

Abstract
A method for the regioselective arylboration of isoprene and its derivatives is presented. These reactions allow for the synthesis of useful building blocks from simple components. Through these studies, an unusual additive effect with DMAP has been uncovered that allows for altered reactivity and the formation of quaternary carbon centers. The utility of this method is demonstrated toward the formal synthesis of mesembrine.
Due to the large-scale production (1 million tons produced/year) and low cost of isoprene, conversion to more complex small molecules with additional functionality represents an attractive process for chemical synthesis.1 In addition, due to the presence of isoprene units in natural products (e.g., terpenes) and biologically active molecules, isoprene-derived small molecules constitute useful building blocks for the construction of these targets. As such, numerous methods exist for the functionalization of isoprene, the majority of which are hydrofunctionalization processes.2 Difunctionalization of isoprene is less common but significant as the products incorporate an additional group (compared to hydrofunctionalization).3–5 At the outset of our studies, a recent report by Sigman et al. represented the current state-of-the-art for three-component couplings of isoprene (Scheme 1A).5e
Scheme 1.

Difunctionalization of Isoprene
Our group has taken an interest in carboboration processes that operate by Pd/Cu cooperative catalysis.6–8 Accordingly, we have developed methods for the arylboration of styrene derivatives (note that Semba and Nakao independently developed a closely related process).9,10 The reactions function by addition of a Cu–Bpin complex across an alkene followed by Pd-catalyzed cross-coupling of the generated Csp3–Cu complex. We became interested in extending this process to the arylboration of isoprene (Scheme 1B, C). Realization of such a process would allow for the formation of synthetically versatile products. It should be noted that during the final stages of this study, two reports appeared for the carboboration of isoprene with alkylidene malonates8a and imines.8e
It was reasoned that selective arylboration for the formation of any regioisomer would be valuable; however, development of such a transformation is challenging due to the potential formation of up to four regioisomeric products (Scheme 1C). The regioselectivity of the reaction is established at two distinct stages. First, the initial addition of Cu–Bpin across isoprene can generate two possible adducts, 5 and 6.11 Even if this process can be controlled with judicious choice of ligand and reaction conditions, the subsequent Pd-catalyzed cross coupling can lead to the formation of up to four regioisomers. This event is undoubtedly controlled by the nature of both the Pd catalyst and Cu complex. Here we disclose a method for the regioselective arylboration of isoprene in which, with different conditions, either a 1,4-product or a 2,1-product can be generated (Scheme 1B). With respect to the latter process, we also have uncovered an unusual additive effect with 4-(dimethylamino)pyridine (DMAP).
Initial studies found that reaction of isoprene (1) under conditions similar to those previously identified for arylboration of styrenes led to the formation of 7, 8, and 9 in a 3:1:2 ratio (Table 1, entry 1).6 It was reasoned that the formation of 7 and 8 resulted from Cu complex 5 whereas the remaining product 9 is formed from the regioisomeric complex 6. It was reasoned that the regioselectivity could be increased if a more sterically encumbered NHC was used such that reaction at the less substituted alkene of isoprene would be favored. This ultimately led to the identification of It-BuCuBr as a catalyst, which allowed for formation of only 7 and 8 suggesting that the borylcupration step could be controlled (Table 1, entry 2). Since the regioselectivity of borylcupration had been overcome, Pd catalysts were evaluated to increase the selectivity of the transmetalation. This initiative led to the discovery that Pd-QPhos-G312 allowed for highly selective synthesis of 7 (vs 8 or 9) as a 9:1 mixture of Z- and E-isomers (Table 1, entry 5).
Table 1.
Optimization of Reactions Conditions
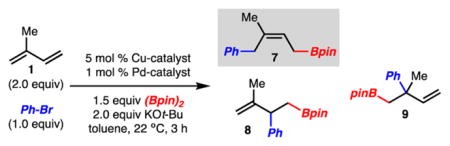
| ||||
|---|---|---|---|---|
| entry | Cu catalyst | Pd catalyst | 7:8:9 | yield of 7 (%)a |
| 1b | IMesCuCl | Pd-Pt-Bu3-G3 | 3:1:2 | 24 |
| 2b | It-BuCuBr | Pd-Pt-Bu3-G3 | 11:1:<0.1 | 42 |
| 3 | It-BuCuBr | Pd-Pt-Bu3-G3 | 11:1:<0.1 | 57 |
| 4 | It-BuCuBr | Pd-Pt-Bu2CH2t-Bu-G3 | 3:1:< 0.1 | 47 |
| 5 | It-BuCuBr | Pd-QPhos-G3 | 15:1:<0.1 | 77 |
| 6c | It-BuCuBr | Pd-QPhos-G3 | 10:1:<0.1 | 58 |
Yield and product ratios determined by 1H NMR analysis of the crude reaction mixture with an internal standard.
Reaction run with 1.5 equiv of KOt-Bu instead of 2 equiv.
Reaction run with NaOt-Bu instead of KOt-Bu.
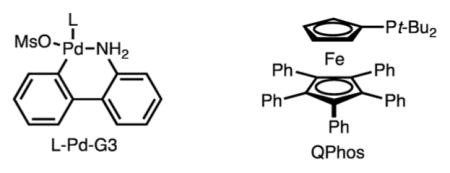
With an optimized set of conditions in hand, the scope of this process was evaluated. Several points are noteworthy (Scheme 2): (1) Electron-rich and electron-poor aryl bromides can be used (products 11, 12 and 10, 13, respectively). (2) Sterically hindered aryl bromides functioned less well in this process, likely due to adverse steric interactions during the putative transmetalation (product 15). (3) Vinyl bromides were found to be competent; however, slightly lower diastereoselectivities (Z:E) are observed relative to those achieved with aryl bromides (product 16). (4) In all cases, while good yields of the allylboronic ester were observed, the reaction products were typically oxidized to allow for more facile purification. (5) The related terpene, myrcene, also functioned well under the reaction conditions (products 17 and 18).
Scheme 2. Substrate Scope.

a Yield and selectivity of the allylboronate determined by analysis of the crude 1H NMR with an internal standard. bYield (two steps from R2Br) of isolated alcohol after oxidation (only a mixture of alkene isomers). c9:1 product:other isomers. d8:1 product:other isomers. e4.4:1 product:other isomers.
During the course of our investigations, the observation was made that reactions involving pyridine derivatives, such as 19, gave rise to small quantities of the 2,1-adduct 22 (Scheme 3). The formation of the 2,1-arylboration product 9 was observed previously with reactions promoted by IMesCuCl (Table 1, entry 1). However, optimization of the reaction for selective formation of 9 was unsuccessful. Since pyridine derivatives were clearly affecting the selectivity of these reactions, further studies were carried out based on the result outlined in Scheme 3.
Scheme 3.

Formation of 2,1-Arylboration Product with Bromopyridine
We reasoned that the Lewis basic nitrogen of the pyridine was allowing for the altered reactivity. To test this hypothesis, reactions with isoprene and PhBr were carried out under standard conditions shown in Scheme 2, except that various pyridine derivatives were added. Initially, when pyridine was used as an additive, the formation of 9 was observed, albeit in low yield as the minor isomer (Table 2, entry 1). Further evaluation of pyridine derivatives led to the finding that DMAP allowed for generation of 9 as the major product. Standard optimization of conditions and assessment of other sterically large phosphine ligands led to improved reaction yields (Table 2, entry 6). Finally, it was found that increased equivalents of DMAP (2.0 vs 1.0) allowed for formation of 9 as the exclusive product in 61% yield as judged by 1H NMR analysis (Table 2, entry 8). Control experiments were also carried out that suggested DMAP is not simply replacing It-Bu or Pt-Bu2CH2t-Bu from either Cu or Pd, respectively (Table 2, entries 9 and 10).
Table 2.
Evaluation of Pyridine Derivatives as Additives
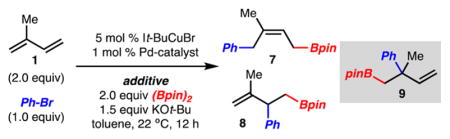
| ||||
|---|---|---|---|---|
| entry | additive/equiv | Pd catalyst | 7:8:9a | yield of 9 (%)a |
| 1 | pyridine/1.0b | Pd-QPhos-G3 | 6:1:1 | 5 |
| 2 | DMAP/1.0b | Pd-QPhos-G3 | 1:1:4 | 17 |
| 3 | DMAP/1.0 | Pd-QPhos-G3 | 2:1:3 | 34 |
| 4 | DMAP/1.0 | Pd-Pt-Bu3-G3 | 4:1:2 | 14 |
| 5 | DMAP/1.0 | Pd-APhos-G3 | 1:1:5 | 27 |
| 6 | DMAP/1.0 | Pd-Pt-Bu2CH2t-Bu-G3 | 1:1:11 | 68 |
| 7 | DMAP/1.0c | Pd-Pt-Bu2CH2t-Bu-G3 | 2:1:13 | 67 |
| 8 | DMAP/2.0c | Pd-Pt-Bu2CH2t-Bu-G3 | 1:1:>20 | 61 |
| 9 | DMAP/2.0c | [Pd-G3]2 | n.d. | <2 |
| 10 | DMAP/2.0c,d | Pd-Pt-Bu2CH2t-Bu-G3 | n.d. | <2 |
Yield and (1,2):(1,4) determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard.
Reaction run with 1.5 equiv of (Bpin)2 and 2.0 equiv of KOt-Bu.
Reaction run at 45 °C instead of 22 °C.
Reaction run with 5 mol% CuBr instead of 5 mol% It-BuCuBr. n.d. = not determined
The scope of the 2,1-arylboration reactions was investigated and found to accommodate electron-rich (products 24, 25, 28), electron-poor (products 26 and 27), and sterically hindered aryl bromides (products 23, 24) (Scheme 4). In addition, myrcene could also be substituted for isoprene with little loss in efficiency (products 30 and 31). In general, the 2,1-arylboration products were formed in high selectivity (>10:1 product:other isomers); they can also be converted to other structures due to the versatility of the C–B bond.13 Illustrated in Scheme 5 are two representative examples of C–C bond formation.
Scheme 4. Substrate Scope.

a Yield of isolated product (>20:1 product:other isomers) after silica gel column chromatography. bYield determined by 1H NMR analysis of the crude reaction mixture with an internal standard. c8:1 product:other isomers. d9:1 product:other isomers. e2:1 product:other isomers. fYield reported for two steps after oxidation to alcohol, 16:1 product:other isomers, see the SI for details.
Scheme 5.

Representative Functionalizations
Cyclic 1,2-disubstituted diene 34 was also tolerated (Scheme 6). In this example, 36 was generated in high selectivity with formation of the anti-diastereomer. Boronic ester 36 could be easily converted to 37 through a hydroboration–oxidation sequence,14 which can be transformed to mesembrine (38) through established protocols.15,16 Several additional examples of arylboration (products 40 and 42) are also shown in Scheme 6. Highly stereoselective synthesis of 40 demonstrates the effect of an existing stereocenter, while preparation of 42 showcases heterocycle functionalization.
Scheme 6.

Stereoselective Arylboration
Preliminary mechanistic investigations of the reaction have been carried out. To probe the regioselectivity of the migratory insertion, isoprene was treated with equimolar quantities of It-BuCuBr, KOt-Bu and (Bpin)2 (Scheme 7A). This reaction led to the formation of 43 and 44 in a 3:1 ratio in 98% yield. It should be noted that 43 and 44 are formed at roughly the same rate.17 Treatment of this mixture with Pd-QPhos-G3 and PhBr gave rise to 7 and 45 in a 3:1 ratio and 75% yield (also note that 7 and 45 are produced at approximately the same rate).17 This result is surprising as under the optimized catalytic reaction conditions, <2% of isomer 45 is formed. The above data suggests that either 43 and 44 undergo interconversion, or 43 is formed selectively under the catalytic reaction conditions. With respect to the former we have not been able to provide evidence of this interconversion through crossover experiments (this scenario is also unlikely as qualitative rate measurements for formation and consumption of 43 and 44 are nearly the same, as mentioned previously).17 Regarding the latter, we probed the transient formation of 43 and 44 by running the catalytic reaction in the presence of t-BuOH, as protonation of allylcopper intermediates is known to be rapid.18 In this case only the expected arylboration product 7 and the protoboration adduct (46) derived from 43 were observed. This suggests that 44 was not generated under the catalytic reaction conditions. The apparent divergence in selectivity for the stoichiometric and catalytic reactions is unclear at this time.
Scheme 7.

Mechanistic Studies
With respect to the mechanism of the 2,1-arylboration reaction, DMAP was found to not inhibit or alter in any way the stoichiometric formation of 43 and 44. Furthermore, the mixture of 43 and 44 could not be converted to the 2,1-arylboration products, despite numerous attempts (Scheme 7A).17 In addition, a protoboration experiment in the presence of DMAP (Scheme 7B) also resulted in the exclusive formation of 46. This data suggests that 43 was generated under the 2,1-arylboration reaction conditions; however, its potential role as a catalytic intermediate on the pathway to product 9 remains unclear. Altogether, it is evident that the mechanisms of the both 1,4- and 2,1-arylboration reactions are quite complex and further investigations are in progress.
Finally, for formation of Z-crotyl metal Cu complexes 43 and 44, two possibilities are proposed: (1) it has been established that Z-crotyl metal complexes are more stable relative to the E-crotyl metal complexes,19 and (2) insertion of 1,3-dienes into organometal bonds occurs preferentially in the s-cis conformation to give rise to Z-crotyl metal complexes.20
In conclusion, a process for the regioselective arylboration of isoprene and its derivatives is presented. DMAP has been shown to alter the normal reactivity of the system. Future efforts aim to further investigate the role of DMAP and develop enantioselective variants.
Supplementary Material
Acknowledgments
We thank Indiana University and the NIH (5R01GM114443) for generous financial support.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b04024.
Experimental procedures and analytical data (PDF)
References
- 1.Morais ARC, Dworakowska S, Reis A, Gouveia L, Matos CT, Bogdał D, Bogel-Łukasik R. Catal Today. 2015;239:38. [Google Scholar]
- 2.For selected recent examples, see: Schmitt DC, Lee J, Dechert-Schmitt AMR, Yamaguchi E, Krische MJ. Chem Commun. 2013;49:6096. doi: 10.1039/c3cc43463j.Parker SE, Börgel J, Ritter T. J Am Chem Soc. 2014;136:4857. doi: 10.1021/ja5008596.Timsina YN, Sharma RK, RajanBabu TV. Chem Sci. 2015;6:3994. doi: 10.1039/c5sc00929d.
- 3.Corey EJ. Angew Chem, Int Ed. 2009;48:2100. doi: 10.1002/anie.200805374. [DOI] [PubMed] [Google Scholar]
- 4.Cho HY, Morken JP. Chem Soc Rev. 2014;43:4368. doi: 10.1039/c3cs60482a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For selected examples, see: Obora Y, Tsuji Y, Kawamura T. J Am Chem Soc. 1995;117:9814. doi: 10.1021/ja010674p.Ishiyama T, Yamamoto M, Miyaura N. Chem Commun. 1996:2073.Cho HY, Morken JP. J Am Chem Soc. 2008;130:16140. doi: 10.1021/ja806113v.Geary LM, Glasspoole BW, Kim MM, Krische MJ. J Am Chem Soc. 2013;135:3796. doi: 10.1021/ja400691t.McCammant MS, Sigman MS. Chem Sci. 2015;6:1355. doi: 10.1039/c4sc03074e.
- 6.(a) Smith KB, Logan KM, You W, Brown MK. Chem - Eur J. 2014;20:12032. doi: 10.1002/chem.201404310. [DOI] [PubMed] [Google Scholar]; (b) Logan KM, Smith KB, Brown MK. Angew Chem, Int Ed. 2015;54:5228. doi: 10.1002/anie.201500396. [DOI] [PubMed] [Google Scholar]; (c) Logan KM, Brown MK. Angew Chem, Int Ed. 2017;56:851. doi: 10.1002/anie.201609844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For reviews regarding carboboration reactions, see: Shimizu Y, Kanai M. Tetrahedron Lett. 2014;55:3727.Semba K, Fujihara T, Terao J, Tsuji Y. Tetrahedron. 2015;71:2183.Lazreg F, Nahra F, Cazin CSJ. Coord Chem Rev. 2015;293–294:48.Neeve EC, Geier SJ, Mkhalid IAI, Westcott SA, Marder TB. Chem Rev. 2016;116:9091. doi: 10.1021/acs.chemrev.6b00193.
- 8.For selected recent examples of Cu-catalyzed carboboration, see: Li X, Meng F, Torker S, Shi Y, Hoveyda AH. Angew Chem, Int Ed. 2016;55:9997. doi: 10.1002/anie.201605001.Semba K, Ohtagaki Y, Nakao Y. Org Lett. 2016;18:3956. doi: 10.1021/acs.orglett.6b01675.Yeung K, Ruscoe RE, Rae J, Pulis AP, Procter DJ. Angew Chem, Int Ed. 2016;55:11912. doi: 10.1002/anie.201606710.Zhao W, Montgomery J. J Am Chem Soc. 2016;138:9763. doi: 10.1021/jacs.6b05216.Jiang L, Cao P, Wang M, Chen B, Wang B, Liao J. Angew Chem, Int Ed. 2016;55:13854. doi: 10.1002/anie.201607493.
- 9.Semba K, Nakao Y. J Am Chem Soc. 2014;136:7567. doi: 10.1021/ja5029556. [DOI] [PubMed] [Google Scholar]
- 10.Jia T, Cao P, Wang B, Lou Y, Yin X, Wang M, Liao J. J Am Chem Soc. 2015;137:13760. doi: 10.1021/jacs.5b09146. [DOI] [PubMed] [Google Scholar]
- 11.η3 complexes and alkene isomers are not shown for clarity. The isomeric C2 and C3 complexes are not considered, as Cu(I)-allyl complexes typically prefer the least substituted position. Russo V, Herron JR, Ball ZT. Org Lett. 2010;12:220. doi: 10.1021/ol902458v.(b) Also see ref 8e.
- 12.Shelby Q, Kataoka N, Mann G, Hartwig J. J Am Chem Soc. 2000;122:10718. [Google Scholar]
- 13.Sandford C, Aggarwal VK. Chem Commun. 2017;53:5481. doi: 10.1039/c7cc01254c. [DOI] [PubMed] [Google Scholar]
- 14.Xie A, Stahl SS. J Am Chem Soc. 2015;137:3767. doi: 10.1021/jacs.5b01036. [DOI] [PubMed] [Google Scholar]
- 15.(a) Kulkarni MG, Rasne RM, Davawala SI, Doke AK. Tetrahedron Lett. 2002;43:2297. [Google Scholar]; (b) Wu X, Chen Z, Bai YB, Dong VM. J Am Chem Soc. 2016;138:12013. doi: 10.1021/jacs.6b06227. [DOI] [PubMed] [Google Scholar]
- 16.Diene 34 did not lead to arylboration products under conditions outlined in Scheme 2.
- 17.See the Supporting Information for details.
- 18.Semba K, Shinomiya M, Fujihara T, Terao J, Tsuji Y. Chem -Eur J. 2013;19:7125. doi: 10.1002/chem.201300443. [DOI] [PubMed] [Google Scholar]
- 19.(a) Bates RB, Beavers WA. J Am Chem Soc. 1974;96:5001. [Google Scholar]; (b) Schleyer PvR, Kaneti J, Wu YD, Chandrasekhar J. J Organomet Chem. 1992;426:143. [Google Scholar]; (c) Liepins V, Bäckvall JE. Eur J Org Chem. 2002;2002:3527. doi: 10.1021/jo016229u. [DOI] [PubMed] [Google Scholar]
- 20.For a seminal report, see: Ojima I, Kumagai M. J Organomet Chem. 1978;157:359.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.