

SUMMARY
A mechanistic understanding of neural computation requires determining how information is processed as it passes through neurons and across synapses. However, it has been challenging to measure membrane potential changes in axons and dendrites in vivo. We use in vivo, two-photon imaging of novel genetically encoded voltage indicators, as well as calcium imaging, to measure sensory stimulus-evoked signals in the Drosophila visual system with subcellular resolution. Across synapses, we find major transformations in the kinetics, amplitude, and sign of voltage responses to light. We also describe distinct relationships between voltage and calcium signals in different neuronal compartments, a substrate for local computation. Finally, we demonstrate that ON and OFF selectivity, a key feature of visual processing across species, emerges through the transformation of membrane potential into intracellular calcium concentration. By imaging voltage and calcium signals to map information flow with subcellular resolution, we illuminate where and how critical computations arise.
Graphical Abstract
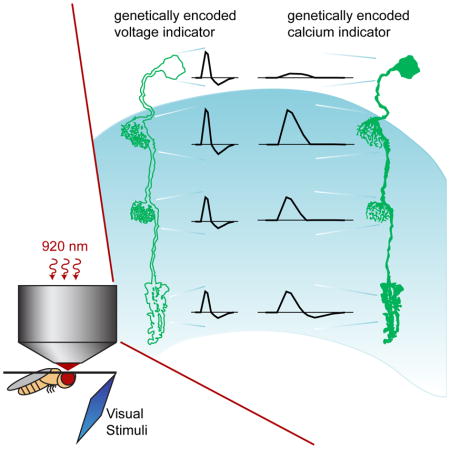
INTRODUCTION
Neuronal circuits perform a diverse array of computations to ultimately guide animal behavior. These circuits encode information in dynamic changes in neuronal membrane potential and intracellular calcium concentration; stepwise transformations of these signals then provide the mechanistic basis for extracting behaviorally relevant features. Mapping these signals as they flow along dendrites and axons and across synapses is thus critical to understanding neural processing. Here we describe a broadly applicable approach for measuring voltage and calcium changes with subcellular resolution in vivo.
Sensory systems link critical information from the environment to behavior. Given their experimental tractability, these systems have long been studied to understand neural computation, and the information extracted by many peripheral sensory circuits has been characterized in detail. For example, in many sensory systems, including vision, chemosensation, audition, and thermosensation, a single input is initially transformed into two representations, an ON pathway for increases in signal intensity, and an OFF pathway for decreases (Chalasani et al., 2007; Kuffler, 1953; Liu et al., 2015; Scholl et al., 2010; Wässle, 2004). This fundamental organizational principle has been associated with a variety of different strategies for efficiently extracting information about the natural world (Clark et al., 2014; Gjorgjieva et al., 2014; Westheimer, 2007). Thus, understanding where and how circuits implement ON and OFF selectivity provides fundamental insights into neuronal computation.
The Drosophila visual system represents a powerful in vivo model for exploring the mechanistic basis of neuronal computation. Experimentally, cell-type specific expression of genetically encoded indicators of neural activity is straightforward, the optic lobes are accessible for live imaging, well-controlled stimuli can be presented, the cell types are well defined, and their synaptic connections have been reconstructed at the ultrastructural level (Borst, 2014; Silies et al., 2014). Biologically, the fly visual system performs computations essential for behavior using algorithms that are broadly conserved across visual systems. For example, like the vertebrate visual system (Wässle, 2004), the Drosophila visual system is split into ON and OFF pathways (Clark et al., 2011; Joesch et al., 2010; Maisak et al., 2013). The lamina monopolar cells L1 and L2 are the strongest synaptic targets of R1-6 photoreceptors in the lamina and provide input into ON circuitry for light increments and OFF circuitry for light decrements, respectively, in the medulla (Figure 1A). L1 makes synapses onto Mi1 and Tm3; L2 makes synapses onto Tm1 and Tm2 (Takemura et al., 2013). Synapsing in the medulla and the lobula, these medulla interneurons then provide input onto deeper circuitry, including the direction selective cells T4 and T5 (Shinomiya et al., 2014; Takemura et al., 2013), and represent central feedforward visual pathways.
Figure 1. Voltage imaging in the Drosophila visual system.

(A) The Drosophila visual system. L1 and L2 receive input from photoreceptors R1-6 in the lamina neuropil. L1 synapses onto Mi1 and Tm3 in layers M1 and M5 of the medulla neuropil. L2 synapses onto Tm1 and Tm2 in medulla layer M2. L1 and its postsynaptic partners are in green; L2 and its postsynaptic partners are in blue. Tm1, Tm2, and Tm3 project axons into the lobula neuropil. In all figures, cell diagrams modified from Fischbach and Dittrich, 1989. (B) In ASAP sensors, changes in membrane potential induce movement of a positively charged transmembrane helix of a voltage sensitive domain (VSD), altering the fluorescence of a circularly permuted GFP (cpGFP). (C) Schematic diagram of ASAP2f, showing the VSD transmembrane domains (S1–S4, blue), cpGFP (green), and the location of the residues changed from ASAP1 (A147S ΔA148). (D) Schematic illustration of the setup for in vivo, two-photon imaging of visually evoked responses in Drosophila. (E) Illustration of L2 with the imaged region, the axon terminal in medulla layer M2, highlighted. Inset: in vivo, two-photon image of L2 axon terminals expressing ASAP2f, averaged across one time series. One axon terminal is highlighted. Scale bar: 5 μm. (F, G) Responses of L2 neurons to alternating 300 ms-long dark and light flashes, as measured with (F) ASAP1 (n = 52 cells, 3 flies) and (G) ASAP2f (n = 170 cells, 8 flies). Top: mean response across all cells. Each cell contributes its average response across 100 trials (1 trial = 1 dark flash and 1 light flash). Bottom: 5 exemplar single-trial responses from a representative L2 cell (gray) and the same cell’s mean response averaged over all trials (black or blue). The solid line is the mean response; the shading is ± 1 SEM. (H–J) Parameters quantifying the response: (H) peak ΔF/F; (I) tpeak; (J) τdecay. The mean ± 1 SEM is plotted. *** p < 0.001 (two-sample t-test, Bonferroni correction for multiple comparisons). See also Figures S1 and S2 and Table S1.
Mapping transformations of visual information onto changes in membrane potential in dendrites and axons critically depends on a genetically encoded voltage indicator that can capture signals in vivo. Here we describe fast, bright, two-photon compatible voltage indicators that capture responses predicted from biophysical neuronal models and previously reported electrophysiological measurements. Using these indicators as well as genetically encoded calcium indicators, we reveal the local changes in voltage and calcium signaling that transform sensory information in the Drosophila visual system.
RESULTS
Benchmarking in vivo voltage imaging of ASAP2f
We began our study of information processing in the visual system by developing a genetically encoded voltage sensor with higher sensitivity and rapid kinetics that is compatible with two-photon imaging in the fly. We chose the voltage sensor ASAP1 as our starting point for further engineering (St-Pierre et al., 2014). In this indicator, a circularly permuted Green Fluorescent Protein (cpGFP) is inserted into a voltage-sensing domain (VSD) from a voltage sensitive phosphatase (Figure 1B). Voltage-induced conformational changes in the VSD perturb GFP fluorescence, such that depolarizations decrease GFP fluorescence and hyperpolarizations increase it. These responses have fast on and off time constants of approximately 2.5 ms (Table S1) (St-Pierre et al., 2014). Based on in vitro characterization of variants with mutations in the linker between the third transmembrane segment of the VSD (S3) and GFP (Figures S1, S2A–S2C, and Table S1), we examined the in vivo performance of ASAP1 and one variant, ASAP1 A147S ΔA148 (renamed ASAP2f; Figure 1C). To do this, we expressed ASAP1 and ASAP2f specifically in the lamina neuron L2 and measured visually evoked responses using two-photon microscopy. We positioned an awake fly in front of a screen displaying visual stimuli and imaged L2 axon terminals through a window cut in the cuticle at the back of the head (Figures 1D and 1E). We acquired frames at 39 Hz and by taking advantage of differences in timing between frame acquisition and visual stimulus presentation, were able to measure responses at an effective sampling rate of 120 Hz (see Supplemental Experimental Procedures).
To robustly stimulate L2, we first presented alternating 300 ms full contrast dark and light flashes covering the entire screen. ASAP1 and ASAP2f both reported that L2 transiently depolarized to light offset and transiently hyperpolarized to light onset, consistent with previous electrophysiological recordings (Figures 1F and 1G) (Zettler and Järvilehto, 1971). The signal amplitudes and signal-to-noise ratios of these indicators were sufficient to record robust single-cell responses; we could also measure responses from single stimulus presentations (Figures 1F and 1G, bottom). ASAP2f produced larger fluorescence changes than ASAP1 (14.0 ± 0.7% increase for depolarization and 20.3 ± 1.4% increase for hyperpolarization), with similar response kinetics (Figures 1H–1J). ASAP2f responses were stable for at least 10 minutes of continuous imaging, facilitating many stimulus presentations (Figures S2D and S2E). We therefore used ASAP2f in subsequent experiments.
ASAP2f allows impulse response measurements
One commonly used approach to characterizing a dynamic system is to measure the impulse response, the response evoked by an extremely brief stimulus. In visual neuroscience, impulses correspond to brief flashes of light and reveal critical aspects of a cellular response, including the kinetics and waveform (Smith, 1997). For linear systems, the impulse response is equivalent to the linear filter that can be extracted using white noise analysis (Chichilnisky, 2001). We therefore presented both 8 ms and 25 ms light and dark flashes off of a uniform gray background to flies in which ASAP2f was expressed in L2. Such short flashes approximately matched the flicker fusion rate of Drosophila photoreceptors (Miall, 1978) and therefore should have been seen by the animal as a single impulse signal instead of a distinct light to dark and dark to light transition for each flash. For both 25 ms and 8 ms flashes, L2 initially hyperpolarized to the light flash and depolarized to the dark flash (Figures 2A and 2B). The response to the light flash was strongly biphasic, with a slower depolarizing phase that followed the initial hyperpolarization. Conversely, the response to the dark flash was only weakly biphasic. Thus, impulse responses to light and dark were not sign-inverted or rescaled versions of the same waveform, suggesting that even at this early stage of visual processing, nonlinearities in voltage responses are significant.
Figure 2. Voltage imaging captures L2 impulse responses.

(A) Response of L2 axon terminals to a 25 ms light flash (left) or a 25 ms dark flash (right), with a 500 ms gray interleave. Contrast = 0.5. n = 125 cells, 11 flies. (B) Response of L2 axon terminals to an 8 ms light flash (left) or 8 ms dark flash (right), plotted with an electrophysiological recording from a lamina monopolar cell responding to similarly brief light flashes of two different intensities (data from Nikolaev et al., 2009). All responses are aligned to the onset of the light flash (red line).
As a critical control, we examined whether the kinetics of these impulse responses were consistent with previous electrophysiological studies. The onset of the impulse response was delayed relative to the onset of light by approximately 17 ms (Figure 2C). This delay was intermediate between the response latencies measured under bright and dim illumination conditions using electrophysiological recordings from lamina monopolar cells (Figure 2C) (Nikolaev et al., 2009). Thus ASAP2f can report rapid changes in neural activity.
ASAP2f reports voltage signals across different cellular compartments
Unlike electrophysiological approaches, monitoring neural activity using voltage imaging allows responses to be easily measured from multiple regions of the same neuron. We therefore expressed ASAP2f in the Mi1 cell type, a small monopolar neuron that is representative of interneurons in the fruit fly brain. Mi1 is approximately 50 μm long, with its main neurite being 200–300 nm in diameter and its arbors being comprised of many processes of 200 nm or less (Figure 3A) (Takemura et al., 2013). We measured robust voltage responses in the cell body and the three dendritic and axonal arbors in medulla layers M1, M5, and M10 (Figure 3B). The amplitude of the responses generally decayed away from the site of primary synaptic input in layer M1, but the time courses of the response waveforms were largely preserved throughout the length of the neuron (Figures 3B and 3C). Moreover, our impulse response, the ASAP2f-measured voltage change to a 25 ms light flash, and the linear filter extracted from white noise analysis using electrophysiological approaches, both measured in the cell body of Mi1, had indistinguishable times to initial response and times to peak (Figure 3D) (Behnia et al., 2014). However, we note that the impulse response we measured was more biphasic than the linear filter, likely because fast white noise stimuli do not drive temporal and spatial surrounds as well as isolated flashes (Simoncelli et al., 2004).
Figure 3. Voltage imaging captures Mi1 response kinetics and the decay in response amplitude along the length of the cell.

(A) Illustration of Mi1 with the imaged regions highlighted. (B) Voltage responses of the Mi1 arbors in layer M1 (blue, n = 79 cells, 4 flies), layer M5 (red, n = 92 cells, 4 flies), layer M10 (green, n = 67 cells, 4 flies), and the cell body (black, n = 37 cells, 2 flies) to a 25 ms light flash with a 500 ms gray interleave, contrast = 0.5. (C) Quantification of the decay in peak response amplitude (peak ΔF/F) as a fraction of the response in layer M1. (D) Impulse responses measured from the cell body of Mi1. Black: voltage response to a 25 ms light flash measured with ASAP2f (data from (B)). Pink: linear filter extracted from white noise analysis measured using electrophysiological recordings (data from Behnia et al., 2014). (E) Morphology of a NEURON model of Mi1. The arrow indicates the site of current injection. The model neuron was given passive membrane properties: specific membrane capacitance (Cm) = 1 μF/cm2, axial resistance (Ri) = 40 to 420 Ω•cm, and specific membrane resistance (Rm) = 1 to 21 kΩ•cm2. (F, G) Peak membrane potential during current injection presented as the fraction decayed from the peak response in layer M1. (F) Layer M5 and (G) layer M10 of the NEURON model. The shaded areas indicate the set of Ri and Rm values that result in decay values within 1 SEM of the decay measured with ASAP2f. (H) The set of Ri and Rm values that result in decay values within 1 SEM of the decay measured with ASAP2f for both layers M5 and M10.
We next asked whether the decay in the amplitude of the responses along the length of Mi1 was to be expected from the electrical properties of the cell or whether ASAP2f was behaving differently in distinct arbors. Electrode recordings are impossible from these small processes, so we instead implemented a biophysical model of the cell using NEURON (Carnevale and Hines, 2006). We used the morphology from the electron microscopic reconstruction of Mi1 to describe the structure of the arbors in the medulla (Takemura et al., 2013), and as this reconstruction lacked a cell body, we added a simple connecting process and cell body to the model (Figure 3E). We gave the model cell only passive membrane properties and simulated synaptic input by injecting current into the arbor in layer M1, where the neuron receives most of its synapses. From this, we asked whether the decay in amplitude from layer M1 to layers M5 and M10 was consistent with a biologically plausible set of passive membrane properties. Critically, the same parameters should be able to explain the decay amplitude in both layers M5 and M10. To do this, we incorporated previous estimates of the specific membrane capacitance and resting membrane potential, and then swept the axial resistance and specific membrane resistance across biologically plausible values (Behnia et al., 2014; Cuntz et al., 2013; Gouwens and Wilson, 2009; Haag et al., 1996). These simulations revealed a range of parameter values that predicted the amplitude decay we measured with ASAP2f in both M5 and M10 (Figures 3F–3H). Thus, ASAP2f appears to accurately report changes in membrane voltage in different neuronal compartments. These simulations also suggest that membrane potential changes spread passively through Mi1, though we do not exclude a role for active conductances.
Voltage responses are transformed in amplitude, sign, and kinetics across synapses
We next sought to monitor how changes in light intensity are progressively transformed across synapses. We therefore imaged presynaptic axon terminals and their corresponding postsynaptic dendrites while presenting 25 ms light and dark flashes. We first examined responses in the OFF pathway, comparing Tm1 and Tm2 voltage dynamics to those of L2, their presynaptic partner (Figure 4A). Like the axon terminals of L2, the dendrites of Tm1 and Tm2 hyperpolarized to light flashes and depolarized to dark flashes (Figures 4B, S3A, and S3B), consistent with previous electrophysiological recordings from the cell body (Behnia et al., 2014). Additionally, we found that the amplitude of the voltage responses was significantly larger in the postsynaptic dendrite than in the presynaptic axon terminal (Figures 4B, 4C, S3B, and S3C). As these are thought to be non-spiking neurons, this increase in amplitude may boost the voltage signal to ensure reliable propagation to the neurons’ distal arbors. Furthermore, the kinetics of the voltage signal were significantly slower in the postsynaptic dendrites: Tm1 and Tm2 responses peaked later and persisted longer than L2 responses (Figures 4B, 4D, 4E, S3B, S3D, and S3E). Mathematically, these responses suggest that Tm1 and Tm2 integrate the L2 voltage signal. As the responses to a 25 ms flash approximate the cells’ impulse responses, the slower kinetics of Tm1 and Tm2 demonstrate that at the synapse between L2 and its postsynaptic partners, the circuit becomes tuned for slower changes in light intensity. Finally, we note that Tm2 responses peaked slightly earlier than Tm1 responses, consistent with previous measurements (Behnia et al., 2014).
Figure 4. Voltage responses are transformed between presynaptic axons and postsynaptic dendrites.

(A) Illustration of L2 and its postsynaptic targets Tm1 and Tm2. The imaged arbors in medulla layer M2 are highlighted. (B) Responses of L2 (black, n = 125 cells, 11 flies), Tm1 (blue, n = 79 cells, 4 flies), and Tm2 (red, n = 89 cells, 4 flies) to a 25 ms light flash with a 500 ms gray interleave, contrast = 0.5. The solid line is the mean response; the shading is ± 1 SEM. (C–E) Quantification of the response: (C) peak ΔF/F; (D) tpeak; (E) full width at half maximum of the initial response. The mean ± 1 SEM is plotted. (F–J) In medulla layer M1, presynaptic cell L1 (black, n = 23 cells, 6 flies) and postsynaptic cells Mi1 (blue, n = 79 cells, 4 flies) and Tm3 (red, n = 153 cells, 8 flies). (K–O) In medulla layer M5, L1 (black, n = 14 cells, 5 flies), Mi1 (blue, n = 92 cells, 4 flies), and Tm3 (red, n = 35 cells, 4 flies). In (H) and (M), the light gray bar is the inverted L1 peak ΔF/F for comparison. * p < 0.05, ** p < 0.01, *** p < 0.001 (two-sample t-test for peak ΔF/F and full width at half maximum, Mann-Whitney U test for tpeak, Bonferroni correction for multiple comparisons). See also Figure S3.
We next asked whether visual information is transformed differently across synapses in the ON pathway compared to the OFF pathway. In the ON pathway, L1 makes synaptic connections with Mi1 and Tm3 via two axon terminals located in medulla layers M1 and M5 (Takemura et al., 2013). In layer M1, L1 responded very similarly to L2, hyperpolarizing to light increments and depolarizing to light decrements (Figures 4F, 4G, S3F, and S3G). However, unlike the OFF pathway, we observed a sign inversion between L1 and its postsynaptic cells (Figures 4G, 4H, S4G, and S4H), as has been previously reported (Behnia et al., 2014). Interestingly, similar to the transformation between L2 and its postsynaptic targets, Mi1 and Tm3 responses were larger in amplitude and slower in kinetics than those of their presynaptic partner L1 (Figures 4G–4J and S3G–S3J). In layer M5, we observed similar changes in response sign, amplitude, and kinetics to those we saw in layer M1 (Figures 4K–4O and S3K–S3O). Thus, the L1 and L2 pathways perform similar transformations in signal amplitude and kinetics even as they employ synapses of opposite sign, consistent with the view that they are parallel ON and OFF channels. Taken together, these results demonstrate that voltage signals undergo profound transformations in amplitude, sign, and kinetics as they are passed across synapses.
Calcium responses, unlike voltage responses, are compartmentalized within a neuron
Chemical synaptic transmission requires changes in membrane potential to be converted into changes in intracellular calcium concentration, which then trigger neurotransmitter release (Fatt and Katz, 1952). We therefore compared voltage and calcium signals in corresponding regions of the same neuron. Specifically, we measured voltage signals with ASAP2f and calcium signals with GCaMP6f, a fast variant of the GCaMP family (Chen et al., 2013), across multiple compartments of Tm3, Mi1, and Tm1. In all three cell types, the temporal waveforms of the voltage responses were similar throughout the length of each neuron while the amplitude of the responses decayed away from the site of primary synaptic input (layer M1 for Tm3 and Mi1 and layer M2 for Tm1; Figures 5A, 5B, 5D, 5F, 5G, 5I, S4A, S4B, and S4D), a result consistent with passive spread of membrane potential changes in Tm3, Mi1, and Tm1.
Figure 5. Mapping voltage and calcium responses in different subcellular compartments of the same neuron.

(A) Illustration of Tm3 with the imaged regions highlighted. (B) Voltage responses of Tm3 arbors in layer M1 (blue, n = 158 cells, 8 flies), layer M5 (red, n = 35 cells, 4 flies), layer M10 (green, n = 100 cells, 9 flies), and the cell body (black, n = 13 cells, 2 flies) to a 25 ms light flash with a 500 ms gray interleave, contrast = 0.5. (C) Calcium responses of Tm3 arbors in layer M1 (blue, n = 126 cells, 8 flies), layer M5 (red, n = 69 cells, 8 flies), layer M10 (green, n = 99 cells, 4 flies), and the cell body (black, n = 71 cells, 4 flies) to a 25 ms light flash with a 1500 ms gray interleave, contrast = 0.5. The solid line is the mean response; the shading is ± 1 SEM. (D) Quantification of the voltage signals. Left: peak ΔF/F. Middle: tpeak. Right: full width at half maximum. The mean ± 1 SEM is plotted. (E) Quantification of the calcium signals with the metrics arranged as in (D). (F–J) Responses of Mi1. (G, I) Voltage responses of the Mi1 arbors in layer M1 (blue, n = 79 cells, 4 flies), layer M5 (red, n = 92 cells, 4 flies), layer M10 (green, n = 67 cells, 4 flies), and the cell body (black, n = 37 cells, 2 flies). (G) is repeated from Figure 3B. (H, J) Calcium responses of the Mi1 arbors in layer M1 (blue, n = 94 cells, 5 flies), layer M5 (red, n = 67 cells, 5 flies), layer M10 (green, n = 89 cells, 5 flies), and the cell body (black, n = 51 cells, 5 flies). ** p < 0.01, *** p < 0.001 (two-sample t-test for peak ΔF/F and full width at half maximum, Mann-Whitney U test for tpeak, Bonferroni correction for multiple comparisons). See also Figure S4.
To our surprise, however, unlike voltage signals, calcium signals were highly compartmentalized. That is, calcium responses were different among distinct regions of the same cell, and the way in which they were compartmentalized differed among cell types. The calcium signal in any compartment could not be predicted from either the voltage response in that compartment nor the calcium responses in other regions of the same cell. For example, in response to a 25 ms light flash, as expected for a depolarization, the calcium concentration increased across all of Tm3 (Figure 5C and 5E). However, inconsistent with the relative amplitudes of the voltage signals in each region, calcium responses in the cell body and layer M5 were much smaller than those in layers M1 and M10, the neighboring regions (Figures 5A–5E). Despite the large differences in the magnitude of intracellular calcium responses, the waveform and kinetics were the same across layers M1, M5, and M10 (Figures 5C and 5E). However, the calcium signals were slower in the cell body. In Mi1, in contrast to Tm3, the relative amplitude of the calcium changes in the different layers of the medulla was consistent with the voltage responses (Figures 5F–5J). However, in this cell, the calcium response waveform in layer M10 was biphasic and relatively fast, while the responses in layers M1 and M5 were monophasic and slower to peak (Figures 5H and 5J). Thus, as Mi1 has synaptic outputs from each of these arbors (Takemura et al., 2013), it could transmit qualitatively different visual information from layer M10 than from layers M1 and M5. Additionally, like Tm3, the calcium responses were the slowest in the Mi1 cell body (Figures 5H and 5J). Finally, in Tm1, unlike Tm3 or Mi1, calcium responses in all arbors were similar in amplitude and kinetics, much like the voltage responses, though the cell body responses were slow and small (Figure S4). Taken together, these results demonstrate that both the amplitude and kinetics of changes in intracellular calcium concentrations have distinct relationships with voltage in different compartments of the same cell and are cell type specifically regulated.
ON and OFF selectivity arises in the transformation between voltage and calcium
Postsynaptic to Mi1 and Tm3, T4 is strongly selective for light stimuli and hence rectified for ON; postsynaptic to Tm1 and Tm2, T5 is strongly selective for dark and hence rectified for OFF (Fisher et al., 2015; Leonhardt et al., 2016; Maisak et al., 2013). However, electrophysiological studies demonstrated that Mi1, Tm3, Tm1, and Tm2 are moderately selective (Behnia et al., 2014), a degree of rectification that cannot account for the nonlinearity of the postsynaptic responses. To map the emergence of rectification across visual circuitry, we presented contrast-matched light and dark flashes spanning six different contrast values to L1 and Tm3, members of the ON pathway, as well as L2 and Tm1, members of the OFF pathway, and measured voltage and calcium responses in their axon terminals.
Voltage signals in L1 and L2 responded strongly to both light and dark, with slightly larger responses to increments of light relative to decrements of equivalent magnitude. Intracellular calcium signals in both cell types responded robustly to both light and dark, but slightly favored decrements, consistent with previous reports (Clark et al., 2011; Juusola et al., 1995) (Figures 6A, 6B, 6D, 6E, 7A, and 7C). Voltage signals in Tm3, like in L1, responded strongly to both light and dark, with a modestly larger response to increments (Figures 6C and 7B). Strikingly, this cell’s calcium signals displayed strong, half-wave rectification, with depolarizing changes in membrane potential producing large increases in calcium concentration but hyperpolarizations producing minimal decreases in calcium concentration (Figure 6C). Calcium signals in Tm3 were therefore selective for light: large calcium increases were evoked by light flashes but much smaller calcium decreases were evoked by dark flashes (Figures 6C and 7B). We obtained similar results from the other ON pathway cell, Mi1 (Figures S5A–S5C). Voltage signals in the OFF pathway neuron Tm1 also responded strongly to both light and dark (Figures 6F and 7D). Remarkably, in this cell, calcium signals evoked by decrements were much stronger than those evoked by increments (Figures 6F and 7D). Again, we observed similar results from the other OFF pathway neuron Tm2 (Figures S5D–S5F). Taken together, these data demonstrate that contrast selectivity emerges at the level of the voltage to calcium transformation in the third order neurons of both the ON and OFF pathways.
Figure 6. Measuring ON and OFF selectivity.

(A) Illustration of the ON pathway with the imaged regions highlighted. (B, C) Voltage (left) and calcium (right) responses of (B) L1 (voltage: n = 43 cells, 9 flies and calcium: n = 68 cells, 9 flies) and (C) Tm3 (voltage: n = 97 cells, 8 flies and calcium: n = 85 cells, 7 flies) to 25 ms light and dark flashes of varying contrasts off of gray (contrast = 0.125, 0.25, or 0.5). The solid line is the mean response; the shading is ± 1 SEM. (D) Illustration of the OFF pathway. (E, F) Voltage (left) and calcium (right) responses of (E) L2 (voltage: n = 116 cells, 7 flies and calcium: n = 88 cells, 8 flies) and (F) Tm1 (voltage: n = 84 cells, 7 flies and calcium: n = 136 cells, 8 flies). See also Figures S5–S7.
Figure 7. ON and OFF selectivity arises in the transformation between voltage and calcium.

(A, B) Quantification of selectivity to light (ON). The line, box, and whiskers are the median, interquartile range, and maximum and minimum values within one interquartile range, respectively. (A) L1 layer M1 axon terminals (voltage: n = 43 cells, 9 flies and calcium: n = 68 cells, 9 flies). (B) Tm3 layer M10 axon terminals (voltage: n = 97 cells, 8 flies and calcium: n = 85 cells, 7 flies). (C, D) Quantification of selectivity to dark (OFF). (C) L2 layer M2 axon terminals (voltage: n = 116 cells, 7 flies and calcium: n = 88 cells, 8 flies). (D) Tm1 layer Lo1 axon terminals (voltage: n = 84 cells, 7 flies and calcium: n = 136 cells, 8 flies). (E) Schematic summarizing the emergence of ON and OFF selectivity, displaying idealized impulse responses, at the level of membrane potential and intracellular calcium. See also Figures S5–S7.
We were concerned that indicator nonlinearities and not underlying biological processes might account for the rectification we observed. To examine this, we measured contrast responses across these cell types using different voltage and calcium indicators. For voltage, we developed a new member of the ASAP family, ASAP1 I67T Q397R, that had larger fluorescence changes to depolarizations and smaller fluorescence changes to hyperpolarizations compared with ASAP2f (Figure S6). Using this indicator, we found that the degree of rectification in L2, Tm1, and Tm3 was consistent with that measured with ASAP2f (Figures S7A–S7F). For calcium, we used GCaMP6m, which has a Kd that is approximately half that of GCaMP6f (Chen et al., 2013). If the observed half-wave rectification reflected the fact that GCaMP6f is less sensitive to low calcium concentrations, then the responses measured with GCaMP6m should not be rectified. However, this was not the case, as the degree of rectification was similar for GCaMP6f and GCaMP6m (Figures S7A–S7F). As an independent approach to exploring the effect of indicator nonlinearities on rectification, we also examined whether measurement of GCaMP6f responses to visually-evoked hyperpolarization was limited by the ability of the indicator to report calcium decreases. Replacing the perfusion saline with one that contained a calcium chelator, we observed that GCaMP6f fluorescence in L2, Tm1, and Tm3 decreased dramatically, much more than observed when presenting visual stimuli (Figures S7G–S7I). Thus, our measurements are well within GCaMP6f’s dynamic range in these cells, a conclusion that is also consistent with our ability to measure the robust calcium increases and decreases evoked by dark and light flashes using this indicator in L1 and L2.
Taken together, these data demonstrate that the degree of linearity of the voltage to calcium transform differs across neurons to implement contrast selectivity (Figure 7E). In L1 and L2, the amplitude of calcium and voltage responses vary largely linearly with respect to contrast. However, in two postsynaptic targets of each cell, calcium responses, but not voltage responses, were strongly half-wave rectified to favor the depolarizing visual stimulus. The upstream synaptic sign inversion between L1 and its postsynaptic partners combined with rectification at the level of intracellular calcium in Mi1 and Tm3 makes the L1 pathway ON selective. The sign preserving synapse between L2 and its postsynaptic partners combined with rectification at the level of calcium in Tm1 and Tm2 makes the L2 pathway OFF selective.
DISCUSSION
The transformation of information by individual neurons and circuits underlies nervous system function. By performing both voltage and calcium imaging of visually evoked responses not only in cell bodies but also in dendrites and axons, we take a novel approach towards understanding information processing. While neurons transmit voltage signals across their processes with high fidelity, we observe large transformations in the sign, kinetics, and amplitude of visually driven changes in voltage across synapses. Thus, critical neuronal computations are performed between synaptically connected neurites. We also find that calcium responses, unlike voltage responses, are compartmentalized within a neuron. This functional specialization allows an individual neuron to transmit distinct information across different synapses, thereby creating a neural substrate for local computation. Finally, we demonstrate that ON and OFF selectivity, a critical split in sensory processing pathways in many systems, emerges at the voltage to calcium transformation in third-order neurons in this circuit.
Validation of in vivo voltage imaging with genetically encoded indicators
There has been considerable interest in applying genetically encoded voltage indicators to in vivo studies of neuronal computation. These tools hold the promise of allowing measurements of membrane potential changes in subcellular compartments; compared to genetically encoded calcium indicators, they have faster temporal resolution and are a direct readout of voltage changes. However, limitations in the dynamic range, brightness, and critically, the ability to be excited by two-photon stimulation have restricted their use in vivo. Using an improved voltage indicator, we demonstrate that in vivo voltage imaging can provide novel biological insights that could not have been obtained through other techniques.
To validate this new indicator, ASAP2f, we compared ASAP2f responses to previous electrophysiological recordings from the same cells (Figures 1–3) (Behnia et al., 2014; Nikolaev et al., 2009). The temporal features revealed by the indicator matched the electrophysiological response profiles well, thereby providing confidence that voltage imaging is an accurate readout of the underlying biological responses. However, a key advantage of imaging is that signals from subcellular regions inaccessible to electrodes can also be measured. To validate these measurements, we took an in silico approach, implementing a simple biophysical model of the cell. These studies revealed that the changes in membrane potential revealed by the indicator are consistent with those predicted by the model cell across physiologically reasonable membrane properties (Figure 3). Thus, ASAP2f appears to accurately report voltage changes across dendrites and axons.
ASAP2f complements other genetically encoded voltage indicators that have been used for in vivo imaging in Drosophila, ArcLight and Ace2N-mNeon (Cao et al., 2013; Gong et al., 2015). Using ArcLight, Cao and colleagues were the first to measure sensory stimulus-evoked voltage signals in living flies (Cao et al., 2013). More recently, Gong and colleagues used Ace2N-mNeon to record similar voltage responses with greatly improved temporal resolution (Gong et al., 2015). Both of these studies used epifluorescence illumination; in vivo, two-photon imaging has not been reported for either indicator. ASAP2f performs well under two-photon excitation, and this afforded us two critical advantages. First, we could image in the visual system where one-photon illumination would stimulate the photoreceptors (Salcedo et al., 1999). Second, more generally, the minimal out-of-plane fluorescence of this imaging modality increases the effective spatial resolution in deep tissue, an advantage for mapping neuronal responses with subcellular resolution (Svoboda and Yasuda, 2006). However, we note that while Ace2N-mNeon signals were captured at 1 kHz (Gong et al., 2015), our signals were sampled at 120 Hz, a difference that reflects the distinct imaging modalities used. Nonetheless, two-photon imaging of ASAP2f will be broadly applicable to experimental systems where the advantages of two-photon excitation are required.
More generally, from the perspective of reporters of neuronal activity, we provide definitive evidence that voltage and calcium changes are distinct signals (Figures 5–7). This demonstrates the need for careful interpretation of responses measured with different indicators but also the need for sensors of different aspects of neuronal activity to provide a richer picture of nervous system function.
Neuronal computations occur between synaptically connected neurites
Taking advantage of cell-type specific indicator expression and sufficient spatial resolution and indicator sensitivity to measure signals from subcellular compartments, we observed information flow between synaptically connected axons and dendrites. Across four different synapses, we found transformations in the amplitude and kinetics of voltage signals evoked by sensory stimuli in vivo (Figure 4). Voltage responses to identical visual stimuli were larger in the postsynaptic dendrite than they were in the axon. This amplification may enable reliable passive propagation of weak input signals to the axon terminal of the postsynaptic cell. Given the relatively poor quantum efficiency of the fly compound eye, this amplification may be critical to the transmission of low contrast visual inputs. In addition, across all four synaptic connections, initial postsynaptic voltage signals emerged with a short latency relative to the presynaptic signal but evolved more slowly and appeared to integrate it. As these cells represent critical inputs to neurons that compute the direction of motion (Shinomiya et al., 2014; Takemura et al., 2013), these temporal transformations tune the speed sensitivity of motion detectors. More broadly, our results emphasize the central role that synaptic connections play in precisely shaping processing of behaviorally relevant information in the brain.
Compartmentalization of calcium responses can support local computation
Individual neurons are powerful computational elements, implementing combinations of arithmetic and logical operations to nonlinearly combine inputs to support behaviorally relevant computations in vivo (London and Häusser, 2005; Stuart and Spruston, 2015). However, most studies have focused on how inputs are transformed, making the implicit assumption that a neuron has one output signal that is captured by its pattern of action potential firing. Theoretical studies have instead proposed that further computation can occur after the action potential is generated, perhaps in the transformation between voltage and calcium or between calcium and neurotransmitter release (Abbott and Regehr, 2004). Significantly, this would allow different axon terminals of the same neuron to send different output signals; a neuron would therefore communicate distinct information to its various postsynaptic partners, increasing the opportunity for local circuit computation.
In this study, we found that the medulla neurons Tm3 and Mi1 have the necessary properties to allow this. While they are not spiking neurons, the voltage responses throughout these cells were fundamentally similar in waveform and kinetics (Figure 5). In contrast, the calcium responses differed in amplitude or kinetics among subcellular compartments in a manner that could not be predicted from the voltage responses. As calcium is required for neurotransmitter release and as these regions all contain presynaptic active zones, Tm3 and Mi1 could be conveying multiple distinct streams of information. We speculate that differential regulation of voltage-gated calcium channel density, threshold, or kinetics across the cell could account for the compartmentalization of calcium responses. For example, CaV2- and CaV3-type voltage-gated calcium channels in Drosophila antennal lobe projection neurons (PNs) mediate sustained and transient calcium currents, respectively (Gu et al., 2009; Iniguez et al., 2013). Differential distribution of these two types of channels within a neuron could produce compartments with distinct calcium response kinetics.
The emergence of ON and OFF selectivity
Vertebrate and invertebrate visual systems have parallel ON and OFF selective pathways that process light and dark, respectively. This fundamental organization parallels the dichotomy between light and dark in natural scenes, and exploitation of this feature of the visual world has been proposed to be computationally advantageous in a variety of ways (Clark et al., 2014; Gjorgjieva et al., 2014; Westheimer, 2007). In this study, we found that the selectivity in neuronal responses for ON and OFF arises specifically at the transformation between voltage and calcium in third-order neurons (Figures 6 and 7). Previous electrophysiological recordings from Tm3, Mi1, Tm1, and Tm2 observed modest half-wave rectification (Behnia et al., 2014). Our voltage imaging experiments revealed a similar degree of rectification in Tm3 and slightly less rectification in the other neurons. Given the demands of two-photon imaging, our stimuli are notably dimmer and have distinct spectral content from those used previously. Light intensity affects response linearity in the fly visual system (Laughlin et al., 1987); as a result, these minor technical differences are likely to account for the observed differences in rectification between the studies. Nonetheless, the rectification in calcium signals that we observed was stronger than the rectification seen in the voltage measurements and was sufficient to account for the strong selectivity reported in these neurons’ postsynaptic partners T4 and T5 (Fisher et al., 2015; Leonhardt et al., 2016; Maisak et al., 2013). The voltage to calcium transformation implements half-wave rectification by producing an increase in calcium concentration upon depolarization but little decrease in calcium concentration upon hyperpolarization. Therefore, to create selectivity for both ON and OFF, there must be an additional sign inversion in one pathway so that both an increase in light and a decrease in light cause depolarization. We observe this sign inversion in the ON pathway (Figure 4). The synaptic sign inversion between L1 and its postsynaptic partners combined with rectification at the level of intracellular calcium signals in Mi1 and Tm3 makes the L1 pathway ON selective. The sign preserving synapse between L2 and its postsynaptic partners combined with rectification at the level of calcium signals in Tm1 and Tm2 makes the L2 pathway OFF selective.
We speculate that thresholding by voltage-gated calcium channels underpins this half-wave rectification. Indeed, distinct low-voltage-activated and high-voltage-activated calcium currents mediated by the CaV2-type voltage-gated calcium channel have been reported in a Drosophila motor neuron (Ryglewski et al., 2012). Intriguingly, in the vertebrate retina, voltage responses in many bipolar cells are linear with respect to contrast (Baccus and Meister, 2002; Rieke, 2001), but at the synapse between bipolar cells and retinal ganglion cells, the output is half-wave rectified (Demb et al., 2001; Enroth-Cugell and Freeman, 1987). These results, combined with our data, suggest that rectification through a transformation between voltage and calcium could be a mechanism conserved across phyla for producing the computationally critical response property of ON and OFF selectivity.
Considering our study more broadly, we directly observe the transformations in signal gain, kinetics, and linearity that represent neural computation within cells and across synapses. This approach of in vivo voltage and calcium imaging in subcellular compartments can be applied to other systems. By identifying where and how elementary transformations occur within a circuit, our results pave the way for future studies determining the molecular implementations of neural computation.
EXPERIMENTAL PROCEDURES
In vivo imaging experiments were performed on adult female Drosophila. UAS-ASAP2f, UAS-GCaMP6f, UAS-ASAP1 I67T Q397R, and UAS-GCaMP6m were expressed cell-type specifically using the R48A08AD; R66A01DBD split Gal4 (L1), 21D-Gal4 (L2), R19F01-Gal4 (Mi1), R13E12-Gal4 (Tm3), R74G01-Gal4 (Tm1), and otd-Gal4; GMR-Gal80 (Tm2) driver lines. Unless otherwise specified, all voltage responses were measured with ASAP2f, and all calcium responses were measured with GCaMP6f. Neurons were imaged with two-photon microscopy (excitation wavelength: 920 nm). Emitted photons were collected with a 525/50 nm bandpass filter. Data was acquired at 38.9 Hz and resampled to 120 Hz during post hoc analysis. Visual stimuli were filtered with a 447/60 nm bandpass filter and projected onto a screen in front of the fly. The screen spanned approximately 80° of the fly’s visual field horizontally and 50° vertically. The stimuli presented were: 300 ms alternating full contrast light and dark flashes; 8.33 ms light and dark flashes interleaved with 500 ms of gray, Michaelson contrast = 0.5; and 25 ms light and dark flashes interleaved with 500 ms or 1500 ms of gray, Michaelson contrast = 0.5, 0.25, or 0.125. All stimuli were presented to cover the entire screen. The quantification metrics for the responses to 300 ms flashes were peak ΔF/F, the largest value of the amplitude of the fluorscence change; tpeak, the time at which the peak ΔF/F occurs; and τdecay, the time constant of the decay from the peak ΔF/F. The quantification metrics for the responses to 25 ms flashes were peak ΔF/F; tpeak; and the full width at half maximum of the initial response phase. Selectivity for ON and OFF was computed as: |peak light response/peak dark response| for the ON pathway and |peak dark response/peak light response| for the OFF pathway. The peak response was quantified as the maximal ΔF/F of the initial response phase minus the average ΔF/F value before the response. See Supplemental Experimental Procedures for detailed methods.
Supplementary Material
Acknowledgments
We thank L. Luo, S. Baccus, and members of the Clandinin lab for comments on the manuscript. We thank Y. Geng, M. Hintze, and S. Ganesan for providing dissociated neurons. We thank C. Desplan for the otd-Gal4 driver line and M. Wernet for building the GMR-Gal80; otd-Gal4 driver line. Cytometry data was collected on an instrument in the Stanford Shared FACS Facility supported by NIH shared instrument grant S10RR027431-01. Two-photon photobleaching data was collected using equipment from the Stanford Neuroscience Microscopy Service, supported by NIH NS069375; training and use of this equipment was supported by a two-photon pilot grant from the Stanford Neurosciences Institute. This work was supported by National Science Foundation grant 1134416 (F.S.-P., M.Z.L.), DARPA grant W911NF-14-1-0013 (M.Z.L.), NIH Brain Initiative grant 1U01NS090600-01 (M.Z.L.), and NIH grants R01 EY022638 (T.R.C.) and R21 NS081507 (T.R.C). H.H.Y. is supported by a Stanford Graduate Fellowship and a Stanford Interdisciplinary Graduate Fellowship. M.Z.L. receives funding from the Rita Allen Foundation and the Burroughs Wellcome Fund. Competing financial interests: F.S.-P. and M.Z.L. have filed a patent application for a voltage sensor design that includes the sensors reported in this paper.
Footnotes
AUTHOR CONTRIBUTIONS
H.H.Y. and F.S.-P. designed and performed experiments, analyzed data, and wrote the manuscript. X.S. and X.D. performed experiments. M.Z.L. provided advice and supervision. T.R.C. wrote the manuscript and provided advice and supervision.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott LF, Regehr WG. Synaptic computation. Nature. 2004;431:796–803. doi: 10.1038/nature03010. [DOI] [PubMed] [Google Scholar]
- Baccus SA, Meister M. Fast and slow contrast adaptation in retinal circuitry. Neuron. 2002;36:909–919. doi: 10.1016/s0896-6273(02)01050-4. [DOI] [PubMed] [Google Scholar]
- Behnia R, Clark DA, Carter AG, Clandinin TR, Desplan C. Processing properties of ON and OFF pathways for Drosophila motion detection. Nature. 2014;512:427–430. doi: 10.1038/nature13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst A. Fly visual course control: behaviour, algorithms and circuits. Nat Rev Neurosci. 2014;15:590–599. doi: 10.1038/nrn3799. [DOI] [PubMed] [Google Scholar]
- Cao G, Platisa J, Pieribone VA, Raccuglia D, Kunst M, Nitabach MN. Genetically Targeted Optical Electrophysiology in Intact Neural Circuits. Cell. 2013;154:904–913. doi: 10.1016/j.cell.2013.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnevale NT, Hines ML. The NEURON Book 2006 [Google Scholar]
- Chalasani SH, Chronis N, Tsunozaki M, Gray JM, Ramot D, Goodman MB, Bargmann CI. Dissecting a circuit for olfactory behaviour in Caenorhabditis elegans. Nature. 2007;450:63–70. doi: 10.1038/nature06292. [DOI] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chichilnisky EJ. A simple white noise analysis of neuronal light responses. Network. 2001;12:199–213. [PubMed] [Google Scholar]
- Clark DA, Bursztyn L, Horowitz MA, Schnitzer MJ, Clandinin TR. Defining the Computational Structure of the Motion Detector in Drosophila. Neuron. 2011;70:1165–1177. doi: 10.1016/j.neuron.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark DA, Fitzgerald JE, Ales JM, Gohl DM, Silies MA, Norcia AM, Clandinin TR. Flies and humans share a motion estimation strategy that exploits natural scene statistics. Nat Neurosci. 2014;17:296–303. doi: 10.1038/nn.3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuntz H, Forstner F, Schnell B, Ammer G, Raghu SV, Borst A. Preserving Neural Function under Extreme Scaling. PLoS One. 2013;8:e71540. doi: 10.1371/journal.pone.0071540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demb JB, Zaghloul K, Haarsma L, Sterling P. Bipolar cells contribute to nonlinear spatial summation in the brisk-transient (Y) ganglion cell in mammalian retina. J Neurosci. 2001;21:7447–7454. doi: 10.1523/JNEUROSCI.21-19-07447.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enroth-Cugell C, Freeman AW. Receptive-field spatial structure of cat retinal Y cells. J Physiol. 1987;384:49–79. doi: 10.1113/jphysiol.1987.sp016443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatt P, Katz B. Spontaneous subthreshold activity at motor nerve endings. J Physiol. 1952;117:109–128. [PMC free article] [PubMed] [Google Scholar]
- Fischbach K-F, Dittrich AP. The optic lobe of Drosophila melanogaster. I: A. Golgi analysis of wild-type structure. Cell Tissue Res. 1989;258:441–475. [Google Scholar]
- Fisher YE, Silies M, Clandinin TR. Orientation Selectivity Sharpens Motion Detection in Drosophila. Neuron. 2015;88:1–14. doi: 10.1016/j.neuron.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjorgjieva J, Sompolinsky H, Meister M. Benefits of Pathway Splitting in Sensory Coding. J Neurosci. 2014;34:12127–12144. doi: 10.1523/JNEUROSCI.1032-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Huang C, Li JZ, Grewe BF, Zhang Y, Eismann S, Schnitzer MJ. High-speed recording of neural spikes in awake mice and flies with a fluorescent voltage sensor. Science. 2015;350:1–11. doi: 10.1126/science.aab0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouwens NW, Wilson RI. Signal Propagation in Drosophila Central Neurons. J Neurosci. 2009;29:6239–6249. doi: 10.1523/JNEUROSCI.0764-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Jiang SA, Campusano JM, Iniguez J, Su H, Hoang AA, Lavian M, Sun X, O’Dowd DK. Cav2-type calcium channels encoded by cac regulate AP-independent neurotransmitter release at cholinergic synapses in adult Drosophila brain. J Neurophysiol. 2009;101:42–53. doi: 10.1152/jn.91103.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag J, Theunissen F, Borst A. The intrinsic electrophysiological characteristics of fly lobula plate tangential cells: I. Passive membrane properties. J Comput Neurosci. 1996;3:313–336. doi: 10.1007/BF00161091. [DOI] [PubMed] [Google Scholar]
- Iniguez J, Schutte SS, O’Dowd DK. Cav3-type α1T calcium channels mediate transient calcium currents that regulate repetitive firing in Drosophila antennal lobe PNs. J Neurophysiol. 2013;110:1490–1496. doi: 10.1152/jn.00368.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joesch M, Schnell B, Raghu SV, Reiff DF, Borst A. ON and OFF pathways in Drosophila motion vision. Nature. 2010;468:300–304. doi: 10.1038/nature09545. [DOI] [PubMed] [Google Scholar]
- Juusola M, Uusitalo RO, Weckström M. Transfer of graded potentials at the photoreceptor-interneuron synapse. J Gen Physiol. 1995;105:117–148. doi: 10.1085/jgp.105.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuffler SW. Discharge Patterns and Functional Organization of Mammalian Retina. J Neurophysiol. 1953;16:37–68. doi: 10.1152/jn.1953.16.1.37. [DOI] [PubMed] [Google Scholar]
- Laughlin SB, Howard J, Blakeslee B. Synaptic limitations to contrast coding in the retina of the blowfly Calliphora. Proc R Soc Lond B Biol Sci. 1987;231:437–467. doi: 10.1098/rspb.1987.0054. [DOI] [PubMed] [Google Scholar]
- Leonhardt A, Ammer G, Meier M, Serbe E, Bahl A, Borst A. Asymmetry of Drosophila ON and OFF motion detectors enhances real-world velocity estimation. Nat Neurosci. 2016;2016 doi: 10.1038/nn.4262. [DOI] [PubMed] [Google Scholar]
- Liu WW, Mazor O, Wilson RI. Thermosensory processing in the Drosophila brain. Nature. 2015;519:353–357. doi: 10.1038/nature14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London M, Häusser M. Dendritic Computation. Annu Rev Neurosci. 2005;28:503–532. doi: 10.1146/annurev.neuro.28.061604.135703. [DOI] [PubMed] [Google Scholar]
- Maisak MS, Haag J, Ammer G, Serbe E, Meier M, Leonhardt A, Schilling T, Bahl A, Rubin GM, Nern A, et al. A directional tuning map of Drosophila elementary motion detectors. Nature. 2013;500:212–216. doi: 10.1038/nature12320. [DOI] [PubMed] [Google Scholar]
- Miall R. The flicker fusion frequencies of six laboratory insects, and the response of the compound eye to mains fluorescent “ripple.” Physiol. Entomol. 1978;3:99–106. [Google Scholar]
- Nikolaev A, Zheng L, Wardill TJ, O’Kane CJ, de Polavieja GG, Juusola M. Network Adaptation Improves Temporal Representation of Naturalistic Stimuli in Drosophila Eye: II Mechanisms. PLoS One. 2009;4:e4306. doi: 10.1371/journal.pone.0004306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieke F. Temporal contrast adaptation in salamander bipolar cells. J Neurosci. 2001;21:9445–9454. doi: 10.1523/JNEUROSCI.21-23-09445.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryglewski S, Lance K, Levine RB, Duch C. Ca(v)2 channels mediate low and high voltage-activated calcium currents in Drosophila motoneurons. J Physiol. 2012;590:809–825. doi: 10.1113/jphysiol.2011.222836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo E, Huber A, Henrich S, Chadwell LV, Chou WH, Paulsen R, Britt SG. Blue- and green-absorbing visual pigments of Drosophila: ectopic expression and physiological characterization of the R8 photoreceptor cell-specific Rh5 and Rh6 rhodopsins. J Neurosci. 1999;19:10716–10726. doi: 10.1523/JNEUROSCI.19-24-10716.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl B, Gao X, Wehr M. Nonoverlapping Sets of Synapses Drive On Responses and Off Responses in Auditory Cortex. Neuron. 2010;65:412–421. doi: 10.1016/j.neuron.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinomiya K, Karuppudurai T, Lin T-Y, Lu Z, Lee C-H, Meinertzhagen IA. Candidate Neural Substrates for Off-Edge Motion Detection in Drosophila. Curr Biol. 2014;24:1062–1070. doi: 10.1016/j.cub.2014.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silies M, Gohl DM, Clandinin TR. Motion-Detecting Circuits in Flies: Coming into View. Annu Rev Neurosci. 2014;37:307–327. doi: 10.1146/annurev-neuro-071013-013931. [DOI] [PubMed] [Google Scholar]
- Simoncelli EP, Paninski L, Pillow J, Schwartz O. Characterization of Neural Responses with Stochastic Stimuli. Cogn Neurosci III, Third Ed. 2004:327–338. [Google Scholar]
- Smith SW. The Scientist and Engineer’s Guide to Digital Signal Processing 1997 [Google Scholar]
- St-Pierre F, Marshall JD, Yang Y, Gong Y, Schnitzer MJ, Lin MZ. High-fidelity optical reporting of neuronal electrical activity with an ultrafast fluorescent voltage sensor. Nat Neurosci. 2014;17:884–889. doi: 10.1038/nn.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart GJ, Spruston N. Dendritic integration: 60 years of progress. Nat Neurosci. 2015;18:1713–1721. doi: 10.1038/nn.4157. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Yasuda R. Principles of Two-Photon Excitation Microscopy and Its Applications to Neuroscience. Neuron. 2006;50:823–839. doi: 10.1016/j.neuron.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Takemura S, Bharioke A, Lu Z, Nern A, Vitaladevuni S, Rivlin PK, Katz WT, Olbris DJ, Plaza SM, Winston P, et al. A visual motion detection circuit suggested by Drosophila connectomics. Nature. 2013;500:175–181. doi: 10.1038/nature12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wässle H. Parallel processing in the mammalian retina. Nat Rev Neurosci. 2004;5:747–757. doi: 10.1038/nrn1497. [DOI] [PubMed] [Google Scholar]
- Westheimer G. The ON-OFF dichotomy in visual processing: From receptors to perception. Prog Retin Eye Res. 2007;26:636–648. doi: 10.1016/j.preteyeres.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Zettler F, Järvilehto M. Decrement-Free Conduction of Graded Potentials Along the Axon of a Monopolar Neuron. Z Vgl Physiol. 1971;75:402–421. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.