

Abstract
CISD2 is a redox-sensitive gene critical for normal development and mitochondrial integrity. CISD2 was known to have aberrant expression in several types of human cancers. However, its relation with lung cancer is still not clear. In this study we found CISD2 mRNA was significantly upregulated in lung adenocarcinoma (ADC) samples, compared with their adjacent normal counterparts, and was correlated with tumor stage, grade, and prognosis based on analysis of clinical specimens-derived expression data in public domain and our validation assay. Cell based assay indicated that CISD2 expression regulated accumulation of reactive oxygen species (ROS), polarization of mitochondrial membrane potential, as well as cell viability, apoptosis, invasiveness, and tumorigenicity. In addition, CISD2 expression was found significantly correlated with stress response/redox signaling genes such as EGR1 and GPX3, while such correlations were also found valid in many public domain data. Taken together, upregulation of CISD2 is involved in an increased antioxidant capacity in response to elevated ROS levels during the formation and progression of lung ADC. The molecular mechanism underlying how CISD2 regulates ROS homeostasis and augments malignancy of lung cancer warrants further investigations.
Introduction
Lung cancers are a major cause of cancer-related deaths globally, mostly attributable to late diagnosis and lack of efficient treatment1. Given such a high mortality, it is critical that we improve our understanding of how lung cancers arise and progress so that cancer prevention strategies can be implemented or biomarkers and therapeutic targets can be developed to improve early detection and treatment.
The most well-known risk factor for lung cancer is cigarette smoking2,3. Many components of cigarette smoke induce oxidative stress by transmitting or generating reactive oxygen species (ROS)4. ROS are also produced endogenously in cells, where mitochondria have been conventionally recognized as one of the sources of intracellular ROS5. Additionally, ROS are generated in response to external stimuli, such as inflammatory cytokines, chemotherapeutic drugs, and ionizing radiation. Having been shown to be genotoxic, ROS also functions as signaling molecules6. ROS are known to play a pivotal role in carcinogenesis by promoting proliferation, invasiveness, and metastasis, and by inhibiting apoptosis. Alternatively, they can also play anticarcinogenic roles, e.g., by inducing cell cycle arrest, apoptosis, and necrosis7,8. As with all other types of human cancer, the role of ROS/oxidative stress in lung cancer seems important but complicated.
In an initiative to scrutinize the roles of oxidative stress-related genes during tumorigenesis or progression of lung cancer, we noticed the upregulation of CISD2 (CDGSH iron sulfur domain 2) in lung adenocarcinoma (ADC). CISD2, also known as NAF-1, ZCD2 or Miner1, belongs to the CDGSH iron sulfur domain protein family. The CISD2 gene is located on chromosome 4q; it encodes a protein contains a CDGSH domain, a transmembrane domain, and a conserved amino acid sequence for iron binding9,10. CISD2 is an essential protein in development. Its dysfunction in humans is known to cause Wolfram syndrome type 2 and CISD2 knockout mice show a phenotype of premature aging11. CISD2 is localized mainly in mitochondria and partially in the endoplasmic reticulum (ER), and has been functionally linked to maintaining mitochondrial integrity and autophagy12,13. Although CISD2 activity is critically required for normal development, overexpression of CISD2 has been linked to several human cancers, including breast cancer13,14, cervical cancer15, gastric cancer16, and laryngeal squamous carcinoma (SQC)17, indicating it plays an oncogenic role. Despite these findings, it is still unclear whether CISD2 is associated with lung cancer.
In the present study, we aimed to understand whether CISD2 expression is associated with the formation of lung ADC, and with the prognosis for patients with this cancer type. We provide several lines of evidence to show that overexpression of CISD2 is oncogenic to lung cancer. The antioxidant but oncogenic roles of CISD2 in lung cancer are discussed.
Results
CISD2 expression is upregulated in lung ADC and linked to poor prognosis
To determine whether CISD2 is aberrantly expressed in human lung ADC and if its expression has any clinical relevance, we first analyzed CISD2 expression profiles in public lung ADC datasets. We analyzed three independent datasets (GSE3121018, GSE2726219, and GSE1918820), which comprise a total of 406 cases, and found repeatedly elevated CISD2 mRNA expression in lung ADC tissue samples compared with their adjacent normal counterparts (Figs 1A and S1A). This upregulation of CISD2 mRNA was also found in our in-house-generated dataset GSE46539 (Supplementary Fig. S1B), and can be confirmed by an independent RT-qPCR assay using our previously collected lung ADC samples (c.f. Materials and Methods) (Fig. 1B). We also examined CISD2 protein expression by Immunohistochemistry (IHC) assay (Fig. 1C) using commercial lung cancer tissue microarrays. The results showed that the CISD2 protein was also significantly upregulated in lung ADC tissues (Fig. 1D). Together, these results suggest that CISD2 expression is increased during the formation of lung ADC.
Figure 1.

CISD2 expression is upregulated in lung ADC and associated with poor prognosis. (A) Box plot showing upregulation of CISD2 mRNA expression levels in lung ADC tumor tissues compared with those in normal lung tissues, based on public domain data GSE31210. Data were downloaded from Oncomine and analyzed directly without further processing. (B) RT-qPCR assay of CISD2 mRNA expression of samples from our previously used cohort of 56 specimens containing 28 lung ADC and 28 normal lung tissues (not matched pair). (C) Representative images of IHC staining of CISD2 protein performed on normal and lung ADC tissues. Scale bar: 50 μm. (D) A summary of IHC analysis of CISD2 protein expression in lung ADC and normal lung tissues in commercial tissue microarrays (c.f. Materials and Methods). (E and F) Survival analysis on CISD2 mRNA expression and recurrence-free survival using public domain data. In each dataset, patients were stratified into low CISD2 (solid line) and high CISD2 (dashed line) groups using median CISD2 expression level as cutoff; significance of difference in survival between groups was estimated using log rank test. For (A), (B) and (D) p values were obtained using Student’s t-test, ***P < 0.001. Detailed information about public domain datasets is available in Supplementary Table S1.
We next examined the associations of CISD2 expression with other clinicopathological features, and found that CISD2 mRNA expression was significantly correlated with tumor stage, grade of differentiation, or smoking status in several datasets (Supplementary Fig. S1C and D), suggesting that the transcript level of CISD2 might also be clinically relevant to cancer progression. By performing further survival analysis using Cox’s regression model, we found that CISD2 expression was significantly associated with prognosis of patients with lung ADC in two independent datasets. In dataset GSE8894, the CISD2 mRNA expression level was significantly associated with recurrence-free survival (hazard ratio (HR): 2.10; 95% confidence interval (CI) of the HR: 1.21–3.64; P = 0.009). In dataset GSE31210, CISD2 expression level was significantly associated with either recurrence-free survival (HR: 1.64, 95% CI: 1.11–2.42; P = 0.013) or overall survival (HR: 2.10, 95% CI: 1.24–3.57; P = 0.006). For each of the above datasets, when patients were stratified into two groups using the median CISD2 mRNA expression level as a threshold, those with higher CISD2 expression levels had significantly shorter recurrence-free survival than those with lower CISD2 expression levels (Fig. 1E and F, P = 0.007 and P = 0.009, respectively). These data further indicate that CISD2 might play a role in the progression of lung ADC.
CISD2 expression positively affects cell proliferation and tumorigenicity
We then utilized in vitro cancer cell line model, in which the expression level of CISD2 was interfered by using siRNA or shRNA techniques, to observe their effects on cancer associated phenotypes. When CISD2 was transiently knocked down in the CISD2-abundant cell line A549 or H1299 (Supplementary Fig. S2), a significant reduction in colony formation capability, as measured by clonogenic assay, was observed (Figs 2A and S3). In addition, using CL1–1, a cell line expressing relatively low level of CISD2 (Supplementary Fig. S2), we also generated a stable transfectant overexpressing CISD2, which was named CISD2(+)-CL1-1. Consistently, the viability of CISD2(+)-CL1-1 was increased compared with the vector control clone when measured by MTT assay (Fig. 2B). These results suggest that CISD2 expression is in general advantageous to the proliferation/viability or survival of lung ADC cells.
Figure 2.

CISD2 expression affects viability of lung ADC cells. (A) Left panel, representative images of clonogenic assay of siCISD2 transfected A549 cells; right panel, result of quantification of corresponding images as in the top panel. (B) Viability assay of CISD2(+)-CL1-1 cells and its vector control clone. (C and D) Western blot analysis of the cell cycle regulator P21 expression in indicated cells. (E) GSEA analysis of CISD2 silencing or overexpression experiments in lung ADC cell lines. For (A) and (B) p values were obtained using Student’s t-test, *P < 0.05, **P < 0.01. All data are mean ± SD of at least triplicate measurements.
Given that CISD2 expression might affect cancer cell proliferation, we tested whether it would influence tumor growth phenotypes in vivo. Compared with experiments using nontargeting control cells, when CISD2-silencing cells were injected subcutaneously into mice (n = 5 for each group), a significant retardation in the growth of xenograft tumors (Supplementary Fig. S4A and B) was observed, as was a reduction in either their mass or volume (Supplementary Fig. S4C). Due to large variation no statistical significance was established in our study, additional in vivo studies will be needed in order to establish the role of CISD2 in tumor development in vivo. Of interest, we noted that p21 (CDKN1A), an important cell cycle regulator, was significantly upregulated in two CISD2 knockdown clones (Figs 2C and S5) and significantly downregulated in CISD2-overexpressing CL1-1 cells (Figs 2D and S6), implying that those CISD2-associated proliferation phenotypes might involve regulation of cell cycle progression.
We also applied bioinformatic approaches to analyze gene sets/pathways affected by forced expression or silencing of CISD2. We compared the differential gene expression profiles of cells having overexpressed or silenced CISD2, versus their own vector control cells. When these differential expression profiles were analyzed using Gene Set Enrichment Analysis (GSEA), many pathways/gene sets related to the abovementioned cellular phenotypes (Fig. 2E)—including cellular respiration, cellular responses to stimulus, anti-apoptosis, response to oxidative stress, mitochondrion organization, and biogenesis—were significantly enriched in the differential expression profiles. These consistent findings consolidate the notion that CISD2 expression in cancer cells is associated with changes in mitochondrial function and cellular oxidative stress.
CISD2 expression regulates cell apoptosis
Since our aforementioned gene expression microarray analysis indicates that CISD2 expression could affect apoptosis (Fig. 2E), we first performed annexin V/PI double staining assays. The results indicated that knocking down CISD2 expression in lung ADC cells caused a significant increase in the apoptotic fraction (annexin V-high/PI-low; Fig. 3A). Accompanying this increase, an obvious elevation in the expression of molecular markers of apoptosis, such as cytochrome c, the cleaved form of caspase 3, and PARP1, were also detected (Figs 3B and S7) in CISD2-silenced cells compared with transfectants of nonsilencing shRNA. These data indicate that CISD2 silencing is cytotoxic, which can, in turn, induce cell apoptosis. Noticeably, in the presence of the anticancer drug cisplatin, there was a doubling in the proportion of apoptotic cells in CISD2-depleted cells compared with the control experiment in which cisplatin was absent and no increase in the apoptotic fraction was observed (Fig. 3C, 48 h). This indicated that knocking down CISD2 expression can enhance or sensitize lung ADC cells to the cytotoxic effects of cisplatin.
Figure 3.

CISD2 expression affects cell apoptosis. (A) Cell apoptosis assay using double staining technique. Cells were seeded overnight, and then labeled with annexin V and PI for flow cytometry analysis. (B) Western blot analysis of cell apoptosis markers in shCISD2 transfectant cells and their vector control cells. (C) Cell apoptosis in the absence or presence of cisplatin. Cells were seeded into six-well plates and incubated overnight, then cultured in serum-free media with or without cisplatin (30 μM). Cells were harvested at indicated time interval, labeled with annexin V and PI for flow cytometry assay. Data are mean ± SD of triplicate measurements. **P < 0.01, Student’s t-test.
CISD2 expression is involved in the regulation cell invasiveness
Our finding that CISD2 expression levels is associated with patient survival implies that CISD2 could be involved in behavioral changes of cancer cells linked with cancer progression. To test this hypothesis, we conducted Transwell invasion assays to measure whether cell invasion would be affected by CISD2 expression. Interestingly, CISD2-silenced H1299 cells exhibited a remarkable decrease in the number of cells that invaded (Fig. 4A), whereas forced expression of CISD2 in CL1-1 cells resulted in a significant increase (Fig. 4B), strongly suggesting that CISD2 expression might promote the invasiveness of lung ADC cells. To explore whether this could involve epithelial-to-mesenchymal transition (EMT), a phenomenon frequently linked to alterations in cell invasiveness, we further examined the expression of EMT markers using RT-qPCR and western blot analyses. In general, in CISD2-silenced H1299 cells, the expression levels of epithelial markers such as E-cadherin (CDH1) and desmoplakin (DSP) were upregulated, whereas mesenchymal markers such as vimentin (VIM) and zinc finger protein SNAI2 were downregulated (Figs 4C,D and S8). By contrast, in CISD2-overexpressing cells, epithelial markers such as tight junction protein (TJP)-1 (also known as zona occludens 1 or ZO1) were downregulated, whereas mesenchymal markers such as VIM, CDH2, and SNAI2 were significantly upregulated (Figs 4E,F and S9). These data indicate that EMT might be involved in the CISD2-induced increase in cell invasion potential.
Figure 4.

CISD2 promotes cell invasion in lung ADC cells. (A and B) Left panel, representative images of cell invasion assay showing invading cells on the bottom side of membrane of transwells; right panel, result of quantification analysis. (C) RT-qPCR analyses of the transcript levels of EMT markers in shCISD2 transfectant cells. (D) Western blot analysis of EMT marker proteins in shCISD2 transfectant cells. (E) RT-qPCR analyses of the transcript levels of EMT markers in CISD2(+)-CL1-1 cell. (F) Western blot analysis of EMT markers in CISD2(+)-CL1-1 cells. For (A), (B), (C) and (E), p values were obtained using Student’s t-test, *P < 0.05, **P < 0.01. All data are mean ± SD of at least triplicate measurements.
CISD2 contributes to the maintenance of mitochondrial function and the suppression of ROS production in lung cancer cells
Previous studies suggested that CISD2 plays a role in maintaining mitochondrial integrity and respiration capacity11,13. Our GSEA analysis of microarray data also identified involvement of mitochondrial, oxidative stress, and ROS signaling gene sets or pathways in CISD2-silencing or CISD2-overexpressing cell lines (Fig. 2E). We therefore hypothesized that the oncogenic features of CISD2 involve its roles in the maintenance of mitochondrial function in lung ADC cells. To test this, we utilized flow cytometry to detect whether mitochondrial membrane potential was affected in CISD2 silencing context. As shown in Fig. 5A and B, a 34% ± 0.7% increase in the fluorescence intensity of the cationic dye JC-1 was observed in CISD2 silencing conditions, indicating a significant drop in the mitochondrial membrane potential. By contrast, a 31% ± 4.2% decrease in JC-1 fluorescence intensity was found in CISD2 forced expression conditions (Fig. 5B). These data strengthen our conjecture that CISD2 might play a critical role in mitochondrial homeostasis in lung ADC cells.
Figure 5.

Silencing of CISD2 causes dysfunction of mitochondria in lung ADC cells. (A) Flow cytometry assay for detection of change in mitochondrial membrane potential using JC-1 dye. (B) JC-1 green fluorescence quantification analyses of experiments of silencing or overexpressing CISD2 in lung ADC cell lines. (C and D) Measurements of ROS levels using DCFH-DA and DHE dyes. CISD2(−)#1-H1299 cells were seeded overnight, refreshed in serum free medium in the absence (left panels) or presences of indicated amount of H2O2 (middle panels) for 20 m, then subjected to flow cytometry assay. Right panels, results of quantification. For (B), (C) and (D), data are mean ± SD of triplicate measurements. *P < 0.05, **P < 0.01, Student’s t-test.
Because mitochondria are one of the major sources of ROS in cells, it is possible that imbalanced homeostasis or damage of mitochondria might result in the release of ROS, leading to oxidative damage and impairing the survival of cancer cells. Therefore, it would be of interest to know whether targeting CISD2 expression could induce the generation of ROS. To address this, we used the redox-sensitive dyes 2′,7′-dichlorofluorescein diacetate (DCFH-DA) and dihydroethidine (DHE) to measure the levels of intracellular ROS and superoxide radicals, respectively. We found that the fluorescence intensities of both DCF and DHE were significantly increased in CISD2-silenced H1299 cells CISD2(−)#1, compared with control cells (left panels of Fig. 5C and D), indicating that CISD2 depletion leads to the accumulation of ROS and superoxide radicals in cells. Markedly, such accumulation became even more obvious in the additional presence of hydrogen peroxide (right panels of Fig. 5C and D), suggesting that under stress conditions, CISD2 is required to an even greater degree for maintenance of ROS homeostasis in lung ADC cells. These consistent findings consolidate the notion that CISD2 expression in lung ADC is associated with changes in mitochondrial function and cellular oxidative stress.
Downregulation of CISD2 induces expression of antioxidant GPX3
In response to increases in ROS accumulation upon perturbation caused by CISD2 depletion, the detection of antioxidant or stress response gene expression would be expected. In this regard, we noted that some genes encoding antioxidant enzymes such as SOD2, GPX1, GPX3, GPX5, and GPX6 showed markedly elevated expression levels when CISD2 was silenced in H1299 cells, as evidenced by RT-qPCR profiling (Fig. 6A and B). Among those genes, GPX3, which encodes a glutathione peroxidase, an antioxidant enzyme and putative tumor suppressor that defends cells against ROS, was the most significantly upregulated. This suggests that knocking down CISD2 might cause upregulation of GPX3 and further implies that CISD2 expression may inhibit GPX3 expression. In support of this, from analyses of public data derived from clinical lung ADC samples, we found that not only was GPX3 expression significantly downregulated in several independent datasets (Figs 6C and S10A), but also there was a significant negative correlation between the mRNA levels of CISD2 and GPX3 in related datasets (Figs 6D and S10B). Furthermore, since CISD2 was shown in a previous session to be positively associated with poor prognosis, it is of interest to find that GPX3 expression exhibited a significantly negative correlation with poor prognosis in two independent datasets (Fig. 6E). Jointly, these in vitro and clinical samples-based data suggest that upregulation of CISD2 might contribute to carcinogenesis and/or cancer progression by regulating ROS homeostasis, and, therefore, offer cells a survival advantage by protecting them from raised oxidative stress, which in turn prevents the production of antioxidant genes, such as GPX3.
Figure 6.

CISD2 negatively regulates mRNA expression of antioxidant gene GPX3 and ROS-mediated EGR1 signaling. (A and B) RT-qPCR analysis of expression levels of selected antioxidant genes using shCISD2 transfectant CISD2(−)#1-H1299 (A) or siCISD2 transfected cells harvested at 48 h post transfection (B). (C) GPX3 mRNA expression profile of lung ADC versus normal lung tissues extracted from public domain dataset GSE31210. (D) Scatter plot showing significantly negative correlation between mRNA expression levels of CISD2 and GPX3 in public dataset GSE31210. Correlation test was used to estimate the significance of correlation. (E) Cox’s regression analyses (age adjusted) of GPX3 mRNA expression and prognostic outcome in patients with lung ADC. PFS, progression-free survival; OS, overall survival. (F) EGR1 mRNA expression profile of lung ADC versus normal lung tissues extracted from public domain dataset GSE19188. (G) Scatter plots showing significant negative correlation between mRNA expression levels of CISD2 and EGR1 in public dataset GSE19188. (H) Survival analysis on EGR1 mRNA expression and recurrence-free survival using public domain data GSE31210. Patients were stratified into low EGR1 (solid line) and high EGR1 (dashed line) groups using median EGR1 expression level as cutoff; significance of difference in survival between groups was estimated using log rank test. (I) EGR1 mRNA expression in siCISD2 transfected lung ADC cell lines as measured by RT-qPCR assay. Cells were harvested at 48 h post transfection. (J) Western blot of CISD2 and EGR1 proteins of cells used in (I). (K) Western blot analysis of CISD2 silencing-induced upregulation of EGR1 in CL1-5 cells was reduced when cells were pretreated with 1 mM of NAC for 1 h before siCISD2 transfection. (L and M) Western blotting (L) and RT-qPCR (M) analyses of CISD2 and EGR1 expression in H1299, CL1-0 and CL1-5 cells treated with 100 μM H2O2 for 1 h. (N) Western blotting of putative targets downstream EGR1 signaling in CISD2 silencing clones. For (A), (B) (C), (F) and (I), p values were obtained using Student’s t-test, *P < 0.05, **P < 0.01, ***P < 0.001. All data are mean ± SD of at least triplicate measurements.
CISD2 silencing-induced EGR1 expression is mediated via ROS and affects AKT signaling
By the same token as in the case of CISD2-GPX3 axis, we hypothesized that upregulation of CISD2 expression in lung ADC cells might prevent the expression of tumor suppressors associated with ROS/oxidative stress response. To test this possibility, we examined the correlation between CISD2 expression and known stress response genes. Among others, we found that EGR1 expression is not only downregulated in many lung ADC datasets (Figs 6F and S10C), but also significantly negatively correlated with CISD2 expression in multiple datasets (Figs 6G and S10D). Again, since that CISD2 expression is associated with poor prognosis and that CISD2 and EGR1 mRNA levels are negatively correlated, we anticipated that EGR1 might be a favorable prognostic factor in the context of lung ADC. As expected, we found that EGR1 expression was positively correlated with either recurrence-free patient survival (Fig. 6H) or overall survival (Supplementary Fig. S10E).
We resorted to cell-based assays again to find possible supporting evidence. Consistent with these clinical sample-based observations, we found that EGR1 expression can be transcriptionally affected by CISD2 expression, as CISD2 silencing triggered EGR1 mRNA (Fig. 6I), no matter EGR1 protein was detectable (as for H1299 and CL1-5 cell lines, Figs 6J and S11) or not (as for A549 cell line, Figs 6J and S11), indicating that CISD2 could repress EGR1 expression. We noted that EGR1 protein expression was undetectable in A549 cells is also consistent to an earlier report by Zhang et al.21; this suggests that in addition to regulation by CISD2, there might exist other unidentified pathway(s) involved in the suppression of EGR1 protein expression. However, when EGR1 protein expression was detectable, silencing of CISD2 expression can also cause a moderate to strong upregulation of EGR1 protein expression (Figs 6J and S11).
To test our conjecture that the negatively correlated expression levels between CISD2 and EGR1 might be mediated by ROS, we performed two experiments. In the first, we examined whether the upregulation of EGR1 upon CISD2 silencing could be attenuated in the presence of a ROS scavenger. As shown in Figs 6K and S12, in the presence of N-acetyl-l-cysteine (NAC), there was no increase in EGR1 protein level upon silencing of CISD2, confirming that the upregulation of EGR1 expression requires ROS. In the second, we explored whether EGR1 or CISD2 expression level would be affected by treatment with hydrogen peroxide in lung ADC cells. As expected, the expression of EGR1 was upregulated significantly (Figs 6L and S13). Intriguingly, levels of the CISD2 protein (Figs 6L and S13) or its mRNA (Fig. 6M) were both downregulated and also seemed inversely correlated with those of EGR1 among the three cell lines used, again confirming that ROS mediates CISD2–EGR1 signaling.
As EGR1 is responsive to ROS-mediated signaling from CISD2, we explored whether the downstream signaling of EGR1 might be affected by CISD2 expression. Interestingly, the protein expression of phosphatase and tensin homolog (PTEN), reported as a downstream factor of EGR122,23, was significantly enhanced in lung ADC cells (Figs 6N and S14). Closely associated with and perhaps attributed to the increase in PTEN, the level of phosphorylation of AKT at serine residue 473 (S473) was significantly attenuated (Figs 6N and S14) as expected. Collectively, these results imply that CISD2 may function as an oncogene by regulating ROS homeostasis and EGR1 induction, which further dysregulate PTEN/AKT signaling in lung ADC cells.
Discussion
In this study, using a combined approach relating evidence from analysis of public gene expression data derived from clinical specimens of lung cancer tissues, cell-based assays, and animal xenograft models, we have identified the oncogenic properties of CISD2 and its clinical significance in lung ADC. To the best of our knowledge, this is the first report to link CISD2 with lung cancer. In addition to establishing an association of important clinical relevance, we also provide some possible molecular explanations of how the upregulation of CISD2 expression could foster cancer cell formation and progression. We propose a model of CISD2–ROS–EGR1/GPX3 axis signaling (Fig. 7), in which increased levels of CISD2 contribute to the neutralization of excessive ROS production, which would otherwise increase antitumor activity mediated at least by the tumor suppressors EGR1 and GPX3. The oncogene nature of CISD2 revealed here could be analogous to that of the NRF2–KEAP1 system in lung cancer, where NRF2, a well-known stress response gene hijacked by cancer cells24, was found to be significantly associated with prognosis25.
Figure 7.
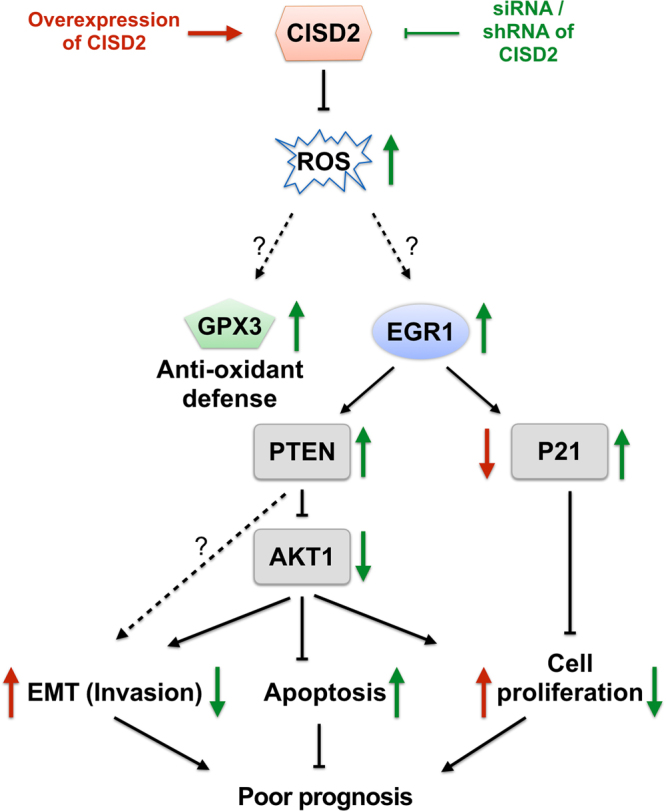
A proposed model relating CISD2, GPX3 and EGR1 signaling in lung ADC cells to poor prognosis of lung ADC patients. Texts/arrows in red represent observations from cell based assays under CISD2 overexpressed condition; texts/arrows in green represent those under CISD2 silencing condition. Arrows pointing upward, upregulation; arrows pointing downward, downregulation.
CISD2 belongs to the CDGH iron sulfur domain-containing protein family, which contains a 2Fe-2S cluster known to function as an active redox-sensitive center26,27. Other proteins that contain a 2Fe-2S cluster are also known to have important roles in redox transition26. Given that CISD2 is a redox-sensitive protein localized in mitochondria11,28,29 and the ER12,30, which are two major cellular sources of ROS, and that depletion of CISD2 in lung cancer cells can lead to the accumulation of ROS as shown in our data, it is highly plausible that CISD2 is an antioxidant protein capable of reducing ROS to maintain redox homeostasis in cancer cells. Therefore, a reasonable scenario emerges: during the process of cancer formation or progression, the upregulation of CISD2 offers cancer cells critical survival and proliferation signals by providing them with greater antioxidant capacity to counteract gradually elevated ROS levels. In such a scenario, the expression level of CISD2 could be a marker reflecting the levels of accumulated oxidative stress in cancer cells. This could explain why the CISD2 level is significantly higher in lung ADC patients who are smokers than in those who are nonsmokers (Supplementary Fig. S1D), reflecting exposure to ROS generated exogenously. It could also explain why the CISD2 level is positively associated with the clinical stage and grade of differentiation (Supplementary Fig. S1D), reflecting an elevated overall ROS level during cancer progression. In the emerging milieu, where targeting the antioxidant capacity of tumor cells could have a positive therapeutic impact31, further investigations are needed into the biochemical properties of CISD2, its interacting partners, and how it is regulated in cancer cells.
Our finding that CISD2 silencing-induced ROS can promote the expression of putative tumor suppressor EGR1 is also novel to lung cancer biology. EGR1 is known as a stress-response gene that plays a protective role when cells suffer starvation, ultraviolet light irradiation, hypoxia, and oxidative stress. It functions as a tumor suppressor gene in various types of cancer by inducing cell cycle arrest and apoptosis32–34 and by promoting other potent tumor suppressors such as P5322, PTEN22,23, and p21 CDK1A32,35. In a colon cancer cell model, drug-induced apoptosis has been shown to be mediated by ROS–EGR1 signaling36. More relevant to lung cancer, EGR1 expression can predict the expression of its putative downstream target PTEN37; this is also in agreement with our data. We showed that knocking down CISD2 expression significantly increased EGR1 and PTEN protein levels, and eventually decreased the level of phospho-AKT (Figs 6N and S14). A similar mechanism may also act in gastric cancer cells, in which CISD2 positively regulates AKT activity16. Downregulation of EGR1 has been implicated in the changes of mobility and migratory ability of lung cancer cells21, or in the process of EMT38. Furthermore, EGR1/PTEN/AKT axis was known to play a key role in inhibiting EMT through downregulation of AKT phosphorylation39,40. Since EMT phenotype observed in cell based study could provide a much likely link to poor prognosis of patients in clinical setting, our findings that CISD2 may regulate EGR1 expression and, at the same time, positively affect EMT phenotype (Fig. 4) become very important. It would be useful to know whether similar phenotypes that are affected by CISD2 are mediated through ERG1.
Overall, we found that the upregulation of CISD2 expression was involved in the tight regulation of redox homeostasis, which fosters the development and progression of lung ADC by enabling escape from apoptosis and the gain of cell proliferation and invasion potential. These findings strongly suggest that it would be worth investigating whether CISD2 or its associated downstream targets can be considered when developing anticancer therapies. For instance, because knocking down CISD2 expression or targeting CISD2 with pioglitazone, as exemplified by Darash-Yahana and colleagues14, can lead to an imbalance in ROS homeostasis and an increase in oxidative stress, methods for inhibiting the action of CISD2 may be developed to assist in the eradication of lung cancer cells, or to function as sensitizers to enhance chemotherapy or radiotherapy, in a similar way to that proposed for the NRF2–KEAP1 scenario in lung cancer treatment41,42.
Although this study has focused on lung ADC in terms of the oncogenic role of the CISD2–ROS–EGR1/GPX3 axis, we found that CISD2 expression was also upregulated and seemed to be inversely correlated with prognosis among patients with lung SQC (unpublished data), another major lung cancer subtype that is etiologically and genetically distinct from lung ADC. It would be worth investigating whether CISD2 also plays an oncogenic role in lung SQC and to explore its underlying mechanisms for identification of therapeutic targets in corresponding pathways for the effective treatment of lung cancers.
Materials and Methods
Public domain data
Several lung cancer datasets, including GSE3121018, GSE2726219, GSE1918820, GSE889443, and GSE1024544 were downloaded from Oncomine (http://www.oncomine.com). In addition to the above datasets generated by others, our in-house-generated dataset GSE4653945 has been deposited in the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) and was also utilized in this study. Information about the probes of genes of interest applied in this study can be found in Supplementary Table S1. For each dataset, gene expression data and associated clinical information of samples belonging to cancer subtype of lung ADC or normal lung tissues were all downloaded without selection. Since data from Oncomine have already been normalized, they were used directly without further processing.
Clinical samples
Fifty-six clinical lung tissue samples, which comprise 28 normal lung samples and 28 lung ADC samples, originated from a cohort we have described previously46. These samples were obtained from surgical resection samples collected at Chang Gung Memorial Hospital, Taoyuan, with approval from the Institutional Review Board of Chang Gung Memorial Hospital and informed consent from all participants. All procedures and methods were achieved in accordance with the relevant guidelines and regulations. Human lung cancer tissue microarrays LC810 and LC1006 were purchased from US Biomax (Rockville, MD, USA); CCA3, CC4, and CCN4 were from Super Bio Chips (Seoul, Korea). Collectively, these arrays contain 47 lung ADC and seven normal lung tissues. Corresponding clinical information of each specimen was provided by the manufacturers.
Cell lines and DNA constructs
Lung ADC cell lines NCI-H1299 and A549 were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). CL1-1 and CL1-5 lung ADC cell lines were gifts from Dr. Cheng-Wen Wu as described previously47. All cell lines were maintained in RPMI 1640 medium (Invitrogen, Grand Island, NY, USA) supplemented with 10% FBS (Invitrogen) and 1% penicillin/streptomycin (Biological Industries, Cromwell, CT, USA). To construct a plasmid expressing the full-length CISD2, the coding sequence of CISD2 in an EST clone (IMAGE:5105935) was amplified by PCR with the forward primer 5′-GGATCCATGGTGCTGGAGA-3′ and reverse primer 5′-GAATTCTTATACTTCTTTCTTCTTCAGT-3′ and then subcloned into a pcDNA3.1 expression vector (Invitrogen). Cell transfection was conducted with Lipofectamine 2000 (Invitrogen) and cells were maintained in selection media containing 2 mg/mL G418 (Calbiochem, San Diego, CA, USA).
siRNA
An siRNA mixture (SMARTpool) targeting human CISD2 gene expression containing the following sequences: 5′-UCAGAAUGGCUUCGGUUAU-3′, 5′-UAUUGUAGGUGUUGGCGUU-3′, 5′-CCUGAAAGCAUUACCGGGU-3′, and 5′-UGGAGAGCGUGGCCCGUAU-3′, was obtained from Dharmacon (GE Healthcare, Pittsburg, PA, USA). The negative control siRNAs (nontargeting pool; Dharmacon) were used according to the manufacturer’s instructions.
shRNA constructs
The shRNA expression vectors targeting CISD2 (CISD2(−)#1 and CISD2(−)#2) were purchased from RNAi Core Lab (Academia Sinica, Taipei, Taiwan) and transOMIC (Huntsville, AL, USA), respectively, as well as their negative controls. Plasmid DNA was transfected into cells according to the manufacturer’s instructions. Cells were maintained in selection media containing 2 μg/mL puromycin (Calbiochem).
GSEA
To analyze biological pathways affected by silencing or forced ectopic expression of CISD2 in lung ADC cell lines, the gene expression profiles of CISD2-silencing H1299 cells and CISD2-overexpressed CL1-1 cells and their nontargeting or vector control cells were obtained by performing Illumina HT12 Beadchip experiments as previously described46. GSEA48 was then used to obtain gene sets or biological pathways enriched in the differential expression profiles using log2R as ranking metric, where R was the ratio of normalized gene expression level of a gene in test cells to that in control cells. GSEA Desktop Application (http://software.broadinstitute.org/gsea/downloads.jsp) was used for GSEA implementation.
RT-qPCR
Total RNA was reserve-transcribed by using SuperScript III (Invitrogen) and oligo(dT) primers per the manufacturer’s instructions. Methods for RT-qPCR and analysis for the quantification of relative mRNA level were as described previously47. The primers and the Universal Probe Library probes (Roche Molecular Diagnostics, Pleasanton, CA, USA) are listed in Supplementary Table S2.
IHC analysis
IHC staining was performed using rabbit anti-CISD2 antibodies (Sigma-Aldrich, St Louis, MO, USA, HPA015914, 1:25) following a procedure described previously49. The immunoreactivity patterns of all tissues were scored by two independent pathologists. Staining intensity was scored as 0 (negative), 1+ (weak), 2+ (moderate), and 3+ (strong), as described elsewhere46.
Immunoblot analysis
Protein separation and western blotting protocols were as described previously47. Briefly, cells were lysed with RIPA solution, succeeded to SDS-PAGE gel electrophoresis, and the proteins transferred to PVDF membranes. The proteins were probed with anti-CISD2 (1:1,000 dilution; HPA015914, Sigma-Aldrich), anti-V5 (1:2,000 dilution; R960, Invitrogen), anti-GAPDH (1:10,000 dilution; MAB374, Millipore, Billerica, MA, USA), anti-E-cadherin (1:1,000 dilution; #3195, Cell Signaling Technology), anti-vimentin (1:1,000 dilution; #5741,Cell Signaling Technology), anti-ZO-1 (1:1,000 dilution; #5406, Cell Signaling Technology), anti-P21 (1:1,000 dilution; #2947,Cell Signaling Technology), anti-cytochrome C (1:1,000 dilution; GTX108585, GeneTex), anti-caspase 3 (1:1,000 dilution; GTX110543, GeneTex), anti-PARP1 (1:1,000 dilution; GTX100573, GeneTex), anti-EGR1 (1:1,000 dilution; #4153, Cell Signaling Technology), anti-PTEN (1:1,000 dilution; #9188, Cell Signaling Technology), anti-phospho-AKT Ser473 (1:1,000 dilution; (#4060, Cell Signaling Technology), and anti-AKT1 (1:1,000 dilution; #2938, Cell Signaling Technology) antibodies, and then further probed by horseradish peroxidase-conjugated secondary antibodies.
Cell viability assay
Cells (~2 × 104) were seeded in 24-well plates for predetermined time intervals. The protocol for MTT assays was as described previously47.
Clonogenic assay
Serially diluted cells (~1 × 102) were seeded with culture medium in six-well plates and cultured for 8 to 12 days. Colonies were stained with 1% crystal violet (Merck, Darmstadt, Germany) and at least 50 cells were counted.
Animal experiments
For xenograft tumor experiments, a total of 1 × 106 cells in 100 μL PBS were injected subcutaneously into five severe combined immunodeficient mice (BALB/cAnN.Cg-Foxn1nu/CrlNarl) for each test (shCISD2) and control (nontargeting shRNA) groups. The volume of tumor formed was measured over a 7-week period post-injection. Tumor volume was calculated according to the formula of length × width × width/2. Tumor mass was evaluated upon sacrifice at the seventh week after injection. All experimental procedures of the animal use protocol was approved by the Institutional Animal Care and Use Committee of National Health Research Institutes (NHRI) and all methods were performed in accordance with the relevant guidelines and regulations.
Apoptosis assay
siRNA-treated cells or stable transfectants of shCISD2 were seeded into six-well plates and incubated overnight. Culture media were replaced by a serum-free medium with or without cisplatin (30 μM), and cells were harvested at 12, 24 and 48 h, labeled with annexin V and PI (Invitrogen) per the manufacturer’s instructions, and then analyzed by flow cytometry.
Invasion assay
siRNA-treated cells or stable transfectants of shCISD2 in serum-free media were seeded into a cell culture insert (Falcon, Corning, NY, USA) at a density of 1 × 104/well and placed into a 24-well plate with regular medium. After incubation for 16 h, Matrigel (BD Biosciences) and cells remaining on the seeding side of the membrane were wiped off with cotton swabs, while cells that invaded the bottom surface of the membrane were fixed with methanol (Merck) and detected using Giemsa stain (Sigma-Aldrich).
Mitochondrial membrane potential assay
Cells were seeded into six-well plates and incubated for 24 h, then transferred to serum-free medium containing 2 μg/mL JC-1 (eBioscience, San Diego, CA, USA), a membrane potential-sensitive cationic dye, incubated at 37 °C for 20 min, washed with PBS, and then analyzed using flow cytometry. Aggregated JC-1 exhibits red fluorescence, indicating a high membrane potential, while monomer JC-1 shows green fluorescence, which indicates a membrane potential collapse.
Measurement of intracellular ROS
To detect the production of intracellular ROS, cell membrane-permeable fluorogenic probes DCFH-DA (Sigma-Aldrich) and DHE (Sigma-Aldrich) were applied. Cells were seeded into six-well plates and incubated for 24 h, and transferred into serum-free medium containing 10 μM DCFH-DA or 10 μM DHE. After incubation at 37 °C for 20 min, cells were harvested and washed with PBS, and then analyzed using flow cytometry.
Statistical analysis
Student’s t-test was used to assess the significance of difference in mRNA or protein levels between conditions of interest. Cox proportional hazard regression model was used to analyze the association between mRNA expression level and patients’ survival. The log-rank test was used to evaluate the significance of difference in survival between patients of stratified groups. Pearson correlation analysis was used to assess the significance of correlation between mRNA expression levels of a pair of genes of interest. In all analyses, P < 0.05 was considered statistically significant.
Electronic supplementary material
Acknowledgements
We thank Dr. Lu-Hai Wang, Dr. Chuang-Rung Chang, and Dr. Alan Yueh-Luen Lee for critical comments. This work was supported by Ministry of Science and Technology (Bioinformatics Core Facility for Translational Medicine and Biotechnology Development,MOST106-2319-B-400-001 to I.S.C. and C.A.H., and MOST104-2314-B-400 -011 -MY2 to S.S.J.), and from Ministry of Health and Welfare (CA-105-SP-01), and National Health Research Institutes (NHRI), NHRI05 A1-PH-PP03-014 to C.A.H.
Author Contributions
C.A.H., I.S.C., S.S.J. jointly supervised the study. C.A.H., I.S.C., S.S.J., C.P.C., S.M.L. conceived and designed the experiment. W.T.F., Y.C.Y. and S.M.L. performed the experiments. I.S.C., C.H.C., F.Y.T., S.F.H., J.L.C. and Y.Y.S. performed data analysis. C.A.H., S.F.H., S.S.J. and Y.W.C. contributed reagents and materials. S.S.J. and S.M.L. wrote the manuscript. All authors reviewed and approved the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-12131-x.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chao A. Hsiung, Email: hsiung@nhri.org.tw
Shih Sheng Jiang, Email: ssjiang@nhri.org.tw.
References
- 1.Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008;83:584–594. doi: 10.1016/S0025-6196(11)60735-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hecht SS. Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 3.Thun MJ, et al. Cigarette smoking and changes in the histopathology of lung cancer. Journal of the National Cancer Institute. 1997;89:1580–1586. doi: 10.1093/jnci/89.21.1580. [DOI] [PubMed] [Google Scholar]
- 4.Valavanidis A, Vlachogianni T, Fiotakis K. Tobacco smoke: involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health. 2009;6:445–462. doi: 10.3390/ijerph6020445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer. 2014;14:709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 7.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat. Rev. Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 8.Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen YF, Wu CY, Kirby R, Kao CH, Tsai TF. A role for the CISD2 gene in lifespan control and human disease. Ann. N. Y. Acad. Sci. 2010;1201:58–64. doi: 10.1111/j.1749-6632.2010.05619.x. [DOI] [PubMed] [Google Scholar]
- 10.Conlan AR, et al. Crystal structure of Miner1: The redox-active 2Fe-2S protein causative in Wolfram Syndrome 2. J. Mol. Biol. 2009;392:143–153. doi: 10.1016/j.jmb.2009.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen YF, et al. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 2009;23:1183–1194. doi: 10.1101/gad.1779509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang NC, et al. Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle. Hum. Mol. Genet. 2012;21:2277–2287. doi: 10.1093/hmg/dds048. [DOI] [PubMed] [Google Scholar]
- 13.Sohn YS, et al. NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proc. Natl. Acad. Sci. USA. 2013;110:14676–14681. doi: 10.1073/pnas.1313198110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Darash-Yahana M, et al. Breast cancer tumorigenicity is dependent on high expression levels of NAF-1 and the lability of its Fe-S clusters. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:10890–10895. doi: 10.1073/pnas.1612736113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu L, et al. CISD2 expression is a novel marker correlating with pelvic lymph node metastasis and prognosis in patients with early-stage cervical cancer. Med. Oncol. 2014;31:183. doi: 10.1007/s12032-014-0183-5. [DOI] [PubMed] [Google Scholar]
- 16.Wang L, et al. Overexpressed CISD2 has prognostic value in human gastric cancer and promotes gastric cancer cell proliferation and tumorigenesis via AKT signaling pathway. Oncotarget. 2016;7:3791–3805. doi: 10.18632/oncotarget.6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang L, et al. A novel prognostic score model incorporating CDGSH iron sulfur domain2 (CISD2) predicts risk of disease progression in laryngeal squamous cell carcinoma. Oncotarget. 2016;7:22720–22732. doi: 10.18632/oncotarget.8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okayama H, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72:100–111. doi: 10.1158/0008-5472.CAN-11-1403. [DOI] [PubMed] [Google Scholar]
- 19.Wei TY, et al. Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer Sci. 2012;103:1640–1650. doi: 10.1111/j.1349-7006.2012.02367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou J, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;5:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang H, et al. EGR1 decreases the malignancy of human non-small cell lung carcinoma by regulating KRT18 expression. Sci. Rep. 2014;4:5416. doi: 10.1038/srep05416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baron V, Adamson ED, Calogero A, Ragona G, Mercola D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFbeta1, PTEN, p53, and fibronectin. Cancer Gene Ther. 2006;13:115–124. doi: 10.1038/sj.cgt.7700896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Virolle T, et al. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat. Cell Biol. 2001;3:1124–1128. doi: 10.1038/ncb1201-1124. [DOI] [PubMed] [Google Scholar]
- 24.Mitsuishi Y, Motohashi H, Yamamoto M. The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front. Oncol. 2012;2:200. doi: 10.3389/fonc.2012.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solis LM, et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res. 2010;16:3743–3753. doi: 10.1158/1078-0432.CCR-09-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lill R. Function and biogenesis of iron-sulphur proteins. Nature. 2009;460:831–838. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- 27.Tamir S, et al. Structure-function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochim. Biophys. Acta. 2015;1853:1294–1315. doi: 10.1016/j.bbamcr.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 28.Colca JR, et al. Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am. J. Physiol. Endocrinol. Metab. 2004;286:E252–260. doi: 10.1152/ajpendo.00424.2003. [DOI] [PubMed] [Google Scholar]
- 29.Wiley SE, Murphy AN, Ross SA, van der Geer P, Dixon JE. MitoNEET is an iron-containing outer mitochondrial membrane protein that regulates oxidative capacity. Proc. Natl. Acad. Sci. USA. 2007;104:5318–5323. doi: 10.1073/pnas.0701078104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amr S, et al. A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am. J. Hum. Genet. 2007;81:673–683. doi: 10.1086/520961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 32.Kim SJ, Kim JM, Shim SH, Chang HI. Shikonin induces cell cycle arrest in human gastric cancer (AGS) by early growth response 1 (Egr1)-mediated p21 gene expression. J. Ethnopharmacol. 2014;151:1064–1071. doi: 10.1016/j.jep.2013.11.055. [DOI] [PubMed] [Google Scholar]
- 33.Liu J, et al. Concurrent down-regulation of Egr-1 and gelsolin in the majority of human breast cancer cells. Cancer Genomics Proteomics. 2007;4:377–385. [PubMed] [Google Scholar]
- 34.Shin DY, et al. Implication of intracellular ROS formation, caspase-3 activation and Egr-1 induction in platycodon D-induced apoptosis of U937 human leukemia cells. Biomed. Pharmacother. 2009;63:86–94. doi: 10.1016/j.biopha.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 35.Ragione FD, et al. p21Cip1 gene expression is modulated by Egr1: a novel regulatory mechanism involved in the resveratrol antiproliferative effect. J. Biol. Chem. 2003;278:23360–23368. doi: 10.1074/jbc.M300771200. [DOI] [PubMed] [Google Scholar]
- 36.Han MH, Kim GY, Yoo YH, Choi YH. Sanguinarine induces apoptosis in human colorectal cancer HCT-116 cells through ROS-mediated Egr-1 activation and mitochondrial dysfunction. Toxicol. Lett. 2013;220:157–166. doi: 10.1016/j.toxlet.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 37.Ferraro B, Bepler G, Sharma S, Cantor A, Haura EB. EGR1 predicts PTEN and survival in patients with non-small-cell lung cancer. J. Clin. Oncol. 2005;23:1921–1926. doi: 10.1200/JCO.2005.08.127. [DOI] [PubMed] [Google Scholar]
- 38.Shan LN, et al. Early Growth Response Protein-1 Involves in Transforming Growth factor-beta1 Induced Epithelial-Mesenchymal Transition and Inhibits Migration of Non-Small-Cell Lung Cancer Cells. Asian Pac J Cancer Prev. 2015;16:4137–4142. doi: 10.7314/APJCP.2015.16.9.4137. [DOI] [PubMed] [Google Scholar]
- 39.Sarver AL, Li L, Subramanian S. MicroRNA miR-183 functions as an oncogene by targeting the transcription factor EGR1 and promoting tumor cell migration. Cancer Res. 2010;70:9570–9580. doi: 10.1158/0008-5472.CAN-10-2074. [DOI] [PubMed] [Google Scholar]
- 40.Wang H, et al. PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res. 2007;67:2922–2926. doi: 10.1158/0008-5472.CAN-06-3598. [DOI] [PubMed] [Google Scholar]
- 41.Abazeed ME, et al. Integrative radiogenomic profiling of squamous cell lung cancer. Cancer research. 2013;73:6289–6298. doi: 10.1158/0008-5472.CAN-13-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tao S, et al. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer research. 2014;74:7430–7441. doi: 10.1158/0008-5472.CAN-14-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee ES, et al. Prediction of recurrence-free survival in postoperative non-small cell lung cancer patients by using an integrated model of clinical information and gene expression. Clin. Cancer Res. 2008;14:7397–7404. doi: 10.1158/1078-0432.CCR-07-4937. [DOI] [PubMed] [Google Scholar]
- 44.Liu J, Yang XY, Shi WJ. Identifying differentially expressed genes and pathways in two types of non-small cell lung cancer: adenocarcinoma and squamous cell carcinoma. Genet. Mol. Res. 2014;13:95–102. doi: 10.4238/2014.January.8.8. [DOI] [PubMed] [Google Scholar]
- 45.Chang, I. S. et al. Genetic modifiers of progression-free survival in never-smoking lung adenocarcinoma patients treated with first-line TKIs. Am J Respir Crit Care Med, doi:10.1164/rccm.201602-0300OC (2016 [Epub ahead of print]). [DOI] [PMC free article] [PubMed]
- 46.Fang WT, et al. Downregulation of a putative tumor suppressor BMP4 by SOX2 promotes growth of lung squamous cell carcinoma. International journal of cancer. 2014;135:809–819. doi: 10.1002/ijc.28734. [DOI] [PubMed] [Google Scholar]
- 47.Jiang SS, et al. Upregulation of SOX9 in lung adenocarcinoma and its involvement in the regulation of cell growth and tumorigenicity. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:4363–4373. doi: 10.1158/1078-0432.CCR-10-0138. [DOI] [PubMed] [Google Scholar]
- 48.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu SC, et al. G(alpha)12-mediated pathway promotes invasiveness of nasopharyngeal carcinoma by modulating actin cytoskeleton reorganization. Cancer Res. 2009;69:6122–6130. doi: 10.1158/0008-5472.CAN-08-3435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.