

Abstract
The Ras proteins are essential GTPases involved in the regulation of cell proliferation and survival. Mutated oncogenic forms of Ras alter effector binding and innate GTPase activity, leading to deregulation of downstream signal transduction. Mutated forms of Ras are involved in approximately 30% of human cancers. Despite decades of effort to develop direct Ras inhibitors, Ras has long been considered ‘undruggable’ due to its high affinity for GTP and its lack of hydrophobic binding pockets. Herein, we report a total chemical synthesis of all L- and all D-amino acid biotinylated variants of oncogenic mutant KRas(G12V). The protein is synthesized using Fmoc-based solid-phase peptide synthesis and assembled using combined native chemical ligation and isonitrile-mediated activation strategies. We demonstrate that both KRas(G12V) enantiomers can successfully fold and bind nucleotide substrates and binding partners with observable enantiodiscrimination. By demonstrating the functional competency of a mirror-image form of KRas bound to its corresponding enantiomeric nucleotide triphosphate, this study sets the stage for further biochemical studies with this material. In particular, this protein will enable mirror-image yeast surface display experiments to identify all-D peptide ligands for oncogenic KRas, providing a useful tool in the search for new therapeutics against this challenging disease target.
Graphical Abstract
INTRODUCTION
The Ras family of GTPase proteins plays a crucial role in cellular signal transduction, acting as a molecular switch to propagate extracellular signals to intracellular phosphorylation cascades that drive cellular growth and survival mechanisms.1 Ras activity is controlled by a bound guanine nucleotide cofactor. Guanosine triphosphate (GTP)-bound (“on”) Ras adopts a conformation that binds to Ras effector proteins, such as B-Raf and PI3 kinase, with high affinity and leads to downstream pathway activation. Guanosine diphosphate (GDP)-bound (“off”) Ras adopts an inactive conformation that does not bind to effectors. The Ras nucleotide state is primarily regulated by guanine exchange factors (GEFs), which favor GTP loading, and GTPase activating proteins (GAPs), which catalyze hydrolysis of GTP to GDP.2 Single point mutations at key residues in the primary sequence of Ras are capable of disrupting GTP hydrolysis by blocking interactions with GAPs and by reducing the intrinsic GTPase activity of Ras, thereby trapping Ras in the GTP-bound “on” state and leading to constitutive pro-growth signaling.3,4 Such inappropriately activated Ras mutants are found in approximately 30% of human cancers, with the KRas isoform representing an overwhelming majority (>80%) of these cancer types.5 Direct inhibition of mutant oncogenic Ras represents a “holy grail” in the development of cancer therapeutics; one which has been met with tremendous difficulty since the initial correlation between the RAS oncogene and human cancers was described in the early 1980’s.6
A number of approaches aimed at modulating oncogenic Ras signaling via small-molecule inhibitors have been only partly successful, and no such molecules have yet reached the clinic.7 Inhibition of the nucleotide binding site of Ras has proven largely futile due to the unusually high affinity of Ras for its GDP and GTP substrates and the high concentrations of these molecules in the cell. Moreover, inhibition of Ras-effector interactions or Ras-GEF interactions with small molecules is challenging due to difficulties associated with perturbing pertinent protein-protein interactions (PPIs), and the limited successes reported thus far have been hampered by moderate potency.8–10 Attempts to chemically perturb Ras post-translational processing and localization have failed thus far in the clinical setting due to the existence of promiscuous and alternative lipidation pathways.11 Recent efforts aimed at the selective inhibition of KRas(G12C) via covalent-binding inhibitors have shown considerable promise.12 However, these approaches target only a small population of Ras-driven cancers (albeit a relatively high proportion of lung cancers) and do not represent a general solution toward inhibiting constitutive Ras activity.
Peptide-derived and peptidomimetic inhibitors have great potential in targeting PPIs.13,14 Of note, α-helical stabilized peptides, including hydrocarbon stapled peptides, represent a promising class of cell-penetrating modulators of PPIs with biological stability and have been successfully applied in targeting Ras.14a–b,15,16 Recently, one of our labs has developed a class of miniproteins that bind to the Ras effector domain in vitro with high affinity and inhibit Ras-effector interactions.17 These miniproteins were discovered by screening unbiased combinatorial libraries using yeast surface display (YSD), demonstrating that it is possible to identify high-affinity Ras ligands from naïve libraries.18 With these results in mind, we wondered if mirror-image YSD could be used to identify high-affinity Ras binders composed of all D-amino acids. Unnatural, D-residue peptides have been shown to possess improved stability and bioavailability over their L-residue peptide counterparts, and mirror-image display has successfully afforded useful peptide ligands in the context of various disease types, including HIV/AIDS, Alzheimer’s Disease, and cancer.19,20 Additionally, synthetic all-D proteins have other biochemical research value, such as racemic protein crystallography.21 Critical to such studies is the total chemical synthesis of an all D-residue version of the naturally occurring all-L residue protein. In this report, we describe the chemical synthesis and folding of all-L residue and all-D residue variants of the oncogenic mutant KRas(G12V)[1–166] (1), which bear a biotin affinity tag for use in YSD and other biochemical experiments. We conducted a series of in vitro assays to demonstrate the biochemical viability of each protein enantiomer, thus establishing this mirror-image protein pair as a potentially valuable tool for the future discovery of anti-Ras therapeutics.
RESULTS AND DISCUSSION
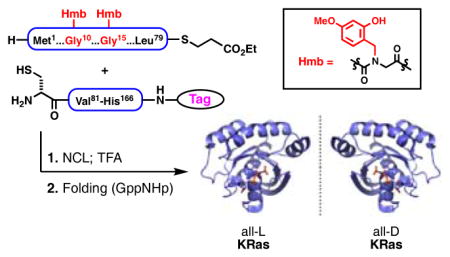
The primary sequence of KRas(G12V), a 166-residue protein, is shown in Figure 1 with highlighted points of assembly. The synthesis of HRas has been previously described utilizing Boc-based solid phase peptide synthesis (SPPS) combined with native chemical ligation (NCL) assembly as well as via a semisynthesis approach.22 Our synthetic approach toward KRas(G12V) relies on the use of Fmoc-based SPPS to generate five peptidyl sequences, which are subsequently assembled via combined NCL and isonitrile-mediated activation strategies to access a biotinylated variant of KRas(G12V).23, 24 Peptide thioesters were synthesized via Sakakibara elongation of protected peptides with pre-formed C-terminal residue thioesters following SPPS or alternatively by C-terminal hydrazide oxidation.25,26 In a retrosynthetic fashion, taking primary advantage of the native cysteine residues within the full sequence reduces the synthetic challenge to three NCLs at assembly points Cys51, Cys80, and Cys118. However, in our hands, preliminary attempts to synthesize KRas[118–166] in its entirety by Fmoc-based SPPS proved inefficient and resulted in poor recovery of the desired peptide. Therefore, this 49-mer sequence was further dissected to enable a more efficient preparation. In the absence of available cysteine residues to permit NCL, two alternative assembly points were hypothesized. Utilizing an established strategy, we envisioned a potential NCL-desulfurization approach at Ala146, requiring the installation, ligation, and subsequent desulfurization of a non-native Cys146.27 Alternatively, the assembly of large peptides via the chemoselective activation of C-terminal thiocarboxylic acids using a hindered isonitrile represents an attractive complementary strategy that is independent of cysteine.24,28–30 We have recently reported the use of this method to generate challenging hydrophobic peptidyl sequences greater than 100 amino acids in length.30 Here, we hypothesized that a C-terminal thioacid at Gly138 could serve as a potential disconnection site to eradicate concern for epimerization during activation. However, this instance would require us to demonstrate the potential of a hindered β-branched nucleophile at Ile139 with a combined strategy to orthogonally protect and deprotect side-chain nucleophiles at Lys128, Lys147, and Lys165. This target sequence and associated challenges present a worthwhile opportunity to expand the utility of this promising strategy.
Figure 1.

Retrosynthetic disconnection sites for KRas(G12V)[1–166]
Our synthesis initially focused on completion of the protein using all-L amino acids, and our optimized synthetic route was repeated using all D-residues. Yields are provided in the schemes for both of these syntheses, distinguished by the color-coding. We began our studies with the challenging C-terminal sequence, KRas[118–166], assembled using independent ligation strategies compared in a side-by-side fashion in Scheme 1A. In each series, a PEGylated biotin label (referred herein as PEG2-Biotin) was appended at the C-terminus for projected biochemical studies. For the isonitrile-mediated activation approach, we chose to orthogonally mask the ε-amines of lysine residues with allyloxycarbonyl (Alloc) groups.
Scheme 1.

aReagents and conditions: (i) tBuNC (3.2 equiv), HOBt (10.2 equiv), DMA, room temperature (r.t.), 48 h. (ii) Piperidine (20 vol%), 30 min r.t.. (iii) PdCl2(dppf) (50 mol%), PhSiH3 (50 equiv), DMF, r.t. (iv) 6 M Gnd HCl, 0.2 M Na2HPO4, 0.2 M MPAA, 40 mM TCEP HCl, pH = 7.0, 6 h, r.t. (v) VA-044, tBuSH, TCEP, Gnd HCl buffer, 4 h, r.t. (vi) AgOAc (100 equiv), 70% aqueous AcOH, 2.5 h, r.t. (vii) 6 M Gnd HCl, 0.2 M Na2HPO4, pH = 7.0, 6 h, r.t.
bReagents and conditions: (viii) 6 M Gnd HCl, 0.2 M Na2HPO4, 0.2 M MPAA, 40 mM TCEP HCl, pH = 7.0, 4 h, r.t. (ix) NH2OMe HCl added, pH = 4.5−5.0, 4.5 h, r.t. (x) 6 M Gnd HCl, 0.2 M Na2HPO4, 0.2 M MPAA, 20 mM TCEP HCl, pH = 7.0, 6.5 h, r.t. (xi) TFA/thioanisole/ethanedithiol/anisole (90:5:3:2), 2 h, r.t. (xii) Solubilization in 1:1 MeCN/H2O (0.1% TFA) with sonication, then TCEP, pH = 8.0, and RP-HPLC purification; HOBt = 1-Hydroxybenzotriazole, DMA = N,N-dimethylacetamide, Gnd = guanidine, MPAA = 4-mercaptophenylacetic acid, TCEP = Tris(2-carboxyethyl)phosphine, VA-044 = 2,2′-Azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride, TFA = trifluoroacetic acid. Yields are reported in the following manner: green = all-L-residue peptides; purple = all-D-residue peptides.
Under previously described conditions, the activation of C-terminal thioacid 3 with tert-butyl isocyanide in the presence of HOBt and N-terminal nucleophilic partner 2 afforded the desired Alloc-protected product.30 Moreover, concentrations comparable to NCL conditions (in this case, 4–5 mM) provided nearly full conversion within 48 hours. Further experimentation found that the N-terminal Fmoc could be removed in the same pot by the subsequent addition of piperidine to the crude reaction mixture, allowing for a clean, two-step, one-pot ligation procedure.31 The intermediate product, KRas[118–166], was isolated in crude form following precipitation with diethyl ether and was directly subjected to reductive Alloc deprotection. Brief exposure of the precipitate to PdCl2(dppf) and PhSiH3 in DMF afforded 4 following purification by HPLC.32
In a direct comparison of strategies, the projected NCL between 6 and 7 proceeded in high yield as anticipated (82% isolated), with the N-terminal cysteine of 7 protected as S-acetamidomethyl (S-Acm) to impart selectivity in the NCL and the subsequent desulfurization step.27b The resultant ligation product (8) was subjected to desulfurization and Acm deprotection to afford KRas[118–166] (5) in 69% isolated yield over two steps following HPLC purification. Overall, the three-step conversion of 6 and 7 to common ligation product KRas[118–166] proceeded in 57% yield. The comparable yields and ease of reaction set-up in each study suggests that in certain cases, isonitrile-mediated activation of C-terminal Gly thioacids can also provide facile access and should be considered as a complementary strategy to NCL-desulfurization, especially for hydrophobic sequences lacking available or median cysteine residues. Moreover, although both of these highlighted approaches require multi-step assembly and deprotection, we contend that the isonitrile-mediated activation approach may be preferable due to intermediary purification ease—requiring only precipitation upon addition of diethyl ether with no desalting or HPLC necessary to isolate intermediate peptides—in cases where the overall yields are similar.
Having validated the synthetic practicality of subtarget KRas[118–166] (4), we turned our attention to the overarching goal of completing a total synthesis of the entire oncogenic KRas sequence (Scheme 1B). We further extended 4 via NCL with thioester, KRas[80–117] (9), followed by direct addition of methoxylamine hydrochloride to remove the N-terminal thiazolidine (Thz), furnishing KRas[80–166] (10). Next focusing on the N-terminal portion of the protein, we found synthesis and handling of KRas[1–50] (11) to be challenging due to its hydrophobic nature. Solubility issues and synthesis efficiency could be drastically improved through incorporation of acid-labile 2-hydroxy-4-methoxybenzyl (Hmb) groups within the amide backbone of 11.33 Presumably, the incorporation of tertiary amides at Gly10 and Gly15 serves to beneficially disrupt secondary structural elements of this hydrophobic sequence, thereby augmenting general solubility to facilitate handling and purification. The Hmb group also holds an advantage in that it can be rendered temporarily acid-stable upon acylation (for synthesis of this peptide, see Supporting Information). Becker and co-workers also note similar challenges associated with hydrophobicity in the synthesis of wild-type HRas.22a,34 In our hands, 11 could be subjected to kinetic chemical ligation with 12 to selectively afford alkyl thioester, KRas(G12V)[1–79] 13.35 Finally, NCL between 13 and 10 was found to be efficient, with near complete consumption of limiting reagent 13 after 6–8 h at neutral pH. The corresponding Hmb-protected sequence, KRas[1–166] (14), could be conveniently isolated via its direct precipitation from the ligation buffer upon the addition of cold water followed by centrifugation of the resultant precipitate. Here, solubility differences of full sequence 14 were exploited. Excess 10 remained largely in the supernatant with minimal solubilization of target 14 (Figure 2). The subsequent exposure of this precipitated material to acidic deprotection conditions (TFA/thioanisole/1,2-ethanedithiol/anisole) afforded 1 following purification by HPLC, with excellent agreement by high-resolution mass analysis (Figure 3).
Figure 2.
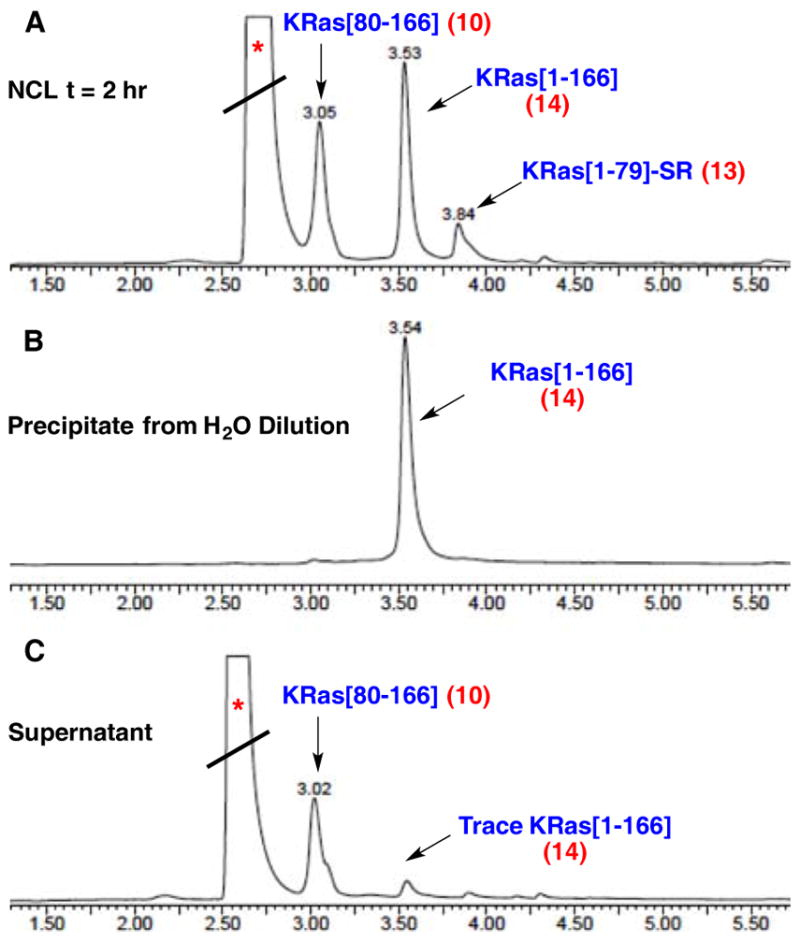
UPLC traces of the NCL between 10 and 13 and isolation of 14. (A) NCL after 2 h. (B) Only 14 is obtained in the precipitate from H2O dilution after the NCL is complete (t = 6.5 h). (C) Only trace 14 remained in the supernatant after dilution of the NCL with H2O, while excess 10 remains entirely dissolved. *denotes absorption peak derived from MPAA.
Figure 3.
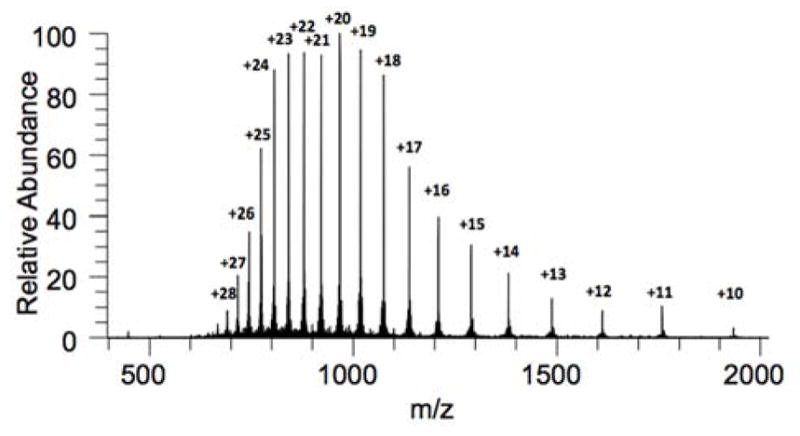
High-resolution mass spectrum of biotinylated, all D-residue KRas(G12V) (1).
With a reproducible synthetic route established, we turned our attention to establishing biochemical competency of the synthetic protein pair. We adopted a previously reported folding protocol, which entailed solubilization by guanidine hydrochloride and rapid folding into denaturant-free buffer.22a After optimization, we found both enantiomers of synthetic KRas to consistently display moderate folding efficiency based on gel filtration analysis (Figure 4A), using either D-GppNHp (for the all L-residue protein) or L-GppNHp (for the all D-residue protein) as folding templates.36,37 In each case, further purification by size-exclusion chromatography afforded a monomeric species with a retention time equivalent to recombinant KRas(G12V), as assessed by analytical gel filtration. Moreover, circular dichroism analysis of this pair of folded proteins showed good agreement with that of recombinant KRas(G12V), with the all D-residue protein exhibiting an opposite sign compared to the all L-residue proteins. (Figure 4B). Comparison of A260/A280 values showed an increase from 0.6–0.7 for the unfolded proteins to 1.0–1.1 for the recombinant and folded proteins, consistent with association with guanine nucleotides, which possess an absorption maximum at 252 nm (Figure 4C).
Figure 4.

Folding analysis of synthetic proteins. A) Crude gel filtration traces and purified synthetic all L- and all D-KRas(G12V) after folding with GppNHp; B) Circular dichroism analysis of folded and purified synthetic proteins and recombinant KRas(G12V); C) Comparison of A260/A280 values for synthetic proteins and recombinant KRas(G12V).
To further characterize the synthetic proteins, we folded both in the presence of fluorescent mant-GppNHp ((2′/3′)-O-(N-methylanthraniloyl)guanosine-5′-O-[(β,γ)-imidotriphosphate]) nucleotide analogs, which exhibit an increase in fluorescence upon binding to Ras and have been used to study Ras-ligand binding interactions.38 The enantiomeric pair of proteins folded with mant-GppNHp nucleotides of corresponding stereochemistry exhibited comparable nucleotide dissociation behavior to recombinant KRas, marked by a decrease in fluorescence upon the addition of excess unlabeled GppNHp of correlated stereochemistry. In contrast, addition of the incorrect antipode of nucleotide showed no significant nucleotide exchange for either material.39 In the case of synthetic L-KRas, the addition of the Ras-binding domain (RBD) of B-Raf, a canonical Ras effector known to inhibit nucleotide dissociation, slowed dissociation in a similar manner to that observed with recombinant KRas.38 As expected, the addition of the B-Raf RBD had no observed effect on nucleotide dissociation from all D-residue KRas. Similarly, addition of the Ras-binding miniprotein 225–44 (all L-amino acid residues) slowed nucleotide dissociation for recombinant and synthetic L-KRas, with no effect observed on all D-KRas.17 Conversely, as anticipated, addition of an all D-residue variant of miniprotein 225-44 does indeed slow nucleotide dissociation for all-D KRas but had no effect on recombinant or synthetic L-KRas (Figure 5).
Figure 5.

Unlabeled nucleotide displacement of mant-GppNHp-loaded KRas(G12V) in the presence of Ras-binding miniproteins. A) Recombinant L-KRas(G12V); B) Synthetic L-KRas(G12V); C) Synthetic D-KRas(G12V).
Finally, we performed surface plasmon resonance (SPR) binding assays to confirm direct binding of synthetic, folded KRas to Ras-binding miniproteins (Figure 6). We found that the 225-44 miniprotein exhibited high nonspecific binding to the SPR sensor chip, so we prepared all-L and all-D variants of the 225-11 miniprotein, which has been previously co-crystallized with KRas(G12V) and shown by SPR to bind with low-nanomolar affinity.17 Recombinant and synthetic all-L KRas bound to all-L 225-11 with Kd values of 6 nM and 7 nM, respectively, comparable to the reported value of 3.6 nM, but did not bind to all-D 225-11. Conversely, synthetic all-D KRas bound to all-D 225-11 with a Kd of 2 nM but did not bind to all-L 225-11. A small amount of non-specific binding of the all-D 225-11 miniprotein to L-KRas was observed in this SPR assay, as were slight differences in the sensogram curvature. These discrepancies may potentially result from the presence of some protein aggregate and/or microheterogeneity within the synthetic materials, or as a consequence of the microscale nature of the refolding procedure. Regardless, selective low-nanomolar binding to all-D KRas is clearly observed. These combined biophysical experiments demonstrate that folding, nucleotide association, and miniprotein-binding profiles of both mirror-images of synthetic KRas(G12V) resemble that of recombinant KRas(G12V) and exhibit the expected enantiodiscrimination.40
Figure 6.

SPR binding analysis of Ras proteins with Ras-binding miniproteins. A) Binding of miniproteins with recombinant KRas; B) Binding of miniproteins with synthetic all-L KRas; C) Binding of miniproteins with synthetic all-D KRas. Kd values are mean ± SEM for triplicate runs.
CONCLUSION
In conclusion, we have described the total chemical synthesis and folding of both mirror-image forms of biotinylated oncogenic KRas(G12V). Overall ligation yields of the linear synthetic route 11 + 12 + 10 were 6.7% and 9.0% for all- L and all-D peptides, respectively. Following assembly of five peptidyl fragments accessible by Fmoc-based SPPS and in vitro folding, the all-L residue variant of KRas(G12V) showed a similar nucleotide and miniprotein binding profile to that of recombinant KRas(G12V). In contrast, the all-D residue variant of KRas(G12V) exhibited similar behavior but with opposite enantiorecognition with respect to nucleotide and miniprotein binding. The synthesis, folding, and biochemical verification of all-D KRas(G12V) provides the crucial reagent needed for the identification of all D-amino acid Ras inhibitors by mirror-image display screening, affording a valuable tool for the ongoing effort to develop direct-acting Ras therapies.
Supplementary Material
Acknowledgments
Funding Sources
Research reported in this publication was supported by the National Institutes of Health (NIH) under award nos. R21 CA187498-02, R01 GM102872, R01 GM109760, as well as Mr. William H. Goodwin and Mrs. Alice Goodwin, the Commonwealth Foundation for Cancer Research, MSK Cancer Center Support Grant/Core Grant no. P30 CA008748, and the Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center. J.H.M and G.L.V. acknowledge funding from the Lustgarten Foundation. A.G.R. acknowledges the NIH National Cancer Institute (NCI) for a Ruth Kirschstein Postdoctoral Fellowship under award no. F32 CA186398. G.S.C. acknowledges postdoctoral support from the NIH National Institute of General Medical Sciences (NIGMS) under award number F32 GM101746. M.T.P. acknowledges postdoctoral support from the NIH NCI under award no. T32 CA062948.
We thank Prof. Derek Tan, Dr. Baptiste Aussedat, and Mr. Adam Trotta (MSKCC) for helpful discussions. Dr. George Sukenick, Ms. Joan Subrath, and Ms. Hui Fang of the Sloan Kettering Institute Core Facility are acknowledged for NMR and mass spectrometry assistance. We thank Dr. Lisa Ambrosini Vadola for editing and proofreading this manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and characterization data for all new compounds and synthetic peptides, analytical characterization of all L- and all D-KRas(G12V), and details of nucleotide displacement studies are included (PDF).
References
- 1.For useful recent reviews and perspectives on the structure, function and targeting of Ras: Lu S, Jan H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R, Zhang J. Chem Rev. 2016;116:6607–6665. doi: 10.1021/acs.chemrev.5b00542.Samatar AA, Poulikakos PI. Nature Rev Drug Disc. 2014;13:928–942. doi: 10.1038/nrd4281.Spiegel J, Cromm PM, Zimmermann G, Grossmann TN, Waldmann H. Nature Chem Biol. 2014;10:613–622. doi: 10.1038/nchembio.1560.Wang Y, Kaiser CE, Frett B, Li H-Y. J Med Chem. 2013;56:5219–5230. doi: 10.1021/jm3017706.Stephen AG, Esposito D, Bagni RK, McCormick F. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017.
- 2.Konstantinopoulos PA, Karamouzis MV, Papavassilou AG. Nat Rev Drug Disc. 2007;6:541–555. doi: 10.1038/nrd2221.(and references therein) Vetter IR, Wittinghofer A. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023.
- 3.Kamata T, Feramisco JR. Nature. 1984;310:147–150. doi: 10.1038/310147a0.McCormick F. Nature. 1993;363:15–16. doi: 10.1038/363015a0.(and references therein). Adari H, Lowy DR, Willumsen BM, Der CJ, McCormick F. Science. 1988;240:518–521. doi: 10.1126/science.2833817.Scheffzek K, Ahmadian MR, Kabsch W, Wismüller L, Lautwein A, Schmitz F, Winttinghofer A. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333.
- 4.(a) Gibbs JB, Sigal IS, Poe M, Scolnick EM. Proc Natl Acad Sci USA. 1984;81:5704–5708. doi: 10.1073/pnas.81.18.5704. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sweet RW, Yokoyama S, Kamata T, Feramisco JR, Rosenberg M, Gross M. Nature. 1984;311:273–275. doi: 10.1038/311273a0. [DOI] [PubMed] [Google Scholar]; (c) McGrath JP, Capon DJ, Goeddel DV, Levinson AD. Nature. 1984;310:644–649. doi: 10.1038/310644a0. [DOI] [PubMed] [Google Scholar]
- 5.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Nature Rev Drug Disc. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Der CJ, Krontiris TG, Cooper GM. Proc Natl Acad Sci. 1982;79:3637–3640. doi: 10.1073/pnas.79.11.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Parada LF, Tabin CJ, Shih C, Weinberg RA. Nature. 1982;297:474–478. doi: 10.1038/297474a0. [DOI] [PubMed] [Google Scholar]; (c) Santos E, Tronick SR, Aaronson SA, Pulciani S, Barbacid M. Nature. 1982;298:343–347. doi: 10.1038/298343a0. [DOI] [PubMed] [Google Scholar]; (d) Taparowsky E, Suard Y, Fasano O, Shimizu K, Goldfarb M, Wigler M. Nature. 1982;300:762–765. doi: 10.1038/300762a0. [DOI] [PubMed] [Google Scholar]
- 7.Gysin S, Salt M, Young A, McCormick F. Genes & Cancer. 2011;2:359–372. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin L, Wang W, Fang G. Annu Rev Pharmacol Toxicol. 2014;54:435–456. doi: 10.1146/annurev-pharmtox-011613-140028.(and references therein) Fletcher S, Hamilton AD. J R Soc Interface. 2006;3:215–233. doi: 10.1098/rsif.2006.0115. (and references therein)
- 9.For selected examples of small molecule-based inhibition of Ras-GEF interactions: Winter JJG, Anderson M, Blades K, Brassington C, Breeze AL, Chresta C, Embrey K, Fairley G, Faulder P, Finlay MRV, Kettle JG, Nowak T, Overman R, Patel SJ, Perkins P, Spadola L, Tart J, Tucker JA, Wrigley G. J Med Chem. 2015;58:2265–2274. doi: 10.1021/jm501660t.Sacco E, Abraham SJ, Palmioli A, Damore G, Bargna A, Mazzoleni E, Gaponenko V, Vanoni M, Peri F. Med Chem Commun. 2011;2:396–401.Hocker HJ, Cho K-J, Chen C-YK, Rambahal N, Sagineedu SR, Shaari K, Stanslas J, Hancock JF, Gorfe AA. Proc Natl Acad Sci USA. 2013;110:10201–10206. doi: 10.1073/pnas.1300016110.Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D, Ludlam MJC, Bowman KK, Wu J, Giannetti AM, Starovasnik MA, Mellman I, Jackson PK, Rudolph J, Wang W, Fang G. Proc Natl Acad Sci USA. 2012;109:5299–5304. doi: 10.1073/pnas.1116510109.Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, Lee T, Rossanese OW, Fesik SW. Angew Chem Int Ed. 2012;51:6140–6143. doi: 10.1002/anie.201201358.
- 10.For selected examples of small molecule-based inhibition of Ras-Effector interactions: Waldmann H, Karaguni I-M, Carpintero M, Gourzoulidou E, Herrmann C, Brockmann C, Oschkinat H, Müller O. Angew Chem Int Ed. 2004;43:454–458. doi: 10.1002/anie.200353089.Shima F, Yoshikawa Y, Ye M, Araki M, Matsumoto S, Liao J, Hu L, Sugimoto T, Ijiri Y, Takeda A, Nishiyama Y, Sato C, Muraoka S, Tamura A, Osoda T, Tsuda K, Miyakawa T, Fukunishi H, Shimada J, Kumasaka T, Yamamoto M, Kataoka T. Proc Natl Adac Sci USA. 2013;110:8182–8187. doi: 10.1073/pnas.1217730110.Kato-Stankiewicz J, Hakimi I, Zhi G, Zhang J, Serebreiiskii I, Guo L, Edamatsu H, Koide H, Menon S, Eckl R, Sakamuri S, Lu Y, Chen Q-Z, Agarwal S, Baumbach WR, Golemis EA, Tamanoi F, Khazak V. Proc Natl Acad Sci USA. 2002;99:14398–14403. doi: 10.1073/pnas.222222699.
- 11.(a) Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, Bishop WR, Pai J-K. J Biol Chem. 1998;272:14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]; (b) Berndt N, Hamilton AD, Sebti SM. Nat Rev Cancer. 2011;11:775–791. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, Carrasco M, Hunter J, Kim ND, Xie T, Sim T, Jänne PA, Meyerson M, Marto JA, Engen JR, Gray NS. Angew Chem Int Ed. 2014;53:199–204. doi: 10.1002/anie.201307387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nevola L, Giralt E. Chem Commun. 2005;51:3302–3315. doi: 10.1039/c4cc08565e. [DOI] [PubMed] [Google Scholar]
- 14.For examples of peptide-derived modulators of PPIs associated with Ras: Patgiri A, Yadav KK, Arora PS, Bar-Sagi D. Nat Chem Biol. 2011;7:585–587. doi: 10.1038/nchembio.612.Leshchiner ES, Parkhitko A, Bird GH, Luccarelli J, Bellairs JA, Escudero S, Opoku-Nsiah K, Godes M, Perrimon N, Walensky LD. Proc Natl Acad Sci USA. 2015;112:1761–1766. doi: 10.1073/pnas.1413185112.Wu X, Upadhyaya P, Villalona-Calero MA, Briesewitz R, Pei D. Med Chem Commun. 2013;4:378–382. doi: 10.1039/C2MD20329D.Sacco E, Metalli D, Spinelli M, Manzoni R, Samalikova M, Grandori R, Morrione A, Traversa S, Alberghina L, Vanoni M. Biotech Adv. 2012;30:233–243. doi: 10.1016/j.biotechadv.2011.05.011.Barnard D, Sun H, Baker L, Marshall MS. Biochem Biophys Res Commun. 1998;247:176–180. doi: 10.1006/bbrc.1998.8746.Zeng J, Nheu T, Zorzet A, Catimel B, Nice E, Maruta H, Burgess AW, Treutlein HR. Protein Eng. 2001;14:39–45. doi: 10.1093/protein/14.1.39.
- 15.(a) Schafmeister CE, Po J, Verdine GL. J Am Chem Soc. 2000;129:2456–2457. [Google Scholar]; (b) Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For recent reviews: Walensky LD, Bird GH. J Med Chem. 2014;57:6275–6288. doi: 10.1021/jm4011675.Cromm PM, Spiegel J, Grossmann TN. ACS Chem Biol. 2015;10:1362–1375. doi: 10.1021/cb501020r.Verdine GL, Hilinski GJ. Meth Enzym. 2012;503:3–33. doi: 10.1016/B978-0-12-396962-0.00001-X. (and references therein)
- 17.McGee JH, Shim S, Lee S-J, Swanson PK, Jiang Y, Durney MA, Verdine GL. 2016 doi: 10.1074/jbc.M117.816348. (in preparation) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Boder ET, Wittrup KD. Nature Biotech. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]; (b) Gera N, Hussain M, Rao BM. Methods. 2013;60:15–26. doi: 10.1016/j.ymeth.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 19.(a) Schumacher TN, Mayr LM, Minor DL, Jr, Milhollen MA, Burgess MW, Kim PS. Science. 1996;271:1854–1857. doi: 10.1126/science.271.5257.1854. [DOI] [PubMed] [Google Scholar]; (b) Funke SA, Willbold D. Mol BioSyst. 2009;5:783–786. doi: 10.1039/b904138a. (and references therein) [DOI] [PubMed] [Google Scholar]
- 20.For selected examples: Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5.Welch BD, Francis JN, Redman JS, Paul S, Weinstock MT, Reeves JD, Lie YS, Whitby FG, Eckert DM, Hill CP, Root MJ, Kay MS. J Virol. 2010;84:11235–11244. doi: 10.1128/JVI.01339-10.Wiesehan K, Stohr J, Nagel-Steger L, van Groen T, Resner D, Willbold D. Protein Eng Des Sel. 2008;21:241–246. doi: 10.1093/protein/gzm054.Liu M, Pazgier M, Li C, Yuan W, Li C, Lu W. Angew Chem Int Ed. 2010;49:3649–3652. doi: 10.1002/anie.201000329.Mandal K, Uppalapati M, Ault-Riché D, Kenney J, Lowitz J, Sidhu SS, Kent SBH. Proc Natl Acad Sci USA. 2012;109:14779–14784. doi: 10.1073/pnas.1210483109.Uppalapati M, Lee D-J, Mandal K, Li H, Miranda LP, Lowitz J, Kenney J, Adams JJ, Ault-Riché D, Kent SBH, Sidhu SS. ACS Chem Biol. 2016;11:1058–1065. doi: 10.1021/acschembio.5b01006.
- 21.Yeates TO, Kent SBH. Ann Rev Biophys. 2012;41:41–61. doi: 10.1146/annurev-biophys-050511-102333. (and references therein) [DOI] [PubMed] [Google Scholar]
- 22.(a) Becker CFW, Hunter C, Seidel R, Kent SBH, Goody RS, Engelhard M. Proc Natl Acad Sci. 2003;100:5075–5080. doi: 10.1073/pnas.0831227100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Milić J, Seidel R, Becker CFW, Goody RS, Engelhard M. Pept Sci. 2007;90:399–405. doi: 10.1002/bip.20797. [DOI] [PubMed] [Google Scholar]
- 23.(a) Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776–778. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]; (b) Dawson PE, Kent SB. Annu Rev Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923. [DOI] [PubMed] [Google Scholar]
- 24.Wilson RM, Stockdill JL, Wu X, Li X, Vadola PA, Park PK, Wang P, Danishefsky SJ. Angew Chem Int Ed. 2012;51:2834–2848. doi: 10.1002/anie.201106628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Sakakibara S. Biopolymers. 1995;37:17–28. doi: 10.1002/bip.360370105. [DOI] [PubMed] [Google Scholar]; (b) Stuhr-Hansen N, Wilbek TS, Strømgaard K. Eur J Org Chem. 2013;24:5290–5294. [Google Scholar]
- 26.(a) Fang G-M, Li Y-M, Shen F, Huang Y-C, Li J-B, Lin Y, Cui H-K, Liu L. Angew Chem Int Ed. 2011;33:7645–7649. doi: 10.1002/anie.201100996. [DOI] [PubMed] [Google Scholar]; (b) Zheng J-S, Tang S, Qi Y-K, Wang Z-P, Liu L. Nat Prot. 2013;8:2483–2495. doi: 10.1038/nprot.2013.152. [DOI] [PubMed] [Google Scholar]
- 27.Yan LZ, Dawson PE. J Am Chem Soc. 2001;123:526–533. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]; b) Pentelute BL, Kent SB. Org Lett. 2007;9:687–690. doi: 10.1021/ol0630144. [DOI] [PubMed] [Google Scholar]; c) Wan Q, Danishefsky SJ. Angew Chem Int Ed. 2007;46:9248–9252. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- 28.Rao Y, Li X, Danishefsky SJ. J Am Chem Soc. 2009;131:12924–12926. doi: 10.1021/ja906005j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu X, Stockdill JL, Wang P, Danishefsky SJ. J Am Chem Soc. 2010;132:4098–4100. doi: 10.1021/ja100517v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wu X, Stockdill JL, Park PK, Danishefsky SJ. J Am Chem Soc. 2012;134:2378–2384. doi: 10.1021/ja2103372. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wang T, Danishefsky SJ. J Am Chem Soc. 2012;134:13244–13247. doi: 10.1021/ja3063452. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wang P, Danishefsky SJ. J Am Chem Soc. 2010;132:17045–17051. doi: 10.1021/ja1084628. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wang T, Danishefsky SJ. Proc Natl Acad Sci USA. 2013;110:11708–11713. doi: 10.1073/pnas.1310431110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Wang P, Li X, Zhu J, Chen J, Yuan Y, Wu X, Danishefsky SJ. J Am Chem Soc. 2011;133:1597–1602. doi: 10.1021/ja110115a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.For a useful review on alternative peptide ligation and amide-formation methods: Pattabiraman VR, Bode JW. Nature. 2011;480:471–479. doi: 10.1038/nature10702.
- 30.For an in-depth discussion of alternative peptide ligation approaches as well as alternative reactivity modes of C-terminal thiocarboxylic acids in the context of peptide synthesis: Roberts AG, Johnston EV, Shieh J-H, Sondey JP, Hendrickson RC, Moore MAS, Danishefsky SJ. J Am Chem Soc. 2015;137:13167–13175. doi: 10.1021/jacs.5b08754.
- 31.Upon workup with piperidine, some C-terminal piperidide from reaction of residual activated 3 (des-Fmoc) with piperidine was observed in the crude mixture. This result indicates that, even after 48 hours, some activated acyl donor is still present in the reaction mixture.
- 32.For detailed monitoring of the isonitrile-mediated activation sequence by UPLC/MS at each reaction step, see Supporting Information.
- 33.(a) Johnson T, Quibell M, Owen D, Sheppard RC. J Chem Soc Chem Commun. 1993:369–372. [Google Scholar]; (b) Johnson T, Quibell M. Tetrahedron Lett. 1994;35:463–466. [Google Scholar]; (c) Simmonds RG. Int J Peptide Protein Res. 1996;47:36–41. doi: 10.1111/j.1399-3011.1996.tb00807.x. [DOI] [PubMed] [Google Scholar]; (d) Nicolás E, Pujades M, Bacardit J, Giralt E, Albericio F. Tetrahedron Lett. 1997;38:2317–2320. [Google Scholar]; (e) Zheng J-S, Yu M, Qi Y-K, Tang S, Shen F, Wang Z-P, Xiao L, Zhang L, Tian C-L, Liu L. J Am Chem Soc. 2014;136:3695–3704. doi: 10.1021/ja500222u. [DOI] [PubMed] [Google Scholar]
- 34.In ref. 22a, the NCL of HRas[1–50] with HRas[51–166] was described as capricious under standard conditions due to aggregation and precipitation. Their experimentation found that ligation efficiency is much improved in the presence of an amphiphillic detergent to solubilize peptide partners; a study which culminated in the total synthesis of HRas[1–166]. Interestingly, the authors reported an unexpected unsassigned dehydration or deamination of the final protein using this ligation approach; one that did not have observable functional consequences.
- 35.Bang D, Pentelute BL, Kent SBH. Angew Chem Int Ed. 2006;45:3985–3988. doi: 10.1002/anie.200600702. [DOI] [PubMed] [Google Scholar]
- 36.Similar folding efficiency of a semisynthetic form of HRas with GTP as substrate has been previously reported. See Ref. 22b
- 37.D-GppNHp was obtained from commercial sources. L-GppNHp was synthesized from commercially available L-Ribose. For details, see Supporting Information.
- 38.John J, Sohmen R, Feuerstein J, Linke R, Wittinghofer A, Goody RS. Biochemistry. 1990;29:6058–6065. doi: 10.1021/bi00477a025. [DOI] [PubMed] [Google Scholar]
- 39.Intriguingly, we found that at higher assay concentration (1mM nucleotide triphosphate), both enantiomers of GppNHp could competitively displace mant-GppNHp-loaded recombinant KRas(G12V).
- 40.The overlay of our combined biophysical data suggests that synthetic KRas(G12V) variants are properly folded and behave similarly to that of recombinant material. We note slight discrepancies in the biophysical comparison of both synthetic all-L and all-D KRas variants with that of recombinant material. Folding and biophysical characterization were generally performed on microscale – thus slight changes in actual protein quantities may have an effect on the overall data. Additionally, the presence of some protein aggregate within the folded synthetic materials cannot be excluded and may be reflected in the experimental results. Nonetheless, the combination of these experiments clearly demonstrates 1) the enantiomeric relationship of the synthetic proteins, 2) nucleotide binding with enantiodiscrimination, and 3) peptide ligand binding with enantiodiscrimination.
- 41.We have demonstrated that little to no epimerization occurs during thioester synthesis of pertinent peptides (For details see Supporting Information), and regard this as an unlikely factor in the observed folding efficiency of these synthetic proteins. For related C-terminal thioester synthesis methods with particular analysis of the potential for epimerization upon C-terminal residue activation, see: Futaki S, Sogawa K, Maruyama J, Asahar T, Niwa M, Hojo H. Tetrahedron Lett. 1997;35:6237–6240.Quibell M, Packman LC, Johnson T. J Chem Soc, Perkin Trans 1. 1996:1219–1225.Bray BL. Nat Rev Drug Discov. 2003;2:587–593. doi: 10.1038/nrd1133.Nagalingam AC, Radfors SE, Warriner SL. Synlett. 2007;16:2517–2520.Flemer S., Jr J Pept Sci. 2009;15:693–696. doi: 10.1002/psc.1181.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.