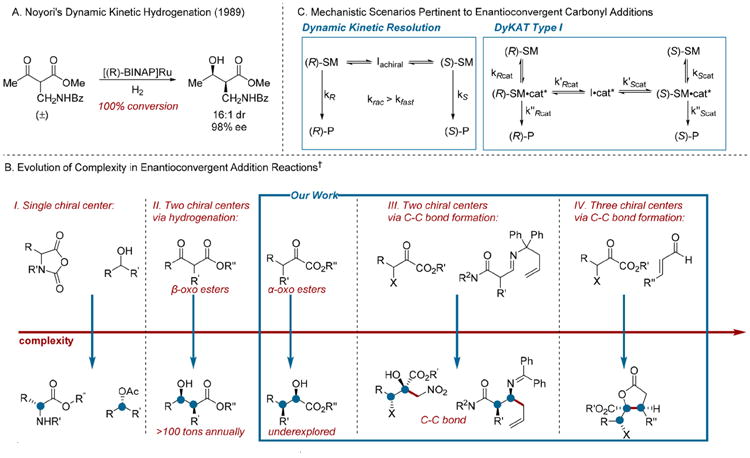
†All transformations shown employ racemic starting materials.
This Account highlights our laboratory's ongoing development of two underexplored subtypes of enantioconvergent transformations: those employing α-keto esters as electrophiles and non-hydrogenative transformations of β-oxo acid derivatives. Enantioconvergent additions are classified as either dynamic kinetic resolutions (DKRs) or Type I dynamic kinetic asymmetric transformations (DyKATs).5,12 In a DKR, the racemization is independent of the chiral catalyst (Scheme 1C), and the stereoselectivity is affected by the rate of racemization, which generally must be greater than the rate of reaction for the fast reacting enantiomer. In a Type I DyKAT, racemization is promoted by the chiral catalyst. The rates of formation and transformation of epimeric catalyst/substrate complexes and their concentrations influence stereoselectivity.
2. Transfer Hydrogenation Dynamic Kinetic Resolutions of α-Keto Esters and Acyl Phosphonates
Our work utilizing silyl glyoxylates as glyoxylate anion equivalents opened the door to our studies of dynamic 1,2-additions.15 During the development of a nucleophilic glyoxylation employing silyl glyoxylate 4 and aldehydes (i.e., benzaldehyde 5), the sensitivity of the derived β-silyloxy-α-keto ester 6 toward base promoted conversion to isomeric β-keto ester 7 was noted. This observation prompted the realization that α-keto esters such as 6 might succumb to facile racemization and thereby hold potential as precursors to diol derivatives such as 8 via dynamic reduction (Scheme 2). Enantioselectivity was obtained with the Noyori-type Ru(II)-diamine catalyst 9 under transfer hydrogenation conditions (Cs2CO3, /HCO2H).16 Unfortunately, conditions could not be identified that both suppressed the intervention of the unreactive α-silyloxy-β-keto ester 7 and engendered high stereoselectivity. Nevertheless, the utilization of α-keto esters in enantioconvergent hydrogenation remained attractive to us as a means to generate diverse glycolate architectures.
Scheme 2. Initial Findings.

We sought to embed functional handles within the α-keto ester framework that would trigger complexity building events in the same flask. For instance, incorporation of γ-diester functionality might trigger a diastereoselective lactonization, providing access to γ-butyrolactone derivatives characterized by three contiguous stereocenters (Scheme 3).11
Scheme 3. Complexity Generating Enantioconvergent Hydrogenation.
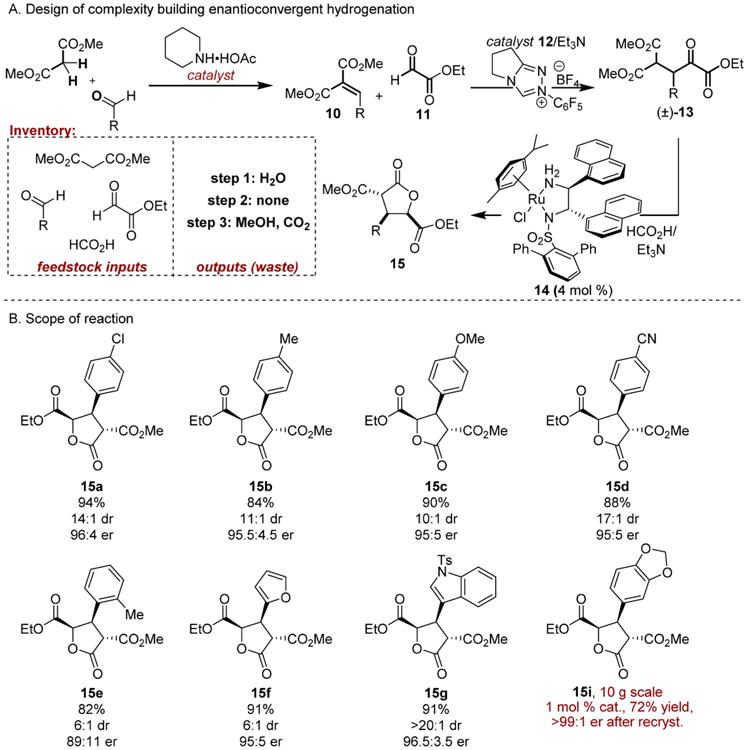
We aimed to develop an efficient route to the α-keto ester starting materials so that the utility of the forecasted complexity-generating hydrogenation/lactonization event would not be undermined by unduly inconvenient or costly procedures.
Accordingly, inspired by a glyoxamidation reaction developed by Rovis and co-workers,4 we developed an N-heterocyclic carbene-catalyzed Stetter addition utilizing benzylidene malonate derivatives 10 and commercially available ethyl glyoxylate 11 (Scheme 3). Notably, the process can produce multigram quantities of α-keto esters 13 and is characterized by 100% atom economy.17 For the key hydrogenation reaction, we returned to Noyori's (arene)RuCl(monosulfinamide-DPEN) framework and employed formic acid/triethylamine azeotrope (5:2) as both the organic reductant and racemization promoter. A new m-terphenyl substituted α-naphthylethylene diamine derived ruthenium(II) complex 14 was developed for optimal performance. At the time, the α-naphthyl ethylene diamine scaffold had rarely been used in asymmetric catalysis,18 while examples of m-terphenyl groups in enantioselective catalysis were even less common.19 These two structural elements operate synergistically to relay asymmetry; corresponding ligands with a DPEN backbone and m-terphenylsulfonamide group or an α-naphthyl ethylene diamine backbone with other sulfonamides failed to provide satisfactory selectivity.
A range of γ-butyrolactones 15a–i, characterized by a syn,anti-configured stereotriad were furnished (Scheme 3). The reduction could be carried out with ease on 10 g scale, using 1 mol % Ru in the synthesis of piperonyl derivative 15i. The overall sequence of three catalytic steps transforms four feedstock starting materials, aldehyde, malonate, ethyl glyoxylate, and formic acid, into stereochemically complex γ-butyrolactone frameworks.
The terphenylsulfonamide of catalyst 14 is a uniquely effective design element in hydrogenations of diverse β-stereogenic-α-keto esters. For instance, α,γ-diketoesters 17, produced by a Stetter reaction between ethyl glyoxylate 11 and chalcones 16, underwent highly chemo- and stereoselective hydrogenation to α-hydroxy-γ-keto esters 18 (Scheme 4).10b The products of this reduction are formal glycolate Michael adducts, a bond construction that is otherwise difficult to achieve without recourse to auxiliary control or protecting group manipulation.20 This methodology was applied to the synthesis of the α-benzylidene-γ-butyrolactone natural products mega-cerotonic acid 19f and shimobashiric acid 19g (Scheme 4B).10e The simplicity of the hydrogenation and the flexibility of substitution on the α,γ-diketoesters 17 should facilitate the preparation of unnatural congeners for biological studies.
Scheme 4. Chemoselective Transfer Hydrogenation DKRs of α-Keto Esters.
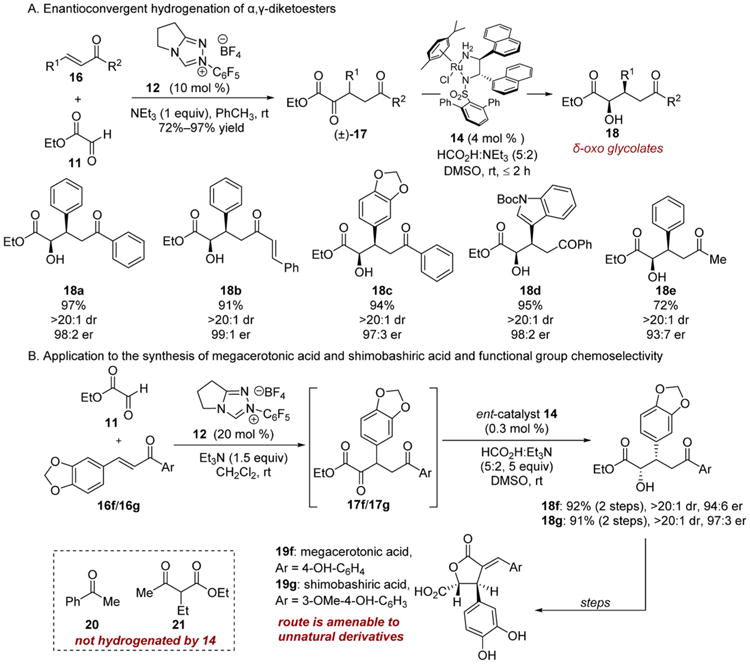
The high chemoselectivity imparted by the m-terphenylsul-fonamide/di-α-naphthylethylenediamine-Ru(II) system 14 toward reduction of α-keto esters is remarkable considering (arene)RuCl(sulfonamide) catalysts are used for reductions of aryl ketones. Acetophenone 20 and ethyl-3-oxo-butanoate 21, archetypal substrates for the development of transfer hydrogenations,17 were not hydrogenated by complex 14. We surmise that the bulky nature of the novel ligand motif suppresses reduction of less activated carbonyls.
β-Aminoglycolates could also be generated by the asymmetric transfer hydrogenation DKR (ATH-DKR, Scheme 5).10d Racemic Mannich adducts 24 were treated with Oxone to provide access to Boc or Cbz protected β-amino-α-keto esters 25. In this instance, the terphenylsulfonamide/ diphenylethylenediamine–Ru(II) complex 26 provided levels of enantioselectivity comparable to 14. The unpurified α-keto ester starting materials could be employed in the transfer hydrogenation reaction, highlighting the robust nature of the catalyst system. An array of β-amino-α-hydroxy esters 27 were prepared in good to high yield with high levels of stereocontrol (Scheme 5, 27a-i). Different amine protecting groups can be employed, although higher selectivities were obtained with Boc-protected substrates (27a vs 27b). As the series of 27c–e shows, the ability of the catalyst to enforce enantiocontrol is marginally influenced by ortho-substitution in this system, although the diastereoselectivity displayed higher sensitivity. An arylpinacol boronate was tolerated in the reduction to β-aminoglycolate product 27h.
Scheme 5. Enantioconvergent Hydrogenation of β-Amino-α-keto Esters.
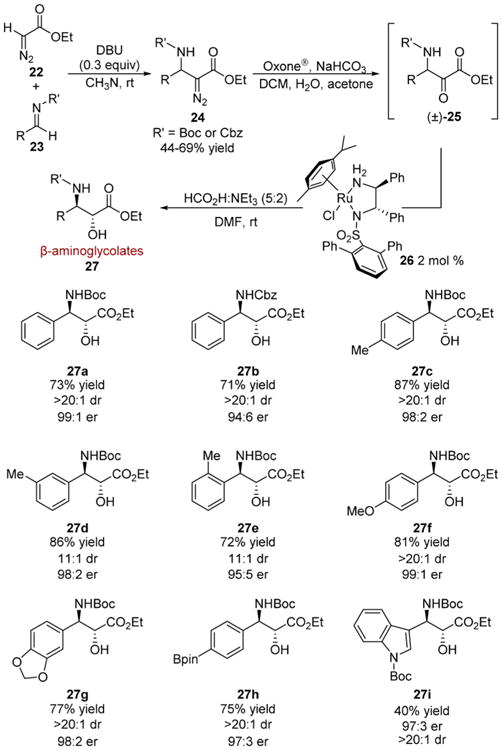
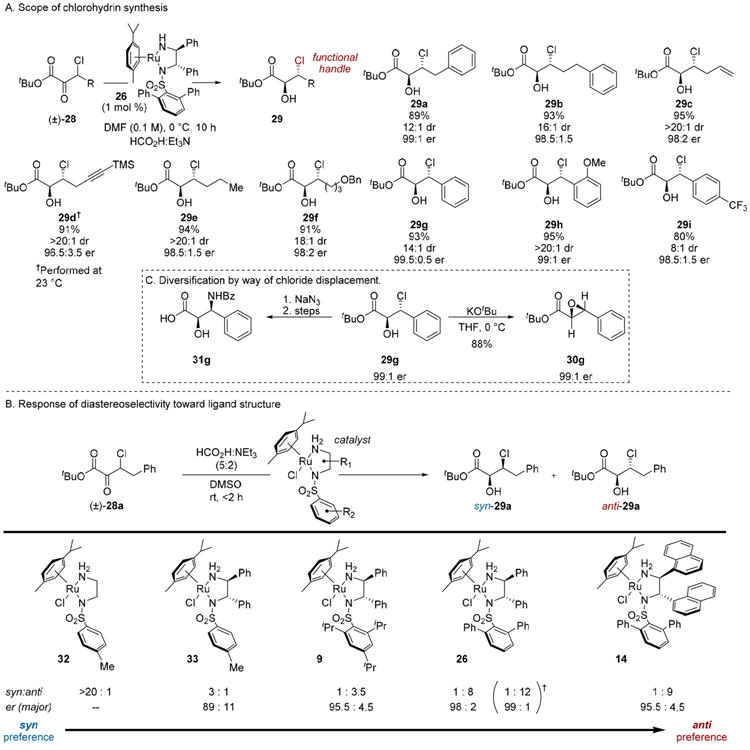
The hydrogenation of a common platform containing a malleable β-functionality was compelling from the standpoint of rapidly generating diverse glycolates from a single hydrogenation product. Thus, the deployment of β-chloro-α-keto esters 28 was undertaken with the assumption chlorohydrins 29 would be amenable to downstream substitution chemistry (Scheme 6).10b
Scheme 6. Enantioconvergent Hydrogenation of β-Chloro-α-keto Esters.
†Experiment conducted in DMF at 0 °C for 10 h.
Ultimately, catalyst 26 was effective for the synthesis of diverse chlorohydrin derivatives 29a–i. In accord with our hypothesis that the β-nucleofuge could be displaced to introduce various heteroatomic substitution, halohydrin 29g was converted to its derived glycidic ester 30g and β-aminoglycolic acid 31g.
The diastereoselectivity of this process exhibits a striking response to ligand structure (Scheme 6). For instance, the ethylene diamine derivative 32 provided >20:1 preference for the syn isomer. In contrast, the novel terphenylsulfonamide scaffolds 14 and 26 were highly anti-selective.
At this juncture we began to consider the extension of the ATH-DKR to other substrate classes. Acyl phosphonates share some characteristics with α-keto esters and can behave analogously in catalytic asymmetric reactions.21 The use of the former in the ATH-DKR was appealing as a means to test the generality of this mechanistic paradigm.10c In analogy to the transfer hydrogenation of β-chloro-α-keto esters, the diastereoselectivity of this transformation could be tuned by altering the ligand structure. Ultimately, we found complex 14 to be optimal in the reduction of acyl phosphonates 34 to a wide array of hydroxy phosphonates 35, but with the opposite facial selectivity relative to analogous β-aryl-substituted α-keto esters: the diastereomeric (1R, 2R) configuration was obtained (Scheme 7B).
Scheme 7. Enantioconvergent Hydrogenation of Acylphosphonates.
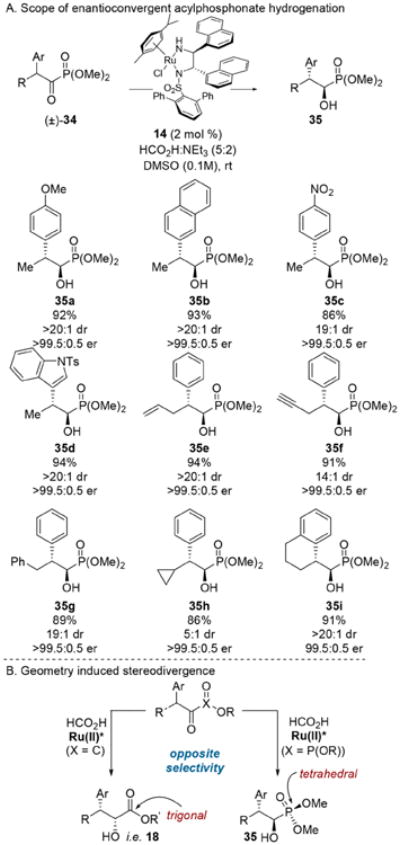
The dichotomy in diastereofacial selectivity observed between acyl phosphonates and α-keto esters stands in contrast to other asymmetric transformations for which both have been employed;21 however, the steric environment surrounding the reactive carbonyl center is different for acyl phosphonates, which are tetrahedral at the phosphorus center, and α-keto esters, which are trigonal at the adjacent carbon. This difference apparently influences the facial selectivity in the (arene)RuCl-(monosulfonamide-diamine) catalyzed transfer hydrogenation.
3. Dynamic Kinetic 1,2-Addition of Carbon-Centered Nucleophiles
3.1. Aminocatalyzed Aldol and Henry Reactions of α-Keto Esters
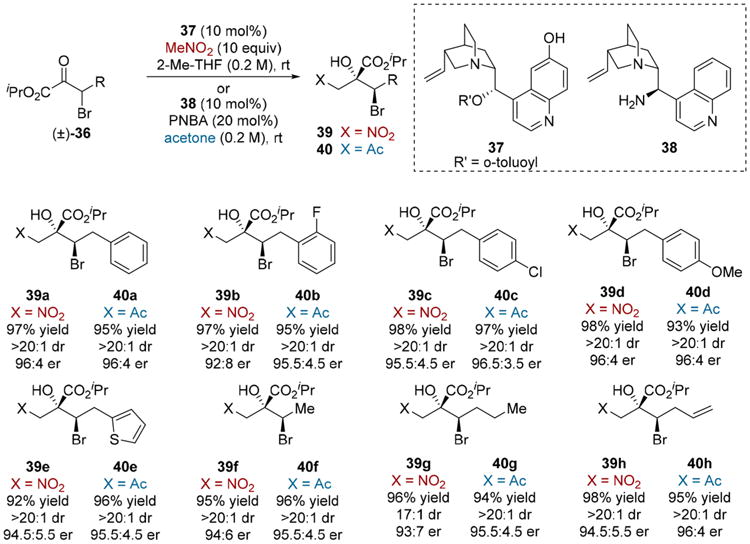
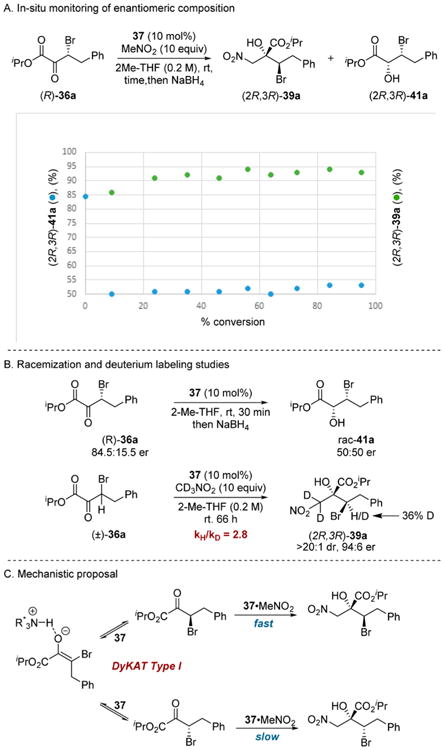
By extending Noyori-type hydrogenations to β-stereogenic α-keto esters and acyl phosphonates, an array of complex glycolate and hydroxyphosphonate frameworks were accessed; however, enantioconvergent processes that proceed via C–H bond formation are limited with respect to the complexity they can generate. In contrast, the addition of carbon-nucleophiles should provide expanded access to diverse scaffolds. Due to our group's interest in complex glycolates,15 the development of enantioconvergent C-centered nucleophile additions to α-keto esters emerged as a compelling means to generate tertiary glycolates. We first targeted the addition of pro-nucleophiles nitromethane and acetone to β-bromo-α-keto esters 36 (Scheme 8).13c Ultimately, a simple, readily accessed quinidine-derived catalyst 37 was found to promote high reactivity and excellent stereoselectivity in the Henry reaction, while the acetone aldol reaction was realized by enamine catalysis using cinchona alkaloid derived primary amine 38. A diverse array of α-keto ester substrates participated in both reactions, providing tertiary glycolates 39a–h (MeNO2 addition) or 40a–h (Me2CO addition).
Scheme 8. Enantioconvergent Aldol Reaction of β-Bromo-α-keto Esters.
To gain a deeper understanding of the mechanism of the dynamic kinetic aldol and Henry reaction, we sought to elucidate the racemization mode. We first measured the enantiomeric composition of enantioenriched α-keto ester 36a and product 39a during the course of the Henry reaction (Scheme 9A). Complete racemization of 36a occurred within 10 min, and the starting material remained racemic throughout the remainder of the reaction, while the product was formed with uniform selectivity throughout the course of the reaction. Notably, the observation that 36a underwent complete racemization in under 30 min in the presence of 37 alone suggests general base catalysis as a mode of substrate racemization in the case of 36 (Scheme 9B). Additionally, when the catalytic Henry addition was conducted with CD3NO2, a kinetic isotope effect of 2.8 and only 36% deuterium incorporation at the β position of 39a was observed, suggesting that racemization of 36a primarily involves an intimate ion process with protonation occurring from the ammonium salt rather than nitromethane and that nitronate formation contributes to the observed reaction rate. In situ monitoring of the reaction by NMR spectroscopy revealed no intermediates, thus confirming the resting state as the neutral amine 37 and suggesting that nitronate formation is endergonic. The observation that 36a undergoes racemization in the presence of the amine catalyst, likely via an enolate– ammonium ion pair intermediate, in combination with the observed moderate deuterium incorporation, suggests that the Henry reaction proceeds via a DyKAT Type I mechanism (Scheme 9C).
Scheme 9. Mechanistic Studies of Enantioconvergent Henry Addition.
Several other aminocatalyzed dynamic aldolizations have been reported in the literature. For instance, prior to our work, Calter reported a highly enantioselective interrupted Feist–Bénary reaction (Scheme 10A).13a Additionally, Ward and co-workers developed an enantioconvergent aldolization of cyclocarbaldeydes (Scheme 10B),14a while Zhang employed β-keto esters in a highly enantioselective dynamic acetone aldol reaction (Scheme 10C).13b Finally, the tandem chlorination/aldol reaction developed by Britton and co-workers was leveraged for synthesis of furanose derivatives (Scheme 10D).14c
Scheme 10. Other Aminocatalyzed Aldol-Type Reactions of α -Keto Esters.

3.2. NHC-Catalyzed Additions to α-Keto Esters
3.2.1. Dynamic Kinetic Cross-Benzoin Additions
Having developed highly enantio- and diastereoselective Henry and aldol reactions of racemic α-keto ester electrophiles, we sought to expand the scope to the addition of other carbon-centered nucleophiles. The benzoin addition is a valuable reaction that furnishes α-hydroxycarbonyl products with 100% atom economy. At the time we began to examine this issue, benzoin reactions that established more than one stereocenter were not commonplace.3d,f Thus, the formation of two stereocenters via dynamic kinetic cross-benzoin reactions of aldehydes 42 and racemic, configurationally labile, α-keto esters (i.e., 28 or 36) was attractive to us as a means to generate complex glycolate frameworks. While highly selective homo-benzoin reactions have been developed, cross-benzoin additions between different donor and acceptor carbonyls are more difficult to implement due to competitive homobenzoin reactions.22 Nevertheless, a highly selective cross-benzoin reaction of aryl aldehydes 42 and β-halo-α-keto esters could be realized when triazolium salt 43 was employed in conjunction with K2CO3 to facilitate formation of the active carbene (Scheme 11A). The β-chloro-α-keto esters 28-Me afforded lower levels of diastereocontrol (i.e., 44a); therefore, the scope of this transformation was studied using the β-bromo-derivatives 36-Me. An array of α-keto esters underwent a highly diastereo- and enantioselective cross-benzoin addition with benzaldehyde leading to products 44a-e. Currently, γ-branched electrophiles represent a limitation, as only 60% conversion was observed in the formation of product 44h, which was isolated in low yield.
Scheme 11. Enantioconvergent Cross-Benzoin Reaction of β-Halo-α-keto Esters.
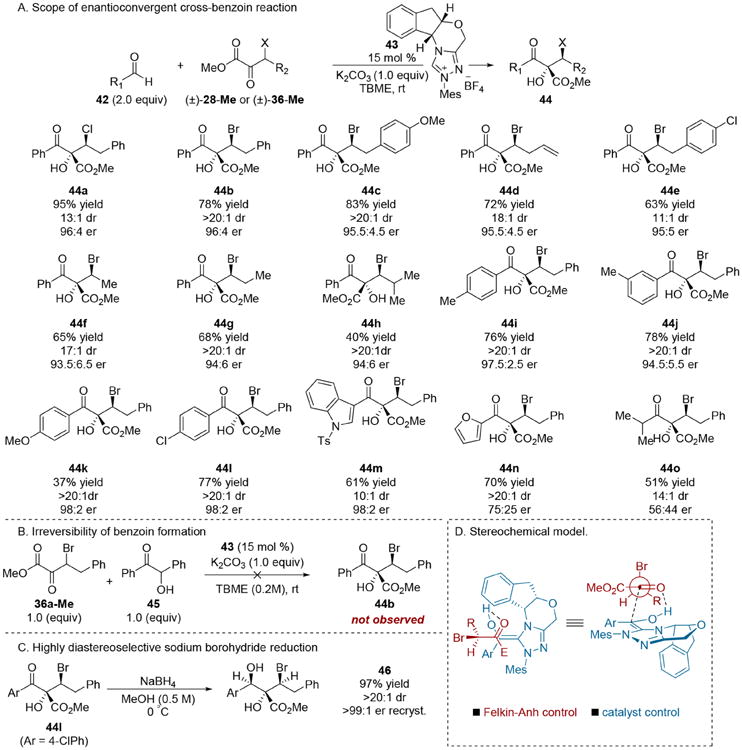
A gram scale benzoin reaction between 36b and 42a revealed a minor amount of the benzoin 45 (9% isolated yield); however, subjecting 45 to the reaction conditions with α-keto ester 36a produced none of the cross-benzoin product 44b (Scheme 11B), indicating that benzoin formation is irreversible under the reaction conditions and that the high selectivity for the cross-benzoin product is likely due to the heightened reactivity of the β-bromo-α-keto esters 36-Me relative to the less activated aryl aldehydes 42. The cross-benzoin product 44l could be reduced by sodium borohydride to complex β-hydroxyglycolate framework 46 bearing three contiguous stereocenters with exquisite levels of diastereoselection (Scheme 11C).
The observed selectivity in the cross-benzoin reaction was rationalized through the interplay of Felkin–Anh23 diastereofacial addition opposite to the large electronegative bromide and approach from the convex face of the Breslow intermediate imposed by the bicyclic framework of the NHC catalyst (Scheme 11D). It should be emphasized that the reactive conformation proposed by the Cornforth model, where the halogen is anti with respect to the carbonyl to achieve dipole minimization, predicts the same stereochemical outcome.24
3.2.2. Dynamic Kinetic Homoenolate Additions
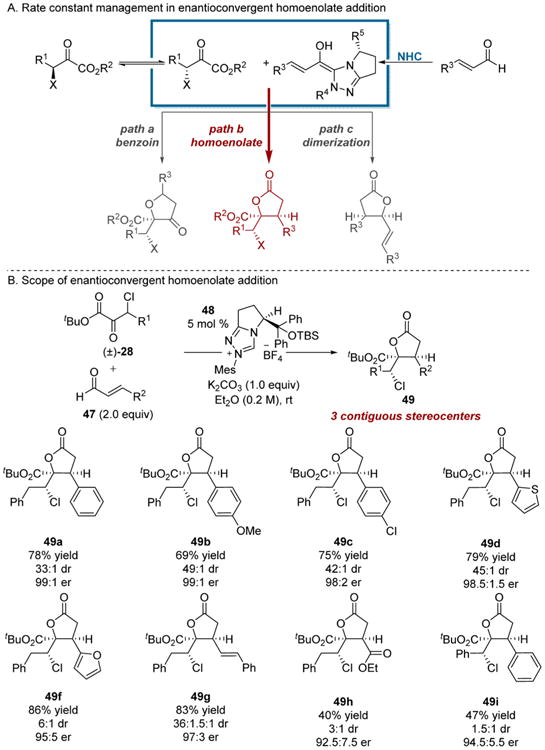
Enantioconvergent reactions that establish three stereogenic centers are unusual. Examples include the transfer hydrogenation/lactonization sequence developed by our laboratory (Scheme 3), processes that involve Michael/retro-Michael induced enantiomerization,25 and the aldol reactions developed by Ward14a and Britton14c (Scheme 10B,D). Building on the theme of enantioconvergent umpolung reactions, the extension to extended Breslow intermediates3e,f was a logical next step. We anticipated that the integration of homoenolates would not be trivial due to the potential formation of eight stereoisomers. Additional perceived challenges to our reaction design included potential acyl anion reactivity and competitive dimerization of the enal (Scheme 12A, paths a and c). Nevertheless, we were compelled by the potential of the dynamic kinetic homoenolate addition of α,β-unsaturated aldehydes 47 and α-keto esters (path b) to furnish γ-butyrolactones bearing three stereogenic centers and a fully substituted glycolic acid motif. The β-chloro substituted α-keto esters 28 employed in previous DKR reactions by our lab were chosen as a model substrate. Concomitant formation of undesired cross-benzoin products was suppressed by employing the pyroglutamic acid derived triazolium precatalyst 48, which allowed the highly chemo-, diastereo-, and enantioselective formation of the desired γ-butyrolactones 49. An array of enals 47 could be employed in the process, but the NHC catalyzed homoenolate addition was sensitive to the steric nature of the nucleophile as o-tolylcinnamaldehyde was not a productive reaction partner. However, thiophene-substituted lactone 49d could be obtained in 84% yield, >30:1 dr, and 99:1 er on 1 g scale. In analogy to the enantioconvergent cross-benzoin addition, the diastereoselectivity of the homoenolate addition reaction is in accord with the Felkin–Anh model.
Scheme 12. Enantioconvergent Homoenolate Addition to β-Chloro-α-keto Esters.
An important precedent for NHC-catalyzed enantioconvergent reactions described here was provided in the form of the intramolecular tandem aldol/lactonization developed by Scheidt and co-workers (Scheme 13A; also relevant to Section 4).14b Additionally, Wang and co-workers have developed an elegant NHC/Lewis acid catalyzed vinylogous aldol/lactonization reaction (Scheme 13B).13f Finally, Fang recently developed an intramolecular enantioconvergent benzoin reaction (Scheme 13C).13h
Scheme 13. Other NHC-Catalyzed Enantioconvergent Additions.

3.3. Enantioconvergent Addition of Nonstabilized Nucleophiles: Chiral(diene)rhodium(I)-Catalyzed Arylboronic Acid Addition
The transition metal-catalyzed addition of nonstabilized carbon nucleophiles to ketones recently emerged as a compelling opportunity to generate complex tertiary alcohols inaccessible through other methods. The enantioselective addition of arylboronic acids to carbonyl derivatives, including α-keto esters, has been widely developed.26 Considering their chemical stability, ease of handling and broad commercial availability, we envisioned the deployment of arylboronic acids in an enantioconvergent addition to racemic α-keto ester electrophiles would facilitate the production of stereochemically complex glycolate architectures.13i The β-alkyl, aryl substituted α-keto ester derivatives 50 were chosen as a model substrates for this process (Scheme 14A). Catalyst screening led to the identification of chiral(diene)rhodium(I) complexes 52a and 52b as highly selective catalysts for the enantioconvergent arylation reaction. The optimal conditions employed CsF to promote transmetalation, and chloroform as solvent at 40 °C. The presence of an amine base was necessary to achieve high levels of enantioselectivity.
Scheme 14. Enantioconvergent Arylation of β-Alkyl-α-keto Esters.
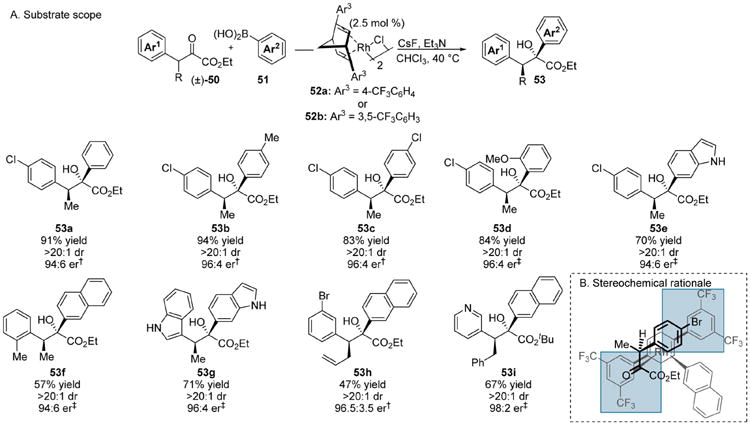
†52a was used as the catalyst. ‡52b was used as the catalyst.
The reaction delivered a diverse array of tertiary arylglycolate products 53a–53i. A notable example involves addition of 6-indoleboronic acid to an indole substituted α-keto ester, producing bis(indole) derivative 53g in good yield with high levels of diastereo- and enantiocontrol. Pyridine containing α-keto esters could also be employed in this process, and product 53i could be obtained in good yield with high levels of enantio- and diastereocontrol. Compact scaffolds containing heterocyclic motifs such as 53g and 53i represent interesting targets in the field of medicinal chemistry.27
The observed stereochemistry can be attributed to the C2-symmetry of (R,R)-catalyst 52b (and 52a), which enforces high levels of enantiocontrol by blocking the shaded quadrants in the model shown in Scheme 14B; the bulky sp3 center is guided to the top left region. In the reaction of the (S)-enantiomer, the less sterically demanding hydrogen projects toward the catalyst assembly.
As noted, the presence of a tertiary amine had a bearing on the enantioselectivity. Low levels of selectivity were observed when Hunig's base was employed; however, simply switching to the more compact base triethylamine allowed the formation of the desired product with high levels of enantioselectivity. Subjecting enantioenriched 50a to the reaction conditions and an array of amine bases revealed that the structure of the basic additive impacted the rate of racemization (Scheme 15). More specifically, less sterically encumbered bases promote faster racemization. In the presence of Hunig's base, α-keto ester 50a is enriched after 100 min, while complete racemization is observed within 2 min in the presence of N-methylpyrrolidine. The observed trend suggests that the kinetic basicity of the amine additive is more important to racemization rate than minute differences in thermodynamic basicity. These findings have since guided the design of other novel enantioconvergent addition reactions in our laboratory.
Scheme 15. Influence of Amine Structure on Racemization Rate.
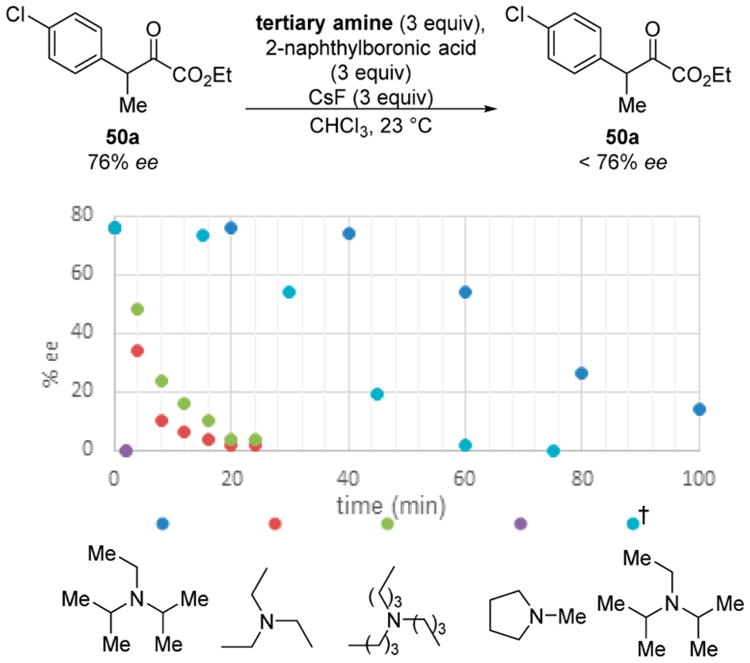
†Trial conducted at 40 °C using 6.0 equiv of Hünig's base.
4. Non-Hydrogenative Enantioconvergent Transformations Of β-Oxo Carboxylic Acid Derivatives
The use of β-oxo carboxylic acid derivatives in dynamic kinetic reactions is attractive, but the markedly diminished carbonyl electrophilicity represents a considerable challenge. This substrate class is routinely employed in dynamic kinetic hydrogenation reactions, but the use of these compounds in non-hydrogenative enantioconvergent reaction manifolds is rare.14b,13h Nevertheless, 1,2-addition products of β-oxocarboxylate derivatives constitute formal aldol (or Mannich) products of carboxylic acid derived nucleophiles; therefore, non-hydrogenative enantioconvergent reactions of such substrates can facilitate access to structures that are difficult to obtain or altogether unknown in enantioenriched form via alternative direct methods.
Initially, we chose to build upon the findings of Rueping and Wulff who disclosed chiral Brønsted acid-catalyzed asymmetric aminoallylations of aldehydes that proceed via a 2-aza-Cope rearrangement (Scheme 16A).28,29 Our attempt to induce rearrangement of the β-formyl ester-derived iminium 55 was unsuccessful due to a strong thermodynamic preference for the inert enamine tautomer 55′. In contrast, β-formyl amide derived 56a underwent a facile 3,3-rearrangement to the desired homoallylamine upon its formation in situ. The manifestation of A1,3 interactions in tautomer 56′ presumably destabilizes this species relative to the analogous ester derivative 55′ resulting in heightened reactivity. Thus, the aminoallylation of β-formylamides 57 in combination with 58 was targeted for the production of homoallyl amines 59. Among the chiral phosphoric acids tested, (S)-TRIP 60 provided optimal levels of enantioselectivity. The reaction tolerated a number of β-formyl amide substrates (Scheme 17). A variety of benzyl-substituted aldehydes could be transformed to the corresponding amine products, including furan- and thiophene-containing 59d and 59e. The alkyl branched products 59f and 59g were formed with similar levels of selectivity. An internally substituted allylamine derivative could be used to generate 59i, albeit with reduced levels of stereocontrol. Finally, in addition to morpholine derived amides, pyrrolidine, piperidine, and dimethylamine derived substrates could also be employed.
Scheme 16. Design of Enantioconvergent 2-Aza-Cope Rearrangement.
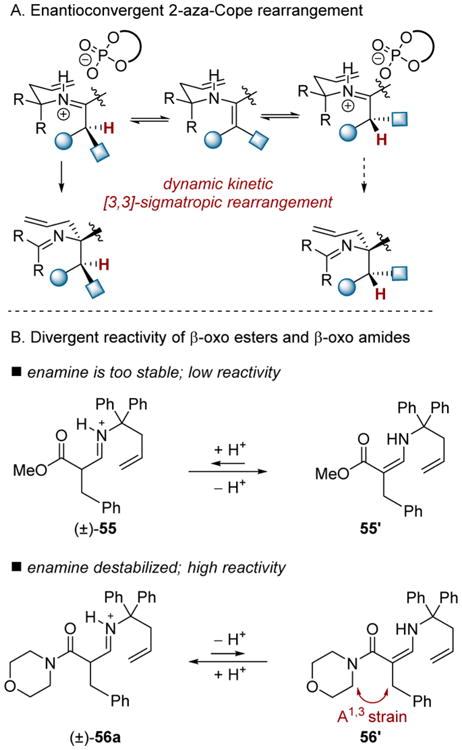
Scheme 17. Scope of 2-Aza-Cope Rearrangement.

5. Summary and Outlook
By capitalizing on the inherent tunability of the basic Ru(II)-sulfonamide framework developed by Noyori, we identified a novel terphenylsulfonamide variant that made possible chemo-and enantioselective reduction of the α-keto ester moiety. This system was applied in the enantioconvergent hydrogenation of a diverse array of α-keto esters as well as related acyl phosphonates. The mechanistic paradigm underlying this chemistry has been extended by us and others for the generation of higher molecular complexity by means of enantioconvergent carbon-centered nucleophile additions to carbonyl derivatives as well as processes that furnish up to three chiral centers in one step. The advancement of this chemistry should occur in tandem with the remarkable advancements that are occurring in the realm of asymmetric 1,2-addition chemistry. The continued exploration of these chemistries in the context of less conventional electrophiles as well as identifying novel or overlooked modes of racemization holds considerable potential for the generation of novel, stereo-chemically complex molecular architectures.
Acknowledgments
We thank the talented co-workers cited herein for their intellectual and experimental contributions to this program in enantioconvergent catalysis. The work described here was supported by Awards R01 GM084927, R01 GM103855, and R35 GM118055 from the National Institute of General Medical Sciences.
Abbreviations
- DKR
dynamic kinetic resolution
- ATH
asymmetric transfer hydrogenation
- DyKAT
dynamic kinetic asymmetric transformation
- Ts
tolylsulfonyl
- DPEN
diphenylethylenediamine
Biographies
Samuel L. Bartlett received his B.S. in chemistry from Oregon State University in 2012 where he conducted research with Prof. Chris Beaudry. He is currently a fifth year Ph.D. candidate in the laboratory of Prof. Jeffrey Johnson. During his doctoral studies, he has also been an NSF EAPSI Fellow with Prof. Mikiko Sodeoka at RIKEN in Japan.
Jeffrey S. Johnson received his B.S. in chemistry from the University of Kansas in 1994 with highest distinction and honors in chemistry. He was a graduate student at Harvard University as an NSF Graduate Fellow in the laboratories of Prof. David A. Evans, receiving his Ph.D. in 1999. He conducted postdoctoral research at the University of California at Berkeley from 1999 to 2001 as an NIH Postdoctoral Fellow under the direction of Prof. Robert G. Bergman. He joined the faculty of the Department of Chemistry at the University of North Carolina at Chapel Hill in 2001 where he is currently the A. Ronald Gallant Distinguished Professor and the department chairperson.
Footnotes
Notes: The authors declare no competing financial interest.
References
- 1.(a) Carreira EM, Kvaerno L. Classics in Stereoselective Synthesis. Wiley-VCH; Weinheim: 2009. [Google Scholar]; (b) Mengel A, Reiser O. Around and Beyond Cram's. Rule Chem Rev. 1999;99:1191–1224. doi: 10.1021/cr980379w. [DOI] [PubMed] [Google Scholar]
- 2.References describing our work on metallophosphite catalysis: Linghu X, Potnick JR, Johnson JS. Metallophosphites as Umpolung Catalysts: The Enantioselective Cross Silyl Benzoin Reaction. J Am Chem Soc. 2004;126:3070–3071. doi: 10.1021/ja0496468.Nahm MR, Potnick JR, White PS, Johnson JS. Metallophosphite-Catalyzed Asymmetric Acylation of α,β-Unsaturated Amides. J Am Chem Soc. 2006;128:2751–2756. doi: 10.1021/ja056018x.Garrett MR, Tarr JC, Johnson JS. Enantioselective Metallophosphite-Catalyzed C-Acylation of Nitrones. J Am Chem Soc. 2007;129:12944–12945. doi: 10.1021/ja076095n..
- 3.Some relevant reviews: Johnson JS. Catalyzed Reactions of Acyl Anion Equivalents. Angew Chem, Int Ed. 2004;43:1326–1328. doi: 10.1002/anie.200301702.Johnson JS. Advances in Acyl Anion and Homoenolate Catalysis. Curr Opin Drug Discovery Dev. 2007;10:691–703.Enders D, Wortmann L, Peters R. Recovery of Carbonyl Compounds from N,N-Dialkylhydrazones. Acc Chem Res. 2000;33:157–169. doi: 10.1021/ar990062y.Enders D, Balensiefer T. Nucleophilic Carbenes in Asymmetric Organocatalysis. Acc Chem Res. 2004;37:534–541. doi: 10.1021/ar030050j.Marion N, Díez-Gonzalez S, Nolan SP. N-Heterocyclic Carbenes as Organo-catalysts. Angew Chem, Int Ed. 2007;46:2988–3000. doi: 10.1002/anie.200603380.Flanigan DM, Romanov-Michailidis F, White NA, Rovis T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem Rev. 2015;115:9307–9387. doi: 10.1021/acs.chemrev.5b00060..
- 4.As an example, during the development of a Stetter reaction of glyoxamides, the Rovis lab noted substantial racemization if a sterically hindered base was not used: Liu Q, Perreault S, Rovis T. Catalytic Asymmetric Intermolecular Stetter Reaction of Glyoxamides with Alkylidenemalonates. J Am Chem Soc. 2008;130:14066–14067. doi: 10.1021/ja805680z..
- 5.Reviews: Bhat V, Welin ER, Guo X, Stoltz BM. Advances in Stereoconvergent Catalysis from 2005 to 2015: Transition-Metal-Mediated Stereoablative Reactions, Dynamic Kinetic Resolutions, and Dynamic Kinetic Asymmetric Transformations. Chem Rev. 2017;117:4528–4561. doi: 10.1021/acs.chemrev.6b00731.Caddick S, Jenkins K. Dynamic Resolutions in Asymmetric Synthesis. Chem Soc Rev. 1996;25:447–456.Huerta FF, Minidis ABE, Backvall JE. Racemization in asymmetric synthesis. Dynamic kinetic resolution and related processes in enzyme and metal catalysis. Chem Soc Rev. 2001;30:321–331.Pellissier H. Dynamic kinetic resolution. Tetrahedron. 2003;59:8291–8327..
- 6.Noyori R, Ikeda T, Ohkuma T, Widhalm M, Kitamura M, Takaya H, Akutagawa S, Sayo N, Saito T, Taketomi T, Kumobayashi H. Stereoselective Hydrogenation via Dynamic Kinetic Resolution. J Am Chem Soc. 1989;111:9134–9135. [Google Scholar]
- 7.Ebbers EJ, Ariaans GJA, Houbiers JPM, Bruggink A, Zwanenburg B. Controlled racemization of optically active organic compounds: Prospects for asymmetric transformation. Tetrahedron. 1997;53:9417–9476. [Google Scholar]
- 8.N-carboxyanhydrides: Hang J, Li H, Deng L. Development of a Rapid, Room-Temperature Dynamic Kinetic Resolution for Efficient Asymmetric Synthesis of α-Aryl Amino Acids. Org Lett. 2002;4:3321–3324. doi: 10.1021/ol026660l..
- 9.Martín-Matute B, Edin M, Bogar K, Kaynak FB, Backvall JE. Combined Ruthenium(II) and Lipase Catalysis for Efficient Dynamic Kinetic Resolution of Secondary Alcohols. Insight into the Racemization Mechanism. J Am Chem Soc. 2005;127:8817–8825. doi: 10.1021/ja051576x. [DOI] [PubMed] [Google Scholar]
- 10.Recent review: Echeverria PG, Ayad T, Phansavath P, Ratovelomanana-Vidal V. Recent Developments in Asymmetric Hydrogenation and Transfer Hydrogenation of Ketones and Imines through Dynamic Kinetic Resolution. Synthesis. 2016;48:2523–2539.. Examples from our research program: Steward KM, Corbett MT, Goodman CG, Johnson JS. Asymmetric Synthesis of Diverse Glycolic Acid Scaffolds via Dynamic Kinetic Resolution of α-Keto Esters. J Am Chem Soc. 2012;134:20197–20206. doi: 10.1021/ja3102709.Corbett MT, Johnson JS. Diametric Stereocontrol in Dynamic Kinetic Reduction of Racemic Acyl Phosphonates: Divergence From α-Keto Ester Congeners. J Am Chem Soc. 2013;135:594–597. doi: 10.1021/ja310980q.Goodman CG, Do D, Johnson JS. Asymmetric Synthesis of Anti-β-Amino-α-Hydroxy Esters via Dynamic Kinetic Resolution of β-Amino-α-Keto Esters. Org Lett. 2013;15:2446–2449. doi: 10.1021/ol4009206.Krabbe SW, Johnson JS. Asymmetric Total Syntheses of Megacerotonic Acid and Shimobashiric Acid A. Org Lett. 2015;17:1188–1191. doi: 10.1021/acs.orglett.5b00140..
- 11.A tandem DKR-ATH/lactonization that furnishes three stereocenters may be between echelon II and III in complexity: Steward KM, Gentry EC, Johnson JS. Dynamic Kinetic Resolution of α-Keto Esters via Asymmetric Transfer Hydrogenation. J Am Chem Soc. 2012;134:7329–7332. doi: 10.1021/ja3027136..
- 12.Walsh PJ, Kozlowski MC. Fundamentals of Asymmetric Catalysis; University Science Books, Sausalito, CA. 2009:272. [Google Scholar]
- 13.Known examples: Calter MA, Phillips RM, Flaschenriem C. Catalytic, Asymmetric, “Interrupted” Feist–Benary Reactions. J Am Chem Soc. 2005;127:14566–14567. doi: 10.1021/ja055752d.Yang J, Wang T, Ding Z, Shen Z, Zhang Y. Highly diastereo- and enantioselective organocatalytic addition of acetone to β-substituted α- ketoesters via dynamic kinetic resolution. Org Biomol Chem. 2009;7:2208–2213. doi: 10.1039/b822127h.Corbett MT, Johnson JS. Dynamic Kinetic Asymmetric Transformations of β-Stereogenic α-Keto Esters by Direct Aldolization. Angew Chem, Int Ed. 2014;53:255–259. doi: 10.1002/anie.201306873.Goodman CG, Johnson JS. Dynamic Kinetic Asymmetric Cross-Benzoin Additions of β-Stereogenic α-Keto Esters. J Am Chem Soc. 2014;136:14698–14701. doi: 10.1021/ja508521a.Wu Z, Li F, Wang J. Intermolecular Dynamic Kinetic Resolution Cooperatively Catalyzed by an N-Heterocyclic Carbene and a Lewis Acid. Angew Chem, Int Ed. 2015;54:1629–1633. doi: 10.1002/anie.201410030.Goodman CG, Johnson JS. Asymmetric Synthesis of β-Amino Amides by Catalytic Enantioconvergent 2-Aza-Cope Rearrangment. J Am Chem Soc. 2015;137:14574–14577. doi: 10.1021/jacs.5b09593.Zhang G, Yang S, Zhang X, Lin Q, Das DK, Liu J, Fang X. Dynamic Kinetic Resolution Enabled by Intramolecular Benzoin Reaction: Synthetic Application and Mechanistic Insights. J Am Chem Soc. 2016;138:7932–7938. doi: 10.1021/jacs.6b02929.Bartlett SL, Keiter KM, Johnson JS. Synthesis of Complex Tertiary Glycolates by Enantioconvergent Arylation of Stereochemically Labile α-Keto Esters. J Am Chem Soc. 2017;139:3911–3916. doi: 10.1021/jacs.7b00943..
- 14.Known examples: Ward DE, Jheengut V, Akinnusi OT. Enantioselective Direct Intramolecular Aldol Reactions With Enantio-topic Group Selectivity and Dynamic Kinetic Resolution. Org Lett. 2005;7:1181–1184. doi: 10.1021/ol050195l.Cohen DT, Eichman CC, Phillips EM, Zarefsky ER, Scheidt KA. Catalytic Dynamic Kinetic Resolutions with N-Heterocyclic Carbenes: Asymmetric Synthesis of Highly Substituted β-Lactones. Angew Chem, Int Ed. 2012;51:7309–7313. doi: 10.1002/anie.201203382.Bergeron-Brlek M, Teoh T, Britton RA. Tandem Organocatalytic α-Chlorination-Aldol Reaction That Proceeds with Dynamic Kinetic Resolution: A Powerful Tool for Carbohydrate Synthesis. Org Lett. 2013;15:3554–3557. doi: 10.1021/ol401370b.Goodman CG, Walker MM, Johnson JS. Enantioconvergent Synthesis of Functionalized γ-Butyrolactones via (3 + 2)-Annulation. J Am Chem Soc. 2015;137:122–125. doi: 10.1021/ja511701j..
- 15.(a) Steward KM, Johnson JS. Catalytic Nucleophilic Glyoxylation of Aldehydes. Org Lett. 2010;12:2864–2867. doi: 10.1021/ol100996w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boyce GR, Greszler SN, Johnson JS, Linghu X, Malinowski JT, Nicewicz DA, Satterfield AD, Schmitt DC, Steward KM. Silyl Glyoxylates. Conception and Realization of Flexible Conjunctive Reagents for Multicomponent Coupling. J Org Chem. 2012;77:4503–4515. doi: 10.1021/jo300184h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Ikariya T, Blacker AJ. Asymmetric Transfer Hydrogenation of Ketones with Bifunctional Transition Metal-Based Molecular Catalysts. Acc Chem Res. 2007;40:1300–1308. doi: 10.1021/ar700134q. [DOI] [PubMed] [Google Scholar]; (b) Noyori R, Hashiguchi S. Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes. Acc Chem Res. 1997;30:97–102. [Google Scholar]
- 17.Trost BM. Atom Economy-A Challenge for Organic Synthesis: Homogenous Catalysis Leads the Way. Angew Chem, Int Ed Engl. 1995;34:259–281. [Google Scholar]
- 18.α,α-Dinaphthylethylenediamine: Denmark SE, Su X, Nishigaichi Y, Coe DM, Wong KT, Winter SBD, Choi JY. Synthesis of Phosphoramides for the Lewis Base-Catalyzed Allylation and Aldol Addition Reactions. J Org Chem. 1999;64:1958–1967. doi: 10.1021/jo9820723.. m-Terphenylsulfonamide: Basak AK, Shimada N, Bow WF, Vicic DA, Tius MA. An Organocatalytic Asymmetric Nazarov Cyclization. J Am Chem Soc. 2010;132:8266–8267. doi: 10.1021/ja103028r.Kim H, Nguyen Y, Yen CPH, Chagal L, Lough AJ, Kim BM, Chin J. Stereospecific Synthesis of C2 Symmetric Diamines from the Mother Diamine by Resonance-Assisted Hydrogen-Bond Directed Diaza-Cope Rearrangement. J Am Chem Soc. 2008;130:12184–12191. doi: 10.1021/ja803951u.. More recent examples: Lu Z, Wilsily A, Fu GC. Stereoconvergent Amine-Directed Alkyl–Alkyl Suzuki Reactions of Unactivated Secondary Alkyl Chlorides. J Am Chem Soc. 2011;133:8154–8157. doi: 10.1021/ja203560q.Ahlin JSE, Cramer N. Chiral N-Heterocyclic Carbene Ligand Enabled Nickel(0)-Catalyzed Enantioselective Three-Component Couplings as Direct Access to Silylated Indanols. Org Lett. 2016;18:3242–3245. doi: 10.1021/acs.orglett.6b01492..
- 19.Kosugi Y, Akakura M, Ishihara K. Kinetic resolution of alcohols catalyzed by minimal artificial acylases derived from L-histidine. Tetrahedron. 2007;63:6191–6203. [Google Scholar]
- 20.Oxazolones have been used as masked hydroxy acid derivatives: Misaki T, Kawano K, Sugimura T. Highly Z-Selective Asymmetric 1,4-Addition Reaction of 5H-Oxazol-4-ones with Alkynyl Carbonyl Compounds Catalyzed by Chiral Guanidines. J Am Chem Soc. 2011;133:5695–5697. doi: 10.1021/ja200283n.Aleman J, Milelli A, Cabrera S, Reyes E, Jørgensen KA. Asymmetric 1,4-Addition of Oxazolones to Nitroalkenes by Bifunctional Cinchona Alkaloid Thiourea Organo-catalysts: Synthesis of α,α-Disubstituted α-Amino Acids. Chem - Eur J. 2008;14:10958–10966. doi: 10.1002/chem.200802030.Trost BM, Hirano K. Highly Stereoselective Synthesis of α-Alkyl-α-Hydroxycarboxylic Acid Derivatives Catalyzed by a Dinuclear Zinc Complex. Angew Chem, Int Ed. 2012;51:6480–6483. doi: 10.1002/anie.201201116.Hynes PS, Stranges D, Stupple PA, Guarna A, Dixon DJ. Organocatalytic Diastereo- and Enantioselective Michael Addition Reactions of 5-Aryl-1,3-Dioxolan-4-ones. Org Lett. 2007;9:2107–2110. doi: 10.1021/ol070532l..
- 21.Relevant examples: Evans DA, Johnson JS. Catalytic Enantioselective Hetero Diels-Alder Reactions of α,β-Unsaturated Acylphosphonates with Enol Ethers. J Am Chem Soc. 1998;120:4895–4896.Evans DA, Johnson JS, Olhava EJ. Enantioselective Synthesis of Dihydropyrans. Catalysis of Hetero Diels-Alder Reactions by Bis(oxazoline) Copper(II) Complexes. J Am Chem Soc. 2000;122:1635–1649..
- 22.The triazolium catalyzed cross-benzoin reaction of aryl and alkyl aldehydes has been achieved with low enantiocontrol: Langdon SM, Wilde MMD, Thai K, Gravel M. Chemoselective N-Heterocyclic Carbene-Catalyzed Cross-Benzoin Reactions: Importance of the Fused Ring in Triazolium Salts. J Am Chem Soc. 2014;136:7539–7542. doi: 10.1021/ja501772m.. Also, by use of chiral aldehyde derived Breslow intermediates: Haghshenas P, Gravel M. Chemo- and Diastereoselective N-Heterocyclic Carbene-Catalyzed Cross-Benzoin Reactions Using N-Boc-α-amino Aldehydes. Org Lett. 2016;18:4518–4521. doi: 10.1021/acs.orglett.6b02123..
- 23.Early references: Cherest M, Felkin H, Prudent N. Torsional strain involving partial bonds. The stereochemistry of lithium aluminum hydride reduction of some simple open-chain ketones. Tetrahedron Lett. 1968;9:2199–2204.Anh NT, Eisenstein O. Tetrahedron Lett. 1976;17:155–158..
- 24.Cornforth JW, Cornforth RH, Mathew KK. A general stereoselective synthesis of olefins. J Chem Soc. 1959:112–117. [Google Scholar]
- 25.Yang W, Yang Y, Du DM. Squaramide–Tertiary Amine Catalyzed Asymmetric Cascade Sulfa-Michael /Michael Addition via Dynamic Kinetic Resolution: Access to Highly Functionalized Chromans with Three Contiguous Stereocenters. Org Lett. 2013;15:1190–1193. doi: 10.1021/ol400025a. [DOI] [PubMed] [Google Scholar]
- 26.Representative examples: Duan HF, Xie JH, Qiao XC, Wang LX, Zhou QL. Enantioselective Rhodium-Catalyzed Addition of Arylboronic Acids to α-Keto Esters. Angew Chem, Int Ed. 2008;47:4351–4353. doi: 10.1002/anie.200800423.Cai F, Pu X, Qi X, Lynch V, Radha A, Ready JM. Chiral Allene-Containing Phosphines in Asymmetric Catalysis. J Am Chem Soc. 2011;133:18066–18069. doi: 10.1021/ja207748r..
- 27.Relevant references include the following: Vitaku E, Smith DT, Njardarson JT. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J Med Chem. 2014;57:10257–10274. doi: 10.1021/jm501100b.Brase S. Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, and Evaluation. The Royal Society of Chemistry; London, U.K.: 2016. .
- 28.Rueping M, Antonchick AP. Catalytic Asymmetric Aminoallylation of Aldehydes: A Catalytic Enantioselective Aza-Cope Rearrangement. Angew Chem, Int Ed. 2008;47:10090–10093. doi: 10.1002/anie.200803610. [DOI] [PubMed] [Google Scholar]
- 29.Ren H, Wulff WD. Direct Catalytic Asymmetric Amino-allylation of Aldehydes: Synergism of Chiral and Nonchiral Brønsted Acids. J Am Chem Soc. 2011;133:5656–5659. doi: 10.1021/ja1110865. [DOI] [PubMed] [Google Scholar]