

Abstract
The total synthesis of both oridamycin A and oridamycin B was accomplished starting from a common synthetic intermediate readily prepared from geranyl acetate. The sequence utilizes an oxidative radical cyclization to construct the trans-decalin ring system, setting three of four contiguous stereocenters in one operation. The carbazole nucleus was forged through a one-pot process entailing acid-promoted dehydration followed by 6π-electrocyclization/aromatization.
Graphical abstract
The xiamycin family of indolosesquiterpenes comprises bioactive compounds recently isolated from several strains of Streptomyces.1 Among the family members are xiamycin A (3), its biosynthetic precursor indosespene (4), and the homodimers dixiamycin A/B (5), isolated as a mixture of atropisomers about the N-N linkage (Figure 1). These compounds display a range of interesting activities, including antiviral and antibacterial properties.1 Notably, the N-linked dimers dixiamycin A/B (5) are potent agents against both methicillin resistant Staphylococcus aureus 134/94 and vancomycin resistant Enterococcus faecalis 1528 R10.1e The closely related monomers oridamycin A (1) and oridamycin B (2) were isolated from Streptomyces sp. KS84 during screening for activity against Saprolegnia spp.—a water mold that infects freshwater fish, including those raised in commercial aquaculture.2 These compounds have elicited significant interest within the synthetic community; efficient total syntheses of xiamycin A (3) and dixiamycin B (5) have been reported by Baran and coworkers, employing a cationic cyclization to construct the monomer and a novel electrochemical dimerization to generate the N-N linkage.3 Elegant syntheses of xiamycin A (3), indosespene (4), sespenine, oridamycin A (1), oridamycin B (2), and dixiamycin C have been reported by the Li group, utilizing powerful radical cascades to construct the trans-decalin ring systems.4
Figure 1.
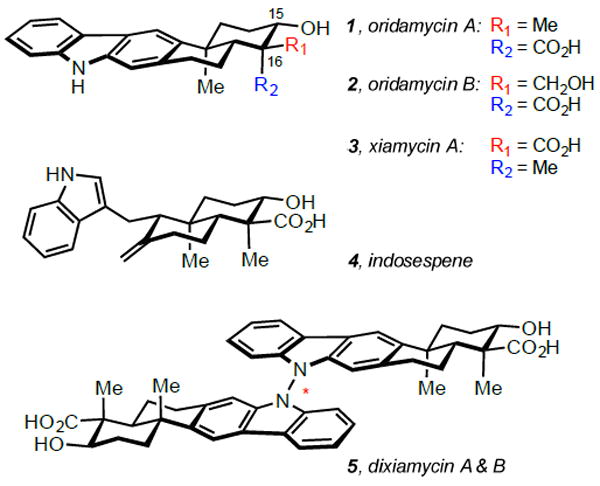
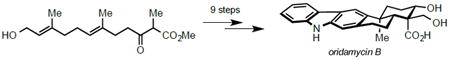
Selected members of the oridamycin and xiamycin families.
Structurally, these pentacyclic molecules possess a carbazole nucleus fused to a trans-decalin ring system containing four contiguous stereocenters, including two quaternary centers. The major difference between the family members manifests at the C16 quaternary center; oridamycin A (1) and oridamycin B (2) each bear an axial carboxylic acid and an equatorial methyl/hydroxymethyl, while xiamycin A (3), indosespene (4), and dixiamycin A/B (5) contain an axially disposed methyl and an equatorial acid (Figure 1). Biosynthetically, it is known that xiamycin A (3) arises from precursor 6 through a selective enzymatic oxidation mediated by XiaM (Figure 2, A).5 It seems plausible that intermediate 6 is also the biosynthetic precursor of the oridamycins, likely proceeding through an analogous methyl oxidation. Drawing inspiration from nature, we anticipated utilizing a common synthetic intermediate to access congeners from both the oridamycin and xiamycin families (Figure 2, B). Anticipating that biomimetic methyl oxidation of intermediate 6 would be difficult to replicate in the flask, we envisioned imparting the requisite oxidation pattern on a linear precursor that would be cyclized under two distinct sets of conditions to yield both of the desired C16 epimers. Employing an oxidative radical cyclization cascade under standard conditions would yield the stereochemistry associated with the oridamycins, proceeding through transition state 10 to produce intermediate 8 (Figure 2, C).6 To access the C16 stereoisomer relevant to the xiamycin family, we envisioned that introduction of a Lewis acid could invert the selectivity via chelation of the β-keto ester, producing transition state 11 en route to intermediate 9.7
Figure 2.
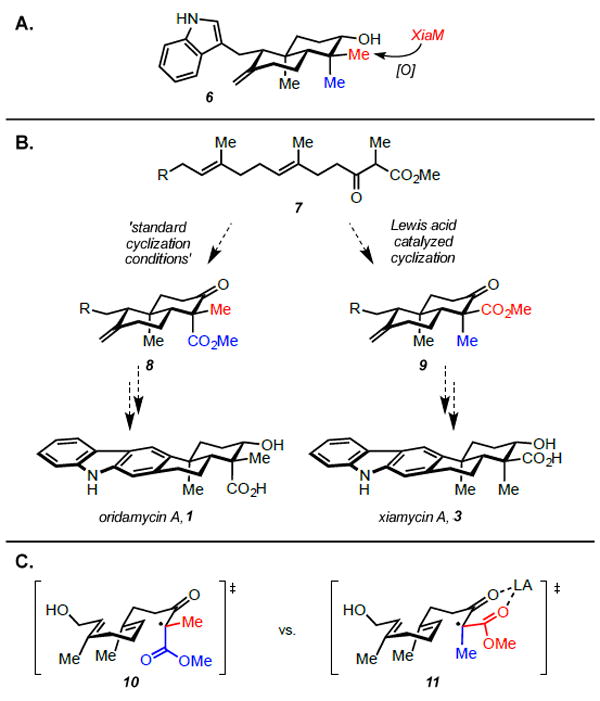
A. Biosynthetic precursor for the xiamycin family, and proposed precursor for the oridamycin family, B. General strategy to access both the xiamycin and oridamycin families from a common linear precursor, C. Proposed radical transition states for the cyclization reactions.
We reasoned that it would be expeditious to initiate our synthetic efforts with the oridamycins, allowing for validation of late-stage transformations before attempting to invert the selectivity of the radical cyclization. Retrosynthetically, we anticipated that the C16 hydroxymethyl of oridamycin B (2) could arise from an oxime-directed, palladium-catalyzed C-H oxidation of substrate 12 (Scheme 1).8 The C15 alcohol would arise via axial hydride delivery onto a suitable ketone precursor. The carbazole nucleus could be forged through 6π-electrocyclization/aromatization of an indole moiety appended to a diene system, which would itself arise from dehydration of a homo-allylic alcohol such as 13.9 Grignard addition of a Boc-protected indole onto a suitable aldehyde would provide compound 13, containing all the requisite carbon atoms of the natural products. Finally, the trans-decalin ring system of 14 would be forged through an oxidative radical cyclization of a linear precursor readily derived from geranyl acetate.10 Oridamycin A (1) would be synthesized in analogous fashion without requiring the C-H oxidation sequence.
Scheme 1.
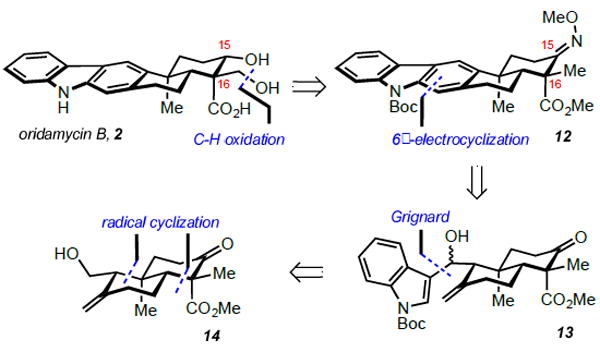
Retrosynthesis of Oridamycin B
In the forward direction, known compound 16 was readily obtained from geranyl acetate in multigram quantities in three operations (Scheme 2).10 Oxidative radical cyclization proceeded smoothly under the standard conditions to give alcohol 14 as a single diastereomer.11 Subsequent oxidation with Dess-Martin periodinane afforded the corresponding aldehyde, which was coupled with the Grignard reagent generated from 17 to produce 13 as an inconsequential mixture of diastereomers.9,12 Next, when considering the dehydration of intermediate 13, we hypothesized that the alcohol would be more prone to elimination if the Boc group were removed, allowing for the electron-rich indole to more readily participate in the process. To that end, substrate 13 was treated with TFA, and, to our surprise, the secondary alcohol was cleanly eliminated to form a triene intermediate with the Boc group still intact. Thus, this interesting result became the foundation of a one-pot process to forge the free carbazole directly from intermediate 13. In the event, treatment of a solution of 13 in CH2Cl2 with TFA formed the desired triene, which was not isolated but concentrated to remove solvent and acid. Next, this material was dissolved in toluene and heated to 135 °C to induce a thermal 6π-electrocyclization/aromatization sequence under aerobic conditions.9 Upon completion, TFA was added at 45 °C to remove the Boc group, yielding free carbazole 18. A stereoselective reduction using NaBH4 proceeded uneventfully, yielding oridamycin A methyl ester (d.r.> 20:1). The final remaining operation entailed conversion of the C16 axial methyl ester into the free acid. Initial attempts at saponification using various hydroxide sources were unproductive, likely due to the 1,3-diaxial relationship between the C12 methyl and the C16 ester—reminiscent of podocarpic acid13—which may impede formation of the requisite tetrahedral intermediate.14 Accordingly, we utilized a nucleophilic cleavage protocol, allowing for successful dealkylation of the ester. Thus, upon treatment with NaCN in DMSO at elevated temperature, oridamycin A (1) was produced in 86% yield, cleanly affording characterization quality material.15
Scheme 2.
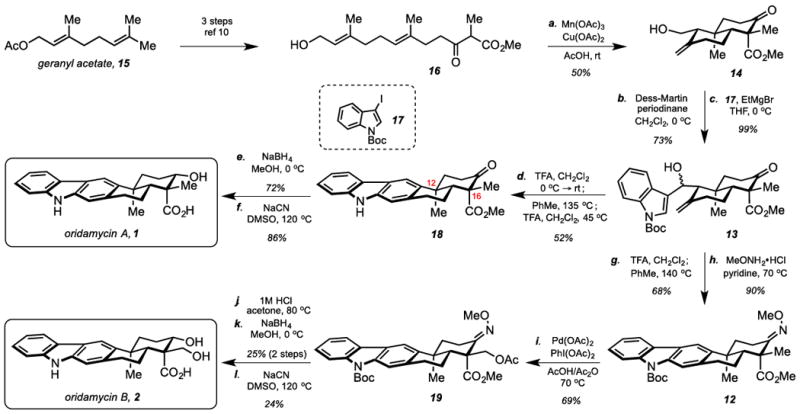
Synthesis of Oridamycin A and B
To access oridamycin B (2), the route to oridamycin A (1) was diverted from intermediate 13. The same sequence of dehydration and electrocyclization was employed, but the Boc group was preserved to produce the protected carbazole. The ketone was smoothly converted into O-methyloxime derivative 12 as a prerequisite for the selective, late-stage C-H oxidation. Upon treatment with PhI(OAc)2 and catalytic Pd(OAc)2 at elevated temperature, compound 19 was cleanly produced in 69% yield. Next, global deprotection using a 3:1 mixture of aq 1M HCl:acetone at 80 oC cleaved the acetate, removed the Boc group, and hydrolyzed the O-methyloxime. A variety of conditions were screened for this transformation, most of which produced a significant amount of a presumed retro-Aldol product, which was largely suppressed through the use of acetone as a cosolvent. However, the elevated temperature required to cleave the O-methyloxime appeared to cause a significant amount of nonspecific decomposition, resulting in diminished recovery. The product of the global deprotection was unstable to silica, requiring reduction of the crude material to produce oridamycin B methyl ester. Finally, treatment of this compound with NaCN produced oridamycin B (2). Interestingly, it was found that the 1H NMR spectrum of 2 was pH dependent (see Supporting Information).
With the successful synthesis of the oridamycins, the next phase of our program is aimed at accessing epimeric intermediate 9 via Lewis acid chelation of the radical intermediate during cyclization (see Figure 2).7 Realization of this strategy could provide access to the xiamycin family from the same linear precursor 7 used to generate the oridamycins. Furthermore, we are optimistic that this transformation could be rendered asymmetric through the use of a suitable chiral ligand.16 Efforts are ongoing in our laboratory to validate these assertions.
In conclusion, both oridamycin A and B were synthesized from known compound 16 in six and nine steps, respectively. The synthesis was diverted from common intermediate 13 harboring all of the carbon atoms contained within each natural product. The successful construction of the oridamycins lays the foundation for a unified synthetic strategy for this entire class of indolosesquiterpenes.
Supplementary Material
Acknowledgments
Prof. Samuel J. Danishefsky (MSKCC) is foremost acknowledged for his intellectual mentorship. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. 2014157713. The MSKCC core facility is also acknowledged for mass spectrometric assistance and for providing the necessary instrumentation.
Footnotes
Supporting Information
Experimental procedures and characterization data (including spectra for new compounds) is available free of charge via the Internet at http://pubs.acs.org.
Notes
The author declares no competing financial interests.
References
- 1.(a) Ding L, Münch J, Goerls H, Maier A, Fiebig HH, Lin WH, Hertweck C. Bioorg Med Chem Lett. 2010;20:6685–6687. doi: 10.1016/j.bmcl.2010.09.010. [DOI] [PubMed] [Google Scholar]; (b) Ding L, Maier A, Fiebig HH, Lin WH, Hertweck C. Org Biomol Chem. 2011;9:4029–4031. doi: 10.1039/c1ob05283g. [DOI] [PubMed] [Google Scholar]; (c) Zhang Q, Mandi A, Li SM, Chen Y, Zhang W, Tian XP, Zhang H, Li HX, Zhang W, Zhang S, Ju JH, Kurtan T, Zhang C. Eur J Org Chem. 2012:5256–5262. [Google Scholar]; (d) Xu ZL, Baunach M, Ding L, Hertweck C. Angew Chem Int Ed. 2012;51:10293–10297. doi: 10.1002/anie.201204087. [DOI] [PubMed] [Google Scholar]; (e) Baunach M, Ding L, Bruhn T, Bringmann G, Hertweck C. Angew Chem Int Ed. 2013;52:9040–9043. doi: 10.1002/anie.201303733. [DOI] [PubMed] [Google Scholar]
- 2.Takada K, Kajiwara H, Imamura N. J Nat Prod. 2010;73:698–701. doi: 10.1021/np1000522. [DOI] [PubMed] [Google Scholar]
- 3.Rosen BR, Werner EW, O’Brien AG, Baran PS. J Am Chem Soc. 2014;136:5571–5574. doi: 10.1021/ja5013323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meng Z, Yu H, Li L, Tao W, Chen H, Wan M, Yang P, Edmonds DJ, Zhong J, Li A. Nat Commun. 2015:6. doi: 10.1038/ncomms7096.. NOTE: This closely related publication appeared just as our own studies were being completed. Sun Y, Chen PX, Zhang DL, Baunach M, Hertweck C, Li A. Angew Chem Int Ed. 2014;53:9012–9016. doi: 10.1002/anie.201404191.
- 5.Zhang Q, Li H, Li S, Zhu Y, Zhang G, Zhang H, Zhang W, Shi R, Zhang C. Org Lett. 2012;14:6142–6145. doi: 10.1021/ol302782u. [DOI] [PubMed] [Google Scholar]
- 6.Snider BB. Chem Rev. 1996;96:339–363. doi: 10.1021/cr950026m.. and references therein.
- 7.Yang D, Ye XY, Xu M, Pang KW, Cheung KK. J Am Chem Soc. 2000;122:1658–1663. [Google Scholar]
- 8.Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]
- 9.Bian M, Wang Z, Xiong X, Sun Y, Matera C, Nicolaou KC, Li A. J Am Chem Soc. 2012;134:8078–8081. doi: 10.1021/ja302765m. [DOI] [PubMed] [Google Scholar]
- 10.(a) González MA, Molina-Navarro S. J Org Chem. 2007;72:7462–7465. doi: 10.1021/jo0712401. [DOI] [PubMed] [Google Scholar]; (b) Jenny L, Borschberg HJ. Helv Chim Acta. 1995;78:715–731. [Google Scholar]; (c) Kitagawa Y, Itoh A, Hashimoto S, Yamamoto H, Nozaki H. J Am Chem Soc. 1977;99:3864–3867. [Google Scholar]
- 11.Yang D, Xu M. Org Lett. 2001;3:1785–1788. doi: 10.1021/ol015722p. [DOI] [PubMed] [Google Scholar]
- 12.Leung JC, Patman RL, Sam B, Krische MJ. Chem Eur J. 2011;17:12437–12443. doi: 10.1002/chem.201101554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Wenkert E, Afonso A, Bredenberg JBs, Kaneko C, Tahara A. J Am Chem Soc. 1964;86:2038–2043. [Google Scholar]; (b) Wenkert E, Jackson BG. J Am Chem Soc. 1958;80:217–219. [Google Scholar]; (c) Sherwood IR, Short WF. J Chem Soc (Resumed) 1938:1006–1013. [Google Scholar]
- 14.Reddy BVS, Reddy LR, Corey EJ. Tetrahedron Lett. 2005;46:4589–4593. [Google Scholar]
- 15.O’Brien EM, Morgan BJ, Kozlowski MC. Angew Chem Int Ed. 2008;47:6877–6880. doi: 10.1002/anie.200800734. [DOI] [PubMed] [Google Scholar]
- 16.(a) Yang D, Zheng BF, Gao Q, Gu S, Zhu NY. Angew Chem Int Ed. 2006;45:255–258. doi: 10.1002/anie.200503056. [DOI] [PubMed] [Google Scholar]; (b) Sibi MP, Ji JG, Wu JH, Gurtler S, Porter NA. J Am Chem Soc. 1996;118:9200–9201. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.