

Abstract
Dosage-dependent upregulation of most of chromosome 21 (Hsa21) genes has been demonstrated in heart tissues of fetuses with Down syndrome (DS). Also miRNAs might play important roles in the cardiac phenotype as they are highly expressed in the heart and regulate cardiac development. Five Hsa21 miRNAs have been well studied in the past: miR-99a-5p, miR-125b-2-5p, let-7c-5p, miR-155-5p, and miR-802-5p but few information is available about their expression in trisomic tissues. In this study, we evaluated the expression of these miRNAs in heart tissues from DS fetuses, showing that miR-99a-5p, miR-155-5p, and let-7c-5p were overexpressed in trisomic hearts. To investigate their role, predicted targets were obtained from different databases and cross-validated using the gene expression profiling dataset we previously generated for fetal hearts. Eighty-five targets of let-7c-5p, 33 of miR-155-5p, and 10 of miR-99a-5p were expressed in fetal heart and downregulated in trisomic hearts. As nuclear encoded mitochondrial genes were found downregulated in trisomic hearts and mitochondrial dysfunction is a hallmark of DS phenotypes, we put special attention to let-7c-5p and miR-155-5p targets downregulated in DS fetal hearts and involved in mitochondrial function. The let-7c-5p predicted target SLC25A4/ANT1 was identified as a possible candidate for both mitochondrial and cardiac anomalies.
1. Introduction
Down Syndrome (DS) is a major cause of congenital heart defects (CHD), mainly represented by atrioventricular canal defect (AVCD), ventricular septal defect (VSD), and tetralogy of Fallot (TOF) [1]. Most of them derive from the abnormal development of the endocardial cushions [1, 2]. Defects of the outflow tract are also frequent.
Attempts to identify chromosome 21 (Hsa21) genes possibly contributing to the DS phenotype have focused in the past on the Down syndrome critical region (DSCR). The DSCR hypothesis assumed that one or more genes in this region may be sufficient to produce the specific DS features when present in three copies [3].
A chromosome segment spanning from D21S3 to PFKL in band 21q22.3 was considered a critical region for cardiac anomalies in DS (DS-CHD) [4]. Later, a 5.4 Mb genomic region was identified in the DS mouse model Dp (16) associated to congenital heart defects similar to that observed in DS subjects [5]. This region, which spans from Tiam1 and Kcnj6 and includes 52 Hsa21 ortholog genes, was further narrowed to 3.7 Mb [6] from Ifnar1 and Kcnj6 (35 Hsa21 ortholog genes). The two CHD critical regions described by Barlow and by Liu, identified according to different criteria, were mapped to very different loci of Hsa21. Discrepancies like this one suggest today that the origins of trisomic phenotypes are more complicated than formerly assumed and that they possibly involve multiple gene interactions.
Another interesting detail is that only about 50% of DS individuals manifest CHD. This means that we cannot hypothesize a simple correlation between gene overexpression and cardiac alterations. Therefore, a more extensive analysis of both transcriptome data and pathway perturbations must be applied to identify the complex molecular defects underlying CHD in DS.
In a previous study, we applied the microarray technology to analyze genome-wide expression profiles of the heart of DS fetuses with and without CHD [7]. The rationale for analyzing fetal tissues was based on the concept that CHD are thought to arise from anomalies in cardiac morphogenesis. By this approach, it was found that Hsa21 gene expression was globally upregulated 1.5 fold in trisomic samples, in general agreement with the gene dosage hypothesis. More than 400 genes located on other chromosomes were also differentially expressed, either upregulated or downregulated, between trisomic and nontrisomic hearts. Functional class scoring and gene set enrichment analyses of these genes revealed a global downregulation of nuclear-encoded mitochondrial genes (NEMGs) and upregulation of genes encoding extracellular matrix proteins. These data indicate that dosage-dependent upregulation of Hsa21 genes causes dysregulation of the genes responsible for mitochondrial function and for the extracellular matrix organization in the fetal heart of trisomic subjects and suggest that these alterations might be a prelude to heart defects. However, no significant differences in gene expression in hearts from DS fetuses (18–22 gestational weeks) with CHD could be found [7].
Throughout that study only the expression of protein-coding RNAs was analyzed even though noncoding RNAs, such as microRNAs (miRNAs), expressed in the heart, might be implicated in determining the CHD as they control protein expression in development, differentiation, and metabolism [8].
Recent findings indicate the involvement of microRNAs in mouse cardiac development and diseases [9–12]. In the context of DS, this can occur by 2 mechanisms: (i) Hsa21 miRNAs could be overexpressed as a consequence of the trisomy and could affect target genes directly or indirectly involved in heart morphogenesis, and (ii) Non-Hsa21 dysregulated miRNAs might affect target genes involved in heart morphogenesis.
In this study, we have investigated the former mechanism in heart tissues from DS fetuses with and without CHD.
Hsa21 encodes several classes of noncoding RNAs, the most enriched being long noncoding RNAs, while miRNAs are the less represented [13]. The most recent annotation of miRNA database (miRBase, release 21) has indicated that the Hsa21 harbors at least 29 miRNAs.
Five of them, namely miR-99a-5p, let-7c-5p, miR-125b-2-5p, miR-155-5p, and miR-802-5p, were first identified (Figure 1). Their involvement in different DS associated phenotypes has been established, and several of their targets have been experimentally validated [14–20].
Figure 1.
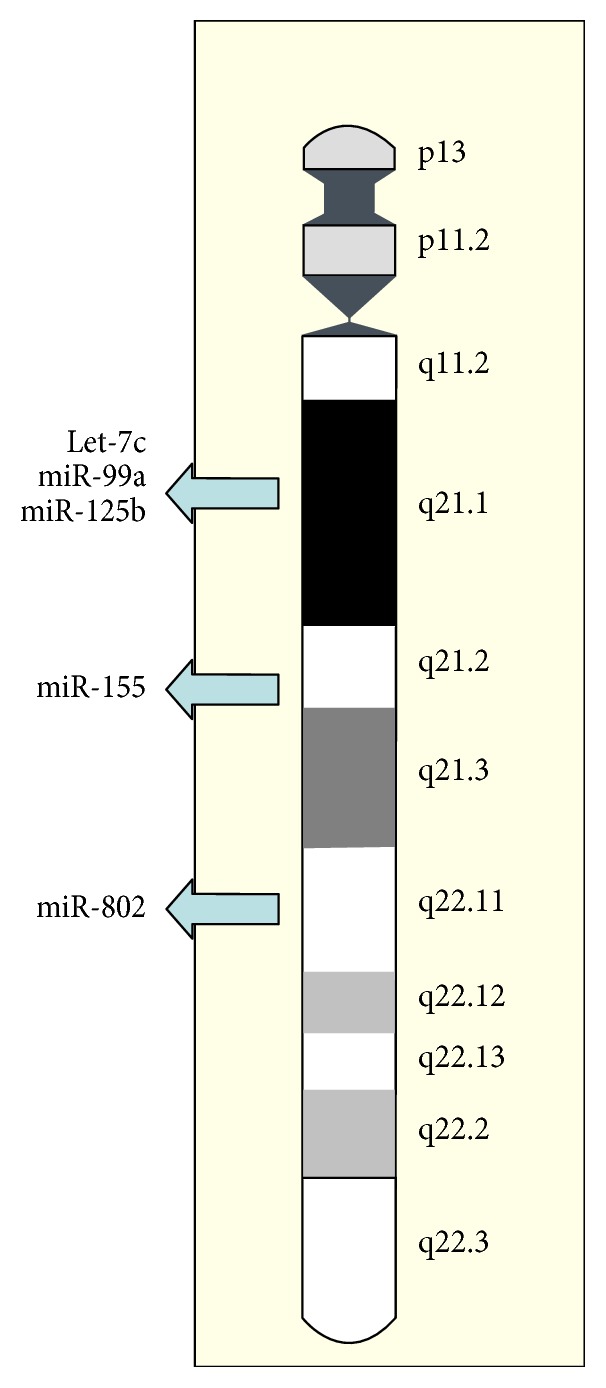
Hsa21 miRNAs. miR-99a-5p, let-7c-5p, and miR-125b-2-5p are located on Hsa21 in the sense orientation within an intron of the C21orf34 gene, located at the beginning of q21.1 band. miR-99a-5p and let-7c-5p are only 659 bp apart, whereas miR-125b-2-5p lies just over 50.000 bp downstream of let-7c. miR-155-5p is located within the BIC gene, almost 9 Mb downstream from the C21orf34 gene at Hsa21 genomic position q21.1. Finally, miR-802-5p is located just over 10 million bp downstream from the BIC/miR-155 gene in the antisense orientation within intron 1 of the RUNX1 gene at position q22.11.
Few information is available about the expression of Hsa21-derived miRNAs in human heart tissues and their possible role in cardiomyogenesis.
We have analyzed the expression of 5 well-studied Hsa21 miRNAs to determine if they are dysregulated as consequence of trisomy and if their dysregulation might affect molecular mechanisms involved in mitochondrial function and heart development. To this aim, a bioinformatic analysis of Hsa21 miRNA target prediction by different databases was performed and cross-validated using the gene expression profiling dataset we previously generated for trisomic fetal hearts.
2. Materials and Methods
2.1. Samples
Cardiac tissues were obtained from fetuses at 18–22 weeks of gestation after therapeutic abortion according to protocols approved by our Institutional Ethical Committee. Tissues and RNAs were stored at the Telethon Bank of Fetal Biological Samples at the University of Naples. For this study, 3 hearts from euploid fetuses (NH), 3 from fetuses with DS without CHD (DH), and 3 from fetuses with DS and CHD (CDH) were analyzed. For let-7c mimic and inhibitor transfection, 2 previously characterized [21, 22] primary lines of euploid (N-HFF) and trisomic (DS-HFF) fetal fibroblasts were, respectively, used. Fibroblasts from biopsies were cultured in T25 flasks (BD Falcon) with Chang Medium B + C (Irvine Scientific) supplemented with 1% penicillin/streptomycin (Gibco) at 37°C in 5% CO2 atmosphere; all the analyses described throughout this study were carried out at cell culture passages 4-5.
2.2. RNA Extraction and Quantitative Real-Time PCR
Total RNA from each sample was extracted using TRIzol Reagent (Gibco/BRL Life Technologies Inc., Gaithersburg, MD) and was reverse-transcribed using iScript cDNA synthesis kit (Bio-Rad). qRT-PCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) on a Bio-Rad iCycler CFX96 Touch Real-Time PCR Detection System according to the manufacturer's protocols. Primer pairs (MWG-Biotech, Ebersberg, Germany) were designed using the Primer 3 software (http://bioinfo.ut.ee/primer3-0.4.0/primer3; date last accessed 2015) to obtain amplicons ranging from 100 to 150 base pairs. In order to test primer efficiency, serial dilutions of cDNAs generated from selected samples, which expressed the target genes at a suitable level, were used to generate standard curves for each gene. qRT-PCR results are presented as relative mRNA levels normalized against reference control values. The GAPDH housekeeping gene was chosen as reference gene. Primer sequences of analyzed genes are the following SLC25A4/ANT1-F: GGGTTTCAACGTCTCTGTCC; SLC25A4/ANT1-R: TCCAGCTCACAAAAATGTGC; DICER-F: CTGATGGAATTAGAAGAAGCACTTAAT; DICER-R: ACCAGGGTCCCAGAACTACC; GAPDH-F: TGCACCACCAACTGCTTAGC; and GAPDH -R: GGCATGGACTGTGGTCATGAG.
For miRNA reverse transcription miScript II RT Kit (Qiagen) was used, and the mature miRNAs, mir-99a-5p, let-7c-5p, mir-125b-2-5p, mir-155-5p, and mir-802-5p, were quantified using miScript Primer Assay system and miScript SYBR Green PCR Kit (Qiagen). RNAUS and RNA5S were used as reference genes.
For end point PCR, assays were performed using the PCR Master Mix (2X) Kit (Thermo Fisher #K0171). Amplification products were visualized on 2% agarose gel and quantified using the Fiji software (http://www.fiji.sc) [23].
2.3. Bionformatic Analysis
Predicted miRNA-mRNA interactions were retrieved from 9 different algorithms (DIANA-microT [24], TargetScan [25], PITA [26], TargetMiner [27], miRDB [28], RNA22 [29], Pictar [30], and MiRanda [31]) using as input the sequences/IDs for the miRNA of interest. Before merging the date, divergent database annotations were unified by converting transcript-wise predictions to the gene level (Ensembl gene ID) using the BiomaRt package [32]. The lists of targets predicted by each database were subsequently marked with Affymetrix probe IDs using the related annotation package and finally merged with the list of probes (n = 279) found significantly downregulated in DS hearts compared to euploid controls [7]. For further analyses, we brought forward miR targets predicted by at least two databases, with a P value ≤ 0.05 and fold change ≤ −1.2 as defined in the original paper [7].
Gene ontology (GO) functional class scoring of the lists of significantly downregulated genes was performed using the Web-based GEne SeT Analysis Toolkit V2 (http://www.webgestalt.org) [33]. Special attention was given to mitochondria-related categories and pathways.
2.4. miRNA Mimic Transfection
For let-7c-5p upregulation, a let-7c miRNA mimic (miScript miRNA Mimics, Qiagen) was transfected in 2 N-HFF lines. Cells were plated in a concentration of 70,000/well on 24 well plates (BD Falcon) and after 24 hours were transfected with a miRNA mimic using the INTERFERin transfection reagent (Polyplus-transfection). A fluorescent siRNA (AllStars Neg. siRNA AF 488, Qiagen) has been used to monitor the efficiency of the chosen transfecting agent. Forty-eight hours after the transfection cells were harvested and ANT1 and DICER expression were evaluated, cells treated with the INTERFERin transfection agent only were used as mock control for all experiments performed after transfection.
2.5. miRNA Inhibitor Transfection
For let-7c-5p downregulation, an Anti-hsa-let-7c miRNA (miScript miRNA Inhibitor, Qiagen) was transfected in 2 DS-HFF lines. Cells were plated in a concentration of 150,000/dish on 3,5 cm petri dishes (BD Falcon) and after 24 hours were transfected with the inhibitor using the INTERFERin transfection reagent (Polyplus-transfection). Forty-eight hours after transfection cells were harvested for RNA collection while 72 hours after transfection protein lysates were obtained.
2.6. Immunoblot Analysis
Cells were lysed in radioimmunoprecipitation assay buffer (1% Triton; 0,5% sodium deoxycholate; 0,1% sodium dodecyl sulfate; 0,15 M NaCl; 0,05 tris-HCl; and pH 7.2) supplemented with protease inhibitors. For western blot analysis, proteins were separated on SDS-PAGE (Mini-Protean TGX gels, Bio-Rad), gels were blotted onto Immobilon-P (Millipore, Bedford, MA, USA) for overnight, and the membranes were blocked in 5% nonfat dry milk in PBS plus 0,05% Tween 20 for 2 h or overnight before the addition of the primary antibody for 2 h. The primary antibodies used were anti-SLC25A4/ANT1 mAb (ab 110322, Abcam) and anti-vinculin (N-19) (sc-7649, Santa Cruz).
The filters were washed three times in PBS plus 0.05% Tween 20 before the addition of horseradish peroxidase-conjugated secondary antibodies for 45 min. Horseradish peroxidase was detected with ECL (Pierce).
2.7. Statistics
The Student's t-test was applied to evaluate the statistical significance of data in this study. The threshold for statistical significance (P value) was set at 0.05.
3. Results
We evaluated, by qRT-PCR, the expression of Hsa21 miRNAs in fetal heart samples from 6 DS individuals, 3 with CHD (CHD samples) and 3 without CHD (DH samples), and from 3 nontrisomic controls (NH samples).
We found that miR-99a-5p, miR-125b-2-5p, let-7c-5p, and miR-155-5p were expressed in fetal hearts at 18–22 gestational weeks. miR-802-5p was not expressed. miR-99a-5p, miR-155-5p, and let-7c-5p were overexpressed in trisomic hearts when compared with euploid ones, whereas miR-125b-2-5p was not dysregulated and was quite variably expressed (Figure 2). The comparison between heart tissues from fetuses with and without CHD indicated that miR-155-5p and let-7c-5p did not show significant differences between DH and CDH samples while miR-99a-5p was differentially expressed even though the sample size is too small to obtain a statistical significance.
Figure 2.

Hsa21 miRNA expression evaluated by qRT-PCR in fetal hearts. Let-7c-5p, miR-99a-5p, and miR-155-5p were upregulated in trisomic hearts without cardiopathy (DH) and with cardiopathy (CDH). miR-125b-2-5p was variably expressed and not upregulated. Results are expressed as relative mean values ± SEM of 3 DH and 3 CDH samples, compared with control hearts (NH) set equal to 1. ∗P < 0.05, ∗∗P < 0.001. P value expresses statistical significance for trisomic versus nontrisomic sample comparisons.
As miRNAs could affect protein expression by either interfering with RNA translation or promoting mRNA degradation [34], we looked at mRNA expression of target genes of overexpressed miRNAs by using the dataset of our previous study by which we investigated gene expression profiling in the same hearts [7].
We considered as target list the sum of all targets predicted by 9 different software from miRBase annotation, and we filtered them according to gene expression microarray results. The rationale of our approach is based on the notion that overexpression of Hsa21 miRNAs can result in downregulation of specific target genes possibly involved in DS phenotype. For each miRNA, we compared target genes predicted at least by 2 different software with the list of genes downregulated in trisomic hearts with a fold change (FC) > |1,2| and P value< 0.05. Using these criteria, we unraveled that 85 targets of let-7c-5p (Supplementary Table 1 available online at https://doi.org/10.1155/2017/8737649), 33 of miR-155-5p (Supplementary Table 2), and 10 of miR-99a-5p (Supplementary Table 3) were expressed in fetal heart and downregulated in trisomic samples.
Over-representation analysis (ORA) of these lists, performed using the Web-based GEne SeT Analysis Toolkit (http://www.webgestalt.org) [33], demonstrated a highly significant enrichment (P < 0.0001) of the cell component gene ontology category “mitochondrion” for let-7c-5p and miR-155-5p downregulated targets, with a cluster of 26 and 8 genes, respectively (Tables 1 and 2). No mitochondria-related genes were found among targets of miR-99a-5p.
Table 1.
Targets of let-7c-5p significantly downregulated in DS hearts and belonging to gene ontology cellular component category “mitochondrion.” Twenty-six genes were observed instead of the expected 6.49 genes with P < 0.00001.
| Gene symbol | Description | FC |
|---|---|---|
| AKAP8 | A-kinase anchoring protein 8 | −1,361 |
| COX10 | COX10, heme A: farnesyltransferase cytochrome c oxidase assembly factor | −1,307 |
| COX5A | Cytochrome c oxidase subunit 5A | −1,263 |
| DLAT | Dihydrolipoamide S-acetyltransferase | −1,678 |
| DLST | Dihydrolipoamide S-succinyltransferase | −1,724 |
| FKBP4 | FK506 binding protein 4 | −1,558 |
| GADD45GIP1 | GADD45G interacting protein 1 | −1,664 |
| GHITM | Growth hormone inducible transmembrane protein | −1,477 |
| HSPB7 | Heat shock protein family B (small) member 7 | −1,422 |
| MECP2 | Methyl-CpG binding protein 2 | −1,22 |
| MPC1 | Mitochondrial pyruvate carrier 1 | −1,339 |
| MRPL33 | Mitochondrial ribosomal protein L33 | −1,333 |
| MRS2 | MRS2, magnesium transporter | −1,416 |
| NDUFS3 | NADH: ubiquinone oxidoreductase core subunit S3 | −1,314 |
| NMT1 | N-myristoyltransferase 1 | −1,443 |
| NOL7 | Nucleolar protein 7 | −1,299 |
| PANK2 | Pantothenate kinase 2 | −1,361 |
| PCCB | Popionyl-CoA carboxylase beta subunit | −1,335 |
| PDHA1 | Pyruvate dehydrogenase (lipoamide) alpha 1 | −1,524 |
| PGS1 | Phosphatidylglycerophosphate synthase 1 | −1,233 |
| SDHC | Succinate dehydrogenase complex subunit C | −1,23 |
| SLC25A12 | Solute carrier family 25 member 12 | −1,408 |
| SLC25A4 | Solute carrier family 25 member 4 | −1,55 |
| TIMM23 | Translocase of inner mitochondrial membrane 23 | −1,508 |
| UQCC1 | Ubiquinol-cytochrome c reductase complex assembly factor 1 | −1,517 |
| UQCRFS1 | Ubiquinol-cytochrome c reductase, Rieske iron-sulfur polypeptide 1 | −1,292 |
Table 2.
Targets of miR-155-5p significantly downregulated in DS hearts and belonging to gene ontology cellular component category “mitochondrion.” Eight genes were observed instead of the expected 2.34 genes with P < 0.00001.
| Gene Symbol | Description | FC |
|---|---|---|
| DLAT | Dihydrolipoamide S-acetyltransferase | −1,678 |
| HSPB7 | Heat shock protein family B (small) member 7 | −1,422 |
| LPIN1 | Lipin 1 | −1,838 |
| MECP2 | Methyl-CpG binding protein 2 | −1,22 |
| NDUFS3 | NADH: ubiquinone oxidoreductase core subunit S3 | −1,314 |
| NT5C | 5′, 3′-nucleotidase, cytosolic | −1,524 |
| SIRT5 | Sirtuin 5 | −1,255 |
| YWHAQ | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein theta | −1,364 |
We focused our attention on the SLC25A4/ANT1 gene (indicated in bold in Table 1), which was predicted by 5 databases as a let-7c-5p target gene and already proposed as a target of both human and murine let-7b [35–37]. Its downregulation in heart tissue was confirmed by qRT-PCR (Supplementary Figure 1). According to the STarMirDB, a database of microRNA binding sites based on cross-linking immunoprecipitation (CLIP) data (http://sfold.wadsworth.org/starmirDB.php) [38], seven binding sites for let-7c-5p are located in the SLC25A4/ANT1 sequence. One of them has been mapped to a conserved 3'UTR seed site (seed type 7mer-A1) [39] with a logistic probability of 0.68.
We were interested in this gene because of its central role in oxidative phosphorylation (OXPHOS). Our hypothesis was that the SLC25A4/ANT1 downregulation could be an effect of the specific interaction between let-7c-5p and this gene at the seed sequence. To validate this hypothesis, we performed a transfection experiment of a let-7c-5p mimic miRNA in euploid fibroblasts in which SLC25A4/ANT1 is expressed. As control, we evaluated also the expression of DICER, since it is a let-7c-5p validated target [40]. We transfected fibroblasts at different concentrations of miRNA mimic for 48 h, and we measured SLC25A4/ANT1, DICER, and GAPDH expression by end point PCR. As shown in Figure 3, at 25 nM miRNA dosage (lanes 13-14), a reduction of target amplification is already appreciable with respect to nontransfected cells (lanes 1-2) or cells transfected with transfection reagent only (lanes 4-5). As expected, GAPDH amplification did not show variations.
Figure 3.

End point PCR of SLC25A4/ANT1, DICER, and GAPDH in euploid fibroblasts transfected with a let-7c-5p mimic. (a) Agarose gel of SLC25A4/ANT1, DICER, and GAPDH amplification in: Not transfected cells (lanes 1–3); cells + INTERFERin (lanes 4–6); cells + 5 nM miRNA mimic (lanes 7–9); cells + 10 nM miRNA mimic (lanes 10–12); cells + 25 nM miRNA mimic (lanes 13–15); cells + 50 nM miRNA mimic (lanes 16–18). Amplification products of SLC25A4/ANT1: lanes 1, 4, 7, 10, 13, and 16; amplification products of DICER: lanes 2, 5, 8, 11, 14, and 17; amplification products of GAPDH: lanes 3, 6, 9, 12, 15, and 18. (b) Densitometric analysis of SLC25A4/ANT1 and DICER amplification products obtained by ImageJ software. At 25 nM miRNA mimic concentration, a reduction of target amplification is appreciable.
We further evaluated SLC25A4/ANT1 and DICER expression by qRT-PCR showing that after let-7c mimic transfection (25 nM), their expression significantly decreased if compared with cells transfected with the transfecting agent only (Figure 4).
Figure 4.
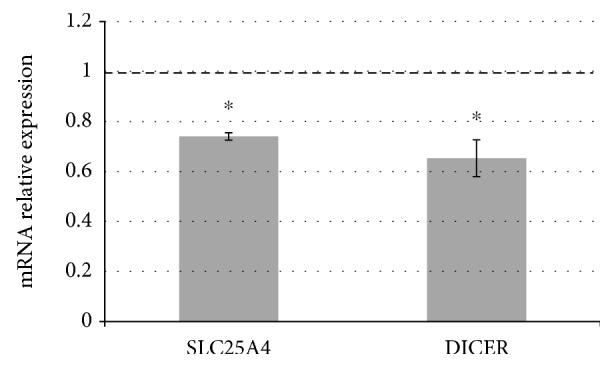
SLC25A4/ANT1 and DICER expression by qRT-PCR. Transfection of let-7c-5p mimic induces a significant decrease of SLC25A4/ANT1 and DICER expression in euploid fibroblasts. Results are expressed as relative mean values ± SEM of three different determination, compared with cells treated only with transfecting agent (set equal to 1). ∗P < 0.05.
Finally, we performed the reciprocal experiment using a let-7c-5p inhibitor in trisomic fibroblasts collecting both RNA and proteins. After treatment with 25 nM inhibitor, which reduced let-7c-5p expression by 35%, the expression of SLC25A4/ANT1 was significantly reduced both at mRNA (a) and protein (b-c) level (Figure 5).
Figure 5.

SLC25A4/ANT1 after let-7c-5p inhibition in trisomic fibroblasts. (a) Transfection of let-7c-5p inhibitor in DS-HFFs induces a significant increase of SLC25A4/ANT1 expression and a decrease of let-7c-5p, as detected by qRT-PCR. Results are expressed as relative mean values ± SEM of three different determination, compared with cells treated only with transfecting agent (set equal to 1). (b) Representative immunoblot of SLC25A4/ANT1 in nontransfected cells (NT), cells transfected only with transfecting agent and cells transfected with 25 nM let-7c-5p inhibitor. Vinculin was used as loading control. (c) Densitometric SLC25A4/ANT1 relative measurement after 72 h treatment. Protein expression was significantly increased after inhibiting treatment if compared with mock transfected cells. The bars show relative mean values ± SEM of two DS-HFFs in duplicate; ∗P < 0.05.
4. Discussion
We have analyzed the expression of 5 Hsa21 miRNAs in trisomic fetal hearts at 18–22 gestational weeks to investigate whether they are affected by the gene dosage effect observed for most of the Hsa21 genes. Three out of five, namely let-7c-5p, miR-99a-5p, and miR-155-5p, were significantly overexpressed, if compared with euploid controls. The upregulation was not higher than 2.5 folds in agreement with a gene dosage effect. In the same samples, miR-802-5p was not expressed and miR-125b-2-5p was normoregulated. This was not surprising as the miR-125b-1-5p, which maps to 11q24.1, encoding an identical miRNA, may mask the eventual overexpression.
Comparison between hearts from fetuses with cardiopathies versus hearts from fetuses without cardiopathies showed that miR-99a-5p was more expressed in the latter samples even though more samples are needed to reach a significant result due to a high variability.
In a recent paper, the role of mmu-let-7c and mmu-miR-99a in cardiomyogenesis was investigated using an overexpression strategy in murine embryonic stem cells. By overexpressing these miRNAs at early stage of differentiation, the authors demonstrated that they are involved in heart development as let-7c induces cardiogenesis while miR-99a appears to repress it by altering the Smad2 signaling [18].
The overexpression of Hsa21 miRNAs can result in downregulation of specific target genes possibly involved in DS phenotype. To select relevant miRNA targets we have used the following criteria:
Bioinformatics prediction
mRNA decreased expression in heart samples from DS fetuses
Inclusion in the category of mitochondria related genes
Involvement in normal and/or abnormal heart development
Among the selected genes, SLC25A4/ANT1, a predicted let-7c-5p target downregulated in trisomic hearts, appeared as a potential candidate for both mitochondrial dysfunction and CHD in DS. SLC25A4 (Solute carrier family 25 member 4) or ANT1 (Adenine nucleotide translocator 1) functions as a gated pore that translocates ADP and ATP between cytoplasm and mitochondria, regulating the intracellular energetic balance. Furthermore, its dysregulation has been associated to mitochondrial cardiomyopathies [41].
Humans have four ANT isoforms that are encoded by four different genes and are distributed in a tissue specific pattern. The human SLC25A4/ANT1 gene is primarily expressed in the heart and in the skeletal muscle [42]. It is downregulated in DS fetal hearts and fibroblasts in which let-7c-5p is upregulated. qRT-PCR after let-7c-5p overexpression showed an inverse relationship with SLC25A4/ANT1, which was downregulated. The reverse experiment using miRNA inhibitor in trisomic cells demonstrated a significant increase of RNA and protein expression of the gene while let-7c-5p expression was decreased if compared with mock controls. These results may be considered an additional experimental support of our hypothesis even though further experiments will be necessary to definitely validate that SLC25A4/ANT1 is a let-7c-5p target, which is beyond the purpose of this paper. To this aim, we plan to perform a luciferase assay to demonstrate a direct interaction.
OXPHOS deficiency and mitochondrial dysfunction have been associated with developmental mechanisms of DS [43]. SLC25A4/ANT1 is one of the 37 NEMGs downregulated after NRIP1 overexpression in the GEO GSE 19836 experiment [38] that we reanalyzed in Izzo et al. [22]. After NRIP1 silencing in trisomic cells [23], SLC25A4/ANT1 expression increased, thus allowing a more efficient exchange of ATP and an improvement of mitochondrial activity in DS samples. Accordingly, genetic inactivation of the heart isoform results in mtDNA damage and increased reactive oxygen species [44].
The above described strategy to select targets relevant for mitochondrial function and heart development was applied also to the miR-155-5p. Thirty-three predicted target genes of miR-155-5p have been found downregulated in trisomic fetal hearts if compared with euploid ones. Eight of them, including MECP2 gene, which is a validated target [37, 45], are included in the cell component GO category “mitochondrion” suggesting an impact of this miRNA on mitochondrial function. MECP2 is included also among the let-7c-5p predicted targets.
It was recently reported that the Hsa21 miR-155-5p regulates mitochondrial biogenesis by targeting mitochondrial transcription factor A (TFAM) [46]. Surprisingly, after studying both DS fibroblasts and heart samples, the authors conclude that the regulation of TFAM by the miRNA impacts mitochondrial biogenesis in the euploid setting but not in the DS setting. We suggest that a possible explanation may lie in the fact that mitochondrial biogenesis is already so decreased in DS cells to mask further defects. TFAM was not identified by our strategy because its heart expression at 18–22 gestational weeks is too low to be analyzed by microarray technology.
Finally miR-155-5p has been hypothesized to be an inducer of cardiac hypertrophy [47]. Its inhibition might have clinical potential to counteract this pathology.
5. Conclusions
In this study, we demonstrated the overexpression of three Hsa21 miRNAs, miR-99a-5p, miR-155-5p, and let-7c-5p, in the heart of trisomic fetuses as a likely consequence of gene dosage effect. Bioinformatic analysis demonstrated that two of these miRNAs, let-7c-5p and miR-155-5p, count several predicted targets among genes involved in mitochondrial function that were found to be downregulated in trisomic fetal hearts. Among the let-7c predicted targets, we identified and partially validated SLC25A4/ANT1, a gene encoding the main translocator of ADP/ATP across the mitochondrial membrane, which plays a major role in mitochondrial function.
Our results also support the hypothesis that the overexpression of miR-155-5p might have a potential impact on mitochondrial biogenesis.
Both miRNAs, let-7c-5p and miR-155-5p, should be taken into account while investigating the molecular mechanisms causing cardiac malformations in DS.
Supplementary Material
Table 1. Targets of let-7c significantly downregulated in DS fetal hearts with a (FC)>|1,2| and p-value<0,05. Number of predictions by different databases are also indicated. Table 2. Targets of miR-155 significantly downregulated in DS fetal hearts with a (FC)>|1,2| and p-value<0,05. Number of predictions by different databases are also indicated. Table 3. Targets of miR-99a significantly downregulated in DS fetal hearts with a (FC)>|1,2| and p-value<0,05. Number of predictions by different databases are also indicated. Figure 1. SLC25A4/ANT1 expression in heart tissue by qRT-PCR. Real-time PCR of SLC25A4/ANT1 in trisomic hearts confirmed the downregulation obtained by microarray analysis. Results are expressed as relative mean values ± SEM of 3 trisomic samples (DSH), compared with control hearts (NH) set equal to 1. ∗∗ p <0.01. P-value expresses statistical significance for trisomic versus non trisomic sample comparisons.
Acknowledgments
The authors thank Mario Senesi for technical support. This work was supported by POR Campania FSE 2007-2013, Campania FSE 2014-2020, and Project CREME from Campania Region to Lucio Nitsch.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
Antonella Izzo and Rosanna Manco are joint first authors.
References
- 1.Park S. C., Mathews R. A., Zuberbuhler J. R., Rowe R. D., Neches W. H., Lenox C. C. Down syndrome with congenital heart malformation. American Journal of Diseases of Children. 1977;131(1):29–33. doi: 10.1001/archpedi.1977.02120140031003. [DOI] [PubMed] [Google Scholar]
- 2.Ferencz C., Neill C. A., Boughman J. A., Rubin J. D., Brenner J. I., Perry L. W. Congenital cardiovascular malformations associated with chromosome abnormalities: an epidemiologic study. The Journal of Pediatrics. 1989;114(1):79–86. doi: 10.1016/S0022-3476(89)80605-5. [DOI] [PubMed] [Google Scholar]
- 3.Korenberg J. R., Chen X. N., Schipper R., et al. Down syndrome phenotypes: the consequences of chromosomal imbalance. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(11):4997–5001. doi: 10.1073/pnas.91.11.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barlow G. M., Chen X. N., Shi Z. Y., et al. Down syndrome congenital heart disease: a narrowed region and a candidate gene. Genetics in Medicine. 2001;3(2):91–101. doi: 10.1097/00125817-200103000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Liu C., Belichenko P. V., Zhang L., et al. Mouse models for Down syndrome-associated developmental cognitive disabilities. Developmental Neuroscience. 2011;33(5):404–413. doi: 10.1159/000329422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu C., Morishima M., Jiang X., et al. Engineered chromosome-based genetic mapping establishes a 3.7 Mb critical genomic region for Down syndrome-associated heart defects in mice. Human Genetics. 2014;133(6):743–753. doi: 10.1007/s00439-013-1407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conti A., Fabbrini F., D'Agostino P., et al. Altered expression of mitochondrial and extracellular matrix genes in the heart of human fetuses with chromosome 21 trisomy. BMC Genomics. 2007;8:p. 268. doi: 10.1186/1471-2164-8-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ambros V., Chen X. The regulation of genes and genomes by small RNAs. Development. 2007;134(9):1635–1641. doi: 10.1242/dev.002006. [DOI] [PubMed] [Google Scholar]
- 9.Zhao Y., Ransom J. F., Li A., et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129(2):303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 10.Yang B., Lin H., Xiao J., et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nature Medicine. 2007;13(4):486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 11.Care A., Catalucci D., Felicetti F., et al. MicroRNA-133 controls cardiac hypertrophy. Nature Medicine. 2007;13(5):613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 12.van Rooij E., Olson E. N. microRNAs put their signatures on the heart. Physiological Genomics. 2007;31(3):365–366. doi: 10.1152/physiolgenomics.00206.2007. [DOI] [PubMed] [Google Scholar]
- 13.Letourneau A., Antonarakis S. E. Genomic determinants in the phenotypic variability of Down syndrome. Progress in Brain Research. 2012;197:15–28. doi: 10.1016/B978-0-444-54299-1.00002-9. [DOI] [PubMed] [Google Scholar]
- 14.Elton T. S., Sansom S. E., Martin M. M. Trisomy-21 gene dosage over-expression of miRNAs results in the haploinsufficiency of specific target proteins. RNA Biology. 2010;7(5):540–547. doi: 10.4161/rna.7.5.12685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sethupathy P., Borel C., Gagnebin M., et al. Human microRNA-155 on chromosome 21 differentially interacts with its polymorphic target in the AGTR1 3′ untranslated region: a mechanism for functional single-nucleotide polymorphisms related to phenotypes. American Journal of Human Genetics. 2007;81(2):405–413. doi: 10.1086/519979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Connell R. M., Rao D. S., Chaudhuri A. A., et al. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. The Journal of Experimental Medicine. 2008;205(3):585–594. doi: 10.1084/jem.20072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ozen M., Creighton C. J., Ozdemir M., Ittmann M. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene. 2008;27(12):1788–1793. doi: 10.1038/sj.onc.1210809. [DOI] [PubMed] [Google Scholar]
- 18.Coppola A., Romito A., Borel C., et al. Cardiomyogenesis is controlled by the miR-99a/let-7c cluster and epigenetic modifications. Stem Cell Research. 2014;12(2):323–337. doi: 10.1016/j.scr.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 19.Wu X., Gong Z., Sun L., Ma L., Wang Q. MicroRNA-802 plays a tumour suppressive role in tongue squamous cell carcinoma through directly targeting MAP2K4. Cell Proliferation. 2017;50(3) doi: 10.1111/cpr.12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malinge S., Izraeli S., Crispino J. D. Insights into the manifestations, outcomes, and mechanisms of leukemogenesis in Down syndrome. Blood. 2009;113(12):2619–2628. doi: 10.1182/blood-2008-11-163501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piccoli C., Izzo A., Scrima R., et al. Chronic pro-oxidative state and mitochondrial dysfunctions are more pronounced in fibroblasts from Down syndrome foeti with congenital heart defects. Human Molecular Genetics. 2013;22(6):1218–1232. doi: 10.1093/hmg/dds529. [DOI] [PubMed] [Google Scholar]
- 22.Izzo A., Manco R., Bonfiglio F., et al. NRIP1/RIP140 siRNA-mediated attenuation counteracts mitochondrial dysfunction in Down syndrome. Human Molecular Genetics. 2014;23(16):4406–4419. doi: 10.1093/hmg/ddu157. [DOI] [PubMed] [Google Scholar]
- 23.Schindelin J., Arganda-Carreras I., Frise E., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reczko M., Maragkakis M., Alexiou P., Grosse I., Hatzigeorgiou A. G. Functional microRNA targets in protein coding sequences. Bioinformatics. 2012;28(6):771–776. doi: 10.1093/bioinformatics/bts043. [DOI] [PubMed] [Google Scholar]
- 25.Nam J. W., Rissland O. S., Koppstein D., et al. Global analyses of the effect of different cellular contexts on microRNA targeting. Molecular Cell. 2014;53(6):1031–1043. doi: 10.1016/j.molcel.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kertesz M., Iovino N., Unnerstall U., Gaul U., Segal E. The role of site accessibility in microRNA target recognition. Nature Genetics. 2007;39(10):1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 27.Bandyopadhyay S., Mitra R. TargetMiner: microRNA target prediction with systematic identification of tissue-specific negative examples. Bioinformatics. 2009;25(20):2625–2631. doi: 10.1093/bioinformatics/btp503. [DOI] [PubMed] [Google Scholar]
- 28.Wong N., Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Research. 2015;43(Database issue):D146–D152. doi: 10.1093/nar/gku1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miranda K. C., Huynh T., Tay Y., et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126(6):1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 30.Krek A., Grun D., Poy M. N., et al. Combinatorial microRNA target predictions. Nature Genetics. 2005;37(5):495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 31.Betel D., Koppal A., Agius P., Sander C., Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biology. 2010;11(8, article R90) doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durinck S., Spellman P. T., Birney E., Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nature Protocols. 2009;4(8):1184–1191. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J., Duncan D., Shi Z., Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Research. 2013;41(Web Server issue):W77–W83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brennecke J., Stark A., Russell R. B., Cohen S. M. Principles of microRNA-target recognition. PLoS Biology. 2005;3, article e85:p. 3. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Helwak A., Kudla G., Dudnakova T., Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153(3):654–665. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chi S. W., Zang J. B., Mele A., Darnell R. B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460(7254):479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chou C. H., Chang N. W., Shrestha S., et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Research. 2016;44(D1):D239–D247. doi: 10.1093/nar/gkv1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rennie W., Kanoria S., Liu C., et al. STarMirDB: a database of microRNA binding sites. RNA Biology. 2016;13(6):554–560. doi: 10.1080/15476286.2016.1182279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bartel D. P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tokumaru S., Suzuki M., Yamada H., Nagino M., Takahashi T. let-7 regulates Dicer expression and constitutes a negative feedback loop. Carcinogenesis. 2008;29(11):2073–2077. doi: 10.1093/carcin/bgn187. [DOI] [PubMed] [Google Scholar]
- 41.Palmieri L., Alberio S., Pisano I., et al. Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Human Molecular Genetics. 2005;14(20):3079–3088. doi: 10.1093/hmg/ddi341. [DOI] [PubMed] [Google Scholar]
- 42.Li K., Warner C. K., Hodge J. A., et al. A human muscle adenine nucleotide translocator gene has four exons, is located on chromosome 4, and is differentially expressed. The Journal of Biological Chemistry. 1989;264(24):13998–14004. [PubMed] [Google Scholar]
- 43.Arbuzova S., Hutchin T., Cuckle H. Mitochondrial dysfunction and Down’s syndrome. BioEssays. 2002;24(8):681–684. doi: 10.1002/bies.10138. [DOI] [PubMed] [Google Scholar]
- 44.Subramaniam V., Golik P., Murdock D. G., et al. MITOCHIP assessment of differential gene expression in the skeletal muscle of Ant1 knockout mice: coordinate regulation of OXPHOS, antioxidant, and apoptotic genes. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2008;1777(7-8):666–675. doi: 10.1016/j.bbabio.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bofill-De Ros X., Santos M., Vila-Casadesus M., et al. Genome-wide miR-155 and miR-802 target gene identification in the hippocampus of Ts65Dn Down syndrome mouse model by miRNA sponges. BMC Genomics. 2015;16:p. 907. doi: 10.1186/s12864-015-2160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quinones-Lombrana A., Blanco J. G. Chromosome 21-derived hsa-miR-155-5p regulates mitochondrial biogenesis by targeting mitochondrial transcription factor A (TFAM) Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2015;1852(7):1420–1427. doi: 10.1016/j.bbadis.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seok H. Y., Chen J., Kataoka M., et al. Loss of MicroRNA-155 protects the heart from pathological cardiac hypertrophy. Circulation Research. 2014;114(10):1585–1595. doi: 10.1161/CIRCRESAHA.114.303784. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table 1. Targets of let-7c significantly downregulated in DS fetal hearts with a (FC)>|1,2| and p-value<0,05. Number of predictions by different databases are also indicated. Table 2. Targets of miR-155 significantly downregulated in DS fetal hearts with a (FC)>|1,2| and p-value<0,05. Number of predictions by different databases are also indicated. Table 3. Targets of miR-99a significantly downregulated in DS fetal hearts with a (FC)>|1,2| and p-value<0,05. Number of predictions by different databases are also indicated. Figure 1. SLC25A4/ANT1 expression in heart tissue by qRT-PCR. Real-time PCR of SLC25A4/ANT1 in trisomic hearts confirmed the downregulation obtained by microarray analysis. Results are expressed as relative mean values ± SEM of 3 trisomic samples (DSH), compared with control hearts (NH) set equal to 1. ∗∗ p <0.01. P-value expresses statistical significance for trisomic versus non trisomic sample comparisons.