

Abstract
Metazoan species maintain oxygen homeostasis through the activity of hypoxia-inducible factors, which are transcriptional activators that regulate the expression of hundreds of genes to match O2 supply and demand. Here, we review the involvement of hypoxia-inducible factors in the molecular physiology and pathophysiology of cellular O2 sensing, O2 delivery, O2 utilization, and systemic O2 sensing.
Graphical abstract
INTRODUCTION
A constant supply of O2 must be delivered to all cells of metazoan organisms for their continued survival. O2 is utilized as the final electron acceptor in the process of oxidative phosphorylation. The function of the respiratory chain is optimized for physiological PO2 levels and sustained deviations from normoxia cause increased production of reactive oxygen species (ROS)* by the electron transport chain (ETC). ROS cause oxidation of lipids, proteins, and nucleic acids that can result in cellular dysfunction or death. Thus, homeostatic mechanisms exist to tightly regulate O2 levels within cells and tissues.
Hypoxia-inducible factors (HIFs) function as master regulators of oxygen homeostasis, which is critical for the survival of all metazoan species.1 As first demonstrated for HIF-1, HIFs are heterodimeric proteins, consisting of an O2-regulated HIF-1α, HIF-2α, or HIF-3α subunit and a constitutively expressed HIF-1β subunit.2 Depending on the context, HIF-1 and HIF-2 activate transcription of target genes that mediate adaptive or maladaptive responses at the systemic level to regulate O2 delivery and at the cellular level to regulate O2 utilization.3 The role of HIF-3 is much less well studied.4 Both systemic and cellular O2 sensing mechanisms are employed to ensure a proper balance between O2 supply and demand, respectively; dysregulation of these mechanisms results in disease pathology. In this brief review article, we will provide illustrative examples regarding the involvement of HIFs in the molecular physiology and pathophysiology of cellular O2 sensing, O2 delivery, O2 utilization, and systemic O2 sensing.
CELLULAR O2 SENSING
The activity of HIFs is regulated by O2-dependent hydroxylation of the HIF-α subunits (Figure 1). Hydroxylation of two proline residues (Pro-402 and Pro-564 in human HIF-1α) is mediated by HIF prolyl hydroxylase domain (PHD) proteins, principally PHD2.5,6 Prolyl hydroxylation is required for binding of the von Hippel-Lindau (VHL) protein, which recruits an ubiquitin-protein ligase complex (consisting of Elongin B, Elongin C, Cullin 2, RBX2, and an E2 ligase) that ubiquitinates HIF-α proteins and thereby targets them for proteasomal destruction.7,8 The PHDs are dioxygenases that utilize O2 and α-ketoglutarate as substrates and contain Fe(II) in their catalytic centers, such that PHD activity may be inhibited by any of the following conditions: reduced availability of O2 or α-ketoglutarate; oxidation of Fe(II) to Fe(III); iron chelation; or administration of an α-ketoglutarate analog.6 HIF-α subunits begin to accumulate as soon as cells are subjected to hypoxia, iron chelators, or α-ketoglutarate analogs, with HIF-1α protein levels peaking after 4 to 8 hours in most cell lines.2 Peak expression of many HIF target gene mRNAs often occurs after 24 hours of continuous hypoxia. Even under conditions of mild hypoxia, increased production of ROS by the mitochondrial ETC may contribute to the inhibition of PHD activity.9
FIGURE 1.
O2-dependent hydroxylation of HIF-α subunits. Hydroxylation of specific proline (Pro) and asparagine (Asn) residues promote VHL binding and block P300/CBP binding, respectively. VHL recruits an E3-ubiquitin protein ligase (BCCRE2) consisting of Elongin B, Elongin C, Cullin 2, RBX1, and an E2 ubiquitin ligase that ubiquitinates HIF-α, thereby targeting the protein for proteasomal degradation.
In addition to prolyl hydroxylation, the HIF-1α and HIF-2α subunits are subject to hydroxylation of an asparagine residue in the transactivation domain (Asn-803 in human HIF-1α), which blocks interaction with the coactivator proteins CBP and P300.10 Asparagine hydroxylation and repression of HIF transcriptional activity is mediated by factor inhibiting HIF-1 (FIH-1), which is also an O2-and α-ketoglutarate-dependent dioxygenase with biochemical properties that are similar to those described above for the PHDs, although FIH-1 activity may be inhibited at higher O2 levels than those required for inhibition of PHD activity.11–13
O2 DELIVERY
Erythropoiesis
A primary means of increasing O2 delivery systemically is to increase erythropoiesis, as red blood cells are responsible for transport of O2, bound to hemoglobin, from the lungs to all body tissues. The synthesis of hemoglobin in erythroid progenitor cells requires iron. Intestinal iron uptake and delivery to the bone marrow for red blood cell production are dependent on the HIF-mediated expression of the following genes: EPO, which encodes erythropoietin, the hormone that stimulates the survival, proliferation, and differentiation of erythroid progenitor cells;14 EPOR, which encodes the EPO receptor on erythroid progenitor cells;15 CYBRD1, which encodes the duodenal cytochrome b (DCYTB; also known as ferric reductase) that reduces Fe(III) to Fe(II) for intestinal uptake;16 SLC11A2, which encodes the apical divalent metal transporter (DMT1; also known as NRAMP2) that allows uptake of Fe(II) from the intestinal lumen into enterocytes;16,17 SLC40A1, which encodes ferroportin (FPN1), the basolateral protein that transports iron out of enterocytes and into blood vessels;18 TF, which encodes transferrin, the protein that transports iron from the intestine to the bone marrow;19 TFRC, which encodes the transferrin receptor on erythroid cells that takes up iron from transferrin;20,21 and FECH, which encodes ferrochelatase, the enzyme that catalyzes the insertion of iron into protoporphyrin to form heme.22 Thus, HIFs act as master regulators that coordinately activate the transcription of multiple genes required for a single physiological response; in this case, HIF-2 regulates at least 8 genes encoding proteins that mediate erythropoiesis (Figure 2).
FIGURE 2.
HIF-2 is a master regulator of erythropoiesis. 8 genes activated by HIF-2 encode protein products that are required for iron absorption, transport, and incorporation into hemoglobin for red blood cell production. CYBRD1, SLC11A2, and SLC40A1 are expressed in duodenal enterocytes; TF in expressed in hepatocytes; EPO is expressed in renal interstitial cells; EPOR, FECH, and TFRC are expressed in erythroid progenitors.
Congenital Polycythemia: A Hereditable Disorder of Oxygen Sensing
Hereditary erythrocytosis or congenital polycythemia is a hereditable disorder of erythropoiesis that is characterized by excess red blood cell production. The increase in hematocrit can lead to occlusion of cerebral vessels and stroke-like symptoms. Remarkably, mutations in VHL, EGLN1, and EPAS1 genes, which encode VHL, PHD2, and HIF-2α, respectively, have been identified in patients with this disorder. Investigation of a form of congenital polycythemia that was endemic to a region of Russia known as Chuvashia revealed that affected individuals were homozygous for a VHL missense mutation in which Arg was replaced by Trp at codon 200.23 The mutant VHLR200W protein was shown to have reduced binding to hydroxylated HIF-1α. As a result, any given O2 concentration, the expression of HIF target genes, including EPO, is higher in cells expressing VHLR200W as compared to cells expressing wild type VHL. As a result, patients with Chuvash polycythemia have higher-than-normal EPO levels despite having significantly increased red blood cell counts. Remarkably, patients with Chuvash polycythemia are not just polycythemic, they also have an increased respiratory rate and increased pulmonary artery pressure, which are physiological responses to hypoxia.24 Thus, homozygosity for the VHLR200W mutation results in a global defect in O2 sensing.
Missense mutations in the EGLN1 gene encoding PHD2 were found to cause polycythemia in several individuals. These missense mutations result in the substitution of evolutionarily conserved residues (P317R or R371H) near the catalytic center of the protein, suggesting that they result in decreased hydroxylase activity.25 Finally, missense mutations in that result in the substitution of evolutionarily conserved residues (M535V, M535I, G537W, G537R) in HIF-2α that are located near a proline residue that is subject to hydroxylation (Pro-532), suggesting that the substitutions reduce the efficiency with which the hydroxylases modify the mutant protein.26 Patients with EPAS1 missense mutations exhibit increased pulmonary artery pressure, cardiac output, heart rate, and respiratory rate.27 Thus, human genetics provides compelling evidence that the HIF-PHD-VHL pathway plays a critical role in the regulation of erythropoiesis as well as many other physiological responses to hypoxia.
Angiogenesis
A primary means of increasing local O2 delivery is by vascular remodeling (increased luminal diameter of existing vessels) and angiogenesis (development of new blood vessels). Again, HIFs function as master regulators by activating the transcription of multiple genes encoding secreted angiogenic growth factors and cytokines that stimulate the proliferation of resident endothelial cells and the recruitment of bone marrow-derived angiogenic cells (BMDACs). The following are some of the HIF target genes that encode angiogenic growth factors and cytokines (Figure 3): VEGFA, which encodes vascular endothelial growth factor A, a protein that binds to VEGFR2 on endothelial cells to stimulate angiogenesis, vascular permeability, and recruitment of VEGFR2+ BMDACs; ANGPT1 and ANGPT2, which encode angiopoietins 1 and 2, proteins that bind to the TIE2 receptor to modulate endothelial-pericyte interactions; ANGPTL4, which encodes angiopoietin-like 4, which stimulates endothelial cell proliferation and vascular permeability; EPO, which encodes a protein (erythropoietin) that binds to its receptor (EPOR) on endothelial cells to stimulate their survival and proliferation; KITL, which encodes kit ligand (also known as stem cell factor, a protein that mediates the recruitment of c-KIT+ BMDACs; PDGFB, which encodes platelet-derived growth factor B, which binds to its receptor (PDGFR) on pericytes to modulate interactions with endothelial cells; PGF, which encodes placental growth factor, a protein that mediates the recruitment of VEGFR1+ BMDACs; and CXCL12, which encodes chemokine (C-X-C motif) ligand 12 (also known as stromal-derived factor 1), a protein that mediates the recruitment of CXCR4+ BMDACs.28
FIGURE 3.
HIF-1 and HIF-2 are master regulators of tissue vascularization. 8 genes activated by HIF-1 and HIF-2 encode secreted proteins that bind to cognate receptors on vascular cells to stimulate angiogenesis and vascular remodeling in response to hypoxia/ischemia.
HIFs Mediate Ischemia-induced Angiogenesis and Vascular Remodeling
HIFs transactivate the target genes described above to mediate coordinate expression of multiple angiogenic cytokines/growth factors that stimulate angiogenesis and vascular remodeling in response to tissue hypoxia/ischemia.29 However, in mice, aging profoundly impairs the accumulation of HIF-1α protein and expression of angiogenic cytokines/growth factors in response to reduced tissue perfusion resulting from femoral artery ligation.30 The incidence of peripheral arterial disease and critical limb ischemia (the state in which perfusion is insufficient to maintain tissue viability) increase with age, which suggests that findings from the mouse model are clinically relevant.29 HIF-1α gene therapy, either alone or in combination with BMDAC cell therapy, improved perfusion following femoral artery ligation in mouse models of aging and diabetes.30–33
Mice that were heterozygous for a HIF-1α knockout allele also had impaired vascular responses to ischemia at all ages,30 suggesting that genetic variation at the HIF1A locus encoding HIF-1α may influence the response to ischemia caused by coronary artery disease (CAD). Among patients with clinical significant stenosis (≥ 70% narrowing of a major coronary artery), approximately two-thirds exhibit remodeling of one or more collateral blood vessels to accept increased blood flow.28,29 Among 100 consecutive patients with CAD, the frequency of a C-to-T single nucleotide polymorphism (SNP) in the HIF1A gene, which changed residue 582 of HIF-1α protein from proline to serine, was significantly increased in patients without collateral vessels on coronary angiography.34 Subsequently, the P582S SNP and several other SNPs at the HIF1A locus were found at increased frequency in patients with CAD, who presented with stable exertional angina, as compared to CAD patients who presented with acute myocardial infarction.35 Taken together, these two clinical studies provide evidence that HIF1A genotype influences vascular phenotype.
Pathological Angiogenesis in Ischemic Retinopathies
One of the major complications of diabetes is the development of proliferative diabetic retinopathy (PDR), in which excess proliferation of blood vessels in the eye leads to hemorrhage and retinal detachment that results in vision loss, which can progress to blindness.36 Increased levels of VEGF were found in fluid taken from the eyes of diabetic patients.37 In mouse models of retinal ischemia, increased HIF-1α production by Mueller cells was found to precede the induction of VEGF mRNA and protein expression in the retina.38 Clinical trials demonstrated that treatment of diabetic patients with anti-VEGF therapy slowed disease progression in some but not all patients: in one study of patients with diabetic retinopathy, the cumulative probability of clinical progression over two years was reduced from 34% to 11%.39 Mouse models revealed that in addition to VEGF, HIF-dependent expression of multiple angiogenic factors is induced by retinal ischemia,40 suggesting that the limited response to anti-VEGF therapy reflected the pathogenic role of other angiogenic factors.
Interrogation of a panel of angiogenic growth factors and cytokines revealed that oxygen-induced retinopathy, which is a mouse model of retinal ischemia, was characterized by dramatically increased expression of two angiogenic factors in the eye: VEGF and ANGPTL4.41 Analysis of aqueous fluid from the eyes of diabetic patients revealed that whereas the mean VEGF level was increased in diabetic patients with PDR as compared to diabetic patients without PDR, there was extensive overlap between groups; in contrast, mean ANGPTL4 levels were increased in PDR and there was virtually no overlap with the non-PDR group.41 Furthermore, aqueous fluid from PDR patients treated with anti-VEGF therapy still activated endothelial cells in vitro and addition of anti-VEGF antibody to the aqueous fluid did not prevent endothelial activation, whereas addition of anti-ANGPTL4 antibody did prevent endothelial activation.41 As in the case of VEGFA, ANGPTL4 promotes permeability as well as proliferation of endothelial cells.42 Thus, anti-ANGPTL4 therapy (either alone or in combination with anti-VEGF therapy) may improve outcome in PDR patients (Figure 4). An alternative strategy is the use of small molecule inhibitors of HIF-1, which were originally shown to inhibit tumor growth and angiogenesis,43–45 to block the expression of ANGPTL4, VEGF, and other angiogenic factors that are induced by retinal ischemia.40,41,46
FIGURE 4.
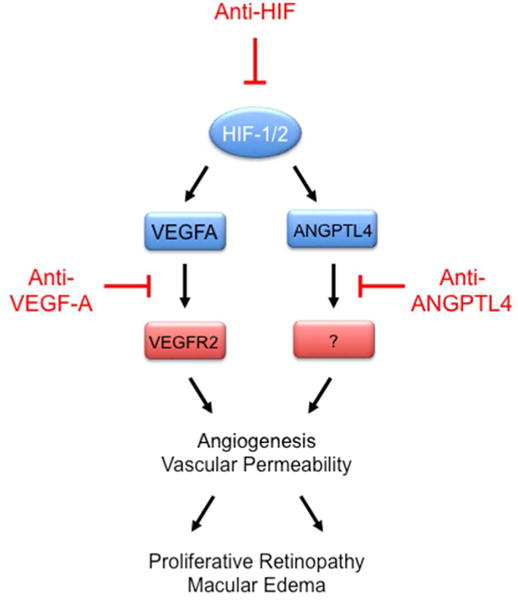
The HIF target genes VEGFA and ANGPTL4 encode secreted proteins that mediate angiogenesis and vascular permeability that leads to proliferative retinopathy and macular edema, respectively. VEGFA binds to VEGFR2 on retinal vascular endothelial cells (VECs); the receptor for ANGPTL4 on retinal VECs has not been identified. Drugs inhibiting VEGFA binding to VEGFR2 on VECs are used to treat ocular diseases; inhibition of HIF activity or ANGPTL4 binding to its receptor on retinal VECs may also have therapeutic utility.
O2 UTILIZATION
All human cells require a constant supply of O2 to carry out oxidative phosphorylation in the mitochondria for ATP generation. O2 is utilized as the final electron acceptor in the mitochondrial ETC, resulting in the generation of water. Under hypoxic conditions, the efficiency of electron transfer is impaired and electrons react with O2 prior to reaching complex IV, to form superoxide radicals, which can be converted to hydrogen peroxide by superoxide dismutase.9 Superoxide and hydrogen peroxide are ROS that oxidize cellular macromolecules and alter their biochemical or physical properties, resulting in cell dysfunction or death. HIFs play a critical role in maintaining redox homeostasis under hypoxic conditions. Many cancers contain hypoxic regions due to high rates of cell proliferation coupled with the formation of vasculature that is structurally and functionally abnormal. Primary tumors with low oxygenation (PO2 < 10 mmHg) are associated with an increased risk of metastasis and patient mortality.47 In order to maintain redox homeostasis in the presence of intratumoral hypoxia, HIFs activate the transcription of target genes that serve to either decrease mitochondrial oxidant production or increase mitochondrial antioxidant production.
HIF-1 Inhibits Mitochondrial Oxidant Production
Cancer cells are characterized by increased glucose and glutamine uptake. In the Embden-Meyerhof pathway, glucose is metabolized to pyruvate, which is then converted to acetyl CoA by pyruvate dehydrogenase (PDH) for entry into the tricarboxylic acid (TCA; also known as Krebs) cycle (Figure 5). Glucose metabolites are also shunted into the pentose phosphate pathway for production of nucleic acids and NADPH or the serine synthesis pathway for production of serine, glycine, and NADPH. Glutamine is converted to glutamate and then to α-ketoglutarate for entry into the TCA cycle. In addition to glucose and glutamine, fatty acids are the other source of energy for cells. Fatty acids can be taken up from the extracellular media, generated by the metabolism of triglycerides from lipid droplets,48 or synthesized de novo from acetyl CoA. De novo synthesis of fatty acids is required for membrane synthesis and therefore for cell growth and proliferation. Fatty acids are catabolized by fatty acid oxidation (FAO; also known as β-oxidation). FAO generates one molecule of acetyl CoA in each oxidation cycle and two in the last cycle (Figure 5). Oxidation of acetyl CoA derived from glucose or fatty acids in the TCA cycle generates NADH and FADH2, which donate their electrons to the ETC, leading to the formation of a proton gradient that drives the production of ATP. Thus, the flux of acetyl CoA metabolized by the TCA cycle determines the flux of electrons delivered to the ETC.
FIGURE 5.
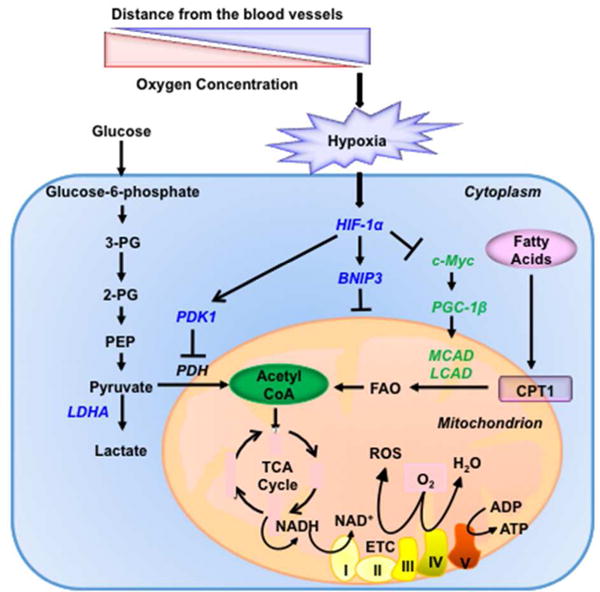
Oxygen-dependent regulation of oxidant production by HIF-1. In hypoxic cells, HIF-1 suppresses glucose oxidation, by activating the transcription of LDHA and PDK1 (blue), and fatty acid oxidation, by repressing the expression of MCAD and LCAD (green). In addition, under hypoxic conditions, HIF-1 activates the transcription of BNIP3 (blue) to induce mitochondrial-selective autophagy and thereby suppress both glucose and fatty acid oxidation. BNIP3, BCL2/adenovirus E1B 19-kDa protein-interacting protein 3; LCAD, long-chain acyl-CoA dehydrogenase; LDHA, lactate dehydrogenase A; MCAD, medium-chain acyl-CoA dehydrogenase; and PDK1, pyruvate dehydrogenase kinase 1.
As in the case of angiogenesis and erythropoiesis, HIF-1 functions as a master regulator of glucose metabolism by activating the transcription of multiple target genes that inhibit the generation of acetyl CoA from glucose, including: PDH kinase, isozyme 1 (PDK1) and PDK3; BCL2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3) and BNIP3-like (BNIP3L); and lactose dehydrogenase A (LDHA) (Figure 5).49 PDKs phosphorylate and inactivate PDH, thereby inhibiting the conversion of pyruvate to acetyl CoA.50–52 LDHA competes with PDH for pyruvate and converts it to lactate, the terminal product of glycolysis.53 BNIP3 and BNIP3L trigger mitochondrial autophagy as a means to reduce oxidative metabolism of glucose, glutamine and fatty acids.54,55
HIF-1 also inhibits FAO, thereby decreasing intracellular generation of acetyl CoA, by repressing the expression of the medium-chain and long-chain acyl-CoA dehydrogenase (MCAD and LCAD) genes under hypoxia.56 MCAD and LCAD are key enzymes catalyzing the first step of FAO in the mitochondria (Figure 5). Under hypoxia, HIF-1α suppresses the expression of C-MYC, which otherwise would activate transcription of the gene encoding PGC-1β, which is a coactivator of MCAD and LCAD transcription. This phenomenon was observed in prostate and liver cancer cells. Consistent with the suppression of FAO, there an increase in lipid accumulation was observed in the cells under hypoxia. The decrease in FAO under hypoxia was necessary to prevent ROS accumulation and cell death under hypoxic conditions.56 Thus, regulation of FAO represents an additional mechanism by which HIF-1 regulates oxidant production as a critical metabolic adaptation to hypoxic conditions. Ranolazine targets carnitine palmitoyl transferase I, which catalyzes fatty acid transport into mitochondria for FAO and is approved for use in cancer patients.57 Cancer cells rely on fatty acids as cellular building blocks for membrane formation, energy storage and production of signaling molecules. Recent preclinical studies suggest that FAO inhibitors may have therapeutic utility in hematological malignancies and triple negative breast cancer with C-MYC overexpression.58,59 Inhibition of HIF-1 represents an alternative therapeutic strategy and has the benefit of blocking oxidative metabolism of both glucose and fatty acids.
HIFs Stimulates Mitochondrial Antioxidant Production
Reduced glutathione is the principal antioxidant in human cells and NADPH is required to maintain glutathione in a reduced form. Two different glycolytic shunt pathways generate NADPH: (ii) the pentose phosphate pathway; and (ii) the combined activity of the serine synthesis pathway (SSP) and one-carbon (folate cycle) metabolism (1CM). In the pentose phosphate pathway, glucose-6-phosphate dehydrogenase (G6PD) converts the glycolytic intermediate glucose-6-phosphate and NADP to 6-phosphogluconate and NADPH in the cytosol (Figure 6). In the SSP, the glycolytic intermediate 3-phosphoglycerate is converted to serine via three reactions, which are catalyzed by phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase 1 (PSAT1), and phosphoserine phosphatase (PSPH). Serine and NADP+ are then utilized for 1CM, either in the cytosol or mitochondria, which generates glycine and NADPH (Figure 6). The enzymes required for mitochondrial 1CM are serine hydroxymethyltransferase 2 (SHMT2), methylene tetrahydrofolate dehydrogenase 2 (MTHFD2), and MTHFD1-like (MTHFD1L), whereas SHMT1 and MTHFD1 carry out these same reactions in the cytosol (MTHFD1 catalyzes the reactions performed by both MTHFD2 and MTHFD1L). The major role of 1CM in NADPH generation and redox regulation has only recently become appreciated.60 Here we will discuss how HIFs facilitate mitochondrial antioxidant production under hypoxic conditions.
FIGURE 6.
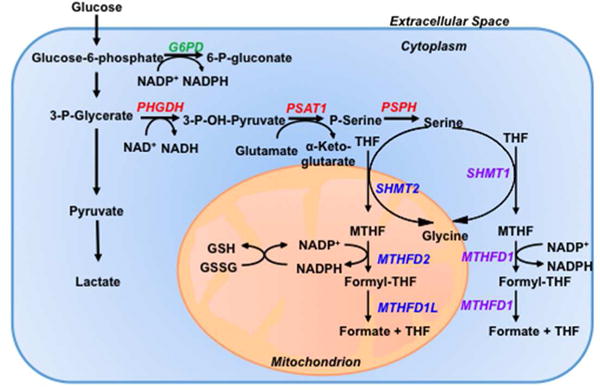
Oxygen-dependent regulation of antioxidant production by HIF1. Under hypoxic conditions, HIFs activate the transcription of PHGDH, PSAT1, and PSPH (red) to increase conversion of glucose to serine (serine synthesis pathway); and SHMT2, MTHFD2, and MTHFD1L (blue) to increase generation of mitochondrial NADPH (mitochondrial one-carbon metabolism), which is required to convert glutathione from oxidized (GSSG) to reduced (GSH) form to protect against increased ROS generated by the ETC. Genes encoding proteins that generate cytosolic NADPH are either not consistently induced [SHMT1, MTHFD1 (purple)] or actively repressed [G6PD (green)] under hypoxic conditions. G6PD, glucose-6-phosphate dehydrogenase; MTHFD, methylene tetrahydrofolate dehydrogenase; MTHFD1L, MTHFD1-like; PHGDH, phosphoglycerate dehydrogenase; PSAT, phosphoserine aminotransferase; PSPH, phosphoserine phosphatase; SHMT, serine hydroxymethyltransferase.
In breast cancer cells, irrespective of the estrogen receptor (ER) status, the expression of all six genes encoding the SSP (PHGDH, PSAT1 and PSPH) and mitochondrial 1CM (SHMT2, MTHFD2 and MTHFD1L) enzymes are induced under hypoxic conditions in a HIF-dependent manner. Expression of the genes encoding cytosolic 1CM enzymes (SHMT1 and MTHFD1) is not induced by hypoxia in most breast cancer cell lines. G6PD gene expression was repressed by hypoxia in all breast cancer cell lines, indicating a reprogramming of glucose metabolism to increase flux through the SSP and decrease flux through the pentose phosphate pathway under hypoxic conditions, which was confirmed by metabolomic analyses.61`
When PHGDH, the first enzyme of the SSP was knocked down in breast cancer cells (irrespective of ER status), mitochondrial ROS levels and apoptosis were increased in the knockdown subclones, as compared to subclones expressing a non-targeting control (NTC) short hairpin RNA (shRNA) under hypoxic conditions.61 The ratio of reduced:oxidized glutathione increased dramatically under hypoxic conditions in NTC but not in PHGDH knockdown subclones, which also had significantly reduced levels of NADPH.61 These results suggest that knockdown of PHGDH in breast cancer cell lines increased oxidant stress. Similarly, knockdown of SHMT2 in neuroblastoma cell lines increased oxidant stress and cell death under hypoxic conditions.62 SHMT2 expression promoted survival of glioblastoma cells in the hypoxic tumor microenvironment.63 SHMT2 was also induced in 6 breast cancer cell lines under hypoxic conditions.61 These results indicate that induction of the SSP and mitochondrial 1CM by HIFs is another critical metabolic adaptation to hypoxia. Presumably knockdown of PSAT1, PSPH, MTHFD2, or MTHFD1L would have similar consequences as observed for knockdown of PHGDH or SHMT2.
PHGDH knockdown in ER+ MCF-7 or ER− MDA-MB-231 breast cancer cells resulted in reduced NADPH levels as well as increased mitochondrial ROS and apoptosis in the bulk cancer cell population.61 However, the effect of PHGDH knockdown on the breast cancer stem cell (BCSC) population was particularly dramatic. Exposure of NTC subclones to hypoxia for 3 days resulted in a greater-than-five-fold increase in the percentage of BCSCs, which was abrogated in PHGDH knockdown subclones. In vivo, PHGDH knockdown subclones exhibited a dramatic impairment in lung metastasis that was consistent with the observed reduction of BCSCs in the primary tumor.61 PHGDH knockdown subclones were also much more sensitive to treatment with doxorubicin or carboplatin, which are chemotherapy drugs that act by generating ROS. Interestingly, whereas HIF-1 alone is responsible for activating transcription of genes required to decrease mitochondrial oxidant production, both HIF-1 and HIF-2 contribute to the regulation of the genes encoding SSP and mitochondrial 1CM enzymes that increase mitochondrial antioxidant capacity. Finally, HIF-1 and HIF-2 also activate the transcription of genes required for the de novo synthesis of glutathione.64
SYSTEMIC OXYGEN SENSING
The Carotid Body Activity Mediates Acute Physiological Responses to Hypoxemia
Nearly 150 years ago, Pflüger reported that hypoxia stimulates breathing.65 Whereas the cell-autonomous O2 sensing that occurs as a result of hydroxylation-dependent modulation of HIF activity enables each cell to respond individually to hypoxia, systemic hypoxia, which occurs in response to blood loss or ascent to high altitude, is sensed as hypoxemia (reduced blood O2 content) by the carotid bodies (CBs), which are tiny bilateral organs located at the bifurcation of the common carotid artery into the internal and external carotid arteries.66,67 The response of the CB to hypoxia is notable for its speed and sensitivity: a 20% reduction in arterial PO2 is sufficient to induce a response within seconds.68 Glomus (type I) cells are excitable cells in the CB that depolarize in response to hypoxemia. Axons from glomus cells course through the carotid sinus nerve (CSN) to the nucleus tractus solitarius, from which reflex arcs project to: the diaphragm, to increase respiratory rate; the heart, to increase heart rate; and via the rostral ventro-lateral medulla and sympathetic nervous system, to the adrenal medulla (AM), to stimulate the secretion of catecholamines (epinephrine and norepinephrine) that increase blood pressure (Figure 7). These cardiovascular and respiratory adaptations increase cardiac output and ventilation, respectively, thereby increasing the capture of O2 by red blood cells for delivery throughout the body.
FIGURE 7.
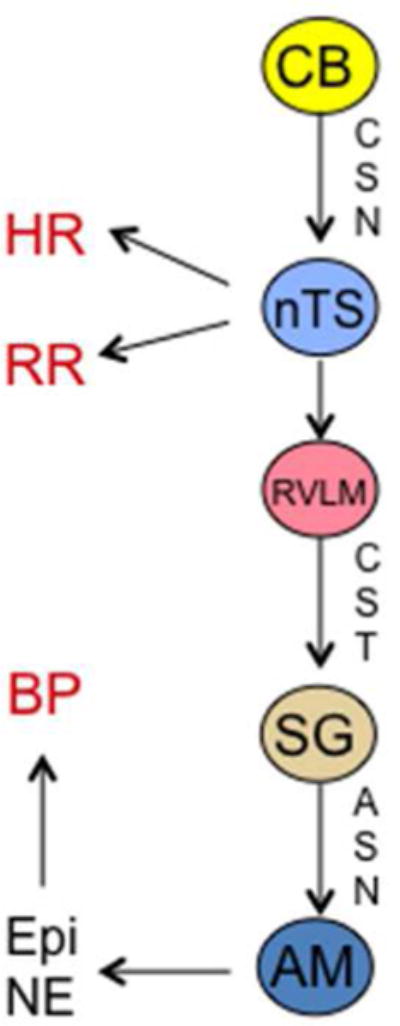
Neural transmission of reflex arcs that result in changes in heart rate (HR), respiratory rate (RR), and blood pressure (BP) in response to depolarization of glomus cells in the carotid body (CB). AM, adrenal medulla; ASN, adrenal sympathetic nerve; CSN, carotid sinus nerve; CST, corticospinal tract; Epi, epinephrine; NE, norepinephrine; nTS, nucleus tractus solitarius; RVLM, rostral ventro-lateral medulla; SG, sympathetic ganglion.
Whereas O2 sensing in non-depolarizable cells occurs via alterations in rates of HIF-α hydroxylation, O2 sensing in the depolarizable glomus cells of the CB involves the generation of carbon monoxide (CO) by the enzyme heme oxygenase 2 (HO2), which utilizes O2 as a molecular substrate (Figure 8). CO binds to the heme moiety of soluble guanylate cyclase, which stimulates its catalytic activity, converting GTP to cyclic GMP (cGMP), which binds to and activates cGMP-dependent protein kinase (protein kinase G [PKG]). Phosphorylation of cystathionine-γ-lyase (CSE) by PKG inhibits CSE catalytic activity, which mediates the generation of hydrogen sulfide (H2S). H2S inhibits K+ channel conductance, triggering increased intracellular Ca2+ and neurotransmitter release.68 Thus, O2 sensing/signal transduction in the CB involves two gas messengers, CO and H2S: under normoxic conditions, CO levels are high, H2S levels are low, and CSN activity is low; in contrast, hypoxemia leads to decreased CO production and increased H2S production, which induces glomus cell depolarization and increased CSN activity.69,70
FIGURE 8.
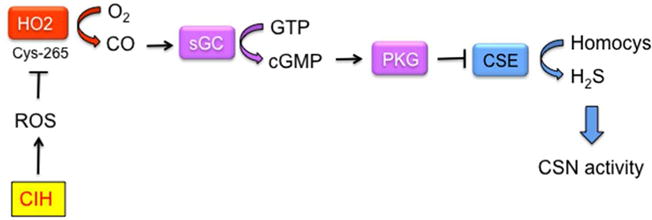
O2 sensing and signaling by gas messengers in the carotid body. cGMP, cyclic GMP; CIH, chronic intermittent hypoxia; CSE, cystathionine-γ-lyase; CSN, carotid sinus nerve; Cys-265, cysteine residue 265; HO2, heme oxygenase 2; Homocys, homocysteine; PKG, cGMP-dependent protein kinase; ROS, reactive oxygen species; sGC, soluble guanylate cyclase. O2 stimulates CO production, which inhibits H2S production, thereby inhibiting glomus cell depolarization. ROS generated by CIH inhibit HO2 activity, probably by oxidation of Cys-265, thereby increasing H2S production and CSN activity.
When CBs from wild type mice are perfused with a hypoxic gas mixture, there is a rapid increase in CSN activity. However, in Cse−/− mice, which lack CSE activity, basal H2S levels were reduced by half and did not increase in response to hypoxia, whereas CB responses to hypercarbia were intact.71 Similarly, CBs from Hif1a+/− mice, which are heterozygous for a null (knockout) allele at the Hif1a locus encoding HIF-1α, exhibited no increase in CSN activity in response to hypoxia, despite normal histological appearance and normal responses to hypercarbia or cyanide, indicating a specific defect in O2 sensing.72
The Carotid Body Mediates Pathological Responses to Intermittent Hypoxia
The classic homeostatic responses of the CB that are described in the preceding section are subverted in obstructive sleep apnea (OSA). In this condition, the upper airway becomes occluded by relaxation of the jaw, tongue, tonsils, and pharyngeal soft tissue during sleep, resulting in apnea (cessation of breathing) for 15–30 sec, which results in hypoxemia that is sensed by the CB. The patient awakens, clears the airway, resulting in reoxygenation, and goes back to sleep. This cycle of hypoxemia and reoxygenation is repeated dozens of times each night. The major pathological consequence of OSA is the development of hypertension, which places the patient at risk for heart failure, myocardial infarction, and stroke.73 Among adult Americans, it is estimated that 1 in 5 males and 1 in 15 females are affected by OSA, which is the major cause of treatment-resistant hypertension. Patients are treated with continuous positive airway pressure to prevent airway occlusion, which lowers blood pressure in some but not all patients.
Although OSA results in both chronic intermittent hypoxemia and chronic intermittent hypercapnea, pioneering studies revealed that exposure of rodents to repeated (8 hours per day) short cycles of ambient hypoxia (5% O2 for 15 sec) and reoxygenation (21% O2 for 5 min), i.e. chronic intermittent hypoxia (CIH), was sufficient to cause hypertension, which was associated by increased plasma levels of norepinephrine.74 Furthermore, transection of the CSN blocked the increase in catecholamine levels and blood pressure in rodents subjected to CIH, demonstrating involvement of the CB.74 Remarkably Hif1a+/− mice were completely protected from increased catecholamine levels and blood pressure in response to CIH.75
CIH generates increased ROS in the CB and AM and treatment of rodents with the radical scavenger manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (MnTMPyP) blocked the development of hypertension in rodents subjected to CIH. Exposure of wild type mice to CIH induced HIF-1α expression in the central nervous system, which was also blocked by MnTMPyP administration, indicating that HIF-1α was downstream of ROS.75 However, CIH induced increased levels of oxidants in the brains of wild type, but not Hif1a+/− mice, indicating that HIF-1α was upstream of ROS.75 This paradox was resolved by the discovery that expression of NADPH oxidase 2 (NOX2), an enzyme that generates superoxide, was induced by CIH in wild type but not in Hif1a+/− mice.76 CIH causes an initial ROS signal, which induces HIF-1α expression. HIF-1 then induces NOX2 activity, which leads to further ROS generation. Thus, a feed-forward loop is established creating a vicious cycle of increased ROS and HIF-1α levels. Further mechanistic studies revealed that increased ROS result in an increase in intracellular calcium that activates protein kinase C and the mammalian target of rapamycin (mTOR), which mediate increased HIF-1α synthesis and stability (Figure 9).77
FIGURE 9.
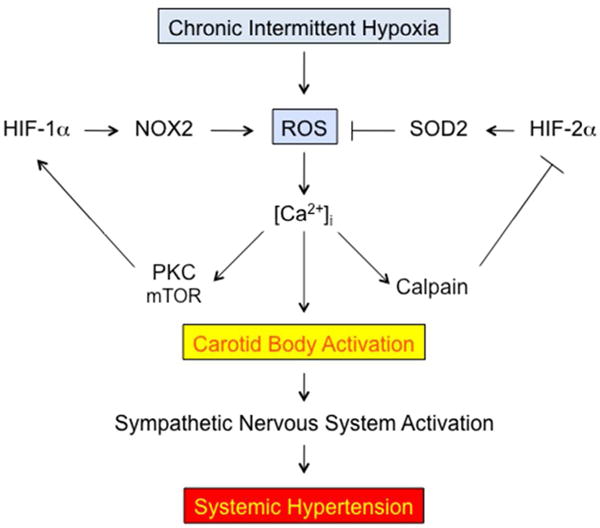
Chronic intermittent hypoxia disrupts redox and HIF balance, leading to sympathetic activation and hypertension. HIF, hypoxia-inducible factor; mTOR, mammalian target of rapamycin; NOX, NADPH oxidase; PKC, Ca2+-dependent protein kinase; SOD, superoxide dismutase.
Remarkably, whereas basal levels of HIF-1α protein in the CB are low and increase after CIH, basal HIF-2α levels are high and decrease after CIH.78 Increased intracellular calcium levels induced increased activity of calpains, which are calcium-dependent proteases, that degraded HIF-2α. CIH repressed expression in the CB and AM of Sod2, which is a known HIF-2 target gene that encodes the mitochondrial superoxide dismutase. Treatment of rodents with the calpain inhibitor ALLM blocked CIH-induced increases in ROS, plasma catecholamines, and blood pressure.78 Thus, CIH increases the expression of HIF-1α and the pro-oxidant enzyme NADPH oxidase 2 (NOX2) but decreases the expression of HIF-2α and the anti-oxidant enzyme superoxide dismutase 2 (SOD2), all of which are required for the development of hypertension (Figure 9).
Epas1+/− mice, which are heterozygous for a knockout allele at the locus encoding HIF-2α, display a phenotype that is in striking contrast to Hif1a+/− mice: under room air conditions, the mice have elevated blood pressure and instability of breathing, and their CB and AM show augmented responses to hypoxia, all of which are observed in wild type mice that have been subjected to CIH.79 Furthermore, SOD2 expression was decreased and ROS were increased in the CB and AM of Epas1+/− mice under room air conditions. Remarkably, treatment of Epas1+/− mice with MnTMPyP for 15 days normalized blood pressure, plasma norepinephrine levels and the CB response to hypoxia.79 HIF-1α protein expression was increased in the CB of Epas1+/− mice and treatment with digoxin, a drug that inhibited the accumulation of HIF-1α protein in the CB, also normalized CB sensitivity and corrected the ROS imbalance (overly oxidized state), plasma norepinephrine levels, breathing, and blood pressure.80 Conversely, treatment of Hif1a+/− mice with 2-methoxyestradiol, a drug that inhibited the accumulation of HIF-2α protein in the CB, corrected the ROS imbalance (overly reduced state) and completely restored responsiveness of the CB and AM to hypoxia.81
The response of Epas1+/− and Hif1a+/− mice to pharmacological inhibitors of HIF-1α and HIF-2α, respectively, suggested that proper functioning of the CB and AM required proper redox balance, which in turn was dependent on a proper balance between HIF-1α and HIF-2α rather than the absolute levels of these proteins. To test this hypothesis, Epas1+/−;Hif1a+/− doubly-heterozygous mice were analyzed.81 Remarkably, despite reduced expression of both HIF-1α and HIF-2α, these mice exhibited normal redox state and sensitivity of the CB and AM to hypoxia, normal blood pressure, and normal breathing --a dramatic confirmation of the model.
Does the dysregulation of redox homeostasis that is induced by CIH impact on the acute carotid body O2 sensing mechanism described in the preceding section? Recent studies have revealed that ROS inhibits HO2 activity (Figure 8), thereby increasing the generation of H2S by CSE.81 Pharmacologic or genetic inhibition of CSE activity blocked carotid body-mediated activation of the sympathetic nervous system and the development of hypertension in rodents subjected to CIH.81 Thus, CIH dysregulates redox balance, HIF-1α:HIF-2α transcriptional balance, and gas messenger signaling in the carotid body to induce sympathetic activation and hypertension.
The remarkable homeostatic mechanism described above can be interpreted in an evolutionary, as well as a physiological, context. The evolution of vertebrates with greatly increased body mass required the elaboration of cardiovascular, respiratory, and nervous systems that ensured the delivery of O2 to every cell of the organism. Whereas HIF-1α is found in all metazoan species with differentiated cell types, HIF-2α is only found in vertebrates. In keeping with the importance of redox homeostasis described earlier in this review in the context of cellular metabolism, mutual antagonism between the pro-oxidant activity of HIF-1α and anti-oxidant activity of HIF-2α generates a redox balance that determines the set point of the CB, sympathetic nervous system, and AM, which in turn establish set points for breathing, heart rate, and blood pressure.80
Supplementary Material
PERSPECTIVE.
Oxygen homeostasis is an organizing principle for understanding development, physiology, and disease pathogenesis. During development, establishment of a functioning circulatory system is required when the growing embryo that can no longer receive adequate O2 by diffusion from maternal tissues; in the mouse, this occurs by embryonic day 8.5, which is when HIF-1α-deficient embryos arrest in their development.82 Systemic or localized hypoxia is a fundamental physiological stimulus, which elicits a homeostatic response — such as HIF-dependent induction of genes mediating erythropoiesis or angiogenesis, respectively — that restores normoxia. However, in some disease states, the response is impaired, as occurs when chronic kidney disease impairs HIF-dependent EPO production. In other disease states, a non-physiological stimulus (such as CIH) leads to a dysregulated response (imbalanced HIF and redox states in the CB) that triggers disease pathogenesis (hypertension and its sequelae). Finally, delineation of the molecular mechanisms by which homeostasis is disrupted by disease provides opportunities to intervene, such as the treatment of chronic kidney disease patients with prolyl hydroxylase inhibitors to induce HIF-dependent EPO production in the liver83 or, perhaps, treatment of OSA patients with HIF-1α inhibitors to block sympathetic activation. Cancer and ischemic cardiovascular disease are also characterized by disordered oxygen homeostasis and, as such, are likely to respond to HIF-targeted therapies.28,84
Acknowledgments
Work in the authors’ laboratories is supported by grants from the American Cancer Society (122437-RP-12-090-01-COUN to G.L.S.), Armstrong Family Foundation (to G.L.S.), Cindy Rosencrans Foundation (to G.L.S.), Department of Defense Breast Cancer Research Program (W81XWH-12-1-0464 to G.L.S.), and National Institutes of Health (P01-HL-90554 to N.R.P.). G.L.S. is an American Cancer Society Research Professor and the C. Michael Armstrong Professor at the Johns Hopkins University School of Medicine.
Abbreviations
- 1CM
one carbon metabolism
- AM
adrenal medulla
- ANGPT
angiopoietin
- ANGPTL
angiopoietin-like
- BCSC
breast cancer stem cell
- BMDAC
bone marrow-derived angiogenic cell
- BNIP3
BCL2/adenovirus E1B 19-kDa interacting protein 3
- BNIP3L
BNIP3-like
- CAD
coronary artery disease
- CB
carotid body
- cGMP
cyclic GMP
- CIH
chronic intermittent hypoxia
- CO
carbon monoxide
- CSE
cystathionine-γ-lyase
- CSN
carotid sinus nerve
- CXCL
chemokine (C-X-C motif) ligand
- CXCR
chemokine (C-X-C motif) receptor
- DCYTB
duodenal cytochrome b
- DMT
divalent metal transporter
- EGLN
egl9 homolog
- EPAS
endothelial PAS domain
- EPO
erythropoietin
- EPOR
EPO receptor
- ER
estrogen receptor
- ETC
electron transport chain
- FAO
fatty acid oxidation
- FECH
ferrochelatase
- FPN
Ferroportin
- FIH-1
factor inhibiting HIF-1
- G6PD
glucose-6-phosphate dehydrogenase
- H2S
hydrogen sulfide
- HIF
hypoxia-inducible factor
- HO2
heme oxygenase 2
- KITL
kit ligand
- LCAD
long-chain acyl-CoA dehydrogenase
- LDHA
lactate dehydrogenase A
- MCAD
medium-chain acyl-CoA dehydrogenase
- MnTMPyP
manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride
- MTHFD
methylene tetrahydrofolate dehydrogenase
- MTHFDL
methylene tetrahydrofolate dehydrogenase-like
- mTOR
mammalian target of rapamycin
- NOX
NADPH oxidase
- NTC
non-targeting control
- OSA
obstructive sleep apnea
- PDGFB
platelet derived growth factor B
- PDGFR
PDGF receptor
- PDH
pyruvate dehydrogenase
- PDR
proliferative diabetic retinopathy
- PGF
placental growth factor
- PHD
proline hydroxylase
- PHGDH
phosphoglycerate dehydrogenase
- PKG
protein kinase G
- PSAT
phosphoserine aminotransferase
- PSPH
phosphoserine phosphatase
- Pro
proline
- ROS
reactive oxygen species
- SHMT
serine hydroxymethyltransferase
- shRNA
short hairpin RNA
- SLC
solute carrier family
- SNP
single nucleotide polymorphism
- SOD
superoxide dismutase
- SSP
serine synthesis pathway
- TCA
tricarboxylic acid cycle
- TF
transferrin
- TFRC
TF receptor
- Trp
tryptophan
- VEGF
vascular endothelial growth factor
- VEGFR
VEGF receptor
- VHL
von Hippel-Lindau
Footnotes
Conflicts of interest: The authors declare no conflicts of interest for this article.
References
- 1.Semenza GL. Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med. 2010;2:336–361. doi: 10.1002/wsbm.69. [DOI] [PubMed] [Google Scholar]
- 2.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev. 2012;92:967–1003. doi: 10.1152/physrev.00030.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duan C. Hypoxia-inducible factor 3 biology: complexities and emerging themes. Am J Physiol Cell Physiol. 2016;310:C260–C269. doi: 10.1152/ajpcell.00315.2015. [DOI] [PubMed] [Google Scholar]
- 5.Berra E, Roux D, Richard DE, Pouysségur J. Hypoxia-inducible factor-1α (HIF-1α) escapes O2-driven proteasomal degradation irrespective of its subcellular localization: nucleus or cytoplasm. EMBO Rep. 2001;2:615–620. doi: 10.1093/embo-reports/kve130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 7.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumor suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 8.Ivan M, Haberberger T, Gervasi DC, Michelson KS, Günzler V, Kondo K, Yang H, Sorokina I, Conaway RC, Conaway JW, et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci USA. 2002;99:13459–13464. doi: 10.1073/pnas.192342099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waypa GB, Smith KA, Schumacker PT. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol Aspects Med. 2016;47–48:76–89. doi: 10.1016/j.mam.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295:858–681. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 11.Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dayan F, Roux D, Brahimi-Horn MC, Pouyssegur J, Mazure NM. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1α. Cancer Res. 2006;66:3688–3698. doi: 10.1158/0008-5472.CAN-05-4564. [DOI] [PubMed] [Google Scholar]
- 14.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoon D, Pastore YD, Divoky V, Liu E, Mlodnicka AE, Rainey K, Ponka P, Semenza GL, Schumacher A, Prchal JT. Hypoxia-inducible factor-1 deficiency results in dysregulated erythropoiesis signaling and iron homeostasis in mouse development. J Biol Chem. 2006;281:25703–25711. doi: 10.1074/jbc.M602329200. [DOI] [PubMed] [Google Scholar]
- 16.Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 2009;9:152–164. doi: 10.1016/j.cmet.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mastrogiannaki M, Matak P, Keith B, Simon MC, Vaulont S, Peyssonnaux C. HIF-2α, but not HIF-1α, promotes iron absorption in mice. J Clin Invest. 2009;119:1159–1166. doi: 10.1172/JCI38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor M, Qu A, Anderson ER, Matsubara T, Martin A, Gonzalez FJ, Shah YM. Hypoxia-inducible factor 2α mediates the adaptive increased of intestinal ferroportin during iron deficiency in mice. Gastroenterology. 2011;140:2044–2055. doi: 10.1053/j.gastro.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rolfs A, Kvietikova I, Gassmann M, Wenger RH. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor 1. J Biol Chem. 1997;272:20055–20062. doi: 10.1074/jbc.272.32.20055. [DOI] [PubMed] [Google Scholar]
- 20.Lok CN, Ponka P. Identification of a hypoxia response element in the transferrin receptor gene. J Biol Chem. 1999;274:24147–24152. doi: 10.1074/jbc.274.34.24147. [DOI] [PubMed] [Google Scholar]
- 21.Tacchini L, Bianchi L, Bernelli-Zazzera A, Cairo G. Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J Biol Chem. 1999;274:24142–24146. doi: 10.1074/jbc.274.34.24142. [DOI] [PubMed] [Google Scholar]
- 22.Liu YL, Ang SO, Weigent DA, Prchal JT, Bloomer JR. Regulation of ferrochelatase gene expression by hypoxia. Life Sci. 2004;75:2035–2043. doi: 10.1016/j.lfs.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 23.Ang SO, Chen H, Hirota K, Gordeuk VR, Jelinek J, Guan Y, Liu E, Sergueeva AI, Miasnikova GY, Mole D, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet. 2002;32:614–621. doi: 10.1038/ng1019. [DOI] [PubMed] [Google Scholar]
- 24.Smith TG, Brooks JT, Balanos GM, Lappin TR, Layton DM, Leedham DL, Liu C, Maxwell PH, McMullin MF, McNamara CJ, et al. Mutation of von Hippel-Lindau tumor suppressor and human cardiopulmonary physiology. PLoS Med. 2006;3:e290. doi: 10.1371/journal.pmed.0030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Percy MJ, Furlow PW, Beer PA, Lappin TR, McMullin MF, Lee FS. A novel erythrocytosis-associated PHD2 mutation suggests the location of a HIF binding groove. Blood. 2007;110:2193–2196. doi: 10.1182/blood-2007-04-084434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Percy MJ, Furlow PW, Lucas GS, Li X, Lappin TR, McMullin MF, Lee FS. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med. 2008;358:162–168. doi: 10.1056/NEJMoa073123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Formenti F, Beer PA, Croft QP, Dorrington KL, Gale DP, Lappin TR, Lucas GS, Maher ER, Maxwell PH, McMullin MF, et al. Cardiopulmonary function in two human disorders of the hypoxia-inducible factor (HIF) pathway: von Hippel-Lindau disease and HIF-2α gain-of-function mutation. FASEB J. 2011;25:2001–2011. doi: 10.1096/fj.10-177378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol. 2014;76:39–56. doi: 10.1146/annurev-physiol-021113-170322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rey S, Semenza GL. Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc Res. 2010;86:236–242. doi: 10.1093/cvr/cvq045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bosch-Marce M, Okuyama H, Wesley JB, Sarkar K, Kimura H, Liu YV, Zhang H, Strazza M, Rey S, Savino L, et al. Effects of aging and hypoxia-inducible factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ Res. 2007;101:1310–1318. doi: 10.1161/CIRCRESAHA.107.153346. [DOI] [PubMed] [Google Scholar]
- 31.Sarkar K, Fox-Talbot K, Steenbergen C, Bosch-Marcé M, Semenza GL. Adenoviral transfer of HIF-1α enhances vascular responses to critical limb ischemia in diabetic mice. Proc Natl Acad Sci USA. 2009;106:18769–18774. doi: 10.1073/pnas.0910561106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rey S, Lee K, Wang CJ, Gupta K, Chen S, McMillan A, Bhise N, Levchenko A, Semenza GL. Synergistic effect of HIF-1α gene therapy and HIF-1-activated bone marrow-derived angiogenic cells in a mouse model of limb ischemia. Proc Natl Acad Sci USA. 2009;106:20399–20404. doi: 10.1073/pnas.0911921106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rey S, Luo W, Shimoda LA, Semenza GL. Metabolic reprogramming by HIF-1 promotes the survival of bone marrow-derived angiogenic cells in ischemic tissue. Blood. 2011;117:4988–4998. doi: 10.1182/blood-2010-11-321190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Resar JR, Roguin A, Voner J, Nasir K, Hennebry TA, Miller JM, Ingersoll R, Kasch LM, Semenza GL. Hypoxia-inducible factor 1α polymorphism and coronary collaterals in patients with ischemic heart disease. Chest. 2005;128:787–791. doi: 10.1378/chest.128.2.787. [DOI] [PubMed] [Google Scholar]
- 35.Hlatky MA, Quertermous T, Boothroyd DB, Priest JR, Glassford AJ, Myers RM, Fortmann SP, Iribarren C, Tabor HK, Assimes TL, et al. Polymorphisms in hypoxia-inducible factor 1 and the initial clinical presentation of coronary disease. Am Heart J. 2007;154:1035–1042. doi: 10.1016/j.ahj.2007.07.042. [DOI] [PubMed] [Google Scholar]
- 36.Campochiaro PA. Ocular neovascularization. J Mol Med. 2013;91:311–321. doi: 10.1007/s00109-013-0993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- 38.Ozaki H, Yu AY, Della N, Ozaki K, Luna JD, Yamada H, Hackett SF, Okamoto N, Zack DJ, Semenza GL, et al. Hypoxia inducible factor-1α is increased in ischemic retina: temporal and spatial correlation with VEGF expression. Invest Ophthalmol Vis Sci. 1999;40:182–189. [PubMed] [Google Scholar]
- 39.Ip MS, Domalpally A, Hopkins JJ, Wong P, Ehrlich JS. Long-term effects of ranibizumab on diabetic retinopathy severity and progression. Arch Ophthalmol. 2012;130:1145–1152. doi: 10.1001/archophthalmol.2012.1043. (2012) [DOI] [PubMed] [Google Scholar]
- 40.Yoshida T, Zhang H, Iwase T, Shen J, Semenza GL, Campochiaro PA. Digoxin inhibits retinal ischemia-induced HIF-1α expression and ocular neovascularization. FASEB J. 2010;24:1759–1767. doi: 10.1096/fj.09-145664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Babapoor-Farrokhran S, Jee K, Puchner B, Hassan SJ, Xin X, Rodrigues M, Kashiwabuchi F, Ma T, Hu K, Deshpande M, et al. Angiopoietin-like 4 is a potent angiogenic factor and a novel therapeutic target for patients with proliferative diabetic retinopathy. Proc Natl Acad Sci USA. 2015;112:E3030–E3039. doi: 10.1073/pnas.1423765112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xin X, Rodrigues M, Umapathi M, Kashiwabuchi F, Ma T, Babapoor-Farrokhran S, Wang S, Hu J, Bhutto I, Welsbie DS, et al. Hypoxic retinal Muller cells promote vascular permeability by HIF-1-dependent up-regulation of angiopoietin-like 4. Proc Natl Acad Sci USA. 2013;110:E3425–E3434. doi: 10.1073/pnas.1217091110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, et al. Digoxin and other cardiac glycosides inhibit HIF-1α synthesis and block tumor growth. Proc Natl Acad Sci USA. 2008;105:19579–19586. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee K, Qian DZ, Rey S, Wei H, Liu JO, Semenza GL. Anthracycline chemotherapy inhibits HIF-1 transcriptional activity and tumor-induced mobilization of circulating angiogenic cells. Proc Natl Acad Sci USA. 2009;106:2353–2358. doi: 10.1073/pnas.0812801106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci USA. 2009;106:17910–17915. doi: 10.1073/pnas.0909353106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Iwase T, Fu J, Yoshida T, Muramatsu D, Miki A, Hashida N, Lu L, Oveson B, Lima e Silva R, Seidel C, et al. Sustained delivery of a HIF-1 antagonist for ocular neovascularization. J Control Release. 2013;172:625–633. doi: 10.1016/j.jconrel.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vaupel P, Hockel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal. 2007;9:1221–1235. doi: 10.1089/ars.2007.1628. [DOI] [PubMed] [Google Scholar]
- 48.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227–232. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 51.Papandreou I, Cairns RA, Fontana L, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 52.Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor 1 promotes metabolic switch and drug resistance. J Biol Chem. 2008:28106–28114. doi: 10.1074/jbc.M803508200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA. 2010;107:2037–2042. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysseégur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang D, Li T, Li X, Zhang L, Sun L, He X, Zhong X, Jia D, Song L, Semenza GL, et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014;25(8):1930–1942. doi: 10.1016/j.celrep.2014.08.028. [DOI] [PubMed] [Google Scholar]
- 57.Currie E, Schulze A, Zechner R, Walther TC, Farese RV., Jr Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153–161. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120:142–156. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, Eyob H, Kajimura S, Krings G, Nomura DK, et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. 2016;22:427–432. doi: 10.1038/nm.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Samanta D, Park Y, Andrabi SA, Shelton LM, Gilkes DM, Semenza GL. PHGDH expression Is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res. 2016;76:4430–4442. doi: 10.1158/0008-5472.CAN-16-0530. [DOI] [PubMed] [Google Scholar]
- 62.Ye J, Fan J, Venneti S, Wan YW, Pawel BR, Zhang J, Finley LW, Lu C, Lindsten T, Cross JR, et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014;4:1406–1417. doi: 10.1158/2159-8290.CD-14-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim D, Fiske BP, Birsoy K, Freinkman E, Kami K, Possemato RL, Chudnovsky Y, Pacold ME, Chen WW, Cantor JR, et al. SHMT2 drives glioma cell survival in ischemia but imposes a dependence on glycine clearance. Nature. 2015;520:363–367. doi: 10.1038/nature14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lu H, Samanta D, Xiang L, Zhang H, Hu H, Chen I, Bullen JW, Semenza GL. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc Natl Acad Sci USA. 2015;112:E4600–E4609. doi: 10.1073/pnas.1513433112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pflüger E. Ueber die ursache der athembewegungen, sowie der dyspnoë und apnoë. Pflügers Arch Gesamte Physiol Meschen Tiere. 1868;1:61–106. [Google Scholar]
- 66.De Castro F. Sur la structure et l’innervation de la glande intercarotidienne (glomus caroticum) de l’homme et des mammiferes et sur un nouveau systeme de l’innervation autonome du nerf glossopharyngien. Trav Lab Rech Biol. 1926;24:365–432. [Google Scholar]
- 67.Heymans J, Heymans C. Sur les modifications directes et sur la regulation reflexe de l’activitie du centre respiratory de la tete isolee du chien. Arch Int Pharmacodyn Ther. 1927;33:273–372. [Google Scholar]
- 68.Prabhakar NR, Semenza GL. Gaseous messengers in oxygen sensing. J Mol Med. 2012;90:265–272. doi: 10.1007/s00109-012-0876-1. [DOI] [PubMed] [Google Scholar]
- 69.Prabhakar NR, Semenza GL. Oxygen sensing and homeostasis. Physiology. 2015;30:340–348. doi: 10.1152/physiol.00022.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yuan G, Vasavda C, Peng YJ, Makarenko VV, Raghuraman G, Nanduri J, Gadalla MM, Semenza GL, Kumar GK, Snyder SH, et al. Protein kinase G-regulated production of H2S governs oxygen sensing. Sci Signal. 2015;8:ra37. doi: 10.1126/scisignal.2005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peng YJ, Nanduri J, Raghuraman G, Souvannakitti D, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci USA. 2010;107:10719–10724. doi: 10.1073/pnas.1005866107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kline DD, Peng YJ, Manalo DJ, Semenza GL, Prabhakar NR. Defective carotid body function and impaired ventilator responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. Proc Natl Acad Sci USA. 2002;99:821–826. doi: 10.1073/pnas.022634199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D’Agostino RB, Newman AB, Lebowitz MD, Pickering TG. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA. 2000;283:1829–1836. doi: 10.1001/jama.283.14.1829. [DOI] [PubMed] [Google Scholar]
- 74.Fletcher EC, Lesske J, Behm R, Miller CC, 3rd, Stauss H, Unger T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol. 1992;72:1978–1984. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- 75.Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1α deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol. 2006;577:705–716. doi: 10.1113/jphysiol.2006.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol. 2011;226:2925–2933. doi: 10.1002/jcp.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR. Induction of HIF-1α expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol. 2008;217:674–685. doi: 10.1002/jcp.21537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng YJ, Kumar GK, Garcia JA, Prabhakar NR. Intermittent hypoxia degrades HIF-2α via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci USA. 2009;106:1199–1204. doi: 10.1073/pnas.0811018106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peng YJ, Nanduri J, Khan SA, Yuan G, Wang N, Kinsman B, Vaddi DR, Kumar GK, Garcia JA, Semenza GL, et al. Hypoxia-inducible factor 2α (HIF-2α) heterozygous-null mice exhibit exaggerated carotid body sensitivity to hypoxia, breathing instability, and hypertension. Proc Natl Acad Sci USA. 2011;108:3065–3070. doi: 10.1073/pnas.1100064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yuan G, Peng YJ, Reddy VD, Makarenko VV, Nanduri J, Khan SA, Garcia JA, Kumar GK, Semenza GL, Prabhakar NR. Mutual antagonism between hypoxia-inducible factors 1α and 2α regulates oxygen sensing and cardio-respiratory homeostasis. Proc Natl Acad Sci USA. 2013;110:E1788–E1796. doi: 10.1073/pnas.1305961110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 81.Yuan G, Peng YJ, Khan SA, Nanduri J, Singh A, Vasavda C, Semenza GL, Kumar GK, Snyder SH, Prabhakar NR. H2S production by reactive oxygen species in the carotid body triggers hypertension in a rodent model of sleep apnea. Sci Signal. 2016;9:ra80. doi: 10.1126/scisignal.aaf3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maxwell PH, Eckardt KU. HIF prolyl hydroxylase inhibitors for the treatment of renal anemia and beyond. Nat Rev Nephrol. 2016;12:157–168. doi: 10.1038/nrneph.2015.193. [DOI] [PubMed] [Google Scholar]
- 84.Semenza GL. The hypoxic tumor microenvironment: a driving force for breast cancer progression. Biochim Biophys Acta. 2016;1863:382–391. doi: 10.1016/j.bbamcr.2015.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.